
Abstract
Heterosis is a main force leading the development of the hybrid seed industry in sunflower. The purpose of this study is to evaluate if heterosis effects for morphological traits among sunflower hybrids can be related to differences in the repetitive component of the genome of parental lines. The assumption is that, at least for certain traits, heterosis results from mutations in the cis-regulatory elements of genes, largely related to retrotransposon insertions and/or removals. Our experimental approach consists of a correlation study between hybrid performance and retrotransposon-related genetic distances between inbreds. Six sunflower inbred lines of different origin were crossed in a half diallel fashion; comparing parental lines and hybrids, mid parent heterosis of F1 hybrids was evaluated for six traits. We estimated the parental genetic distances between the six inbreds on data gathered by the inter-retrotransposon-amplified-polymorphism (IRAP) protocol. Different retrotransposons previously isolated in sunflower were targeted by 11 primer pairs designed on conserved LTR domains. As a control, genetic distances were also calculated using 86 genic SNPs. We analysed the correlation between the mid-parent heterosis for each of the six traits analysed and the genetic distance (calculated on data obtained by SNP or IRAP analyses) between the parental lines. Differences between parents showed to be largely related to variations in the retrotransposon component of the genome. Retrotransposon-related genetic distance between parents resulted to be larger than that related to genic SNPs, and significantly correlated to seed yield and, at a lesser extent, to plant height and stem diameter in hybrids. The hypothesis that variations in the repetitive component of the genome, especially LTR-retrotransposons, affect the displaying of heterosis is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heterosis, or hybrid vigour, refers to the phenomenon in which the hybrid F1 offspring exhibit phenotypic characteristics that are superior to the mean of the two parents (mid-parent heterosis), or the best of the two parents (best-parent heterosis) (Springer and Stupar 2007). Heterosis has been exploited in the breeding and production of many crop and animal species (Janick 1998; Melchinger and Gumber 1998). For example, in maize, it is estimated that the use of hybrids and heterosis increases yields around 15 % per annum (Duvick 1999) and a number of heterotic groups have been proposed for this species (Dubreuil et al. 1996).
Heterosis has been the subject of intense research and speculation for well over a century; however, the basic mechanisms that cause or contribute to heterosis remain unclear. The two main genetic hypotheses advanced to explain this phenomenon are dominance (Davenport 1908; Jones 1917) and overdominance (East 1908; Shull 1908). Also epistasis can have a role (see for example Melchinger et al. 2007). According to the dominance model of heterosis, each of the inbred lines contains slightly deleterious alleles that reduce their fitness. The hybrid will benefit from the complementation of these deleterious alleles and will display a superior phenotype. The overdominance model suggests that the heterozygous combination of alleles at a given locus is phenotypically superior to either of the homozygous combinations for that locus; contributions of overdominant loci thereby result in a superior hybrid.
The cultivated sunflower (Helianthus annuus L.) is a major oil crop species and, along with soybean and rapeseed account for approximately 78 % of the world vegetable oil (Ahmad et al. 2005). Heterosis in sunflower has been exploited especially after the discovery of cytoplasmic male sterility and efficient male fertility restoration system during 1970s. Since many years, hybrid cultivars account for the majority of worldwide sunflower production (see Miller 1992).
Heterosis in sunflower has been largely investigated (see for example Hongtrakul et al. 1997; Cheres et al. 2000; Ahmad et al. 2005; Kaya 2005). Some authors (Hongtrakul et al. 1997; Cheres and Knapp 1998; Cheres et al. 2000) proposed a series of heterotic groups for public inbred lines developed in the US. The heterotic groups found in sunflower do not seem to be as rigid as those found in corn. Within-group genetic distances seem to be greater in sunflower than corn whereas the opposite is true for between-group genetic distances (Cheres and Knapp 1998; Cheres et al. 2000).
Genetic distance between inbreds has been proposed as a parameter for predicting hybrid performance. Positive trends between inbreds genetic distance and hybrid seed yield were reported in many species (Godshalk et al. 1990; Melchinger et al. 1990; Martin et al. 1995; Zhang et al. 1995; Diers et al. 1996; Cheres et al. 2000), although hybrid seed yields were not accurately predicted by genetic distance in any of them. In sunflower, the correlation between genetic distance and general combining ability for seed yield was weak (0.24) in the only predictive study reported thus far (Tersac et al. 1994).
The significance of correlation between inbreds genetic distance and hybrid seed yield depends on DNA markers used in the analysis. Molecular markers based on DNA analysis are independent of environmental factors and exhibit a high degree of polymorphism. Different molecular markers have been used in this respect, namely RFLP, RAPD, SSAP, AFLP, and SSR. They have been applied in the prediction of heterosis in maize (Boppenmaier et al. 1992), rice (Zhang et al. 1996), wheat (Martin et al. 1995), rape (Sheng et al. 2002) and oat (Moser and Lee 1994).
In maize, Charcosset et al. (1991) showed that genetic distance does not accurately predict hybrid performance unless the DNA markers used in the analysis are associated with (linked to) genes affecting the trait. In sunflower a significant correlation between genetic distances and hybrid seed yield was reported only if data gathered by neutral multi-locus markers were analysed, as AFLP fingerprints (Cheres et al. 2000; Darvishzadeh 2012), capable of evaluating a large number of loci in the genome.
Genetic distance between inbreds is related to genetic diversity and allelic variation. In maize, genomic sequencing and transcriptome analysis of maize germoplasm has provided a wealth of information. Maize intraspecific variation includes sequence polymorphism and structural diversity at several loci resulting in extensive sequence non-homologies because of variation in the number and position of transposons and repetitive elements and variation in the presence of gene sequences (Ching et al. 2002; Brunner et al. 2005; Springer et al. 2009; Lai et al. 2010). These variations might affect maize phenotype through a variety of mechanisms. For example, the variation in repetitive elements may influence the expression level over large regions, ultimately resulting in altered phenotypes, as observed at the teosinte branched1 (tb1) locus, which controls maize architecture, and is influenced in cis by a region of repetitive elements located at >50 kb upstream of the gene (Clark et al. 2006).
In sunflower, many retroelements have been recently described (Natali et al. 2006; Cavallini et al. 2010; Buti et al. 2009; Buti et al. 2011; Staton et al. 2012) and analysis of intraspecific variation related to retrotransposons has been performed (Vukich et al. 2009a). With the aim of evaluating if variations in retrotransposons can be correlated to heterosis effects for morphological traits among sunflower hybrids, we have estimated correlations between hybrid performance and retrotransposon-related genetic distances between parental inbreds.
Materials and methods
Plant materials
This research was conducted at Department of Agricultural, Food, and Environmental Sciences (formerly known as Department of Crop Plant Biology), University of Pisa. Six sunflower inbred lines of different origin, hereafter called by their codes (reported in Table 1 along with their characteristics), were crossed in a half diallel fashion to get F1 seeds in 2009. At least 15 individuals per inbred/cross were planted and then evaluated during spring–summer 2010. Experimental plot comprised three 30 m long rows with row to row distance of 1.00 m and plant to plant distance of 0.30 m. Seeds were germinated on moistened paper and then transferred to plastic cups (8 cm height × 8 cm diameter) containing a commercial soil (Technic Gromix, Free Peat B.V., Vriezenveen, Netherland), initially enriched with a dose of complete fertilizer (Osmocote 14–14–14, Sierra Ltd, UK). After 2 weeks, seedlings were transplanted to open field. Fertilizer was applied in the form of NPK 11–22–16 at the rate of 250 kg per hectare before transplantation and in the form of ammonium nitrate 26–0–0 at the rate of 200 kg per hectare 14 days after transplantation. Water was given through a micro-irrigation system only when needed to avoid drought stress. Standard cultural practices were carried out from sowing until harvesting.
Analysis of heterosis
Data were recorded on sunflower plants starting at the stage of 3–4 leaves, i.e. 2–3 weeks after transplantation, to avoid the influence of transplantation stress. The hybrid nature of each plant was controlled by PCR analysis (not shown) and some plants were discarded. For each trait examined, at least five plants per genotype were used.
The following characteristics were evaluated: (i) growth rate from 25 to 32 days after germination (9–16 days after transplantation); (ii) final height of the plant; (iii) final stem diameter; (iv) final head diameter; (v) two-hundred seed weight; (vi) number of seeds per plant.
Data about parents and F1 hybrids were analysed separately for each trait using Graph-pad Prism software. Mid parent heterosis for F1 hybrids (see for example Meyers et al. 2004) was estimated using the following formula:
where F1 and MP are, respectively, the phenotypic values of the hybrid and of the mean of its parental lines. Significant mid-parent heterosis values were identified by t tests.
SNP analysis
SNPs were analysed in eight genes of sunflower previously isolated (Ouvrard et al. 1996; Natali et al. 2003; Giordani et al. 2010). In brief, seeds of the six sunflower inbreds were germinated and, for each genotype, leaflets were collected from one plantlet. DNA was extracted from leaf tissues according to Giordani et al. (1999).
DNA sequences allelic to eight genes of sunflower were isolated by PCR on genomic DNAs obtained from the different genotypes. PCR was performed using oligonucleotides designed on published DNA sequences of sunflower and reported in Table 2. Sequences were amplified using 60 ng of genomic DNA as a template; thermocycling was performed at 94 °C for 30 s, 58 °C for 30 s and 72 °C for 60 s, for 30 cycles, using Thermostable DNA polymerase, FIREPol (Solis BioDyne).
Each of the amplified fragments was purified and directly sequenced by the dideoxy chain termination method using the PRISM dye terminator cycle sequencing kit (Perkin-Elmer, Foster City, CA, USA) according to the manufacturer’s instructions; sequences were analysed using the SEQUENCING ANALYSIS 2.1.2 (Perkin-Elmer) and SEQUENCHER 3.0 analysis programs (Gene Codes Corporation). All sequences are deposited in the GenBank database (accession numbers FR670620, FR670622, FR670623, FR670626, FR671161, FR671163-64, FR671167, FR671177, FR671179-80, FR671183, FR671185, FR671187, FR671188, FR671191, FR671193, FR671195-96, FR671199, FR671351, FR671353-54, FR671357, FR671359, FR671361-62, FR671365, KC286130-49).
Sequences were aligned using CLUSTAL W (Thompson et al. 1994). Alignments were manually edited and SNPs among allelic sequences were identified.
IRAP analysis
Helianthus annuus LTR sequences were obtained by sequencing three BAC clones obtained from a H. annuus library by CUGI (Clemson University) (Buti et al. 2011). Primers were designed using OLIGO 4.0 software (Rychlik and Rhoads 1989) and are reported in Table 2 together with primers used in these experiments and corresponding to sunflower LTRs described by Vukich et al. (2009a, b). Genomic DNAs from the six sunflower inbreds were used as templates. Primers were used in the combinations reported in Table 2. PCR reactions for IRAP analyses were performed as in Vukich et al. (2009b) in a 20 μl reaction mixture containing: 20 ng genomic DNA, 2.5 mM MgCl2, 0.25 μM primers, 1 U Taq FirePol (Biodyne) DNA polymerase. Thermocycling was performed at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 150 s. PCR products were visualized on EtBr-stained 2 % agarose gel.
Each electrophoresis was repeated three times and fingerprints were scored to prepare binary matrices.
Measurement of genetic distances and statistical analyses
Genetic distances between inbreds were calculated on data obtained analysing both gene sequences and IRAP fingerprints.
Within gene sequences, exons, introns, and UTRs were analysed separately. Intron delimitation within genomic sequences was carried out by comparing of the genomic sequences versus the published cDNAs and confirmed by using the program FEX (Baylor College of Medicine, TX, USA).
Relationships among gene sequences from different inbreds were investigated using the DNADIST program of the PHYLIP program package version 3.572 (Felsenstein 1989) on the gene alignments. Genetic distances were measured using the Kimura two parameters method (Kimura 1980).
IRAP fingerprints were scored by comparing the occurrence of amplification products among the genotypes analysed. The value of “1” or “0” was assigned to each amplification product (visualised on the IRAP-fingerprint as a single band) respectively if present or not in the analysed genotype. In this approach each band was assumed to be representative of a single locus.
Jaccard’s (1908) genetic similarity index was used to calculate genetic similarity, employing the software NTSYS (Rohlf 2008). Given two genotypes, A and B, M11 represents the total number of bands where they both have a value of 1, M01 represents the total number of bands whose values are 0 in A and 1 in B, M10 represents the total number of bands whose values are 1 in A and 0 in B, M00 represents the total number of bands where A and B both have a value of 0. The Jaccard’s similarity index (J), is defined as
Genetic similarity was also evaluated by means of Simple Matching index (Sokal and Michener 1958) that, for multilocus markers is defined as
Jaccard similarity index and Simple Matching index were calculated using the NTSYS software (Rohlf 2008).
The genetic distance or dissimilarity (GD) was calculated as
In order to compare levels of diversity detected by different primer combinations, the various parameters were also calculated for each primer combination separately. One-way ANOVA, Tukey’s tests, correlation statistics were performed using GraphPad Prism software.
Results
Phenotypic values of the parental lines
Mean phenotypic values of inbred lines used in crosses for the analysed morphological traits are reported in Table 3. A significant variation between lines may be observed in mean values of all traits (P < 0.0001). The largest variation, as indicated by ANOVA, was observed for final height (R = 0.95), the smallest was found for growth rate (R = 0.61).
Taking the lowest value for all characteristics as 100, the percentage of variation ranged from 35.7 % for stem diameter to 130.2 % for seed number per head.
Line 821 showed the highest values for growth rate, stem diameter and seed number per head; line EF for growth rate and stem diameter; line GI for head diameter and 200-seed weight; line GB for final height and stem diameter. In some cases, seed weight and seed number were unpaired (for example, line GI showed the largest seed weight and the lowest seed number), in other cases they were paired (line 821 showed high values for both seed weight and seed number). The performance of inbred lines fully matched that observed in the previous years, measured at each cycle of selfing (not shown).
Analysis of heterosis
The six sunflower inbred lines were used in a half diallel cross to obtain fifteen F1 hybrids. The results of the phenotypical analysis in the fifteen hybrids are reported in Table 4. As indicated by the number of classes distinguished by the Tukey’s test, the largest variation was observed for the stem diameter, the lowest for the head diameter and the 200 seed weight.
Heterosis effects were evaluated as the difference (in absolute value) between the hybrid phenotype and the mid-parent value (D) and are presented in Table 5. The significance of such difference was estimated by t tests. Mid-parent heterosis effects were observed in all the analysed traits but the degree of heterosis showed variation from trait to trait (Table 5). The lowest mean value (24.6 %) of heterosis effects was observed for final height. The highest value of heterosis effect (117 %) was found for seed number per head.
All significant heterosis effects were positive. Significant heterosis effects for head diameter and seed number per head were observed in 12 over 15 analysed hybrids; on the other hand, significant heterosis for growth rate was found in only six out of 15 hybrids.
The inbred 383 showed the highest general combining ability, estimated by counting the significant heterosis effects for all traits in the five hybrids obtained by crossing line 383 to the other inbreds. The lowest general combining ability was shown by the inbred 821.
Genetic distances between parents
In a first set of experiments, we isolated and sequenced eight gene fragments (accounting for a total of 4,305 bp of aligned sequences per genotype) from the six inbreds used in the half diallel cross. Gene fragments lengths varied from 338 to 737 bp. We found a total of 86 SNPs, of which 68 in the coding portions and 18 in the non-coding (intron and/or UTR) regions. Considering whole alignments of eight genes six different haplotypes are observed, confirming that the six inbreds are different. Gene alignments are available as additional file one.
Then, we estimated the parental genetic distances between the six inbreds. A distance matrix was deduced from pairwise comparisons of sequences (Fig. 1). The largest distance was observed between inbreds EF and GI (0.0156), the smallest between inbreds EF and R8 (0.0028).

Triangular matrix with genetic distances between the inbreds used in the half diallel cross calculated on data obtained by SNP analysis of eight single copy genes of sunflower
In a second set of experiments, we performed the IRAP protocol and calculated genetic distances between inbreds based on retrotransposon-related variability. The IRAP protocol (Kalendar et al. 1999) can detect genomic loci bounded by retrotransposon LTRs if elements lie close enough to be amplified by Taq polymerase. In IRAP, PCR products may be produced from a single primer if two similar elements are oriented head-to-tail or from two primers designed to two different head-to-head oriented LTRs. Recently, the application of the IRAP protocol has shown large intraspecific variability in wild and cultivated genotypes of H. annuus (Vukich et al. 2009a).
Primers involved in this study were designed on putative LTRs belonging respectively to one Copia and one unknown element (Vukich et al. 2009a; b), and to three Gypsy, one Copia, and two LARD elements (Buti et al. 2011) (see Table 2). Polymorphic fingerprints with bands ranging from 100 up to 2,000 bp were obtained. Nearly identical patterns were obtained in three independent experiments. The rare non-reproducible bands were excluded from subsequent analyses. Primer pairs RLG-des2_for/RLG-des2_rev, U105/U106, and CR/G-LTR2, respectively designed on one Gypsy element, one SURE element (of unknown nature, Vukich et al. 2009a), and on a combination of Copia/Gypsy retrotransposons produced the largest number of easily scorable bands. The electrophoretic patterns using primer pairs U105/U106 and RLG-des2_for/RLG-des2_rev are reported in Fig. 2 as an example. With the exception of primer pairs RLGC3 +/RLGC3- and RLCC2 +/RLCC2-, each primers combination produced a number of bands ≥9. This large number of bands indicated the repetitiveness of the retrotransposons related to the primers; the observed polymorphisms evidenced variability in their insertion sites.

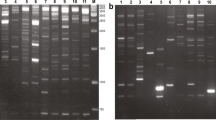
Two examples of IRAP fingerprints produced by primer combinations RLGA2+/RLGA2− and U105/U106 in the six sunflower inbreds used in the half diallel cross. M is the kilobase molecular weight marker (Invitrogen S.R.L., Life Technologies, Milan, Italy); molecular weights are indicated in bp
Among the six inbreds, IRAP fingerprints produced 166 polymorphic bands out of 265 (Table 6). The number of polymorphic bands per primer combination ranged from 1 to 22, and was higher for the U105–U106 primer pair. The average Jaccard’s and Simple Matching dissimilarity indices are also reported in Table 6.
Pairwise comparisons of Jaccard’s and Simple Matching dissimilarity indices produced two triangular distance matrices (Fig. 3).

Rectangular matrix with genetic distances between inbreds calculated on data obtained by IRAP analyses. In the upper part dissimilarities according to Jaccard index, in the lower part according to Simple matching index were reported
Correlation between parental genetic distances and hybrid performance
We analysed the correlation between the mid-parent heterosis for each of the six traits and the genetic distance (calculated on data obtained by SNP and IRAP analyses) between the parental lines, using absolute mid parent heterosis as heterosis measure (Table 7). The correlation calculated using SNP was significant (P < 0.05) only for the stem diameter.
IRAP-based distances showed higher correlation to heterosis than SNP-based distances: correlation was positive and highly significant for seed number per head (P < 0.001 for both J and SM indices) and significant for the final plant height (P < 0.05). The index for the stem diameter was significant (P < 0.05) only when calculated according to Jaccard (1908). The correlation between seed number per head mid parent heterosis and IRAP-based genetic distance between inbred parents is reported in Fig. 4.

Correlation between number of seeds per head mid-parent heterosis in 15 hybrids and genetic dissimilarity between parents calculated on data obtained by IRAP analyses according to Jaccard
To explore if significant correlations could depend on particular primer pairs and, therefore, on the variability of particular retrotransposons, correlation was calculated between mid parent heterosis and the genetic distance based on IRAP fingerprints obtained with the three primer pairs giving the highest number of polymorphic bands (RLG-des2_for/RLG-des2_rev, U105/U106, and CR/G-LTR2), using one primer pair at once. Positive and significant correlations were observed only for genetic distances measured after using primer pair U105-U106, that addresses a SURE retrotransposon, versus the final plant height (P < 0.05, for both J and SM indices) and the number of seeds per head (P < 0.01, only for SM index).
Discussion
According to recent theories on molecular bases of heterosis in maize (Springer and Stupar 2007), our experiments were designed to verify if the variability in the retrotransposon component of the genome can be related to heterosis in hybrids of sunflower.
The study was carried out using inbred lines showing phenotypic variability. Though the number of lines is small, however they were chosen because showing different origin and large phenotypic variations in order to ensure a consistent genetic variability to the experiment. Heterosis was evaluated in 15 hybrids, a number allowing a statistical treatment of data. We decided to evaluate heterosis growing plants in controlled, optimal field conditions as to irrigation and fertilization. The half diallel cross produced hybrids that showed heterosis although, as expected, the extent of heterosis was different depending on the hybrid and the analysed trait. This variability confirms the well-known fact that the occurrence of the heterotic effect in relation to one of the traits does not have to be equivalent to the occurrence of the heterosis in relation to other traits (Krystkowiak et al. 2009).
Our data confirm the occurrence of heterosis in sunflower for certain traits as head diameter and seed weight (see Ahmad et al. 2005). It is worth noting that in a previous work (Cheres et al. 2000), lines 383 and 821 have been described as belonging to the same heterotic group, i.e. their hybrid should show reduced heterosis. Actually, in our experiments, the hybrid 383 × 821 showed heterosis effects for some traits, confirming that, in sunflower, the heterotic groups do not seem to be as rigid as those found in corn and that heterosis can be obtained also crossing inbreds belonging to the same heterotic group.
To verify if genetic distances between parental inbreds can be used as predictive of heterosis, we have calculated the genetic distances between inbreds based on both genic SNPs and IRAP. Obviously, the efficiency of a marker in measuring genetic distances between genotypes depends on the number of loci that are surveyed. The SNP revealed in this study were 86. Though this number should ensure a correct estimation of genetic distances between genotypes, they are referred only to eight different genes (loci), hence such genetic distances should be subjected to certain reservations. The IRAP markers used in this study are specifically related to variations occurring in loci occupied by LTR retrotransposons. IRAP markers should be especially efficient for studies of genetic diversity, being multilocus markers and given the large redundancy of retrotransposons in sunflower genome. In fact, retrotransposons account for more than 50 % of the sunflower genome (Cavallini et al. 2010; Staton et al. 2012) and, even if no major retrotransposon subfamilies occur in the genome, the retrotransposons selected for this study are medium redundant (Vukich et al. 2009a; Buti et al. 2011).
On the whole 265 IRAP bands were surveyed, that can be considered as 265 loci, i.e., considering sunflower genome size of 3.3 Gbp, near one locus per 12 Mbp. Of these loci, 166 proved to be polymorphic, i.e. 1 every 20 Mbp. Such a large number of loci should ensure a valid estimation of genetic distances between inbreds.
Our results on sunflower hybrids indicate that predictive value of genetic distances for heterosis is low, because correlation was significant (P < 0.05) only for one to three over six characteristics analysed, depending on the method used to measure genetic distance. However, our goal was to evaluate if variations in the retrotransposon component of the genome can correlate mid parent heterosis. This proved to be true, at least for three traits, the seed number per head, the final plant height, and the stem diameter.
Cheres et al. (2000) reported that genetic distance was significantly correlated with seed yield and plant height of sunflower hybrids (Cheres et al. 2000). However, genetic distance was a poor predictor of hybrid vigour because hybrid performance varied tremendously among hybrids with similar genetic distances (Cheres et al. 2000). This result was expected and was consistent with results reported in other crop species (see Charcosset and Gallais 2003).
Charcosset and Gallais (2003) reviewed numerous studies of correlation between heterosis and genetic distances, and concluded that results are largely dependent on materials evaluated (see also Melchinger 1999). In general, the knowledge on the relationship between genetic distance based on molecular markers and heterotic effects is insufficient for the development of a methodology of the choice of parents for crossing.
Springer and Stupar (2007) proposed a general model to explain the genetic and molecular bases of heterosis. According to these authors (i) the level of heterosis varies in different species, for example, heterosis has much stronger effects in maize than in Arabidopsis (Zanoni and Dudley 1989; Meyers et al. 2004); (ii) the heterosis levels for specific traits vary and are not correlated in different hybrids of the same species, suggesting that heterosis is not the result of action at a single locus; on the contrary, many of the complex phenotypes that are often assessed for heterosis, such as yield, height and other, are influenced by many genes (see for example Frascaroli et al. 2007); (iii) there appears to be a relationship between genetic diversity and heterosis; however, the correlation is not strong enough to be used as an accurate predictive tool (Melchinger 1999); within a certain threshold, as the genetic distance between the parental inbreds increases, there is generally an increase in heterosis (Moll et al. 1962).
Our data sunflower as experimental system is in accordance to the above described model: (i) the extent of heterosis in sunflower appears reduced compared to maize; (ii) heterosis level for various characteristics is different among hybrids; (iii) the relationship between genetic distance and heterosis is more significant using IRAP-based distances (that account for multiple loci and are referred to sequences that are prone to insert in different loci in the genome) than SNP-based ones (that are referred to a relatively low number of loci, that might not be directly involved in the analysed traits); moreover, the correlation is significant only for some characteristics (seed number per head, final plant height, and stem diameter), therefore it cannot be considered as predictive of “general” hybrid vigour.
Concerning the molecular bases of heterosis, recent data have pointed out that hybrid vigour is at least in part related to differences in transcriptome content: in fact, the suite of expressed genes in heterotic hybrids is greater than that of either of the parental inbred lines (see Buckler et al. 2006; Messing and Dooner 2006). In analyses of gene expression in two maize lines and their F1 hybrid, a number of genes that are differentially expressed between parents were identified, and, all of these genes were expressed in the hybrid leading to a larger number of transcripts in hybrids than in the inbred parents (Stupar and Springer 2006; Guo et al. 2006; Paschold et al. 2012). The combination of inbred-specific sequences in the hybrids allows novel trans-interactions that would not occur in either parent, potentially leading to non-additive expression levels that could be related to heterosis (Springer et al. 2009; Paschold et al. 2012).
Different gene expression levels in hybrids compared to either parents, are related to variations in genome structure between inbreds (Brunner et al. 2005) especially in non-coding, regulatory regions. There is evidence that a primary source of variation in genome structure (and, consequently, in transcriptome content) is the activity of transposable elements. For example, helitrons, DNA transposons and retrotransposons can include genic fragments and increase gene copy number (Morgante et al. 2007). Transposable element insertion events may also cause a reduction in the expression level of genes through disruption or alteration of coding regions but also non-coding, regulatory regions as reported for example in Vitis vinifera (Kobayashi et al. 2004). Alternatively, transposons and retrotransposon activity can determine increase or decrease of gene expression levels providing novel cis-regulatory sequences (Stam et al. 2002; Guo et al. 2004; Clark et al. 2006; Stupar and Springer 2006).
The polymorphisms observed in our analyses are actually related to the occurrence of LTR-retrotransposon activity by which new sequences are inserted in the genome. When these insertions occur in the intergenic regions, they possibly modify the regulatory pattern of neighbour genes. Sunflower appears especially prone to retrotransposon activity as shown by the extremely large IRAP variability observed in both wild and cultivated genotypes (Vukich et al. 2009a). If such structural, retrotransposon-related variability parallels with variation in gene activity, then the genetic distance between lines, calculated on data obtained by IRAP, would be related to differences in gene expression, and, on its turn, on the occurrence of heterosis in hybrids.
In conclusion, there are several potential mechanisms through which genomic variation could combine to produce a heterotic phenotype; each of these mechanisms could occur at a subset of genes, at both gene sequence and gene expression level, and the combination of effects will result in heterosis (Springer and Stupar 2007). Our data in sunflower support the hypothesis that variations in the retrotransposon component of the genome can be involved in generating heterosis, as proposed for maize.
References
Ahmad S, Khan MS, Swati MS, Shah GS, Khalil IH (2005) A study on heterosis and inbreeding depression in sunflower (Helianthus annuus L.). Songklanakarin J Sci Technol 27:1–8
Boppenmaier J, Melchinger AE, Brunklaus-Jung E, Geiger HH, Herrmann RG (1992) Genetic distance for RFLPs in European maize inbreds: I. Relation to performance of flint dent crosses for forage traits. Crop Sci 32:895–902
Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A (2005) Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17:343–360
Buckler ES, Gaut BS, McMullen MD (2006) Molecular and functional diversity of maize. Curr Opin Plant Biol 9:172–176
Buti M, Giordani T, Vukich M, Gentzbittel L, Pistelli L, Cattonaro F, Morgante M, Cavallini A, Natali L (2009) HACRE1, a recently inserted copia-like retrotransposon of sunflower (Helianthus annuus L.). Genome 52:904–911
Buti M, Giordani T, Cattonaro F, Cossu RM, Pistelli L, Vukich M, Morgante M, Cavallini A, Natali L (2011) Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor Appl Genet 123:779–791
Cavallini A, Natali L, Zuccolo A, Giordani T, Jurman I, Ferrillo V, Vitacolonna N, Sarri V, Cattonaro F, Ceccarelli M, Cionini PG, Morgante M (2010) Analysis of genome composition and organization in sunflower (Helianthus annuus L.) and related species. Theor Appl Genet 120:491–508
Charcosset A, Gallais A (2003) Application of markers in selection. In: de Vienne D (ed) Molecular markers in plants genetics and biotechnology. Science Publishers, Enfield, pp 53–176
Charcosset AM, Lefort-Buson M, Gallais A (1991) Relationship between heterosis and heterozygosity at marker loci: a theoretical computation. Theor Appl Genet 81:571–575
Cheres MT, Knapp SJ (1998) Ancestral origins and genetic diversity of cultivated sunflower: analysis of the pedigrees of public germplasm. Crop Sci 38:1476–1482
Cheres MT, Miller JF, Crane JM, Knapp SJ (2000) Genetic distance as a predictor of heterosis and hybrid performance within and between heterotic groups in sunflower. Theor Appl Genet 100:889–894
Ching A, Caldwell KS, Jung M, Dolan M, Smith OS, Tingey S, Morgante M, Rafalski AJ (2002) SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet 3:19
Clark RM, Wagler TN, Quijada P, Doebley J (2006) A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat Genet 38:594–597
Darvishzadeh R (2012) Phenotypic and molecular marker distance as a tool for prediction of heterosis and F1 performance in sunflower (Helianthus annuus L.) under well-watered and water-stressed conditions. Aust J Crop Sci 6:732–738
Davenport CB (1908) Degeneration, albinism and inbreeding. Science 28:454–455
Diers BW, Mac Vetty PBE, Osborn TC (1996) Relationship between heterosis and genetic distance based on restriction fragment length polymorphism markers in oilseed rape (Brassica napus L.). Crop Sci 36:79–83
Dubreuil P, Dufour P, Krejci E, Causse M, de Vienne D, Gallais A, Charcosset A (1996) Organization of RFLP diversity among inbred lines of maize representing the most significant heterotic groups. Crop Sci 36:790–799
Duvick DN (1999) Heterosis: feeding people and protecting natural resources. In: Coors JG, Pandey S (eds) The genetics and exploitation of heterosis in crops. Crop Science Society of America, Madison, pp 19–30
East EM (1908) Inbreeding in corn. Rep Conn Agric Exp Stn 1907:419–429
Felsenstein J (1989) PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5:164–166
Frascaroli E, Canè MA, Landi P, Pea G, Gianfranceschi L, Villa M, Morgante M, Pè ME (2007) Classical genetic and quantitative trait loci analyses of heterosis in a maize hybrid between two elite inbred lines. Genetics 176:625–644
Giordani T, Natali L, D’Ercole A, Pugliesi C, Fambrini M, Vernieri P, Vitagliano C, Cavallini A (1999) Expression of a dehydrin gene during embryo development and drought stress in ABA-deficient mutants of sunflower (Helianthus annuus L.). Plant Mol Biol 39:739–748
Giordani T, Buti M, Natali L, Pugliesi C, Cattonaro F, Morgante M, Cavallini A (2010) An analysis of sequence variability in eight genes putatively involved in drought response in sunflower (Helianthus annuus L.). Theor Appl Genet 122:1039–1049
Godshalk EB, Lee M, Lamkey KR (1990) Relationship of restriction fragment length polymorphisms to single-cross hybrid performance in maize. Theor Appl Genet 80:273–280
Guo M, Rupe MA, Zinselmeier C, Habben J, Bowen BA, Smith OS (2004) Allelic variation of gene expression in maize hybrids. Plant Cell 16:1707–1716
Guo M, Rupe MA, Yang X, Crasta O, Zinselmeier C, Smith OS, Bowen B (2006) Genome-wide transcript analysis of maize hybrids: allelic additive gene expression and yield heterosis. Theor Appl Genet 113:831–845
Hongtrakul V, Huestis GM, Knapp SJ (1997) Amplified fragment length polymorphisms as a tool for DNA fingerprinting sunflower germplasm: genetic diversity among oilseed inbred lines. Theor Appl Genet 95:400–407
Jaccard P (1908) Nouvelles recherches sur la distribution florale. Bull Soc Vaud Sci Nat 44:223–270
Janick J (1998) Hybrids in horticultural crops. In: Lamkey KR, Staub JE (eds) Concepts and breeding of heterosis in crop plants. Crop Science Society of America, Madison, pp 45–56
Jones DF (1917) Dominance of linked factors as a means of accounting for heterosis. Genetics 2:466–479
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman A (1999) IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet 98:704–711
Kaya Y (2005) Hybrid vigor in sunflower (Helianthus annuus L.). Helia 28:77–86
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kobayashi S, Goto-Yamamoto N, Hirochika H (2004) Retrotransposon-induced mutations in grape skin color. Science 304:982
Krystkowiak K, Adamski T, Surma M, Kaczmarek Z (2009) Relationship between phenotypic and genetic diversity of parental genotypes and the specific combining ability and heterosis effects in wheat (Triticum aestivum L.). Euphytica 165:419–434
Lai J, Li R, Xu X, Jin W, Xu M, Zhao H et al (2010) Genome-wide patterns of genetic variation among elite maize inbred lines. Nature Genet 42:1027–1030
Martin JM, Talbort LE, Lanning SP, Blake NK (1995) Hybrid performance in wheat as related to parental diversity. Crop Sci 35:104–108
Melchinger AE (1999) Genetic diversity and heterosis. In: Coors JG, Pandey S (eds) The genetics and exploitation of heterosis in crops. Crop Science Society of America, Madison, pp 99–118
Melchinger AE, Gumber RK (1998) Overview of heterosis and heterotic groups in agronomic crops. In: Lamkey KR, Staub JE (eds) Concepts and breeding of heterosis in crop plants. Crop Science Society of America, Madison, pp 29–44
Melchinger AE, Lee M, Lamkey KR, Woodman WW (1990) Genetic diversity for restriction fragment length polymorphisms: relation to genetic effects in maize inbreds. Crop Sci 30:1033–1040
Melchinger AE, Utz HF, Piepho HP, Zeng ZB, Schön CC (2007) The role of epistasis in the manifestation of heterosis: a systems-oriented approach. Genetics 177:1815–1825
Messing J, Dooner HK (2006) Organization and variability of the maize genome. Curr Opin Plant Biol 9:157–163
Meyers BC, Scalabrin S, Morgante M (2004) Mapping and sequencing complex genomes: let’s get physical. Nat Rev Genet 5:578–588
Miller JF (1992) Registration of five oilseed sunflower germplasm restorer lines (RHA 373 to 377) and two nuclear male sterile populations (NMS 274 and 801). Crop Sci 32:1298
Moll RH, Salhuana WS, Robinson HF (1962) Heterosis and genetic diversity in variety crosses of maize. Crop Sci 2:197–198
Morgante M, De Paoli E, Radovic S (2007) Transposable elements and the plant pan-genomes. Curr Opin Plant Biol 10:149–155
Moser H, Lee M (1994) RFLP variation and genealogical distance, multivariate distance, heterosis, and genetic variance in oats. Theor Appl Genet 87:947–956
Natali L, Giordani T, Cavallini A (2003) Sequence variability of a dehydrin gene within Helianthus annuus. Theor Appl Genet 106:811–818
Natali L, Santini S, Giordani T, Minelli S, Maestrini P, Cionini PG, Cavallini A (2006) Distribution of Ty3-gypsy- and Ty1-copia-like DNA sequences in the genus Helianthus and other Asteraceae. Genome 49:64–72
Ouvrard O, Cellier F, Ferrare K, Tousch D, Lamaze T, Dupuis J-M, Casse-Delbart F (1996) Identification and expression of water stress- and abscisic acid-regulated genes in a drought-tolerant sunflower genotype. Plant Mol Biol 31:819–829
Paschold A, Jia Y, Marcon C, Lund S, Larson NB, Yeh CT, Ossowski S, Lanz C, Nettleton D, Schnable PS, Hochholdinger F (2012) Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res 22(12):2445–2454. doi:10.1101/gr.138461.112
Rohlf FJ (2008) NTSYSpc: numerical taxonomy system, ver. 2.00. Exeter Publishing Ltd., Setauket
Rychlik W, Rhoads RE (1989) A computer program for choosing optimal oligonucleotides for filter hybridization, sequencing and in vitro amplification of DNA. Nucl Acids Res 17:8543–8551
Sheng JX, Lu GY, Fu TD, Yang GS (2002) Relationships between genetic diversity and hybrid performance in oilseed rape (Brassica napus). Acta Agron Sin 28:622–627
Shull GH (1908) The composition of a field of maize. Am Breed Assoc Rep 4:296–301
Sokal RR, Michener CD (1958) A statistical method for evaluating systematic relationships. Univ Kans Sci Bull 38:1409–1438
Springer NM, Stupar RM (2007) Allelic variation and heterosis in maize: how do two halves make more than a whole? Genome Res 17:264–275
Springer NM, Ying K, Fu Y, Ji T, Yeh CT, Jia Y, Wu W, Richmond T et al (2009) Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet 5:e1000734
Stam M, Belele C, Dorweiler JE, Chandler VL (2002) Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev 16:1906–1918
Staton SE, Bakken BH, Blackman BK, Chapman MA, Kane NC, Tang S, Ungerer MC, Knapp SJ, Rieseberg LH, Burke JM (2012) The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J 72:142–153
Stupar RM, Springer NM (2006) Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173:2199–2210
Tersac M, Blanchard P, Brunel D, Vancourt P (1994) Relations between heterosis and enzymatic polymorphisms in populations of cultivated sunflower (Helianthus annuus L.). Theor Appl Genet 88:49–55
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22:4673–4680
Vukich M, Schulman AH, Giordani T, Natali L, Kalendar R, Cavallini A (2009a) Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor Appl Genet 119:1027–1038
Vukich M, Giordani T, Natali L, Cavallini A (2009b) Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol 9:150
Zanoni U, Dudley JW (1989) Comparison of different methods of identifying inbreds useful for improving elite maize hybrids. Crop Sci 29:577–582
Zhang Q, Gao YJ, Saghai Maroof MA, Yang SH, Li JX (1995) Molecular divergence and hybrid performance in rice. Mol Breed 1:133–142
Zhang G, Angeles ER, Abenes MLP, Khush GS, Huang N (1996) RAPD and RFLP mapping for the bacterial blight resistance gene xa-13 in rice. Theor Appl Genet 93:65–70
Acknowledgments
Thanks are due to Dr. Elisabetta Frascaroli (University of Bologna) for her useful suggestions during the investigation and to Dr. Andrea Cini (Kayser Italia) for revising the manuscript. This work was supported by PRIN-MIUR, Projects “Variabilità di sequenza ed eterosi in piante coltivate” and “SUNREP: caratterizzazione molecolare della componente ripetitiva del genoma di girasole”.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Buti and T. Giordani contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Buti, M., Giordani, T., Vukich, M. et al. Retrotransposon-related genetic distance and hybrid performance in sunflower (Helianthus annuus L.). Euphytica 192, 289–303 (2013). https://doi.org/10.1007/s10681-013-0883-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-013-0883-5