
Abstract
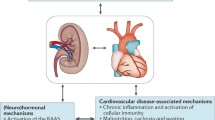
Heart failure (HF) is a pervasive clinical challenge characterized by compromised cardiac function and reduced quality of life. The kinin-kallikrein system (KSS), a multifaceted peptide cascade, has garnered substantial attention due to its potential role in HF. Through activation of B1 and/or B2 receptors and downstream signaling, kinins modulate various physiological processes, including inflammation, coagulation, pain, blood pressure control, and vascular permeability. Notably, aberrations in KKS components have been linked to HF risk. The elevation of vasodilatory bradykinin (BK) due to kallikrein activity reduces preload and afterload, while concurrently fostering sodium reabsorption inhibition. However, kallikrein’s conversion of prorenin to renin leads to angiotensinsII upregulation, resulting in vasoconstriction and fluid retention, alongside increased immune cell activity that fuels inflammation and cardiac remodeling. Importantly, prolonged KKS activation resulting from volume overload and tissue stretch contributes to cardiac collagen loss. The conventional renin-angiotensin-aldosterone system (RAAS) inhibitors used in HF management may inadvertently intensify KKS activity, exacerbating collagen depletion and cardiac remodeling. It is crucial to balance the KKS's role in acute cardiac damage, which may temporarily enhance function and metabolic parameters against its detrimental long-term effects. Thus, KKS blockade emerges as a promising strategy to impede HF progression. By attenuating the link between immune system function and tissue damage, KKS inhibition can potentially reduce cardiac remodeling and alleviate HF symptoms. However, the nuanced roles of BK in various acute conditions necessitate further investigation into the sustained benefits of kallikrein inhibitors in patients with chronic HF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a complicated clinical condition characterized by the inability of the heart to adequately circulate blood, resulting in various symptoms and a lower quality of life. The kinin-kallikrein system (KKS), constituting a complex peptide cascade involved in several physiological processes such as inflammation, coagulation, pain, blood pressure control, and vascular permeability, is one such mechanism that has received much attention and may be involved in HF progression [1].
In the KKS, kinins originate from kininogens through the action of tissue and plasma kallikreins. Some impacts of kinins are induced via activation of B1 and/or B2 receptors and downstream signaling such as nitric oxide. For instance, the KKS releases vasoactive kinins, such as bradykinin (BK), which is implicated in vasodilation, vascular leakage, and pain [1]. Notably, several studies have suggested a key role for KKS in the pathogenesis of HF, evidenced by increased BK levels in both animal and human model studies [2, 3]. Elevated BK levels have been frequently related to increased inflammation, oxidative stress, endothelial dysfunction, and fibrosis, all of which are major clinical hallmarks of HF [4, 5]. Additionally, genetic abnormalities within KKS components have been associated with an increased risk of developing HF or affecting its prognosis [6].
The current study aims to provide a comprehensive overview of the KKS components in pathophysiology of HF. Exploring the involvement of the KKS in the pathogenesis of HF can provide valuable insights into potential treatment targets that may improve patient outcomes. The information discussed in this review will help advance ongoing investigations into the intricate processes underlying HF, ultimately paving the way for more potent therapeutic approaches.
An overview of heart failure
Cardiovascular diseases, which account for 31% of all global fatalities, are the most fatal diseases worldwide resulting in 17.9 million deaths each year [7, 8]. In 2017, the global age-standardized prevalence rates of HF and years lived with disability due to HF were 831.0 and 128.2 per 100,000 people, respectively [9]. HF continues to pose a significant public health challenge globally, particularly in countries with relatively low socio-demographic index [9]. The American College of Cardiology Foundation and the American Heart Association (ACCF/AHA) guideline defined HF as “a complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood” [10]. The new classification of HF according to left ventricular (LV) ejection fraction (LVEF) is as follows: HF with reduced ejection fraction (HFrEF) if the LVEF ≤ %40, HF with mildly reduced ejection fraction (HFmrEF) if the LVEF = %41–49, and HF with preserved ejection fraction (HFpEF) if the LVEF ≥%50 [11].
Patients with HF may experience a wide range of symptoms, with the prevalent ones being shortness of breath, weariness, reduced tolerance to physical activity, orthopnea, dizziness, nausea, vomiting, diarrhea, loss of appetite, and fluid retention [12]. Notably, HF can be a consequence of a variety of heart problems, genetic abnormalities, and systemic disorders. Patients with HF may have a combination of etiologies, including ischemic heart disease, hypertensive heart disease, valvular dysfunction, and autoimmune diseases [13, 14].
Mechanism
Cardiac function overview
Understanding the function of the cardiovascular system is vital for comprehending the body’s circulatory processes. Several key parameters play pivotal roles in this context. Cardiac output (CO) represents the heart’s blood-pumping capacity, typically ranging 4–8 L/min. CO is affected by synergistic ventricular contraction, ventricular wall integrity, and valvular competence. In addition, stroke volume (SV) denotes the blood volume ejected by the ventricle during per heartbeat, usually 1 cc/kg or 60–100 cc. Notably, SV is influenced by preload (fiber stretch at diastole end), afterload (resistance for blood ejection), and contractility (heart’s inotropic state). Further, mean arterial pressure (MAP) is regulated by CO and total peripheral resistance (TPR) [15].
In heart pathogenesis, such as HF, reduced CO leads to lower MAP and reduced tissue perfusion. The body tries to restore MAP through the Frank-Starling mechanism, ventricular remodeling, and neurohormonal activation, which will be briefly discussed here.
Frank-Starling mechanism and HF
The Frank-Starling relationship represents how the LV responds to increased preload under normal conditions. As passive tension increases, active contraction strengthens, resulting in bigger SV and CO [16]. As the ventricular contraction strength increases, the heart muscle gradually becomes hypertrophic, followed by a shrinking ventricular space. To compensate for the decrease in the impact volume, the heart rate increases and the diastole time decreases. Blood supply to the heart muscle itself occurs during diastole. Increased heart demand for blood and decreased blood supply, as observed in HF, ultimately lead to heart muscle damage, fibrosis, and apoptosis [17, 18]. In return, it is also possible to disrupt the interconnection of actin and myosin filaments with excessive ventricular dilation, which decreases the strength of the heart muscle contraction [19].
Heart remodeling and HF
Following cardiac injury or stress stimulation, various multifactorial systemic mechanisms involving structural, neurohumoral, cellular, and molecular factors, are triggered and act together to maintain physiological function. These intricate and coordinated processes result in fluid overload, sympathetic nervous system (SNS) hyperactivity, and circulatory redistribution, leading to significant concomitant and progressive clinical signs and symptoms. This process of structural and functional alterations of the heart after injury is referred to as remodeling, which can be categorized into physiological/pathological and adaptive/maladaptive, depending on the nature of the changes and their impact on the heart’s health and function [20].
Regardless of the underlying pathologic cause, remodeling impacts all cells and components of the heart, resulting in various cellular changes such as cardiomyocyte hypertrophy, myocyte apoptosis, and necrosis, along with fibroblast proliferation, accumulation of proinflammatory mediators, and reorganization of extracellular matrix [21].
Fibrosis, characterized by abnormal formation of collagen and extracellular matrix components, significantly contributes to HF progression [22]. It impacts cardiac structure and function, which leads to reduced contractility, increased stiffness, and electrical disruptions in the myocardium [23]. Key cellular processes include cardiac fibroblast activation, inflammation, and endothelial dysfunction.
Furthermore, the transforming growth factor (TGF) signaling, matrix metalloproteinases (MMPs), and tissue inhibitors of metalloproteinases (TIMPs) are among the molecular pathways that regulate fibroblast activation, collagen formation, and extracellular matrix remodeling [23]. MicroRNA dysregulation has also been linked to cardiac fibrosis by influencing fibroblast activation, collagen production, and extracellular matrix remodeling [24].
Moreover, left ventricular reverse remodeling (LVRR) is a compensatory mechanism that improves cardiac function after HF. This is structurally defined by decreased ventricular volume and improved adrenergic sensitivity and is associated with decreased inflammatory mediators [25].
Neurohormonal mechanism and HF
Neurohormonal activation is the dysregulation of hormonal systems that maintain cardiovascular homeostasis, such as the SNS, renin-angiotensin-aldosterone system (RAAS), vasopressin system, and natriuretic peptides [26]. In patients with HF, the overactivation of the SNS leads to increased release of norepinephrine (NE) [27]. Notably, elevated NE levels in patients with HF have been linked with increased mortality. NE causes vasoconstriction, increased heart rate, and cardiac remodeling [27]. Furthermore, it promotes inflammation, oxidative stress, and apoptosis in the heart muscle, contributing to HF progression [28].
The RAAS is an important regulator of blood volume and systematic vascular resistance. In RAAS, renin is released into the circulation in response to low blood pressure or inadequate sodium levels. This occurs when arterial baroreceptors detect low pressure and the kidneys sense low sodium levels [29]. The SNS triggers renin release through the β-adrenoreceptor-cAMP pathway [30]. In this process, kallikrein (KAL) converts proreninin to renin, which, in turn, converts angiotensinogen to angiotensin I. Angiotensin-converting enzyme (ACE) then converts angiotensin I into angiotensin II, a potent vasoconstrictor that stimulates the release of aldosterone from the adrenal glands. Aldosterone leads to sodium retention and potassium excretion, resulting in fluid overload and electrolyte imbalances [31]. Furthermore, angiotensin II directly influences cardiac remodeling by promoting fibrosis and hypertrophy [32]. Thus, the RAAS plays a crucial role in the development and progression of HF, mainly by promoting vasoconstriction, fluid retention, and cardiac remodeling.
Importantly, angiotensin II can stimulate the release of vasopressin, also called antidiuretic hormone (ADH), which inhibits the secretion of renin, regulates kidney water reabsorption, and maintains fluid. In patients with HF, vasopressin levels tend to rise due to reduced CO and renal hypoperfusion, leading to fluid retention and worsening symptoms such as congestion and edema, resulting in clinical symptoms like dyspnea and peripheral edema [33].
In addition, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and natriuretic peptide precursor-C (NPP-C) play roles in the pathophysiology of HF [34]. These natriuretic peptides are released into the bloodstream in response to pressure, strain, and specific proinflammatory cytokines. Actions of these hormones cause vasodilation, diuresis/natriuresis, inhibition of RAAS, reduction of sympathetic activity, and prevent the progression of heart hypertrophy. BNP levels can also predict outcomes in HF, with higher levels indicating a greater risk of death. Monitoring BNP levels over time can guide treatment decisions and assess treatment effectiveness [35].
Inflammation and HF
While conventional risk factors, genetic cardiomyopathy, and mechanical valve dysfunction are important contributors to HF, the possible role of immune activation should be considered as a significant factor in the development and progression of HF. Even if the initial trigger of HF may not be immunological, the immune system can become activated in the acute setting following an injury, which may also predict clinical outcomes [36].
It is widely acknowledged that inflammation is critical in cardiac hypertrophy and HF. For instance, increased serum pro-inflammatory cytokine levels are often observed in all types of HF, suggesting that chronic low-grade inflammation might be an important mediator contributing to the maintenance or exacerbation in patients with established HF [37]. Yet, the causality of inflammation and disease progression requires further investigations [36, 37].
Inflammatory cytokines have been shown to reduce muscle contractility and promote apoptosis of cardiomyocytes. They can activate a substrate degradation program, induce substrate metalloproteinases, and cause extracellular matrix degradation. Several studies have shown that pro-inflammatory cytokines such as C-reactive protein (CRP), tumor necrose factor-alpha (TNF-α), and members of the interleukin 1 (IL-1) and interleukin 6 (IL-6) family are elevated in patients with HF [37, 38].
In addition, myocardial ischemia-reperfusion injury causes the infarcted heart to produce more inflammatory cytokines [39]. This inflammatory response following ischemia-reperfusion involving toll-like receptor signaling and activation of complement and reactive oxygen species (ROS) generation is also implicated in developing postinfarction ventricular remodeling and HF [40].
It is important to mention that endothelium normally has both anti-inflammatory and antithrombotic functions. The endothelium controls vascular tone in healthy people by balancing the release of vasodilators such as nitric oxide (NO) with endothelium-derived constrictors such as endothelin. Notably, the involvement of several pathways, including TGFβ1/Smad, mitogen-activated protein kinases (MAPKs), and nuclear factor-B (NFκB) signaling in the regulation of endothelial nitric oxide synthase (eNOS) and NO bioavailability is implicated in endothelial function and cardiac chamber remodeling [41, 42]. Understanding the interplay between TGFβ1, MAPKs, NFκB, and inflammatory responses is crucial for developing targeted therapies to modulate these pathways and mitigate adverse cardiac remodeling and inflammation in cardiovascular diseases.
Vascular permeability is crucial in inflammation, allowing immune cells into damaged myocardium, intensifying the inflammatory response. Controlling vascular permeability is critical for preventing excessive leakage, which can cause tissue injury or edema development. Endothelial cells regulate vascular permeability through contraction, intercellular gaps, and transcytosis and thus play critical roles during inflammation [43].
Additionally, increased vascular permeability disrupts the endothelial barrier integrity and impairs heart’s microvascular circulation, oxygen supply, and waste removal, compromising cardiac function [44]. Excess vascular permeability may lead to fibrosis by allowing pro-fibrotic factors such as TGF-β to invade the myocardium, promoting fibroblast activation, and collagen production. Fibrosis is a key aspect of heart remodeling, with excessive deposition of extracellular matrix proteins in the myocardium [22].
Moreover, excess vascular permeability can promote angiogenesis by enhancing endothelial cell movement and growth, which is a compensatory mechanism to improve oxygen supply to the enlarged heart [43]. However, it can also lead to abnormal vessel growth and leakage, further worsening heart function [45]. Gaining insights into the role of vascular permeability in heart remodeling provides opportunities for therapeutic interventions. Modulating inflammation or fibrosis pathways related to vascular permeability may offer new treatments for cardiovascular diseases.
The activated inflammatory pathways are also observed in people at high risk for HF, including obese individuals [46] and cigarette smokers [47], and in the absence of congestive heart failure (CHF) clinical syndromes [48]. On the other hand, protective factors such as exercise have anti-inflammatory effects [49]. All these cases indicate the high importance of inflammation in HF. Nonetheless, the role of inflammation in HF is complicated. Chronic inflammation causes structural and functional changes in the heart, leading to unfavorable remodeling and impaired contractility. Activating various inflammatory pathways, such as cytokines, chemokines, and immune cells, is important in sustaining this inflammatory effect [50].
Kinin-kallikrein system (KSS)
An overview of KKS
The KSS is a complex regulatory system that coordinates various physiological processes, including inflammation, coagulation, pain, cell proliferation, vasodilation, and blood pressure [1, 51]. The KKS contains two pathways including plasma KKS and tissue KKS.
Plasma KKS
The plasma KKS, as part of the intrinsic coagulation system, involves the autoactivation of factor XII when blood encounters negatively charged or neutral surfaces. The activated factor XII (FXIIa) then catalyzes the conversion of prekallikrein (PK) to its activated form, plasma KAL, by cleaving off a small peptide fragment from PK in a process known as contact activation. Accordingly, the plasma KKS is often used as synonymous with the “contact activation system (CAS) [1, 51].
The activated KAL, in turn, cleaves high molecular weight kininogen (HMWK) into the potent inflammatory mediator, nonapeptide BK. BK generated from HMWK acts as the ligand for the G-protein coupled B2-receptor (B2R). Des-Arg9-BK is a biologically active peptide formed when BK undergoes enzymatic cleavage by the carboxypeptidase enzyme kinases I. Des-Arg9-BK is a ligand for the G-protein coupled B1-receptor (B1R). Both receptors help release mediators such as NO, arachidonic acid, prostaglandins, leukotrienes, and endothelium-derived hyperpolarizing factors [52]. While B2R activation results in a transient release of NO in endothelial cells, B1R activation leads to very high and sustained NO production [53].
Notably, the majority of the effects of the plasma KKS on inflammation, vascular function, blood pressure control, and nociceptive response are attributed to the activation of B2R and B1R by BK and des-Arg9-BK, respectively [54]. Additionally, the cleaved HMWK-a binds to neutrophils and monocytes, inhibiting their adhesion to fibrinogen and/or vitronectin. HMWK-a binding to monocytes stimulates the production and release of inflammatory cytokines and chemokines [1].
The KAL is part of both the CAS and KKS, resulting in reciprocal acts. While the CAS is involved in thrombin formation and inflammation, the KKS mainly plays an important part in inflammation and lacks a specific role in blood coagulation. It is worth mentioning that the activated factor XIIa plays a multifaceted role in the plasma KKS and the intrinsic coagulation pathway. The factor XIIa can also initiate the complement system, fibrinolysis, and may regulate cellular response [55].
Tissue KKS
Unlike the plasma KKS, the tissue KKS is independent of factor XII and involves different components, including low molecular weight kininogen (LMWK) and tissue KAL [51]. In the tissue KKS, the enzyme tissue KAL produced and released by various tissues, including kidneys, salivary glands, and pancreas, acts on LMWK to produce a peptide called kallidin or Lys-BK. Kallidin is a vasoactive substance and, similar to BK, interacts with B2R and mediates various physiological responses, including vasodilation, increased vascular permeability, smooth muscle contraction, and inflammation. Kallidin can also be converted to BK by aminopeptidase.
KKS and RAAS
The KKS acts as a natural counter-regulatory system to the RAAS in the body [30]. As discussed, the RAAS is an important hormonal system that regulates blood pressure and fluid balance in the body. In response to low blood pressure or low sodium levels, the RAAS induces vasoconstriction, sodium and water retention, and vascular tone. This primarily achieved through the action of a potent vasoconstrictor named angiotensin II, which is generated by ACE. Angiotensin II stimulates the release of plasminogen activator inhibitor 1 (PAI1) from endothelial cells.
On the other hand, the KKS plays a role in vasodilation and fluid balance by producing kinins, such as BK and kallidin, which are potent vasodilators [51]. They increase vascular permeability, leading to the relaxation of blood vessels, decreased systemic blood pressure, and decreased production of ROS to protect the heart and the kidney from organ damage [56].
Once released, kinin peptides (BK, KAL, and kallidin-like peptide) circulate in the blood and interact with their respective receptors (B1R and B2R) to exert various physiological effects. ACE, which is primarily found on the surface of endothelial cells and other tissues, rapidly cleaves and inactivates these peptides. This regulatory action of ACE on kinin peptides serves as an essential regulatory mechanism to prevent excessive or prolonged effects of kinin in the body [30, 51]. These kinins are crucial in activating endothelial cells during various processes, including inflammation, vasodilation, increased vascular permeability, and smooth muscle contraction within blood vessels. Consequently, disruption in this system can lead to hypotension, angioedema, and heart and kidney disorders [57]. Notably, the KKS is regulated by serpins and has a complex distribution of components, along with numerous interactions with other essential metabolic pathways.
ACE inhibitors and angiotensin receptor blockers (ARBs) are two classes of medications commonly used to manage hypertension and other cardiovascular conditions. It is worth noting that changes in ACE levels have a more significant impact on kinin levels than on angiotensin II levels [58]. ACE inhibitors are the most widely used agents to increase KKS activity [56, 58]. Their primary role is to upregulate kinins rather than to inhibit ACE. Additionally, ARBs are a class of widely used medications that are effective in protecting the heart and kidneys by selectively blocking the angiotensin II type 1 (AT1) receptor, which are specific receptors for angiotensin II [59].
KKS and the heart
KKS in acute cardiac pathological conditions
In acute conditions such as acute coronary syndrome (ACS), KAL activity causes vasodilation due to the production of BK, followed by NO. A study explored the impact of KAL on heart remodeling and apoptosis in post-myocardial infarction (MI) [60]. Rats injected with adenovirus containing human tissue KAL or luciferase gene displayed enhanced cardiac responses during dobutamine-induced stress. Notably, somatic gene delivery improved cardiac responses to stress, reduced myocardial apoptosis, and enhanced cell survival. This study demonstrates the KKS’s pivotal role in mitigating the progression of HF by modulating the Akt-mediated signaling pathway, which reduces cardiac hypertrophy, fibrosis, endothelial dysfunction, and myocardial apoptosis [60].
In another study, intramyocardial infusion of purified tissue KAL following an MI led to the reduction of infarct size and inhibition of cardiomyocyte apoptosis associated with elevated NO levels and Akt signaling [61] as well as reduced caspase-3 activation [62]. Importantly, icatibant, a B2R antagonist, inhibited the effects of KAL. This suggests that via B2R activation, KAL may inhibit apoptosis, inflammation, and ventricular remodeling by enhancing the formation of NO and suppressing oxidative stress pathways. Additionally, KAL may protect the heart against reperfusion injury and vascular injury [63, 64].
KKS in HF
As explained, kinins are active peptides released as a product of KAL’s enzymatic action on kininogen. The cumulative effects include vasodilation, hypotension, endothelial relaxing factor release, and natriuresis. Endogenous BK is rapidly inactivated by kininase I and kinase II, known as ACE.
It has been demonstrated that KAL causes pro-inflammatory reactions by stimulating immune cells, including neutrophils and monocytes/macrophages. TNF-α and interleukins (IL-1, IL-6) are two pro-inflammatory cytokines released due to this activation, leading to cardiac inflammation. Chronic inflammation causes maladaptive remodeling, and fibrosis and worsens heart function [61]. Additionally, KAL activity increases TGF-β production, a profibrotic cytokine [65]. TGF-β promotes the synthesis of extracellular matrix proteins, causing myocardial fibrosis [66]. KAL also generates ROS through interaction with nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes, leading to oxidative damage to cellular components and apoptosis, which may worsen HF progression [56].
In the Second Northwick Park Heart Study (NPHSII) of 2706 middle-aged Caucasian men, 175 events occurred during follow-up, including 124 (70.8) acute MIs, 33 coronary surgeries (18.9%), and 18 silent Mis (10.3%) [67]. The study found that common polymorphisms in the genes encoding the kinins B1R and B2R influence prospective hypertensive coronary risk, suggesting that the B1R and B2R may play the same function in human coronary vascular diseases.
It is worth mentioning that the B1R is more expressed during LV dysfunction and ACE inhibition [68]. An in vitro study discovered that endotoxin-induced kinin B1R induction in pig coronary arteries caused concentration-dependent, endothelium-independent contraction [69]. A B1R antagonist, SSR240612, prevented these contractions, while the B2R antagonist, HOE140, had no effects [69]. Accordingly, the induction of B1R during inflammation could be of clinical concern in the vasculature, especially in coronary arteries with dysfunctional endothelial cells.
Notably, patients with CHF (NYHA class II) may exhibit elevated plasma BK levels and endothelial markers associated with inflammation during long-term ACE-inhibitor therapy [70]. However, those patients treated with ACE-inhibitor may not be able to respond adequately to ischemic and exercise-induced stimuli.
A previous study showed that acute VO in rats increases both angiotensis II and BK levels in the interstitial fluid (ISF) [71]. While the treatment with ACE inhibitors decreased angiotensin II levels, the level of ISF BK was elevated, reducing LV hypertrophic response. Although adding B2R antagonists to ACE inhibitors did not yield a better outcome, B2R blockade produced more concentric hypertrophy as it led to a thicker wall and smaller chamber diameter [71]. These results indicate that the cardioprotective effects of ACE inhibitors are mostly due to their reducing effect on angiotensin II levels. They also found a significant interaction between mast cells and BK in influencing the impact of ACE inhibition on LV remodeling during the initial phase of VO [71].
Furthermore, the ACE inhibitor-induced increase in BK may exacerbate matrix loss. In an animal study, rats underwent either sham surgery or artocaval fistula (ACF) to stimulate VO [72]. ACF rates were treated with either a 2-day B2R blockade or a 4-week ACE inhibition. It was found that the primary mechanism for LV remodeling in response to ACF-induced VO was BK-mediated collagen matrix dissolution [72]. ACE inhibitors, which raise antifibrotic BK, did not reduce LV remodeling in VO. In contrast, B2R blockade prevented eccentric LV remodeling and improved its function.
In another study, LV ISF collection and echocardiography were performed in sham and ACF rats [73]. ACF rats exhibited LV dilatation, higher LV end-diastolic pressure, and elevated LV ISF BK levels. Mast cell numbers increased, while interstitial collagen decreased at 4 and 15 weeks post-ACF. Aprotinin, a KAL inhibitor, preserved interstitial collagen, prevented mast cell increase, and improved LV systolic function in ACF rats. A 24-h LV interstitial BK infusion increased mast cell numbers by twofold and reduced interstitial collagen by 30%, but this effect was reversed by a B2R antagonist [73]. The findings show that VO triggers KKS upregulation, leading to mast cell infiltration, extracellular matrix loss, and LV dysfunction. KAL inhibition may counteract these effects.
It is worth noting that the B1R blockade with BI113823 seems to be as effective as ACE inhibition with lisinopril in attenuating post-infarction LV remodeling and HF in rats; however, the effects of the combination of both compounds may not be additive [74].
Conclusion
In the KKS, KAL activity increases the level of the vasodilator BK, which reduces preload and afterload and directly inhibits sodium reabsorption from renal tubules. On the other hand, KAL converts prorenin to renin, increasing the level of angiotensin II, resulting in vasoconstriction and fluid retention through increasing the permeability of blood vessels. It facilitates the activity of immune system cells, including neutrophils and macrophages, which ultimately increases inflammation and heart remodeling.
VO and tissue stretch cause long-term KKS activation, leading to heart collagen loss. RAAS-blocking drugs currently used in managing HF can increase KKS activity, exacerbating cardiac collagen reduction and, ultimately, cardiac remodeling. Although the KKS function in heart damage can temporarily improve cardiac function and metabolic parameters, it also causes tissue destruction and long-term cardiac remodeling.
Thus, KKS-blocking treatments may play a significant role in mitigating the progression of HF. Through breaking bridges between the immune system function and tissue damage, KKS blockade can reduce cardiac remodeling. Further, reducing the influence of BK contributes to a partial alleviation of HF symptoms. Nevertheless, considering the vital and beneficial roles of BK in various acute conditions, such as stroke, it is anticipated that its deletion could lead to adverse consequences in the long term. The authors propose that partial inhibition of KAL may yield positive outcomes in patients with HF. However, further research is required to elucidate the long-term effects of KAL inhibitors in patients with chronic HF.
References
Bryant JW, Shariat-Madar Z (2009) Human plasma kallikrein-kinin system: physiological and biochemical parameters. Cardiovasc Hematol Agents Med Chem 7:234–250. https://doi.org/10.2174/187152509789105444
Heitsch H (2003) The therapeutic potential of bradykinin B2 receptor agonists in the treatment of cardiovascular disease. Expert Opin Investig Drugs 12:759–770. https://doi.org/10.1517/13543784.12.5.759
Liesmaa I, Kuoppala A, Shiota N et al (2005) Increased expression of bradykinin type-1 receptors in endothelium of intramyocardial coronary vessels in human failing hearts. Am J Physiol Heart Circ Physiol 288:H2317-2322. https://doi.org/10.1152/ajpheart.00815.2004
Sorop O, Heinonen I, van Kranenburg M et al (2018) Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc Res 114:954–964. https://doi.org/10.1093/cvr/cvy038
Hu J, Cheng P, Huang G-Y et al (2018) Effects of Xin-Ji-Er-Kang on heart failure induced by myocardial infarction: role of inflammation, oxidative stress and endothelial dysfunction. Phytomedicine 42:245–257. https://doi.org/10.1016/j.phymed.2018.03.036
Hamid S, Rhaleb IA, Kassem KM, Rhaleb N-E (2020) Role of kinins in hypertension and heart failure. Pharmaceuticals (Basel) 13:347. https://doi.org/10.3390/ph13110347
Hasanpour Dehkordi A, Zare Dehabadi E, Rezaei MR et al (2023) Empowerment and self-efficacy in patients with chronic disease; a systematic review study. J Nephropharmacol 12:e10596. https://doi.org/10.34172/npj.2023.10596
Jiang X, Ming W-K, You JH (2019) The cost-effectiveness of digital health interventions on the management of cardiovascular diseases: systematic review. J Med Internet Res 21:e13166. https://doi.org/10.2196/13166
Bragazzi NL, Zhong W, Shu J et al (2021) Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur J Prev Cardiol 28:1682–1690. https://doi.org/10.1093/eurjpc/zwaa147
Yancy CW, Jessup M, Bozkurt B et al (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62:e147-239. https://doi.org/10.1016/j.jacc.2013.05.019
Hajouli S, Ludhwani D (2023) Heart failure and ejection fraction. StatPearls. StatPearls Publishing, Treasure Island (FL)
National Clinical Guideline Centre (UK) (2010) Chronic Heart Failure: National Clinical Guideline for Diagnosis and Management in Primary and Secondary Care: Partial Update. Royal College of Physicians (UK), London. PMID: 22741186
Ziaeian B, Fonarow GC (2016) Epidemiology and aetiology of heart failure. Nat Rev Cardiol 13:368–378. https://doi.org/10.1038/nrcardio.2016.25
Meijers WC, de Boer RA (2019) Common risk factors for heart failure and cancer. Cardiovasc Res 115:844–853. https://doi.org/10.1093/cvr/cvz035
Mohrman DE, Heller LJ (2018) Overview of the cardiovascular system. Cardiovascular physiology, 9th edn. McGraw-Hill Education
LaCombe P, Jose A, Basit H, Lappin SL (2023) Physiology, starling relationships. StatPearls. StatPearls Publishing, Treasure Island (FL)
Bonow RO, Carabello BA, Chatterjee K et al (2008) 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 118:e523-661. https://doi.org/10.1161/CIRCULATIONAHA.108.190748
Piek A, de Boer RA, Silljé HHW (2016) The fibrosis-cell death axis in heart failure. Heart Fail Rev 21:199–211. https://doi.org/10.1007/s10741-016-9536-9
Alamo L, Ware JS, Pinto A et al (2017) Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 6:e24634. https://doi.org/10.7554/eLife.24634
Azevedo PS, Polegato BF, Minicucci MF et al (2016) Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol 106:62–69. https://doi.org/10.5935/abc.20160005
Kehat I, Molkentin JD (2010) Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122:2727–2735. https://doi.org/10.1161/CIRCULATIONAHA.110.942268
Wight TN, Potter-Perigo S (2011) The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol 301:G950–G955. https://doi.org/10.1152/ajpgi.00132.2011
Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71:549–574. https://doi.org/10.1007/s00018-013-1349-6
van Rooij E, Sutherland LB, Thatcher JE et al (2008) Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 105:13027–13032. https://doi.org/10.1073/pnas.0805038105
Tanai E, Frantz S (2015) Pathophysiology of heart failure Compr Physiol 6:187–214. https://doi.org/10.1002/cphy.c140055
Hartupee J, Mann DL (2017) Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol 14:30–38. https://doi.org/10.1038/nrcardio.2016.163
Lymperopoulos A, Rengo G, Koch WJ (2013) Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113:739–753. https://doi.org/10.1161/CIRCRESAHA.113.300308
Rotariu D, Babes EE, Tit DM et al (2022) Oxidative stress - complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomed Pharmacother 152:113238. https://doi.org/10.1016/j.biopha.2022.113238
Schweda F, Friis U, Wagner C et al (2007) Renin release. Physiology 22:310–319. https://doi.org/10.1152/physiol.00024.2007
Schmaier AH (2002) The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. J Clin Invest 109:1007–1009. https://doi.org/10.1172/JCI0215490
Fountain JH, Kaur J, Lappin SL (2023) Physiology, renin angiotensin system. StatPearls. StatPearls Publishing, Treasure Island (FL)
Schnee J (2000) Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res 46:264–268. https://doi.org/10.1016/S0008-6363(00)00044-4
Rodriguez M, Hernandez M, Cheungpasitporn W et al (2019) Hyponatremia in heart failure: pathogenesis and management. CCR 15:252–261. https://doi.org/10.2174/1573403X15666190306111812
Potter LR, Yoder AR, Flora DR et al (2009) Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol 341–366. https://doi.org/10.1007/978-3-540-68964-5_15
Wright GA, Struthers AD (2006) Natriuretic peptides as a prognostic marker and therapeutic target in heart failure. Heart 92:149–151. https://doi.org/10.1136/hrt.2003.018325
Dick SA, Epelman S (2016) Chronic heart failure and inflammation: what do we really know? Circ Res 119:159–176. https://doi.org/10.1161/CIRCRESAHA.116.308030
Halade GV, Lee DH (2022) Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine 79:103992. https://doi.org/10.1016/j.ebiom.2022.103992
Hanna A, Frangogiannis NG (2020) Inflammatory cytokines and chemokines as therapeutic targets in heart failure. Cardiovasc Drugs Ther 34:849–863. https://doi.org/10.1007/s10557-020-07071-0
Li M, Georgakopoulos D, Lu G et al (2005) p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation 111:2494–2502. https://doi.org/10.1161/01.CIR.0000165117.71483.0C
Frangogiannis NG (2014) The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11:255–265. https://doi.org/10.1038/nrcardio.2014.28
Tran N, Garcia T, Aniqa M et al (2022) Endothelial nitric oxide synthase (eNOS) and the cardiovascular system: in physiology and in disease states. Am J Biomed Sci Res 15:153–177
Moens U, Kostenko S, Sveinbjørnsson B (2013) The role of mitogen-activated protein kinase-activated protein kinases (MAPKAPKs) in inflammation. Genes (Basel) 4:101–133. https://doi.org/10.3390/genes4020101
Nagy JA, Benjamin L, Zeng H et al (2008) Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 11:109–119. https://doi.org/10.1007/s10456-008-9099-z
Lavin B, Phinikaridou A, Lorrio S et al (2015) Monitoring vascular permeability and remodeling after endothelial injury in a murine model using a magnetic resonance albumin-binding contrast agent. Circ Cardiovasc Imaging 8:e002417. https://doi.org/10.1161/CIRCIMAGING.114.002417
Claesson-Welsh L (2015) Vascular permeability—the essentials. Upsala J Med Sci 120:135–143. https://doi.org/10.3109/03009734.2015.1064501
Battineni G, Sagaro GG, Chintalapudi N et al (2021) Impact of obesity-induced inflammation on cardiovascular diseases (CVD). Int J Mol Sci 22:4798. https://doi.org/10.3390/ijms22094798
Gopal DM, Kalogeropoulos AP, Georgiopoulou VV et al (2012) Cigarette smoking exposure and heart failure risk in older adults: the Health, Aging, and Body Composition Study. Am Heart J 164:236–242. https://doi.org/10.1016/j.ahj.2012.05.013
Raymond RJ, Dehmer GJ, Theoharides TC, Deliargyris EN (2001) Elevated interleukin-6 levels in patients with asymptomatic left ventricular systolic dysfunction. Am Heart J 141:435–438. https://doi.org/10.1067/mhj.2001.113078
Paolucci EM, Loukov D, Bowdish DME, Heisz JJ (2018) Exercise reduces depression and inflammation but intensity matters. Biol Psychol 133:79–84. https://doi.org/10.1016/j.biopsycho.2018.01.015
Riehle C, Bauersachs J (2019) Key inflammatory mechanisms underlying heart failure. Herz 44:96–106. https://doi.org/10.1007/s00059-019-4785-8
Pathak M, Wong SS, Dreveny I, Emsley J (2013) Structure of plasma and tissue kallikreins. Thromb Haemost 110:423–433. https://doi.org/10.1160/TH12-11-0840
Couture R, Harrisson M, Vianna RM, Cloutier F (2001) Kinin receptors in pain and inflammation. Eur J Pharmacol 429:161–176. https://doi.org/10.1016/S0014-2999(01)01318-8
Moreau ME, Garbacki N, Molinaro G et al (2005) The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci 99:6–38. https://doi.org/10.1254/jphs.srj05001x
Marceau F, Regoli D (2004) Bradykinin receptor ligands: therapeutic perspectives. Nat Rev Drug Discov 3:845–852. https://doi.org/10.1038/nrd1522
Didiasova M, Wujak L, Schaefer L, Wygrecka M (2018) Factor XII in coagulation, inflammation and beyond. Cell Signal 51:257–265. https://doi.org/10.1016/j.cellsig.2018.08.006
Kayashima Y, Smithies O, Kakoki M (2012) The kallikrein-kinin system and oxidative stress. Curr Opin Nephrol Hypertens 21:92–96. https://doi.org/10.1097/MNH.0b013e32834d54b1
Zito F, Lowe GDO, Rumley A et al (2002) Association of the factor XII 46C>T polymorphism with risk of coronary heart disease (CHD) in the WOSCOPS study. Atherosclerosis 165:153–158. https://doi.org/10.1016/s0021-9150(02)00196-x
Erdös EG, Tan F, Skidgel RA (2010) Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension 55:214–220. https://doi.org/10.1161/HYPERTENSIONAHA.109.144600
Hill RD, Vaidya PN (2023) Angiotensin II receptor blockers (ARB). StatPearls. StatPearls Publishing, Treasure Island (FL)
Agata J, Chao L, Chao J (2002) Kallikrein gene delivery improves cardiac reserve and attenuates remodeling after myocardial infarction. Hypertension 40:653–659. https://doi.org/10.1161/01.hyp.0000036035.41122.99
Sydykov A, Mamazhakypov A, Petrovic A et al (2018) Inflammatory mediators drive adverse right ventricular remodeling and dysfunction and serve as potential biomarkers. Front Physiol 9:609. https://doi.org/10.3389/fphys.2018.00609
Yao Y-Y, Yin H, Shen B et al (2007) Tissue kallikrein infusion prevents cardiomyocyte apoptosis, inflammation and ventricular remodeling after myocardial infarction. Regul Pept 140:12–20. https://doi.org/10.1016/j.regpep.2006.11.020
Huang M, Du J, Wang Y et al (2019) Tissue kallikrein-related peptidase8 protects rat heart against acute ischemia reperfusion injury. Int J Biol Macromol 140:1126–1133. https://doi.org/10.1016/j.ijbiomac.2019.08.195
Chao J, Chao L (2005) Kallikrein-kinin in stroke, cardiovascular and renal disease. Exp Physiol 90:291–298. https://doi.org/10.1113/expphysiol.2004.028464
Hara M, Kirita A, Kondo W et al (2014) LAP degradation product reflects plasma kallikrein-dependent TGF-β activation in patients with hepatic fibrosis. Springerplus 3:221. https://doi.org/10.1186/2193-1801-3-221
Noble NA, Harper JR, Border WA (1992) In vivo interactions of TGF-β and extracellular matrix. Prog Growth Factor Res 4:369–382. https://doi.org/10.1016/0955-2235(92)90017-C
Dhamrait SS, Payne JR, Li P et al (2003) Variation in bradykinin receptor genes increases the cardiovascular risk associated with hypertension. Eur Heart J 24:1672–1680. https://doi.org/10.1016/S0195-668X(03)00441-X
Marin-Castaño ME, Schanstra JP, Neau E et al (2002) Induction of functional bradykinin b(1)-receptors in normotensive rats and mice under chronic angiotensin-converting enzyme inhibitor treatment. Circulation 105:627–632. https://doi.org/10.1161/hc0502.102965
More AS, Kim HM, Khang G et al (2014) Des-Arg9-bradykinin causes kinin B1 receptor mediated endothelium-independent contractions in endotoxin-treated porcine coronary arteries. Pharmacol Res 90:18–24. https://doi.org/10.1016/j.phrs.2014.09.001
Cugno M, Agostoni P, Mari D et al (2005) Impaired bradykinin response to ischaemia and exercise in patients with mild congestive heart failure during angiotensin-converting enzyme treatment. Relationships with endothelial function, coagulation and inflammation. Br J Haematol 130:113–120. https://doi.org/10.1111/j.1365-2141.2005.05569.x
Wei C-C, Lucchesi PA, Tallaj J et al (2003) Cardiac interstitial bradykinin and mast cells modulate pattern of LV remodeling in volume overload in rats. Am J Physiol Heart Circ Physiol 285:H784-792. https://doi.org/10.1152/ajpheart.00793.2001
Ryan TD, Rothstein EC, Aban I et al (2007) Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol 49:811–821. https://doi.org/10.1016/j.jacc.2006.06.083
Wei C-C, Chen Y, Powell LC et al (2012) Cardiac kallikrein-kinin system is upregulated in chronic volume overload and mediates an inflammatory induced collagen loss. PLoS ONE 7:e40110. https://doi.org/10.1371/journal.pone.0040110
Lin X, Bernloehr C, Hildebrandt T et al (2016) Kinin B1 receptor blockade and ACE inhibition attenuate cardiac postinfarction remodeling and heart failure in rats. Toxicol Appl Pharmacol 305:153–160. https://doi.org/10.1016/j.taap.2016.06.005
Author information
Authors and Affiliations
Contributions
Keivan Mohammadi wrote the first draft of the manuscript. All authors commented on previous versions of the manuscript, contributed to the study conception and drafting and revising the manuscript, and they approved the final submitted version.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohammadi, K., Shafie, D., Ghomashi, N. et al. Kinin-kallikrein system: New perspectives in heart failure. Heart Fail Rev 29, 729–737 (2024). https://doi.org/10.1007/s10741-024-10393-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-024-10393-y