
Abstract
Heart failure (HF) is a complex syndrome with cardiac, renal, neurohormonal and sympathetic nervous system’s manifestations, the pathogenesis of which among others is connected to inflammation. PAF has local and systemic effects pertaining to HF progression since it causes a negative inotropic effect, it induces arrhythmias, it induces apoptosis and it is involved in inflammation and atherosclerosis. In the present review the role of PAF in HF will be thoroughly presented along with the relevant data on PAF enzymes and the potential role of PAF metabolic circuit as a novel pharmacological target.
Similar content being viewed by others

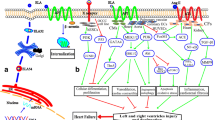
Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a complex syndrome, in which cardiac, renal, neuro-hormonal and sympathetic nervous system’s manifestations are present [1••]. Despite the development of state-of-the art pharmacological agents, the incidence of HF increases at alarming rates [2], implying that several aspects of its pathophysiology remain undertreated. Indeed, the activation of the inflammatory cascade orchestrated by leukocytes, platelets, endothelial cells, myocardium together with other sources consists a key player in HF, which may have been overlooked [3]. Inflammatory markers differentiate along with the severity and progression of the disease [3] and may be modulated by HF-targeted drugs [4].
Although the isolation of the primary etiology of HF is not always easy, the leading cause of HF is atherosclerosis manifested as coronary artery disease (alone or in combination with hypertension) [1••]. Other causes include familial or acquired cardiomyopathy (due to viral infection, alcohol, heavy metals, chemotherapy, selenium deficiency, amyloidosis, etc.), valvular heart disease, pericardial heart disease, endocardial heart disease and arrhythmia [1••]. Moreover, hemodynamic disturbances such as those observed in renal failure or post-operative fluid infusion as well as high-output states such as anemia and thyrotoxicosis can lead to heart failure [1••].
Platelet-activating factor (PAF), (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) [5], is a potent inflammatory phospholipid mediator implicated in atherosclerosis [6, 7] and several mechanisms of HF [6, 8]. For example, PAF causes a negative inotropic effect, it induces arrhythmias, it induces apoptosis and it is involved in leukocyte recruitment [6, 8]. Moreover, recent data suggest that PAF metabolic enzymes may participate in HF development [9, 10•]. With respect to PAF metabolism (Fig. 1), two biosynthetic pathways are responsible for its biosynthesis, namely the remodeling and the de novo pathway [11]. In the remodeling pathway a cytoplasmic phospholipase A2 converts the ether analogs of phosphatidylcholine to lyso-PAF, which is then acetylated to PAF by the action of at least two isoforms of acetyl-CoA: lyso-PAF acetyltransferases (lyso-PAF ATs), namely LPCAT1 and LPCAT2 [12, 13]. Recent data support that production of PAF by the action of LPCAT2 is activated under inflammatory conditions while LPCAT1 is calcium independent and does not participate in inflammatory processes [13]. The de novo pathway is considered to be responsible for the constitutive production of PAF. A key reaction in this route is the final one, in which PAF is produced by 1-O-alkyl-2-acetyl-glycerol through the action of a specific dithiothreitol-insensitive CDP-choline: 1-alkyl-2-acetyl-sn-glycerol cholinephosphotransferase (PAF-CPT, EC 2.7.8.16) [14]. As far as PAF catabolism is concerned, a PAF-specific acetylhydrolase (PAF-AH, EC 3.1.1.47) removes the acetyl chain (or short acyl chain) from sn-2 position and converts PAF to lyso-PAF [15]. The plasma isoform of PAF-AH is known as lipoprotein-associated phospholipase A2 (Lp-PLA2) due to its attachment to lipoproteins and mainly LDL-particles [15].

The metabolic circuit of PAF
In the present review, the local and systemic effects of PAF pertaining to HF will be presented, particularly focusing on PAF effects on myocardium, its hemodynamic actions and its implication in atherosclerosis. Moreover, the relevant data on PAF enzymes will be discussed and the role of PAF circuit as a novel pharmacological target will be examined.
PAF and Myocardium
Inflammation of myocardium and myocardial necrosis caused by prolonged ischemia and hypoxia can lead to HF despite the existing compensating remodeling mechanisms (e.g., ventricular remodeling and neurohormonal stimulation). Moreover, contractile dysfunction and myocardial electrical instability constitute a central feature of HF [1••].
Metabolism of PAF in Myocardium
The exact cellular source of PAF in myocardium remains unknown. Many cell types may be responsible for PAF production under physiological or pathological conditions, i.e., endothelial cells, platelets, monocytes and other cells types including myocardial cells [16]. Myocardial cells produce PAF in vitro and in vivo under appropriate stimuli [6]. For example, immunological agents in guinea pig heart [17], ischemia in baboon and rabbit heart [18, 19] and injury in rat myocytes [20] can lead to PAF production. Confirming its role as an autacoid, PAF in turn exerts direct effects on myocardium, which are presented below.
PAF and Heart Contraction
PAF can influence heart contractility as a result of its hemodynamic effects (see below) or by directly acting on cardiac cells. PAF has been found to reduce heart’s contractility in several models, such as guinea pig [21], dog [22] and rabbit hearts [23]. PAF infusion in isolated perfused guinea pig heart induces changes in cardiac cell structure such as myocardium oedema, decrease of matrix density, rapture of mitochondria crest and decreases of mitochondrial enzyme activities [24]. By this way, PAF impairs the generation of ATP through oxidative metabolism in the myocardium. The aforementioned changes are absent if treatment of myocardial tissue with the PAF antagonist BN 52021 precedes [24]. Moreover, trace elements such as zinc may reduce PAF’s negative inotropic effect in low doses (1.5 μΜ) propably through modulation of PAF-receptor interactions [25].
Moreover, cell cultures demonstrate a direct effect of PAF on calcium [26] and potassium channels [27], which participate in myocardial contraction. Other hypotheses support the implication of leukotrienes [22], phosphatidyl inositol and PKC [28] as mediators of PAF-induced impairement of myocardial contraction. Indeed, genetically modified rats which do not express phosphoinositide 3-kinase-γ (PI3K-γ) are “resistant” to the negative inotropic effects of PAF [29]. Moreover, PAF induces the production of atrial natriuretic peptide [30]. Several data also suggest that PAF’s actions in myocardium may be indirectly exerted through the production of reactive oxygen species by the recruited neutrophils [31]. Indeed, PAF inhibitors lead to reduction in PMNs in models of ischemia in rabbits [32].
Electrophysiological Effects of PAF in Heart
PAF can lead to changes in electrocardiograph in rats [33], guinea pigs [34] and rabbits [35]. PAF induces alterations in the transmembrane potential, i.e., increased duration of the action potential, early afterdepolarizations, transient arrest of repolarization [36] and abnormal automacity [37].
Other studies suggest that PAF induces anomalies in Purkinje cells function [38] and mice ventricular cells [37]. PAF seems to activate its specific receptors in the ventricle and induces arrhythmias [35], which are reduced in the presence of its inhibitors such as BN 52021, WEB 2086 [39, 40], kadsurenone [35] and Ginkgo biloba extract [41]. PAF induced arrhythmias are believed to be connected to the closing of potassium channels [37]. An additional route through which PAF may exert the above actions is the production of eicosanoids, since thromboxane Α2 inhibitors also reduce PAF effects [42].
Hemodynamic Effects of PAF
The first indication of PAF’s hemodynamic effects coincide with the identification of a polar lipid with antihypertensive properties isolated from rat kidney (antihypertensive polar renomedullary lipid, APRL), which has proven to be PAF [43]. Intravenous or oral intake of PAF (or APRL) leads to a dose-dependent reduction in blood pressure in various animal species, which reaches a maximum within 30–60 seconds [6]. At concentrations 1–10 nmol/L PAF reduces coronary flow exerting a negative inotropic effect [44••]. In parallel hypertensive rats have increased Lp-PLA2 activity [45] while in a model of renal clip hypertension PAF acts as a mediator of blood pressure fall after unclipping [46].
The underlying mechanism of hypotensive effects of PAF has not been fully elucidated. Potential mechanisms through which the hypotensive effect of PAF is exerted are the following: (i) it reduces venous blood return, (ii) it produces a right ventricular overload as the result of an increase in pulmonary vascular resistance, (iii) it has a direct negative inotropic effect, and (iv) it affects heart’s conductive system [6]. In line with the observations in animal species is the fact that increased right atrial pressure has been connected to increased PAF levels in patients undergoing coronary angioplasty [47]. PAF’s hemodynamic effects may be exerted through PAF receptors, since selective PAF inhibitors such as Ginkgo biloba extract and ΒΝ 52021 extenuate the reduction of blood flow [48, 49].
Moreover, PAF may exert vasoconstriction effects by producing cyclo- and lipoxygenase metabolites, or by activating platelets and mononuclear cells. The vasoconstriction properties of PAF depend on its concentration range, the integrity of the endothelium and the animal model [44••].
PAF and Atherosclerosis
Atherosclerosis is a slow process orchestrated by oxidative stress, thrombosis and inflammation [50]. Chronic atherosclerotic burden in coronary and peripheral arteries leads to cardiovascular disease which is the most common cause of HF, as mentioned above [1••].
A crucial role of PAF in atherosclerosis has been proposed [7], since it constitutes an important mediator of inflammation [51] and is implicated in several stages of the disease. More particularly, it induces oxidative stress [52, 53], it participates in LDL oxidation [54] and it is produced during LDL oxidation upon Lp-PLA2 inactivation [55]. According to recent data oxidized LDL also interacts with PAF receptor in macrophages to increase oxidized LDL uptake [56] and stimulate chemokine release [57]. Moreover, PAF contributes to the adhesion of leukocytes [58] and their chemotactic entrance in endothelium, since it increases endothelial permeability [59].The PAF mediated activation of leukocytes also results in the secretion of chemokines and growth factors such as MCP-1 [60] and vascular endothelial growth factor (VEGF), respectively [61]. In parallel, PAF causes platelet aggregation [62] and stimulates the release of the stored cytokines and growth factors from platelets [63]. Moreover, it contributes to protease release from leukocytes, such as elastase, which disrupts vessel’s extracellular matrix [64] and may be a risk factor for plaque rupture.
It is thus obvious that PAF acts on various cells which participate in the atherosclerotic process such as platelets, endothelial cells, neutrophils and monocytes [7]. The secretion of PAF from some inflammatory cells, like monocytes and neutrophils can in turn result in the amplification of the inflammatory response [65]. The implication of PAF in atherosclerosis is also underlined by the observations that PAF inhibitors such as BN 52021 inhibit cholesterol deposition in arteries of animals fed an atherogenic diet [66]. A detailed review of PAF’s contribution in atherosclerosis is provided by Demopoulos et al. [7].
PAF and Ischemia
Ischemia, which may be a result of the atherosclerotic process, can also exert its effects on myocardium through PAF. Several data suggest that PAF can play a role in cardiac myocyte death resulting from ischemia/ reperfusion injury by inducing apoptosis [8], which in turn can lead to cardiac dysfunction and HF [67]. Indeed, in concentrations of 0.2 to 20 μM PAF can cause apoptosis in cultured cardiac myocytes through a Ca2+-dependent mechanism. More particularly, PAF results in p38 MAPK phosphorylation, which in turn leads to cytochrome c/caspase-3 signaling activation and apoptosis [8].
However, in ischemia/ reperfusion states, PAF can also play a protective role as it is involved in ischemic preconditioning. Ischemic preconditioning refers to the fact that the myocardium adapts to brief periods of sublethal ischemia and is protected in case of a potential lethal ischemic injury [68]. Treatment with low concentrations of PAF, in the range of pM, before ischemia does not affect cardiac performance but exerts a protective effect, since it reduces infarct’s extension and improves heart’s recovery during reperfusion. Indeed, PAF activates kinases (such as PKC1, PKB/Akt,GSK-3b and ERK1/2), produces NO and affects calcium channels, all of which are implicated in the mechanisms of ischemic preconditioning [44••]. Supportive evidence of the PAF’s protective role also involves the observation that post-ischemic performance is reduced in case of targeted deletion of the PAF receptor or if PAF receptor antagonists are used [69].
PAF’s Metabolic Circuit and HF
Limited evidence exists for the role of PAF’s metabolic enzymes in HF and cardiovascular diseases with the exception of Lp-PLA2. In a study of patients with newly diagnosed HF, we identified a possible relation of the remodeling and the de novo biosynthetic enzymes of PAF in leukocytes, since lyso-PAF-AT and PAF-CPT were correlated [9]. Moreover, both enzymes were related to inflammatory biomarkers [9], which are increased in HF [3]. More particularly, lyso-PAF-AT was positively related to CRP and IL-6 [9], which is in line with the fact that inflammatory stimuli are activators of this enzyme [70]. PAF-CPT was correlated to CRP and IL-6, suggesting that it may be implicated to pathophysiological processes involving inflammation [9], despite the proposed role for its contribution to basal PAF levels production [14]. Interestingly, PAF-CPT was also positively correlated with immunologic markers, i.e., CD40L and sCD14, while PAF-AH correlated to TNF-α [9]. Therefore, it seems that PAF’s biosynthetic enzymes were depressed in HF patients and it was hypothesized that medical treatment affected PAF metabolic profile [10•]. Moreover, PAF levels seem to be low in patients with myocardial infarction at admission [71] and the expression of its receptor is upregulated [72].
Since PAF participates in atherogenesis, it can be assumed that its catabolic enzyme Lp-PLA2 may inhibit its atherogenic actions. Indeed, hyper-expression of Lp-PLA2 gene reduces atheromatous plaque [73]. However, Lp-PLA2 can also act as a pro-inflammatory molecule as it contributes to lyso-PC generation, which in turn leads to macrophage growth, non-esterified fatty acids and endothelial dysfunction [15]. Epidemiological studies have shown that Lp-PLA2 is a risk factor for cardiovascular disease. It is not certain, however, if Lp-PLA2 acts etiologically in cardiovascular disease or if it is increased as a response to increased PAF levels. It is noteworthy that several studies measure only the mass of the enzyme, which is not always indicative of its activity. Although the correlation coefficient between Lp-PLA2 mass and activity is 0.51 (0.47-0.56) [74••], almost 40 % of subjects in the highest quartile of the enzyme mass are in the lowest quartile of enzymatic activity [75]. The recently developed Lp-PLA2 inhibitors have shown some promising protective evidence [76–79] but their exact role remains to be determined [80]. As far as HF is concerned, Lp-PLA2 has been characterized as a prognostic biomarker for HF development [81, 82], is higher in HF patients than healthy controls [10•] and is associated with mortality in HF patients [83]. Moreover, it is higher in HF patients with preserved ejection fraction than in HF with reduced ejection fraction [84] and is not correlated with New York Heart Association (NYHA) status [85].
PAF as a Novel Target for HF Therapy
It is obvious from the aforementioned data that PAF is a crucial mediator of almost all pathophysiological mechanisms that lead to heart failure. Therefore, either its receptor or the enzymes of its metabolism seem to be attractive targets for the treatment of HF. Moreover, several lines of evidence suggest that drugs designed to reduce inflammatory burden in HF, and mainly anti-TNF-a therapy have not always beneficial effects [86]. This observation underlines the necessity of drugs targeting to other molecules in order to further refine the therapy of HF patients.
Many HF drugs such as verapamil [87] and digoxin [88] reduce the PAF induced cardiac and circulatory alterations. In addition, the protective effects of the Mediterranean diet on HF patients [89] may be in part explained by the presence of PAF antagonists in foods. In fact, extracts of several traditional Mediterranean foods which are inversely related to atherosclerosis, such as olive oil, olive mill wastes, wine, fish, honey, milk and yogurt, as well as garlic and onion, contain PAF antagonists [90, 91]. Polar extract of olive oil, which acts as a PAF inhibitor, led to reduction of atheromatous plaque after 45 days in rabbits [92]. In patients with type 2 diabetes consumption of Mediterranean meals with high in vitro PAF inhibitory activity led to reduced platelet aggregation after PAF stimuli [93].
Recent data supports that newly diagnosed HF patients under drug treatment also have an affected profile of PAF biosynthetic enzymes and especially lyso-PAF-AT[10•]. Indeed, aldosterone antagonists, angiotensin-converting enzyme inhibitors, antiarrhythmic agents, statins and diuretics used in HF possess anti-inflammatory effects [94] although there are some studies not showing such effects [95]. Statins decrease Lp-PLA2 and PAF-CPT activities and have a neutral effect on lyso-PAF-AT activity in healthy volunteers [96]. Other possible interactions between PAF and cardiovascular drugs include nitrates and calcium channel blockers, which reduce PAF production in endothelial cells [97], and human umbilical vein endothelial cells [98], angiotensin-converting enzyme inhibitors which partly inhibit PAF effects and may lead to reduced PAF synthesis [99] and salicylates, which inhibit lyso-PAF-AT [100]. Thus, the existing medical treatment for HF may affect PAF biosynthetic enzymes. Whether the design of novel pharmaceutical products targeting on PAF enzymes would have beneficial effects on HF progression is not known.
It is therefore obvious that drugs commonly used for the treatment of HF affect PAF metabolism or its actions. Whether this is a direct effect of the drugs on PAF’s enzymatic machinery/signal transduction pathway, or a secondary effect resulting from the anti-inflammatory properties of the HF treatment, is currently not known. However, in order to establish the pathogenetic role of PAF in HF, and therefore the necessity for its pharmacological modulation novel, well-designed, clinical trials should be conducted, wherein a putative reduction of PAF levels could be linked with an improvement of the HF clinical phenotype.
Another dilemma that arises from studies conducted so far is whether a candidate drug aiming on PAF metabolism/actions should be a PAF receptor antagonist or a modulator of PAF metabolism (inhibitor of its biosynthetic enzymes, activator of its degradation enzymes or an indirect modulator of its metabolism). With the current knowledge, the design of a molecule aiming on the PAFR/signal transduction axis is easier given that both PAF receptors and most of the components of their signal transduction pathways have already been characterized in the molecular level. On the other hand, such drugs may also inhibit PAF’s physiological, homeostatic roles as well as its protective roles as in the case of preconditioning.
Alternatively, the designing of drugs aiming on the modulation of PAF metabolism may confer better specificity especially if they target enzymes of PAF metabolism that are upregulated under conditions that favor HF progression. As previously mentioned, the group of T. Shimizu characterized two lyso-PAF acetylating activities (LPCAT1 and 2) from which only one isoform was activated by inflammatory stimuli [12, 13]. The designing of specific inhibitors for this isoform would prevent the synthesis of PAF only under pathological conditions. However, the molecular details of the enzymatic pathways and the regulatory mechanisms of PAF metabolism are still obscure and only after their clarification the scientific community would be able to identify the best potential drug targets.
Finally, another aspect which deserves attention is the recent development of Lp-PLA2 inhibitor, namely darapladib. More particularly, darapladib reduces Lp-PLA2 activity, IL-6 and CRP in cardiovascular patients [76], prevents necrotic core expansion in atherosclerotic plaques [77] and decreases atherosclerotic plaque formation in ApoE-deficient [78] and LDL-R deficient mice [79]. However, the usefulness of this inhibitor in cardiovascular patients remains to be verified from ongoing clinical trials [80], while no evidence exists on the role of darapladib in HF patients.
Conclusions
In conclusion, PAF is a key player in HF progression since it causes a negative inotropic effect, it induces arrhythmias, it induces apoptosis and it is involved in inflammation and atherosclerosis. Recent data support that PAF metabolic enzymes may participate in atherosclerosis and HF development. However, the use of PAF and/ or its enzymes as pharmacological targets should be critically viewed and cautiously designed in the light of evidence that low PAF concentrations may have a cardioprotective role in ischemic preconditioning.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–847. This publication is of major clinical importance since it presents the guidelines for diagnosis and treatment of heart failure.
Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41.
Dixon DL, Griggs KM, Bersten AD, De Pasquale CG. Systemic inflammation and cell activation reflects morbidity in chronic heart failure. Cytokine. 2011;56:593–9.
Liu L, Zhao SP. The changes of circulating tumor necrosis factor levels in patients with congestive heart failure influenced by therapy. Int J Cardiol. 1999;69:77–82.
Demopoulos CA, Pinckard RN, Hanahan DJ. Platelet-activating factor. Evidence for 1-O-alkyl-2-acetyl-sn-glyceryl-3-phosphorylcholine as the active component (a new class of lipid chemical mediators). J Biol Chem. 1979;254:9355–8.
Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80:1669–99.
Demopoulos C, Karantonis H, Antonopoulou S. Platelet activating factor - a molecular link between atherosclerosis theories. Eur J Lipid Sci Technol. 2003;105:705–16.
Zhao D, Chu WF, Wu L, et al. PAF exerts a direct apoptotic effect on the rat H9c2 cardiomyocytes in Ca2+-dependent manner. Int J Cardiol. 2010;143:86–93.
Detopoulou P, Nomikos T, Fragopoulou E, et al. Platelet activating factor (PAF) and activity of its biosynthetic and catabolic enzymes in blood and leukocytes of male patients with newly diagnosed heart failure. Clin Biochem. 2009;42:44–9.
• Detopoulou P, Fragopoulou E, Nomikos T, et al. Baseline and 6-week Follow-Up Levels of PAF and Activity of its Metabolic Enzymes in Patients With Heart Failure and Healthy Volunteers--A Pilot Study. Angiology 2012. doi:10.1177/0003319712458536. This publication presents the whole metabolic circuit of PAF in heart failure patients along with the progression of the disease.
Snyder F. Platelet-activating factor and its analogs: metabolic pathways and related intracellular processes. Biochim Biophys Acta. 1995;1254:231–49.
Shindou H, Hishikawa D, Nakanishi H, et al. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells: cloning and characterization of acetyl-CoA:lyso-PAF acetyltransferase. J Biol Chem. 2007;282:6532–9.
Harayama T, Shindou H, Ogasawara R, Suwabe A, Shimizu T. Identification of a novel noninflammatory biosynthetic pathway of platelet-activating factor. J Biol Chem. 2008;283:11097–106.
Snyder F. CDP-choline:alkylacetylglycerol cholinephosphotransferase catalyzes the final step in the de novo synthesis of platelet-activating factor. Biochim Biophys Acta. 1997;4:111–6.
Stafforini DM. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2). Cardiovasc Drugs Ther. 2009;23:73–83.
Burghardt C, Janero D. The anoxic rat-heart myocyte produces and releases platelet activating (PAF) as a component of its ischemia-like pathology. J Mol Cell Cardiol. 1987;19:pS69.
Levi R, Burke JA, Guo ZG, et al. Acetyl glyceryl ether phosphorylcholine (AGEPC). A putative mediator of cardiac anaphylaxis in the guinea pig. Circ Res. 1984;54:117–24.
Annable C, McManus L, Carey K, Pinckard R. Isolation of platelet-activating factor (PAF) from ischemic baboon myocardium. Fed Proc. 1985;44:1271.
Montrucchio G, Alloatti G, Tetta C, et al. Release of platelet-activating factor from ischemic-reperfused rabbit heart. Am J Physiol. 1989;256:H1236–46.
Janero DR, Burghardt C. Production and release of platelet-activating factor by the injured heart-muscle cell (cardiomyocyte). Res Commun Chem Pathol Pharmacol. 1990;67:201–18.
Stahl GL, Lefer DJ, Lefer AM. PAF-acether induced cardiac dysfunction in the isolated perfused guinea pig heart. Naunyn Schmiedeberg’s Arch Pharmacol. 1987;336:459–63.
Kenzora JL, Perez JE, Bergmann SR, Lange LG. Effects of acetyl glyceryl ether of phosphorylcholine (platelet activating factor) on ventricular preload, afterload, and contractility in dogs. J Clin Invest. 1984;74:1193–203.
Loucks EB, Godin DV, Walley KR, et al. Role of platelet activating factor in cardiac dysfunction, apoptosis and nitric oxide synthase mRNA expression in the ischemic-reperfused rabbit heart. Can J Cardiol. 2003;19:267–74.
Kecskemeti V, Balogh I. Cardiac ultrastructural effects of the platelet-activating factor and its antagonist BN 52021. Exp Toxicol Pathol. 1995;47:463–70.
Evangelou A, Kalfakakou V, Benveniste J, Arnoux B. Inhibition of PAF-acether effects on isolated guinea pig hearts by zinc ions (Zn2+). Biol Trace Elem Res. 1995;50:43–55.
Camussi G, Alloatti G, Montrucchio G, Meda M, Emanuelli G. Effect of platelet activating factor on guinea-pig papillary muscle. Experientia. 1984;40:697–9.
Wahler GM, Coyle DE, Sperelakis N. Effects of platelet-activating factor on single potassium channel currents in guinea pig ventricular myocytes. Mol Cell Biochem. 1990;93:69–76.
Massey CV, Kohout TA, Gaa ST, Lederer WJ, Rogers TB. Molecular and cellular actions of platelet-activating factor in rat heart cells. J Clin Invest. 1991;88:2106–16.
Alloatti G, Levi R, Malan D, et al. Phosphoinositide 3-kinase gamma-deficient hearts are protected from the PAF-dependent depression of cardiac contractility. Cardiovasc Res. 2003;60:242–9.
Church DJ, van der Bent V, Vallotton MB, Capponi AM, Lang U. Calcium influx in platelet activating factor-induced atrial natriuretic peptide release in rat cardiomyocytes. Am J Physiol. 1994;266:E403–9.
Gupta JB, Prasad M, Kalra J, Prasad K. Platelet-activating-factor-induced changes in cardiovascular function and oxyradical status of myocardium in presence of the PAF antagonist CV-6209. Angiology. 1994;45:25–36.
Montrucchio G, Alloatti G, Mariano F, et al. Role of platelet-activating factor in polymorphonuclear neutrophil recruitment in reperfused ischemic rabbit heart. Am J Pathol. 1993;142:471–80.
Tselepis AD, Evangelou A, Tsoukatos D, Demopoulos CA, Kapoulas VM. Electrocardiographic alterations induced by AGEPC in Wistar rats in relation to its hypotensive and hematologic effects. Comp Biochem Physiol C. 1987;87:41–6.
Robertson DA, Wang DY, Lee CO, Levi R. Negative inotropic effect of platelet-activating factor: association with a decrease in intracellular sodium activity. J Pharmacol Exp Ther. 1988;245:124–8.
Montrucchio G, Alloatti G, Mariano F, Tetta C, Emanuelli G, et al. Cardiovascular alterations in the rabbit infused with platelet activating factor (PAF): effect of kadsurenone, a PAF-receptor antagonist. Int J Tissue React. 1986;8:497–504.
Hoffman BF, Guo SD, Feinmark SJ. Arrhythmias caused by platelet activating factor. J Cardiovasc Electrophysiol. 1996;7:120–33.
Barbuti A, Ishii S, Shimizu T, Robinson RB, Feinmark SJ. Block of the background K(+) channel TASK-1 contributes to arrhythmogenic effects of platelet-activating factor. Am J Physiol Heart Circ Physiol. 2002;282:H2024–30.
Nakaya H, Tohse N. Electrophysiological effects of acetyl glyceryl ether phosphorylcholine on cardiac tissues: comparison with lysophosphatidylcholine and long chain acyl carnitine. Br J Pharmacol. 1986;89:749–57.
Mest HJ, Riedel A, Braquet P, Meyer E. The arrhythmogenic effect of platelet activating factor (PAF) is inhibited by PAF antagonist and by substances influencing eicosanoids. Biomed Biochim Acta. 1988;47:S219–23.
Cakici I, Mataraci N, Ersoy S, et al. Effects of platelet-activating factor antagonists WEB 2086 and BN 50730 on digoxin-induced arrhythmias. Pharmacol Toxicol. 1995;76:343–7.
Guillon JM, Rochette L, Baranes J. Effects of Ginkgo biloba extract on 2 models of experimental myocardial ischemia. Presse Med. 1986;15:1516–9.
Riedel A, Mest HJ. The effect of PAF (platelet-activating factor) on experimental cardiac arrhythmias and its inhibition by substances influencing arachidonic acid metabolites. Prostaglandins Leukot Med. 1987;28:103–9.
Muirhead EE, Byers LW, Desiderio DM, Brooks B, Brosius WM. Antihypertensive lipids from the kidney: alkyl ether analogs of phosphatidylcholine. Fed Proc. 1981;40:2285–90.
•• Penna C, Bassino E, Alloatti G. Platelet activating factor: the good and the bad in the ischemic/reperfused heart. Exp Biol Med (Maywood). 2011;236:390–401. The present review provides a thorough presentation of the role of PAF in ischemia/ reperfusion with a plethora of experimental examples.
Blank ML, Hall MN, Cress EA, Snyder F. Inactivation of 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine by a plasma acetylhydrolase: higher activities in hypertensive rats. Biochem Biophys Res Commun. 1983;113:666–71.
McGowan HM, Vandongen R, Kelly LD, Hill KJ. Increased levels of platelet-activating factor (1-O-alkyl-2-acetylglycerophosphocholine) in blood after reversal of renal clip hypertension in the rat. Clin Sci (Lond). 1988;74:393–6.
Eldar M, Lysko PG, Schulhoff N, et al. Effects of coronary angioplasty on plasma platelet-activating factor in man. J Lipid Mediat. 1992;5:313–9.
Piper PJ, Stewart AG. Coronary vasoconstriction in the rat, isolated perfused heart induced by platelet-activating factor is mediated by leukotriene C4. Br J Pharmacol. 1986;88:595–605.
Piper PJ, Stewart AG. Antagonism of vasoconstriction induced by platelet-activating factor in guinea-pig perfused hearts by selective platelet-activating factor receptor antagonists. Br J Pharmacol. 1987;90:771–83.
Levi M, van der Poll T, Schultz M. Infection and inflammation as risk factors for thrombosis and atherosclerosis. Semin Thromb Hemost. 2012;38:506–14.
Stafforini DM, McIntyre TM, Zimmerman GA, Prescott SM. Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit Rev Clin Lab Sci. 2003;40:643–72.
Stewart AG, Dubbin PN, Harris T, Dusting GJ. Platelet-activating factor may act as a second messenger in the release of eicosanoids and superoxide anions from leukocytes and endothelial cells. Proc Natl Acad Sci U S A. 1990;87:3215–9.
Rouis M, Nigon F, Chapman MJ. Platelet activating factor is a potent stimulant of the production of active oxygen species by human monocyte-derived macrophages. Biochem Biophys Res Commun. 1988;156:1293–301.
Gaut JP, Heinecke JW. Mechanisms for oxidizing low-density lipoprotein. Insights from patterns of oxidation products in the artery wall and from mouse models of atherosclerosis. Trends Cardiovasc Med. 2001;11:103–12.
Liapikos TA, Antonopoulou S, Karabina SP, et al. Platelet-activating factor formation during oxidative modification of low-density lipoprotein when PAF-acetylhydrolase has been inactivated. Biochim Biophys Acta. 1994;1212:353–60.
Rios FJ, Gidlund M, Jancar S. Pivotal role for platelet-activating factor receptor in CD36 expression and oxLDL uptake by human monocytes/macrophages. Cell Physiol Biochem. 2011;27:363–72.
Beaudeux JL, Said T, Ninio E, et al. Activation of PAF receptor by oxidised LDL in human monocytes stimulates chemokine releases but not urokinase-type plasminogen activator expression. Clin Chim Acta. 2004;344:163–71.
Prescott SM, McIntyre TM, Zimmerman GA, Stafforini DM. Inflammation as an early component of atherosclerosis and vascular damage–a role for P-selectin and platelet-activating factor. Jpn Circ J. 1996;60:137–41.
Handley DA, Arbeeny CM, Lee ML, Van Valen RG, Saunders RN. Effect of platelet activating factor on endothelial permeability to plasma macromolecules. Immunopharmacology. 1984;8:137–42.
Weyrich AS, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J Clin Invest. 1995;95:2297–303.
Takahashi T, Nishizawa Y, Hato F, et al. Neutrophil-activating activity and platelet-activating factor synthesis in cytokine-stimulated endothelial cells: reduced activity in growth-arrested cells. Microvasc Res. 2006;73:29–34.
McManus LM, Hanahan DJ, Pinckard RN. Human platelet stimulation by acetyl glyceryl ether phosphorylcholine. J Clin Invest. 1981;67:903–6.
Klinger MH. Platelets and inflammation. Anat Embryol (Berl). 1997;196:1–11.
Rouis M, Nigon F, Lafuma C, Hornebeck W, Chapman MJ. Expression of elastase activity by human monocyte-macrophages is modulated by cellular cholesterol content, inflammatory mediators, and phorbol myristate acetate. Arteriosclerosis. 1990;10:246–55.
Antonopoulou S, Nomikos T, Karantonis HC, Fragopoulou E, Demopoulos CA. PAF, a potent lipid mediator In: Bioactive Phospholipids. Role in Inflammation and Atheroslerosis. Edited by Tselepis AD. India: Research Signpost; 2009:85–134.
Feliste R, Perret B, Braquet P, Chap H. Protective effect of BN 52021, a specific antagonist of platelet-activating factor (PAF-acether) against diet-induced cholesteryl ester deposition in rabbit aorta. Atherosclerosis. 1989;78:151–8.
Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure: focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol. 2006;48:56–66.
Sadat U. Signaling pathways of cardioprotective ischemic preconditioning. Int J Surg. 2009;7:490–8.
Leary PJ, Rajasekaran S, Morrison RR, et al. A cardioprotective role for platelet-activating factor through NOS-dependent S-nitrosylation. Am J Physiol Heart Circ Physiol. 2008;294:H2775–84.
Valone FH, Epstein LB. Biphasic platelet-activating factor synthesis by human monocytes stimulated with IL-1-beta, tumor necrosis factor, or IFN-gamma. J Immunol. 1988;141:3945–50.
Zhang GQ, Tao YK, Li XL, et al. Investigation of platelet activating factor (PAF) in acute myocardial infarction. Chin J Emerg Med. 2010;19:1304–7.
Szmit S, Jank M, Maciejewski H, et al. Gene expression profiling in peripheral blood nuclear cells in patients with refractory ischaemic end-stage heart failure. J Appl Genet. 2010;51:353–68.
Quarck R, De Geest B, Stengel D, et al. Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;103:2495–500.
•• The Lp-PLA2 Studies Collaboration. Lipoprotein-associated phospholipase A2 and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–44. The present paper presents the combined results of several large studies concerning the role of Lp-PLA2 in cardiovascular disease.
Persson M, Hedblad B, Nelson JJ, Berglund G. Elevated Lp-PLA2 levels add prognostic information to the metabolic syndrome on incidence of cardiovascular events among middle-aged nondiabetic subjects. Arterioscler Thromb Vasc Biol. 2007;27:1411–6.
Mohler 3rd ER, Ballantyne CM, Davidson MH, et al. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2008;51:1632–41.
Serruys PW, Garcia-Garcia HM, Buszman P, et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–82.
Wang WY, Zhang J, Wu WY, et al. Inhibition of lipoprotein-associated phospholipase A2 ameliorates inflammation and decreases atherosclerotic plaque formation in ApoE-deficient mice. PLoS One. 2011;6:e23425.
Hu MM, Zhang J, Wang WY, et al. The inhibition of lipoprotein-associated phospholipase A2 exerts beneficial effects against atherosclerosis in LDLR-deficient mice. Acta Pharmacol Sin. 2011;32:1253–8.
White H, Held C, Stewart R, et al. Study design and rationale for the clinical outcomes of the STABILITY Trial (STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY) comparing darapladib versus placebo in patients with coronary heart disease. Am Heart J. 2010;160:655–61.
van Vark LC, Kardys I, Bleumink GS, et al. Lipoprotein-associated phospholipase A2 activity and risk of heart failure: the Rotterdam study. Eur Heart J. 2006;27:2346–52.
Suzuki T, Solomon C, Jenny NS, et al. Lipoprotein-associated phospholipase A(2) and risk of congestive heart failure in older adults: the Cardiovascular Health Study. Circ Heart Fail. 2009;2:429–36.
Gerber Y, Dunlay SM, Jaffe AS, et al. Plasma lipoprotein-associated phospholipase A2 levels in heart failure: association with mortality in the community. Atherosclerosis. 2009;203:593–8.
Moldoveanu E, Serban M, Marta DS, Serban I, Huica R. Lipoprotein-associated phospholipase A2 activity in patients with preserved left ventricular ejection fraction. Biomarkers. 2011;16:587–9.
Charniot JC, Khani-Bittar R, Albertini JP, et al. Interpretation of lipoprotein-associated phospholipase A2 levels is influenced by cardiac disease, comorbidities, extension of atherosclerosis and treatments. Int J Cardiol 2012. doi:10.1016/j.ijcard.2012.09.054.
Gullestad L, Ueland T, Vinge LE, et al. Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 2012;122:23–35.
Alloatti G, Montrucchio G, Mariano F, et al. Protective effect of verapamil on the cardiac and circulatory alterations induced by platelet-activating factor. J Cardiovasc Pharmacol. 1987;9:181–6.
Kelefiotis D, Lanara E, Vakirtzi-Lemonias C, et al. Study of digoxin as inhibitor of the in vivo effects of acetyl glyceryl ether phosphorylcholine (AGEPC) in mice. Life Sci. 1988;42:623–33.
Chrysohoou C, Pitsavos C, Metallinos G, et al. Cross-sectional relationship of a Mediterranean type diet to diastolic heart function in chronic heart failure patients. Hear Vessel. 2012;27:576–84.
Nomikos T, Fragopoulou E, Antonopoulou S. Food ingredients and lipid mediators. Curr Nutr Food Sci. 2007;3:255–76.
Fragopoulou E, Demopoulos CA, Antonopoulou S. Lipid minor constituents in wines. A biochemical approach in the French paradox. Int J Wine Res. 2009;1:131–43.
Karantonis HC, Antonopoulou S, Perrea DN, et al. In vivo antiatherogenic properties of olive oil and its constituent lipid classes in hyperlipidemic rabbits. Nutr Metab Cardiovasc Dis. 2006;16:174–85.
Antonopoulou S, Fragopoulou E, Karantonis HC, et al. Effect of traditional Greek Mediterranean meals on platelet aggregation in normal subjects and in patients with type 2 diabetes mellitus. J Med Food. 2006;9:356–62.
Matsumori A. Anti-inflammatory therapy for heart failure. Curr Opin Pharmacol. 2004;4:171–6.
Godfrey V, Farquharson C, Macdonald JE, Yee CM, Struthers AD. Effect of spironolactone on C-reactive protein levels in patients with heart disease. Int J Cardiol. 2007;117:282–4.
Tsantila N, Tsoupras AB, Fragopoulou E, et al. In vitro and in vivo effects of statins on platelet-activating factor and its metabolism. Angiology. 2011;62:209–18.
Heller R, Bussolino F, Ghigo D, et al. Nitrovasodilators inhibit thrombin-induced platelet-activating factor synthesis in human endothelial cells. Biochem Pharmacol. 1992;44:223–9.
Tolins JP, Melemed A, Sulciner D, Gustafson KS, Vercellotti GM. Calcium channel blockade inhibits platelet activating factor production by human umbilical vein endothelial cells. Lipids. 1991;26:1218–22.
Schror K, Felsch A. Ramiprilat prevents PAF-induced myocellular and endothelial injury in a neutrophil-perfused heart preparation. Agents Actions Suppl. 1992;38(Pt 3):209–16.
White HL, Faison LD. Inhibition of lyso-PAF: acetyl-CoA acetyltransferase by salicylates and other compounds. Prostaglandins. 1988;35:939–44.
Disclosure
Paraskevi Detopoulou declares that she has no conflict of interest.
Tzortzis Nomikos declares that he has no conflict of interest.
Elizabeth Fragopoulou declares that she has no conflict of interest.
Christina Chrysohoou declares that she has no conflict of interest.
Smaragdi Antonopoulou declares that she has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Detopoulou, P., Nomikos, T., Fragopoulou, E. et al. Platelet Activating Factor in Heart Failure: Potential Role in Disease Progression and Novel Target for Therapy. Curr Heart Fail Rep 10, 122–129 (2013). https://doi.org/10.1007/s11897-013-0131-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-013-0131-2