
Abstract
Retrotransposons represent a major component of plant genomes; however, large-scale studies on their expression are rare. Massively parallel sequencing offers new analytical possibilities enabling a comprehensive study of retrotransposon RNA transcription. We evaluated the expression of long terminal repeat-retrotransposons in leaves of two sister hybrids Populus × canadensis (P. deltoides × P. nigra), subjected to moderate or severe water deprivation by mapping Illumina RNA-Seq reads onto a set of 958 unique full-length retrotransposons of P. trichocarpa. Detectable levels of transcription were ascertained for 140 retrotransposons in 1 hybrid and 182 in the other. The two hybrids showed different retrotransposon expression levels, and these differences reduced at increasing drought levels. The number of expressed Gypsy elements in control and water-deprived plants was higher than those of Copia, as were their expression levels. The two hybrids showed different retrotransposon expression patterns following water deprivation. Such variations between hybrids were related to differential expression of a few genes involved in chromatin methylation and remodeling. Overall, our data indicate that even in genetically close individuals, large differences can occur in retrotransposon expression, with possible consequences for genome differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) form the mobile component of the genome, as they can change their location in chromosomes (transposition) through mechanisms operated by enzymes encoded by the transposable element itself. TEs are distinguished between class I retrotransposons (REs) and class II/DNA transposons, depending on their transpositional mechanism. Class I elements transpose through a replicative mechanism that involves an RNA intermediate (“copy and paste”). REs can be autonomous in transposition because of the presence of open reading frames (ORFs) that encode for transposition-related enzymes. Conversely, non-autonomous elements do not possess these ORFs; nevertheless, they can use enzymes encoded by other elements to transpose (Wicker et al. 2007). The replicative mechanism of REs has allowed these elements, often accounting for hundreds of thousands of copies to become the largest portion of genomes during eukaryote evolution (SanMiguel et al. 1998; Vicient et al. 1999). A striking example of RE amplification, which doubled genome size (without polyploidization), has been reported in Oryza australiensis (Piegu et al. 2006).
Based on the presence of long terminal repeats (LTRs) at their ends, retrotransposons can be subdivided into LTR- and non-LTR-REs. Promoter elements, polyadenylation signals, and enhancers are found in the LTRs and regulate RE transcription (Bennetzen 2000). The coding portion of LTR-REs is composed of the gag and the pol domains. The former encodes virus-like particles (VLPs), while the latter encodes enzymes necessary for processing RE transcripts, producing a double-stranded DNA and integrating such DNA into the host genome. LTR-REs have been further divided into two superfamilies, Gypsy and Copia (Voytas et al. 1992; Suoniemi et al. 1998), depending on the order of genes within the pol domain. In the last decade, LTR-REs lacking internal coding domains were described and defined as terminal-repeat retrotransposons in miniatures (TRIMs; Witte et al. 2001) or large retrotransposons derivatives (LARDs; Kalendar et al. 2004). TRIMs and LARDs can be identified only when the complete genome sequences, or at least long DNA sequences, are available. Their species-specific sequence and the absence of coding regions can explain their relative rarity in the literature. However, when surveying complete genomes and using structural features as diagnostics (for example, the occurrence of LTRs, a primer binding site [PBS], and a poly-purine tract [PPT]), they have been shown to form a sizeable component in the TE fraction of the genome (Buti et al. 2011; Natali et al. 2013; Barghini et al. 2014, 2015).
Genome sequences of grasses showed that retrotransposition has determined extensive variations in genome structure, even within one and the same species (Brunner et al. 2005; Scherrer et al. 2005). It was suggested that these structural variations are involved in gene expression regulation, consequently affecting the phenotype, and are subjected to selection. In this sense, the old definition of repeated sequences as “junk” is considered obsolete. For example, allelic cis-regulatory non-genic variation might have a role in heterosis, defined as the superior performance of hybrids in comparison to their parents (Brunner et al. 2005; Stupar and Springer 2006; Morgante et al. 2007; Buti et al. 2013).
The transcription of retrotransposons has been reported in a number of plant species, especially after exposure to various stresses (Grandbastien 2015). A survey of EST libraries of grass species indicates that most LTR-REs are poorly constitutively transcribed (Meyers et al. 2001; Vicient et al. 2001; Vicient and Schulman 2002; Ishiguro et al. 2014). Most of the studies relating to LTR-RE expression refer to specific elements (Vicient et al. 2001; Rico-Cabanas and Martinez-Izquierdo 2007; Ramallo et al. 2008; Buti et al. 2009; Kawakami et al. 2011) and/or are limited to the discovery of LTR-RE sequences in transcriptomes (see for example Parchman et al. 2010; Lu et al. 2013) and only a few comprehensive studies are specifically devoted to this class of sequences (Meyers et al. 2001; Marcon et al. 2015; Jiang et al. 2016).
In a few cases, LTR-RE transcription has determined new insertions in the genome. For example, Tnt1 and Tto1 in Nicotiana and Tos17 in rice are not transcribed in standard culture conditions and are induced to transpose by tissue culture (Hirochika 1993; Hirochika et al. 1996; Grandbastien 1998). More recently, retrotransposition was reported for a Copia element of sunflower for which RNA expression and subsequent insertion in the genome was shown (Vukich et al. 2009).
It has been shown that barley Copia REs produce three distinct classes of transcripts (Chang et al. 2013), of which two are capped and polyadenylated and can be translated or spliced (to produce a large amount of GAG-encoding transcripts); the third is not capped or polyadenylated, and is destined for packaging and ultimate reverse transcription. Conversely, Gypsy elements of Drosophila destined for reverse transcription have been shown to be capped and polyadenylated or not, indifferently (Meignin et al. 2003).
Presumably, the activity of LTR-REs is limited by the host genome because of their potential mutagenic action. The first mechanism of control of mobile elements relies on chromatin structural alterations, since heterochromatin comprises “silent” DNA. Mechanisms underlying chromatin packing in plants act through methylation of histones and cytosine residues in CG and CHG combinations (Dieguez et al. 1998). The importance of epigenetic control of TEs is emphasized by the role of RNA silencing, which determines chromatin specific methylation and RNA degradation (Slotkin and Martienssen 2007; Lisch 2009; Ito 2013). For example, a silencing pathway driven by anti-sense small RNAs is responsible for REs silencing in a Drosophila germline (Vagin et al. 2006).
Retrotransposon dynamics have been mainly investigated in grasses and other monocotyledons. Dicotyledons have in general received less attention, despite their great economic importance. Recently, we performed a survey of LTR-REs in the genome of Populus trichocarpa (Natali et al. 2015). Based on the identification of conserved structural features, building multiple alignments and similarity searches, a number of putative full-length LTR-REs were collected. LTR-RE fragments were by far more abundant in the genome than full-length elements, suggesting that, during poplar evolution, amplification of these elements was followed by DNA depletion. Retrotransposition occurred with increasing frequency following the separation of Populus sections, with different waves of retrotransposition activity between Ty3-Gypsy and Ty1-Copia elements.
Massively parallel sequencing procedures provide unprecedented levels of sequencing coverage in a short time and at relatively low cost, allowing whole genome expression analyses. We have applied such techniques to study the transcription of the entire set of poplar full-length LTR-REs in two sister hybrids (i.e., obtained from the same parental trees) of Populus × canadensis (P. deltoides × P. nigra) in control conditions and in plants subjected to water deprivation treatment. As a reference, we used the available set of full-length LTR-REs of P. trichocarpa (Natali et al. 2015), which is phylogenetically closely related to both P. deltoides and P. nigra, having diverged from them only 8–13 million years ago (Sterck et al. 2005).
Materials and methods
Plant materials
The materials used in this work were the same as those described in Cossu et al. (2014), where gene expression in water-deprived poplar plants was evaluated. Rooted cuttings of two sister hybrids (DxN661200585 and DxN661200589, hereafter, called hybrids 85 and 89, respectively) of Populus × canadensis (P. deltoides × P. nigra), produced from the very same parents at INRA, Orleans (France) in 1998, were cultivated in 20 × 20 cm2 pots in the greenhouse, under natural daylight conditions (750 μm m−2 s−1, maximal photon flux density), with air temperature maintained at 17–29 °C and relative humidity from 55 to 90 %.
In late spring 2011, some plants, 50–70 cm in height, were watered normally and others were subjected to water deprivation by suspending watering for 8 or 13 days. As transcriptome responses to drought are shaped by the time of the day in a diverse range of plant species, including Populus (Wilkins et al. 2009; Hamanishi and Campbell 2011), all materials were sampled at the same time of day (11.00 a.m.). One fully expanded leaf per plant, 6–8 internodes from the apex, was collected per plant. Each leaf was then divided into two portions; one portion was used for RNA isolation, and the other to measure tissue hydration by determining the relative water content [RWC = 100 (FW − DW) / (TW − DW)], where FW is the fresh weight, DW the dry weight, and TW the turgid weight. The experimental design was as follows: 2 plants (biological replicates) × 3 treatments (control [C] and moderate [D1] and severe [D2] drought treatments) × 2 hybrids.
DNA and RNA isolation and preparation of Illumina libraries
Genomic DNA was extracted from leaves (0.5 g fresh weight) of single control plants of hybrid 85 and hybrid 89 as described by Doyle and Doyle (1989). Genomic libraries were prepared from 5 μg of genomic DNA using the Illumina PE DNA Sample Prep kit according to the manufacturer’s instructions. After spin column extraction and quantification, libraries were loaded on Cluster Station to create CSMA (clonal single molecular array) and sequenced at ultra-high throughput on the Illumina HiSeq2000 to produce 51-nt reads. Low quality bases, empty reads, and adapter sequences were removed by using CLC-BIO Genomic Workbench, version 7.0.3 (CLC-BIO). All collected sequences were 50 nt long.
Total RNA was isolated from leaves (of single plants) with different RWC, according to the method described by Logemann et al. (1987) followed by DNAse I (Roche) treatments according to the manufacturer’s instructions to completely remove genomic DNA contamination. RNA was then purified by phenol/chloroform extraction and precipitated following standard procedures.
RNA-Seq libraries were generated using the TruSeq RNA-Seq Sample Prep kit according to the manufacturer’s protocol (Illumina Inc., San Diego, CA). Poly-A RNA was isolated from total RNA and chemically fragmented. First and second strand complementary DNA (cDNA) syntheses were followed by end repair and adenosines were added to the 3′ ends. Adapters were ligated to the cDNA, and 200 ± 25 bp fragments were gel purified and enriched by PCR. The library was quantified using a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA) and run on an Illumina HiSeq2000 (Illumina Inc.) using version 3 reagents. Single read 50 nt sequences were collected. Low quality bases, adapter sequences, and empty reads were removed by using CLC-BIO Genomic Workbench, version 7.0.3 (CLC-BIO).
Alignment of Illumina reads to the P. trichocarpa full-length LTR-REs and analysis of RE abundance
The occurrence and abundance of LTR-REs in the genome were analyzed by aligning two random subsamples of Populus × canadensis DNA reads of hybrids 85 and 89 (generated using an in-house script, each subsample made by the same number of reads, corresponding to two genome-equivalents) to P. trichocarpa full-length LTR-REs (Natali et al. 2015). Alignment to the complete dataset was carried out using CLC-BIO Genomic Workbench, version 7.0.3, with the following parameters: mismatch cost 1, deletion cost 1, insertion cost 1, similarity 0.9, and length fraction 0.9. Alignments were also performed on a set of 33 well-conserved unique cDNAs of P. trichocarpa (Cossu et al. 2012) as a control.
Since CLC-BIO Genomic Workbench 7.0.3 distributes multireads (i.e., those reads that match multiple distinct sequences) randomly, if all sequences of a lineage are taken together, the total number of mapped reads (with respect to total genomic reads) reveals the effective redundancy of that lineage. By contrast, the number of mapped reads to a single sequence cannot indicate its abundance. For this reason, in other analyses, alignments were carried out to the same full-length LTR-REs, one by one, using alignment via Burrows-Wheeler transformation (BWA) version 0.7.5a-r405 (Li and Durbin 2009) with the following parameters: alignment (bwa aln), seed (-l 12), maximum edit distance (-n 4), maximum number of gap opens (-o 3), maximum number of gap extensions (-e 3), mismatch penalty (-M 2), gap open penalty (-O 6), and gap extension penalty (-E 3). The resulting single-end mappings were resolved via the “samse” module of BWA, and the output was converted into a “bam” file using SAMtools version 0.1.19 (Li et al. 2009). SAMtools was used to calculate the number of mapped reads for each alignment using the following parameters: view, counting only (−c), and filtering alignments (−F 4).
Analysis of expression of LTR-REs
CLC-BIO Genomic Workbench 7.0.3 was used to generate cDNA sequence alignments to the set of 958 P. trichocarpa full-length LTR-REs. The parameters used for alignments were the following: mismatch cost 1, deletion cost 1, insertion cost 1, similarity 0.7, length fraction 0.7. The software assigns randomly non-uniquely matching reads (i.e., reads that align at more than one position with an equally good score) to different sequences.
We considered all LTR-REs mapped by at least one read per million of reads in at least one sample as expressed (Lu et al. 2013). The expression level of each sequence was calculated and expressed as RPKM, as described in Mortazavi et al. (2008).
Expression profiles were evaluated considering RPKM values in C, D1, and D2 plants using Baggerly’s test (Baggerly et al. 2003). Weighted proportion fold changes among severely droughted plants, moderately droughted plants, and control plants were treated as follows: when values were higher in droughted than in control plants (or in D2 than in D1 plants), they were reported as positive; when they were higher in control than in droughted plants (or in D1 than in D2 plants) they were reported as negative, thus leading to a “+” value in the case of above-control average expression levels and a “−” value in the case of below control average expression levels.
The weighted proportion fold changes between treatments were considered as significant when the weight of a sample was at least twofold higher or lower than another, with a false discovery rate (FDR) corrected p value ≤0.05, according to Baggerly’s test. Such conservative parameters allowed us to establish significant differences between samples and between treatments, in spite of the low number of non-specific matches assigned randomly by CLC-BIO Genomic Workbench 7.0.3 to different matching sequences and of the small sample size (two replicates) used. Gene expression profiles were subdivided into nine groups: those remaining constant; those increasing their expression in D1 or in D2, or in both treatments; those reducing their expression in D1 or D2, or in both treatments; those increasing their expression in D1 and reducing in D2; and vice versa.
Isolation and analysis of consensus Populus × canadensis LTRs
Consensus 5′-LTR sequences of Populus × canadensis hybrids 85 and 89 were obtained as follows. First, 5′-LTRs were extracted from the P. trichocarpa full-length LTR-REs using an internally developed perl script. Then, consensus LTR sequences were obtained by mapping (using CLC-BIO Genomic Workbench 7.0.3) Illumina DNA reads of the two hybrids to P. trichocarpa LTRs, with the following parameters: similarity 0.7, length fraction 0.7, insertion cost 1, deletion cost 1, and mismatch cost 1.
Consensus 5′-LTR sequences of both hybrids were subjected to motif search by CLC-BIO Genomic Workbench 7.0.3 using a list of putative drought responsive motifs (Supplementary Material 1) selected from the motif list downloaded from the PLACE website (http://www.dna.affrc.go.jp/PLACE/index.html; Higo et al. 1999).
Analysis of gene expression
The P. trichocarpa unigene model database version 9.1, available at the Phytozome site (http://www.phytozome.net/poplar) (Tuskan et al. 2006), was annotated using the web tool BLAST2GO (http://www.blast2go.com/b2ghome). Eighty-two unigenes related to chromatin methylation and remodeling were selected (Supplementary Material 2). CLC-BIO Genomic Workbench 7.0.3 was used to generate cDNA sequence alignments to these genes using the same parameters as above, and RPKM was calculated for each gene.
Those genes mapped by at least one read per million of reads in at least one sample were considered to be expressed. The weighted proportion fold changes between RPKM in control leaves of the two hybrids and in leaves of moderately or severely droughted plants and control plants were considered as different when the RPKM was at least twofold higher or lower in one treatment than in the other, with an FDR corrected p value ≤0.05, according to Baggerly’s test (Baggerly et al. 2003).
Reverse transcription polymerase chain reaction (RT-PCR) experiments were carried out on specific genes, selected according to RNA-Seq data. First-strand cDNA synthesis was performed with 3 μg of purified total RNA using MMLV reverse transcriptase RNase H− (Solis Biodyne, Tartu, Estonia), according to the manufacturer’s instructions. Forward- and reverse-specific primers were designed for four genes involved in chromatin remodeling (Potri.007G026700.1, Potri.013G006000.1, Potri.018G138000.1, Potri.T046100.1) and one gene with constant expression level (Potri.004G152900.1) (Supplementary Material 3). Equal template amounts used in these experiments were verified by RT-PCR performed with this last gene by stopping cycle amplification during the exponential phase of PCR and checking the amounts of products by separating them in 2 % agarose and GelRed™ (Biotium, Hayward, CA) staining.
The PCR amplifications were carried out in a 15 μl volume by using 4 μl of 1:4 dilution of RT reaction as template and a HOT FIREPol DNA Polymerase (Solis Biodyne) in non-saturating conditions. PCR involved a 95 °C step hold for 15 min, followed by 33 cycles (27 for the reference gene) at 95 °C for 30 s, 62 °C (58 °C for the reference gene) for 30 s, and 72 °C for 10 s (30 s for the reference gene). The PCR products were separated in 2 % agarose and GelRed stained. The RT-PCR experiments were repeated three times.
Results
LTR-retrotransposons in two Populus × canadensis hybrids
Two samples of 23,121,470 DNA reads of hybrids 85 and 89, each covering two genome equivalents (Table 1), were aligned to the P. trichocarpa set of full-length LTR-REs (Natali et al. 2015). All P. trichocarpa LTR-REs had DNA reads that mapped to them. The percentages of matched nucleotides resulting were similar in the two hybrids, i.e., 14.76 vs. 13.05 % (Table 1). As a control, the same read packages were aligned to a set of 33 unique, well-conserved genes of P. trichocarpa, and the percentage of matched nucleotides were also similar, i.e., 0.008 vs. 0.007 % (data not shown). Mapping Illumina DNA reads of P. trichocarpa (Slavov et al. 2012) showed 13.38 % nucleotides matching the set of LTR-REs and 0.008 % matching the 33 genes (data not shown).
Figure 1 shows the correlation between the number of reads matching LTR-REs in the hybrids 85 and 89. Only 6/546 Gypsy REs and no Copia RE showed a ratio higher than 2.0 between the two hybrids.

Pairwise comparison of number of mapped reads on each of the 958 P. trichocarpa full-length LTR-REs between Illumina read sets of hybrids 85 and 89. The same number of reads was used for both hybrids
Expression level of LTR-REs in leaves of Populus × canadensis hybrids
We generated 76,635,449 Illumina cDNA reads, encompassing 3.9 Gb of sequence data (Table 2). Each condition (control, moderate and severe drought) was represented by at least 3.96 million reads, a tag density sufficient for quantitative analysis of gene expression (Rai et al. 2013).
We observed that a small portion (0.09–0.16 %) of each RNA-seq read set matched the retrotransposons included in the dataset (Table 2). Separating Gypsy and Copia REs, the mean percentages were 0.112 % (0.072–0.153 %) and 0.010 % (0.005–0.022 %), respectively (data not shown). This indicated that Gypsy elements were far more highly expressed than Copia elements.
Expression analysis of LTR-REs in leaves of normally watered plants
We considered all those LTR-REs mapped by at least one read per million of reads in at least one sample as transcribed (Lu et al. 2013). The correspondence between RNA-Seq data and expression analyses by RT-PCR had already been established for these experiments (Cossu et al. 2014).
It is to be noted that CLC-BIO assigns randomly non-specifically matched reads, i.e., reads that align at more than one position with an equally good score. However, in our experiments, the number of non-specific reads was low. For example, the average percentage of non-specific matches in control leaves was 7.15 ± 1.41 for LTR-REs. Similar percentages were observed for all treatments (data not shown).
Fifty-five LTR-REs out of 958 (5.7 %) were expressed in both hybrids; these belonged to the Gypsy (35 elements) and Copia (16 elements) superfamilies and to a group of unknown elements (4 elements). Forty-five LTR-REs (4.7 %) were expressed in control leaves of one or other of the hybrids (35 in hybrid 85 and 10 in hybrid 89). Finally, 858 LTR-REs (89.6 %) were transcriptionally inactive in control leaves of both hybrids.
The distribution of active LTR-REs in relation to their expression in normally watered plants is reported in Fig. 2, keeping the two hybrids separate. Gypsy REs were by far the most active, considering the 30 most expressed LTR-REs in both hybrids; twenty-one REs belonged to the Gypsy superfamily and six to the Copia; three of the most active elements were unclassified (data not shown). The majority of LTR-REs transcribed in at least one hybrid (83 out of 100) were expressed at comparable levels in both hybrids (42) or were more expressed in hybrid 85 than in hybrid 89 (41) (Fig. 3). Only 12 LTR-REs were more highly expressed in hybrid 89 than in hybrid 85 and showed the highest mean expression levels (expressed as reads per kilobase per million reads mapped [RPKM]) and the largest RPKM variability. No LTR-RE expression specific to hybrid 89 was found and only five LTR-REs were expressed specifically in hybrid 85 (Fig. 3).

Distribution of 206 LTR-REs (transcribed in hybrids 85 and/or 89) in leaves of control plants of hybrids 85 and 89, according to their expression value (RPKM)

Box and whisker plots of LTR-RE expression differences (calculated as RPKM) between control leaves of hybrids 85 (H85) and 89 (H89). Transcribed retrotransposons of the two hybrids were subdivided into five classes: those more highly expressed in hybrid 85 than in hybrid 89 or vice versa; those equally expressed in both hybrids; and those expressed only in hybrid 85 or on hybrid 89. LTR-REs were considered more highly expressed in one hybrid than in the other when the RPKM ratio was higher than 2. The boxes represent the 25 to 75 %; whiskers represent the range of RPKM; and lines in the box represent the mean values of the distribution. For each class, the number of LTR-REs is reported
Since in certain cases the occurrence of LTR-RE sequences in cDNA libraries can be related to genomic DNA contamination, we analyzed the RPKM value of each retroelement in relation to its abundance in the two hybrids (Fig. 4). The most abundant LTR-REs were not (or were slightly) expressed, and correspondingly, the most expressed LTR-REs were poorly represented in the genomes of both hybrids. This indicated that contamination by genomic DNA in the cDNA libraries could be largely ruled out.

Relationship between RPKM expression values (in control plants) of each element of the set of 958 P. trichocarpa LTR-REs and the respective number of mapped DNA reads in hybrids 85 and 89
Expression analysis of LTR-REs in leaves of plants subjected to water deprivation
One hundred and forty and 182 LTR-REs were expressed in at least one culture condition in hybrids 85 and 89, respectively. A number of active LTR-REs were expressed in leaves of control and moderately and severely droughted plants in both hybrids (Table 3). In addition to this group, the other major group was represented by LTR-REs expressed only in severely droughted plants (D2), especially in hybrid 89 (86 vs. 36, i.e., 47.3 vs. 25.7 %).
In all cases, the most abundant LTR-REs were not (or only slightly) expressed (data not shown), indicating that contamination by genomic DNA in the cDNA libraries was not present, as for control plants.
Only 63 LTR-REs were expressed in leaves of droughted plants of both hybrids (Table 3), i.e., the expression of all other LTR-REs were hybrid-specific. Forty-seven out of 86 elements expressed in D2 in hybrid 89 were not expressed in hybrid 85, and 8 out of 36 elements expressed in D2 in hybrid 85 were not expressed in hybrid 89, indicating large differences in LTR-RE transcription between hybrids in droughted plants.
As the parents were heterozygous, the two poplar interspecific hybrids were expected to be genetically different. Modulation of LTR-RE transcription in response to water deprivation was different between the two hybrids (Table 4). The total number of expressed LTR-REs was similar throughout the experiment in hybrid 85 and progressively increased in hybrid 89. The number of transcribed Copia REs slightly reduced in hybrid 85 and, by contrast, progressively increased in hybrid 89. The number of transcribed Gypsy REs increased during the experiment, especially in hybrid 89. Unknown elements were few, and their expression was apparently constant during the experiment (Table 4).
Concerning the expression level in hybrid 85, the mean RPKM value increased after moderate drought and showed a marked reduction in severely droughted plants for Copia and Gypsy elements; similar patterns of expression was observed in hybrid 89 (Table 4). Unknown REs showed higher expression levels than Copia and Gypsy elements in control plants, their expression reduced in moderately droughted plants of hybrid 85 and maintained their expression levels in hybrid 89 (Table 4).
The expression profiles of transcriptionally active LTR-REs in the two hybrids were subdivided into nine clusters (Fig. 5) based on their expression modulation pattern. The majority of expressed LTR-REs were transcribed at constant rates in control and droughted plants (cluster e, 66.4 % in hybrid 85 and 53.3 % in hybrid 89). It is worth noting that LTR-REs activated by water deprivation (clusters a and b) were far more numerous in hybrid 89 than in hybrid 85 (34.6 vs. 2.9 %, respectively); conversely, LTR-REs repressed by water deprivation (clusters h and i) were found only in hybrid 85.

Schematic representation of the expression patterns of nine LTR-REs (indicated by letters (a)–(i)) observed in leaves of Populus × canadensis hybrids (85 and 89) comparing control conditions (C) to moderate (D1) and to severe drought (D2). For each pattern, the number of LTR-REs is reported in hybrids 85 and 89. Only expressed LTR-REs were counted. The total number of expressed LTR-REs per hybrid is reported in parentheses
The expression of single elements was evaluated by pairwise comparisons of mean log RPKMs for each retrotransposon in leaves of C, D1, and D2 plants (Fig. 6). The correlation was highly significant in each condition tested, indicating the same retrotransposons were expressed at comparable levels in different genetic backgrounds. Keeping Gypsy and Copia elements separate, the correlation coefficient progressively increased from control plants to D1 through to D2 plants, especially for Gypsy elements. Interestingly, the correlation coefficient for Copia REs was not significant in the controls and at both levels of water deprivation, whereas they were highly significant for Gypsy REs (Fig. 6).

Two-dimensional representations of LTR-RE expression estimated by log RPKM in leaves from plants of two Populus × canadensis hybrids normally watered (control) and moderately (drought 1) and severely droughted (drought 2). For each treatment, the correlation coefficient between hybrids 85 and 89 is reported. Data are reported for all expressed LTR-REs or keeping separated Copia and Gypsy elements
Retroelements, whose expression was induced, repressed, or unchanged (in D1 and/or D2), were analyzed for the occurrence of sequence motifs putatively recognizable by transcription factors activated by drought in their 5′-LTR (Supplementary Material 1). For this analysis, Populus × canadensis consensus 5′-LTRs were obtained by mapping Illumina DNA reads of hybrids 85 and 89 to P. trichocarpa 5′RE-LTRs. We observed that the mean number of drought-related cis-regulatory motifs was similar in LTRs of all expressed REs, whether activated or repressed (Table 5).
Expression analysis of genes involved in chromatin methylation and remodeling
A collection of 82 P. trichocarpa genes involved in chromatin methylation and remodeling (Supplementary Material 2) was prepared to study possible relationships between the activation of these genes and the different response of the two hybrids in terms of RE expression. Of these genes, 73 were expressed, although generally at low level.
In the two hybrids under control conditions, 18 genes (24.7 %, of which 5 with p < 0.05) showed expression levels twofold higher in hybrid 85 than in hybrid 89 and none were twofold more expressed in hybrid 89, indicating a generally stronger activation trend of this group of genes in the hybrid 85 (Fig. 7a). Conversely, many genes were activated in leaves of droughted plants of hybrid 89, while small changes in the expression levels were observed in droughted leaves of hybrid 85 (Fig. 7b).

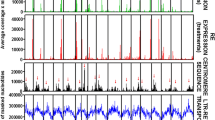
Distribution of RPKM fold changes between hybrids 89 (H89) and 85 (H85) of 82 poplar genes involved in chromatin methylation and remodeling. When values were higher in H89 than in H85, they were reported as positive; when they were higher in H85 than in H89, they were reported as negative. a Distribution of RPKM fold changes between droughted (moderately, D1 or severely, D2) and control (C) plants of the same poplar genes as above in H85 and H89. b When values were higher in droughted than in control plants, they were reported as positive; when they were higher in control than in droughted plants, they were reported as negative. Fold change values significant at p ≤ 0.05 are indicated in red. The lines in the distributions represent the mean values for each fold change
We also specifically studied four genes whose involvement in DNA methylation and activation/repression of REs in Arabidopsis has been reported in the literature, i.e., three decrease in DNA methylation (DDM1, DDM2.1, and DDM2.2) genes and one Morpheus (MOM) gene (Iwasaki 2014) (Fig. 8). In control leaves, DDM1 was more highly expressed in hybrid 89 than in hybrid 85; MOM was more highly expressed in hybrid 85. Concerning DDM1, RT-PCR experiments confirmed that this gene was more highly expressed in hybrid 89 than in hybrid 85 (Fig. 8), while the other three genes did not show appreciable differences in expression (data not shown). Concerning water deprivation treatments, no significant expression variability was observed in both hybrids for the four selected genes, compared to control leaves, except for DDM2 and MOM which were more highly expressed in D1 in hybrid 89 (data not shown).

a Mean and standard deviation of RPKM values of decrease in DNA methylation (DDM) 1.1, 2.1, 2.2 and Morpheus in leaves of normally watered plants of hybrids 85 (H85/3 and H85/4) and 89 (H89/6 and H89/8). The Phytozome code for each gene is reported in parentheses. b RT-PCR analysis of DDM1.1. The CLC protease—proteolytic subunit 6 encoding gene (Potri.004G152900.1)—was used as control as in Cossu et al. (2014). The RPKM value is reported below each sample
Discussion
We used an LTR-RE dataset of P. trichocarpa containing 958 full-length retroelements (Natali et al. 2015) for the evaluation of LTR-RE expression in Populus × canadensis (P. deltoides × P. nigra). We used the LTR-REs of P. trichocarpa because this species can be crossed with both P. deltoides and P. nigra and diverged from them 8–13 million years ago (Sterck et al. 2005). P. trichocarpa LTR-REs could differ in sequence and in abundance from elements of P. deltoides and P. nigra. However, previous data from the literature showed that sequence differences are rare among poplar species, at least for genes (Maestrini et al. 2009; Cossu et al. 2012, 2014).
Mapping Illumina DNA reads to the P. trichocarpa LTR-REs showed nearly the same percentage of matched nucleotides for the two hybrids (27.22 vs. 29.23 %). All 958 LTR-REs were shown to be present in the genomes of the two hybrids, as well as in that of P. trichocarpa. The abundance of each LTR-RE was similar in the two genotypes analyzed in this study. Differences in abundance were observed only for a few elements. It is possible that such differences are, in part, related to LTR-RE copy number variation between homologous chromosomes in the two parents. Alternatively, it is possible that, after interspecific hybridization, a few elements have been subjected to amplification, as observed in newly formed interspecific hybrids of other species (e.g., Feldman and Levy 2012). It is known that interspecific hybridization causes so-called genomic shock (McClintock 1984), leading to activation of transposons (Senerchia et al. 2015).
Generally, the expression of LTR-REs in plant species has been argued, especially surveying EST and cDNA libraries available in public databases (Vicient et al. 2001). In these databases, most elements resulted transcribed at low levels. In poplar also, our data showed low expression levels of LTR-retrotransposons in all analyzed plants. In fact, in the various RNA-Seq samples, the percentages of cDNA nucleotides that mapped to LTR-REs were around 0.1 %, i.e., LTR-RE RNAs represented only a small fraction of the transcriptome.
Low levels of transcription of repeated sequences are often attributed to DNA contamination of RNA samples. In the experiments described here, LTR-RE abundance and transcription were totally uncorrelated. Abundant LTR-REs were not or were only slightly expressed, whereas rare REs were actively transcribed. This suggests that the presence of RE sequences in the cDNA library was not due to DNA contamination. The lack of correlation between LTR-RE abundance and transcription is not surprising because it is known that abundant elements are more easily recognized and subjected to RNA silencing (Meyers et al. 2001; Yamazaki et al. 2001; Lisch 2009).
In our experiments, Copia and Gypsy REs were transcribed at different levels, the least active belonging to the Copia superfamily in both hybrids and in all culture conditions tested in our experiments. It is known that diversified silencing mechanisms occur in plants (Xie et al. 2004). These results indicate that different silencing mechanisms may differentially affect the two RE superfamilies.
Hybrids 85 and 89 showed differences in the expression level of a number of LTR-REs in control plants. In fact, although the correlation between LTR-REs expression in the two hybrids were highly significant, a number of retroelements were differentially transcribed in the two hybrids. In general, more LTR-REs were constitutively transcribed (and at higher rates) in hybrid 85 than in hybrid 89. These differences were accompanied by differences in the expression of a number of genes involved in chromatin methylation and remodeling. It is presumed that differences were related to genetic differences between hybrids, for example, to trans-acting factors more active in one hybrid than in the other. For example, the DDM1 gene was more highly expressed in hybrid 89 than in hybrid 85. It is known that in Arabidopsis, DDM1 allows the methylation status to be maintained, resulting in transposon inactivation (Mirouze and Paszkowski 2011). The lower DDM1 transcript accumulation in hybrid 85 could explain the generally higher LTR-RE expression in this hybrid.
If the different patterns of RE expression in the leaf of the two sister hybrids were to also occur in the vegetative meristem, this would imply that, during evolution, the same hybridization event could determine different results in terms of genome structure of the resulting interspecific hybrid. In fact, if RE expression was followed by retrotranscription and insertion in a new site, copy number variations of those elements would be produced during evolution, contributing to genome structure alteration and consequently to the generation of new species. For example, it is known that Helianthus anomalus, H. deserticola, and H. paradoxus were derived from interspecific hybridization between H. annuus and H. petiolaris, but the LTR-RE portions of their genome are very different (Ungerer et al. 2009).
It remains to be determined whether the occurrence of LTR-RE transcripts in poplar leaves has any biological function. LTR-RE transcripts could be involved in the insertion of new copies in the genome. If the transposition mechanisms (involving three types of transcripts) reported in barley (Chang et al. 2013) were general, the polyadenylated transcripts analyzed in our experiments could lead to retrotransposition only in the presence of other uncapped and not polyadenylated transcripts. Much data obtained in other species suggests that LTR-RE transcripts have acquired specific cell functions during their evolution, especially in the production of small interfering RNAs (siRNAs) involved in transcriptional silencing of REs and, eventually, nearby genes through the RNA-directed DNA methylation pathway (Slotkin and Martienssen 2007; Lisch 2009). In maize, siRNAs, 21 and 22 nt in length, are derived from distinct retrotransposon families and are differentially accumulated between genotypes (Barber et al. 2012). In soybean, another study indicated the 22 nt long siRNAs resulting from different retrotransposon families, as a major component in the silencing of most REs (Zabala et al. 2012).
Concerning retrotransposon transcription in response to drought treatments, it appeared stable for most elements. Many elements were constitutively expressed in the leaves of both control and water-deprived plants. Interestingly, the two hybrids showed a different pattern of expression during water deprivation treatment, i.e., the number of expressed LTR-REs increased in hybrid 89 and remained stable (apart from a small reduction in D1) in hybrid 85. Moreover, many retroelements were differentially transcribed in the two hybrids during drought treatments. Different expression modulation patterns are presumably related to differences in regulatory sequences or silencing mechanisms between hybrids. Another hypothesis is that different RE expression patterns are related to their co-localization with genes involved in drought response.
Differences between the hybrids were also seen in the number of expressed elements during water deprivation between Copia and Gypsy superfamilies. In fact, the number of expressed Copia REs increased in hybrid 89 and decreased in hybrid 85; the number of expressed Gypsy elements increased in D2 in both hybrids.
We showed differences in transcript accumulation of genes involved in chromatin methylation and remodeling between the two hybrids, analogous to those observed for LTR-RE transcription, with a number of genes activated in droughted plants of hybrid 89 only. The activated genes in droughted hybrid 89 plants included DDM2 and MOM. It was hypothesized that activation of these genes constituted a response mechanism to the drought-related activation of LTR-REs because their activity should increase DNA methylation of LTR-REs (Mirouze and Paszkowski 2011). Our data for control leaves indicated that at least one DDM gene (DDM1) is involved in the general maintenance of the chromatin status and its expression level is related to differences in LTR-RE expression between the two hybrids. On the other hand, probably via different molecular pathways, other DDM genes (DDM2) are activated in response to drought to counteract LTR-RE activation. This confirms the existence of different mechanisms of controlling LTR-RE transcription acting over the long term (for some generations) or transiently, as a stress response (Pecinka and Mittelsten Scheid 2012).
Searching for drought-related cis-regulatory motifs in the consensus LTRs of REs showed the occurrence of many ABRE and DRE motifs in the consensus LTRs of both hybrids. It is known that these motifs are crucial for expression of genes activated by dehydration (Yamaguchi-Shinozaki and Shinozaki 2006). However, a similar number of these drought-related cis-regulatory elements were present in LTRs of all expressed elements in both induced and repressed REs. This result suggests that activity of retrotransposons should be related to the presence in LTRs of cis-regulatory motifs other than ABRE and DRE or to differences in condensation/decondensation of chromatin (determined by siRNAs).
In conclusion, our data provides evidence of the low expression in poplar hybrids of a large set of LTR-REs, especially belonging to the Copia superfamily, suggesting that LTR-RE activity is strongly controlled at the transcriptional level. However, the observed small differences in LTR-RE abundance between hybrids and the enhanced transcription of a few elements suggest that LTR-REs contributed and are probably still contributing to the genome differentiation in Populus species. This study also shows that differences in LTR-RE expression occur between genetically close individuals (two sister hybrids) and suggests that they can be related to the activity of a few genes, involved in chromatin remodeling.
Abbreviations
- RE:
-
Retrotransposon
- LTR-RE:
-
Long terminal repeat-retrotransposon
- RPKM:
-
Reads per kilobase per million reads mapped
References
Baggerly KA, Deng L, Morris JS, Aldaz CM (2003) Differential expression in SAGE: accounting for normal between-library variation. Bioinformatics 19:1477–1483
Barber WT, Zhang W, Win H, Varala KK, Dorweiler JE, Hudson ME, Moose SP (2012) Repeat associated small RNAs vary among parents and following hybridization in maize. Proc Natl Acad Sci U S A 109:10444–10449
Barghini E, Natali L, Cossu RM, Giordani T, Pindo M, Cattonaro F, Scalabrin S, Velasco R, Morgante M, Cavallini A (2014) The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol Evol 6:776–791
Barghini E, Natali L, Giordani T, Cossu RM, Scalabrin S, Cattonaro F, Šimková H, Vrána J, Doležel J, Morgante M, Cavallini A (2015) LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Res 22:91–100
Bennetzen JL (2000) Transposable elements contributions to plant gene and genome evolution. Plant Mol Biol 42:251–269
Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A (2005) Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17:343–360
Buti M, Giordani T, Vukich M, Gentzbittel L, Pistelli L, Cattonaro F, Morgante M, Cavallini A, Natali L (2009) HACRE1, a recently inserted copia-like retrotransposon of sunflower (Helianthus annuus L.). Genome 52:904–911
Buti M, Giordani T, Cattonaro F, Cossu RM, Pistelli L, Vukich M, Morgante M, Cavallini A, Natali L (2011) Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor Appl Genet 123:779–791
Buti M, Giordani T, Vukich M, Pugliesi C, Natali L, Cavallini A (2013) Retrotransposon-related genetic distance and hybrid performance in sunflower (Helianthus annuus L.). Euphytica 192:289–303
Chang W, Jääskeläinen M, Li S, Schulman AH (2013) BARE retrotransposons are translated and replicated via distinct RNA pools. PLoS One 8:e72270
Cossu RM, Buti M, Giordani T, Natali L, Cavallini A (2012) A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Genet Genomes 8:61–75
Cossu RM, Giordani T, Cavallini A, Natali L (2014) High-throughput analysis of transcriptome variation during water deficit in a poplar hybrid: a general overview. Tree Genet Genomes 10:53–66
Dieguez MJ, Vaucheret H, Paszkowski J, Mittelsten Scheid O (1998) Cytosine methylation at CG and CNG sites is not a prerequisite for the initiation of transcriptional gene silencing in plants, but it is required for its maintenance. Mol Gen Genet 259:207–215
Doyle JJ, Doyle JL (1989) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Feldman M, Levy AA (2012) Genome evolution due to allopolyploidization in wheat. Genetics 192:763–774
Grandbastien MA (1998) Activation of plant retrotransposons under stress conditions. Trends Plant Sci 3:181–189
Grandbastien MA (2015) LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim Biophys Acta 1849:403–416
Hamanishi ET, Campbell MM (2011) Genome-wide responses to drought in forest trees. Forestry 84:273–283
Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database. Nucleic Acids Res 27:297–300
Hirochika H (1993) Activation of tobacco retrotransposons during tissue culture. EMBO J 12:2521–2528
Hirochika H, Sugimoto K, Otsuki Y, Tsugawa H, Kanda M (1996) Retrotransposons of rice involved in mutations induced by tissue culture. Proc Natl Acad Sci U S A 93:7783–7788
Ishiguro S, Ogasawara K, Fujino K, Sato Y, Kishima Y (2014) Low temperature-responsive changes in the anther transcriptome’s repeat sequences are indicative of stress sensitivity and pollen sterility in rice strains. Plant Physiol 164:671–682
Ito H (2013) Small RNAs and regulation of transposons in plants. Genes Genet Syst 88:3–7
Iwasaki M (2014) Identification of genes preventing transgenerational transmission of stress induced epigenetic states. Proc Natl Acad Sci U S A 111:8547–8552
Jiang S, Cai D, Sun Y, Teng Y (2016) Isolation and characterization of putative functional long terminal repeat retrotransposons in the Pyrus genome. Mob DNA 7:1
Kalendar R, Vicient CM, Peleg O, Anamthawat-Jonsson K, Bolshoy A, Schulman AH (2004) LARD retroelements: conserved, non-autonomous components of barley and related genomes. Genetics 166:1437–1450
Kawakami T, Morgan TJ, Nippert JB, Ochltree TW, Keith R, Dhakal P, Ungerer MC (2011) Natural selection drives clinal life history patterns in the perennial sunflower species, Helianthus maximiliani. Mol Ecol 20:2318–2328
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Lisch D (2009) Epigenetic regulation of transposable elements in plants. Annu Rev Plant Biol 60:43–66
Logemann J, Schell J, Willmitzer L (1987) Improved method for the isolation of RNA from plant tissues. Anal Biochem 163:16–20
Lu X, Chen D, Shu D, Zhang Z, Wang W, Klukas C, Chen LL, Fan Y, Chen M, Zhang C (2013) The differential transcription network between embryo and endosperm in the early developing maize seed. Plant Physiol 162:440–455
Maestrini P, Cavallini A, Rizzo M, Giordani T, Bernardi R, Durante M, Natali L (2009) Isolation and expression analysis of low temperature-induced genes in white poplar (Populus alba). J Plant Physiol 166:1544–1556
Marcon HS, Domingues DS, Costa Silva J, Junqueira Borges R, Filippi Matioli F, de Mattos Fontes MR, Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
Meignin C, Bailly JL, Arnaud F, Dastugue B, Vaury C (2003) The 5′ untranslated region and gag product of Idefix, a long terminal repeat retrotransposon from Drosophila melanogaster, act together to initiate a switch between translated and untranslated states of the genomic mRNA. Mol Cell Biol 23:8246–8254
Meyers BC, Tingey SV, Morgante M (2001) Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res 11:1660–1676
Mirouze M, Paszkowski J (2011) ) Epigenetic contribution to stress adaptation in plants. Curr Opin Plant Biol 14:267–274
Morgante M, De Paoli E, Radovic S (2007) Transposable elements and the plant pan-genomes. Curr Opin Plant Biol 10:149–155
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods 5:621–628
Natali L, Cossu RM, Barghini E, Giordani T, Buti M, Mascagni F, Morgante M, Gill N, Kane NC, Rieseberg L, Cavallini A (2013) The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genomics 14:686
Natali L, Cossu RM, Mascagni F, Giordani T, Cavallini A (2015) A survey of Gypsy and Copia LTR-retrotransposon superfamilies and lineages and their distinct dynamics in the Populus trichocarpa (L.) genome. Tree Genet Genomes 11:107
Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA (2010) Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11:180
Pecinka A, Mittelsten Scheid O (2012) Stress-induced chromatin changes: a critical view on their heritability. Plant Cell Physiol 53:801–808
Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, Collura K, Brar DS, Jackson S, Wing RA, Panaud O (2006) Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res 16:1262–1269
Rai HD, Mock KE, Richardson BA, Cronn RC, Hayden KJ, Wright JW, Knaus BJ, Wolf PG (2013) Transcriptome characterization and detection of gene expression differences in aspen (Populus tremuloides). Tree Genet Genomes 9:1031–1041
Ramallo E, Kalendar R, Schulman AH, Martínez-Izquierdo JA (2008) Reme1, a Copia retrotransposon in melon, is transcriptionally induced by UV light. Plant Mol Biol 66:137–150
Rico-Cabanas L, Martinez-Izquierdo JA (2007) CIRE1, a novel transcriptionally active Ty1-copia retrotransposon from Citrus sinensis. Mol Gen Genomics 277:365–377
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL (1998) The paleontology of intergene retrotransposons of maize. Nat Genet 20:43–45
Scherrer B, Isidore E, Klein P, Kim JS, Bellec A, Chalhoub B, Keller B, Feuillet C (2005) Large intraspecific haplotype variability at the Rph7 locus results from rapid and recent divergence in the barley genome. Plant Cell 17:361–374
Senerchia N, Felber F, Parisod C (2015) Genome reorganization in F1 hybrids uncovers the role of retrotransposons in reproductive isolation. Proc R Soc B Biol Sci 282. doi:10.1098/rspb.2014.2874
Slavov GT, DiFazio SP, Martin J, Schackwitz W, Muchero W, Rodgers-Melnick E, Lipphardt MF, et al. (2012) Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol 196:713–725
Slotkin RK, Martienssen R (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8:272–285
Sterck L, Rombauts S, Jansson S, Sterky F, Rouzé P, Van de Peer Y (2005) EST data suggest that poplar is an ancient polyploid. New Phytol 167:165–170
Stupar RM, Springer NM (2006) Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173:2199–2210
Suoniemi A, Tanskanen J, Schulman AH (1998) Gypsy-like retrotransposons are widespread in the plant kingdom. Plant J 13:699–705
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, et al. (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Ungerer MC, Strakosh SC, Stimpson KM (2009) Proliferation of Ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol 7:40
Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD (2006) A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313:320–324
Vicient CM, Schulman AH (2002) Copia-like retrotransposons in the rice genome: few and assorted. Genome Lett 1:35–47
Vicient CM, Suoniemi A, Anamthawat-Jonsson K, Tanskanen J, Beharav A, Nevo E, Schulman AH (1999) Retrotransposon BARE-1 and its role in genome evolution in the genus Hordeum. Plant Cell 11:1769–1784
Vicient CM, Jaaskelainen MJ, Kalendar R, Schulman AH (2001) Active retrotransposons are a common feature of grass genomes. Plant Physiol 125:1283–1292
Voytas DF, Cummings MP, Konieczny A, Ausubel FM, Rodermel SR (1992) Copia-like retrotransposons are ubiquitous among plants. Proc Natl Acad Sci U S A 89:7124–7128
Vukich M, Giordani T, Natali L, Cavallini A (2009) Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol 9:150
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Wilkins O, Waldron L, Nahal H, Provart NJ, Campbell MM (2009) Genotype and time of day shape the Populus drought response. Plant J 60:703–715
Witte CP, Le QH, Bureau T, Kumar A (2001) Terminal-repeat retrotransposons in miniature (TRIM) are involved in restructuring plant genomes. Proc Natl Acad Sci U S A 98:13778–13783
Xie Z, Johansen LK, Gustafson AM, Kasschaul KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC (2004) Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2:642–652
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yamazaki M, Tsugawa H, Miyao A, Yano M, Wu J, Yamamoto S, Matsumoto T, Sasaki T, Hirochika H (2001) The rice retrotransposon Tos17 prefers low-copy-number sequences as integration targets. Mol Gen Genomics 265:336–344
Zabala G, Campos E, Varala KK, Bloomfield S, Jones SI, Win H, Tuteja JH, Calla B, Clough SJ, Hudson M, Vodkin LO (2012) Divergent patterns of endogenous small RNA populations from seed and vegetative tissues of Glycine max. BMC Plant Biol 12:177
Acknowledgments
This research work was supported by PRIN-MIUR, Italy, project “Verso la delucidazione delle basi molecolari dell’eterosi nelle piante coltivate: variazione cis-regolatoria ed espressione genica in ibridi di pioppo” and, in part, by European Community’s Seventh Framework Program (FP7/2007-2013), under grant agreement number 211917 (ENERGYPOPLAR).
Thanks are due to Dr. Catherine Bastien (INRA-UAGPF, Orleans, France) for providing the hybrid plants, to Pierfrancesco Miscali (DiSAAA-a, Pisa, Italy) for technical collaboration on data handling and treatment, and to Dr. John A. Walsh (Warwick University, UK) for critical reading of the manuscript.
Data archiving statement
All LTR-RE sequences are available in the Department of Agriculture, Food, and Environment of the University of Pisa repository website (http://www.agr.unipi.it/ricerca/plant-genetics-and-genomics-lab/sequencerepository.html).
All genomic DNA and cDNA raw Illumina sequences used in this work are available at the NCBI Sequence Read Archive under the accession numbers SRP078030 (submission: Hybrid Populus deltoides × Populus nigra Raw sequence reads) and SRP024267 (submission: Populus × canadensis RNAseq).
The global analysis of LTR-RE and gene expression is available in an excel file in the Department of Agriculture, Food, and Environment of University of Pisa repository website (http://www.agr.unipi.it/Sequence-Repository.358.0.html), in which each LTR-RE and each gene were represented by its absolute expression level in control, moderately dehydrated and severely dehydrated leaves.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Brunner
Tommaso Giordani and Rosa Maria Cossu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Giordani, T., Cossu, R.M., Mascagni, F. et al. Genome-wide analysis of LTR-retrotransposon expression in leaves of Populus × canadensis water-deprived plants. Tree Genetics & Genomes 12, 75 (2016). https://doi.org/10.1007/s11295-016-1036-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-016-1036-5