
Abstract
An endo-1,4-β-d-glucanase gene was cloned from the thermophilic archaea Sulfolobus shibatae and expressed in E. coli. The recombinant enzyme was purified by heat denaturation and affinity chromatography prior to characterisation. The purified recombinant enzyme exhibited maximum activity at 95–100 °C and displayed a broad pH profile with over 91% of its maximum activity observed at pH 3–5. Upon assessment of enzyme thermal stability, full activity was observed after 1 h incubation at 75, 80 and 85 °C while 98%, 90% and 84% of original activity was detected after 2 h at 75, 80 and 85 °C, respectively. Maximum activity was observed on barley β-glucan and lichenan and the purified enzyme also hydrolysed CMC and xylan. Endoglucanase activity was confirmed by viscometric assay with a rapid decrease in substrate viscosity observed immediately upon incubation with barley β-glucan or CMC. The crude enzyme released reducing sugars from acid-pretreated straw at 75–85 °C. The thermophilic nature and biochemical properties of the enzyme indicate its potential suitability in industrial applications undertaken at high temperature, such as the production of second-generation bioethanol from lignocellulosic feedstocks and in the brewing industry. This is the first known report of an endoglucanase from S. shibatae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enzymes produced by thermophilic microorganisms are of increasing interest in industrial processes and their activity and stability at high temperature can be exploited in the development of a bio-based economy. Chief amongst these industrial applications is the use of lignocellulose-degrading enzymes in the production of second generation biofuel from readily available lignocellulosic feedstocks [1]. The widespread deployment of cellulosic bioethanol as a renewable transport fuel is of global interest as it can contribute to reducing greenhouse gas emissions, decreasing fossil fuel dependency and improving energy security without competing with food production [2,3,4]. Process improvements are however required to achieve competitive cellulosic bioethanol production [5,6,7].
Efficient enzymatic hydrolysis of the cellulose and hemicellulose components of lignocellulose is necessary to maximise the yield of fermentable sugars and achieve viable, cost effective bioethanol production. Improvement of the enzymatic hydrolysis step, currently undertaken at circa. 50 °C using mesophilic enzymes, can potentially be achieved by using higher temperatures which result in improved substrate accessibility, increased substrate solubility, decreased viscosity, higher reaction rates, shorter residence times, higher substrate loadings and reduced risk of microbial contamination [8,9,10]. High temperature enzymatic hydrolysis is also more compatible with existing high temperature pretreatment processes, reducing the need for costly cooling steps prior to enzymatic hydrolysis and reducing process complexity [11, 12]. Currently used mesophilic enzymes are unsuitable for such high temperature enzymatic hydrolysis due to poor catalytic activity and stability under these conditions, necessitating the identification of appropriate thermophilic enzymes exhibiting enhanced stability and activity at high temperature [11]. Optimum hydrolysis of lignocellulosic biomass requires multiple enzymes with different specificities acting in synergy. One of the main enzymes required is endo-β-glucanase activity (EC 3.2.1.4) which randomly hydrolyses accessible internal β-1,4-glucosidic bonds in cellulose chains and acts synergistically with exo-glucanase and β-glucosidase activities in the degradation of cellulose [5, 13].
In addition to their application in high temperature enzymatic hydrolysis, thermophilic endo-β-glucanases also potentially facilitate alternative process configurations aimed at optimising the lignocellulose to bioethanol process. These include a proposed partial pre-hydrolysis or liquefaction step at higher temperature during which thermostable endoglucanase activity is used to decrease the viscosity of the substrate prior to the hydrolysis step resulting in improved mixing properties and facilitating high solids loading which is required to achieve an economically feasible final ethanol concentration [13,14,15]. Additional possibilities include the proposed use of thermophilic endoglucanase activity to reduce the severity of pretreatment and hence the production of toxic by-products [16]. Thermophilic simultaneous saccharification and fermentation, during which hydrolysis and fermentation are undertaken at high temperature in a single reactor using thermostable enzymes and a fermentative thermophile, has also been proposed to achieve more cost-effective processing [5].
Thermophilic endoglucanases are also of interest in other industrial applications [2, 3, 7], including the brewing industry where exogenous glucanase and xylanase enzymes are added during malting to hydrolyse non-starch polysaccharides hence reducing mash viscosity and turbidity and improving filtration rate and brewing quality. Enzymes exhibiting good thermal stability and activity at pH 5–6 are desirable for this application which is associated with continuously rising temperatures which inactivate endogenous cereal enzymes [17, 18].
Screening of thermophilic microorganisms for the production of novel, naturally tailored thermophilic enzymes is an alternative to genetic engineering based approaches aimed at improving the relevant biochemical properties of available mesophilic enzymes. Archaea have been shown to be a promising source of thermophilic lignocellulose-degrading enzymes and screening of selected archaea undertaken in our research group resulted in the identification of a novel thermophilic endo-1,4-β-glucanase from Sulfolobus shibatae (DSM 5389). This strain was originally isolated from a geothermal mud hole [19]. While three thermophilic endoglucanases (SSO1354, SSO1949 and SSO2534) have been reported from Sulfolobus solfataricus [16], no previous reports on the production, purification or characterisation of an endoglucanase from S. shibatae were found in published scientific literature. This paper describes the cloning, expression, purification and characterisation of this industrially-relevant enzyme from this distinct species of the archaebacterial genus Sulfolobus.
Materials and methods
Strains and reagents
The archaea S. shibatae B12 (DSM 5389) was purchased from the DSMZ German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany, and was routinely grown at 75 °C and pH 3–3.5. E. coli DH5α and Rosetta-gami™ B(DE3) were routinely grown at 37 °C in Luria–Bertani medium. Carboxymethyl cellulose-4M (CMC), barley β-glucan, CM-curdlan and cello-oligosaccharides were obtained from Megazyme, Ireland. Restriction enzymes were from Roche Diagnostics Ltd., UK. T4 DNA Ligase and Mark12 unstained protein standard were from Invitrogen. All other chemicals were from Sigma-Aldrich Ireland Ltd. or Fisher Scientific Ireland Ltd.
Cloning and expression of S. shibatae endo-1,4-β-glucanase
Genomic DNA was isolated from S. shibatae using QIAGEN DNeasy Blood and Tissue Kit.
The gene encoding S. shibatae endo-1,4-β-glucanase was amplified by PCR using TaKaRa Ex Taq and the following primers: 5′-CAGCAGGAATTCATGGCTATTTACCTACACATC-3′ and 5′-GGGCAGCAGAAGCTTATATTGTTTAGAGGAGAG-3′. The primers incorporated restriction sites for EcoRI and HindIII, respectively (underlined), and were designed using S. solfataricus P2 nucleotide sequence. The resulting PCR product was purified using QIAGEN Qiaquick PCR Purification Kit, digested with EcoRI and HindIII and ligated into the vector pProEX HTb (facilitating attachment of an N-terminal His tag). Ligation was undertaken using T4 DNA ligase with the resulting construct subsequently referred to as pProEX-5389.7. The ligation mixture was initially transformed into E. coli DH5α competent cells by the CaCl2 method [20]. Positive clones were identified by restriction enzyme analysis and sent to Eurofins Genomics for sequencing. The nucleotide sequence of S. shibatae endoglucanase was deposited in the European Nucleotide Archive under accession number LT221867. Database searches, deduced amino acid sequence analysis and sequence alignments were undertaken using the National Center for Biotechnology Education BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi), EXPASY tools (http://www.expasy.org/) and Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/), respectively. The construct pProEX-5389.7 was transformed into E. coli Rosetta-gami™ B(DE3) for expression. For expression, a culture grown overnight at 37 °C in Luria–Bertani medium containing 100 µg/ml ampicillin, 15 µg/ml kanamycin, 12.5 µg/ml tetracycline and 34 µg/ml chloramphenicol was diluted 1 in 100 in Terrific broth supplemented with 1% glucose and grown to an optical density of 0.6 at 600 nm, prior to induction with 0.05 mM IPTG at 37 °C, 100 rpm for 17 h. The cells were harvested by centrifugation (4500×g, 10 min, 4 °C).
Enzyme purification
The cells were resuspended in 50 mM Tris–HCl, 100 mM NaCl, pH 7.5 and disrupted by sonication. Following removal of cell debris by centrifugation (12,000×g, 20 min, 4 °C), 1 M sodium acetate buffer, pH 4 and 5 M sodium chloride were added to the cell-free extract to a final concentration of 250 mM and 100 mM, respectively, prior to heating at 90 °C for 10 min. After heat treatment, precipitated proteins were removed by centrifugation (15,000×g, 30 min, 4 °C). The pH of the resulting supernatant was adjusted to ~ pH 6.4 by addition of 1 M disodium hydrogen phosphate prior to loading on a Chelating Sepharose Fast Flow column charged with copper and pre-equilibrated with 20 mM sodium phosphate, 500 mM NaCl, pH 7 (Buffer A). The column was washed successively with 70% and 100% Buffer B (20 mM sodium phosphate, 500 mM NaCl, pH 4.3). Fractions containing β-glucanase activity, collected during the second wash step, were pooled and concentrated using a 10,000 MWCO Amicon Ultra centrifugal filter device. Determination of β-glucanase activity was undertaken as described below. Determination of protein concentration was carried out using the method of Bradford [21]. SDS-PAGE was undertaken on a 10% gel, as described by Laemmli [22], with subsequent protein staining using EZBlue Gel Staining Reagent. Zymogram analysis was undertaken by running samples, previously heated at 80 °C for 5 min, on a 10% gel containing 0.1% CMC. After washing in buffer and isopropyl alcohol as previously described [23], the gel was incubated at 95 °C in 50 mM sodium acetate buffer, pH 4 for 2 h. Bands of substrate hydrolysis were observed by staining with 0.1% Congo Red for 30 min prior to destaining with 1 M NaCl.
Endoglucanase assay
Endoglucanase activity was determined by quantifying the release of reducing sugars from 1% (w/v) barley beta-glucan (medium viscosity) in 0.2 M sodium acetate-acetic acid buffer (pH 4) at 95 °C, using dinitrosalicyclic acid (DNS) [24]. One unit of enzyme activity was defined as the amount of enzyme that catalyses the production of 1 µmol reducing sugar (as glucose equivalents) per minute under the assay conditions.
Biochemical characterisation of the purified enzyme
The effect of temperature on enzyme activity was assessed by undertaking the endoglucanase assay, as outlined above, at 75–110 °C. For determination of the effect of pH on enzyme activity, the assay was undertaken at 95 °C using 1% barley beta-glucan in 0.5 M glycine–HCl buffer (pH 2.1–3.4), 0.5 M sodium acetate-acetic acid buffer (pH 3.6–5.4) and 0.5 M KH2PO4–NaOH buffer (pH 5.7–6.3). The effect of various reagents on enzyme activity was determined by undertaking the endoglucanase assay with inclusion of each reagent at the concentration shown in Table 1. Enzyme activity was expressed relative to a control sample assayed without reagent. Enzyme thermal stability was determined by quantifying residual enzyme activity after heating the enzyme in 50 mM sodium acetate buffer (pH 4) at 75, 80, 85 or 90 °C, for the indicated time periods. Substrate specificity was assessed by measuring the release of reducing sugars from the following substrates [1% (w/v)] in 0.5 M sodium acetate buffer, pH 4 at 95 °C: CMC, lichenan (Cetraria islandica), barley β-glucan, beechwood xylan, birchwood xylan, laminarin (Laminaria digitata), starch, CM-curdlan, avicel and filter paper. Determination of 1,4-β-d-xylosidase, β-d-glucosidase and cellobiohydrolase activities was undertaken by measuring the release of p-nitrophenol from p-nitrophenyl β-d-xylopyranoside, p-nitrophenyl β-d-1,4-glucopyranoside and p-nitrophenyl β-d-cellobioside, respectively, at 95 °C and pH 4. One unit of enzyme activity is defined as the amount of enzyme that releases 1 µmol p-nitrophenol per min under the assay conditions. Kinetic constants were determined at 95 °C, pH 4 with 1–10 mg/ml barley β-glucan as substrate and Km and Vmax were calculated using GraphPad Prism Version 6.05 (GraphPad Software, Inc.).
Thin-layer chromatography analysis of hydrolysis products
Reaction mixtures containing equal volumes of purified enzyme (1.47 units/ml) and substrate (1% CMC, 1% β-glucan or 8.25 mg/ml cello-oligosaccaride in 0.2 M sodium acetate buffer, pH 4) were incubated for 2 h at 95 °C. After cooling on ice, aliquots of each reaction mixture were spotted onto a silica gel plate and separated by migration in butanol/ethanol/water (5:3:2). The plates were sprayed with sulphuric acid/ethanol (1:9) and heated at 100 °C for visualisation of hydrolysis products.
Measurement of reaction viscosity and production of reducing ends
Reaction mixtures containing the purified enzyme (0.064 units) and substrate [CMC or barley β-glucan at a final concentration of 1.2% (w/v)] were incubated at 90 °C and pH 4 for 60 min. At the indicated time points, the viscosity of the reaction mixture and the amount of reducing sugars produced were measured. The viscosity of the reaction mixture was measured using a miniature U-tube viscometer. The amount of reducing sugars produced was quantified using the DNS method of Miller [24].
Hydrolysis of pretreated straw
Straw was washed, dried and chopped and pretreated at 10% (w/v) in 0.75% (w/v) sulphuric acid at 121 °C for 15 min and washed extensively with distilled water prior to enzymatic hydrolysis. For hydrolysis the straw was incubated at a concentration of 5% (w/v) with the crude enzyme (2 units/g straw, where activity was quantified at 95 °C using CMC as substrate) in 0.2 M sodium acetate buffer, pH 4 at 75, 80 or 85 °C for 5 h. Parallel control hydrolysis reactions were undertaken at each temperature using distilled water instead of enzyme. The amount of reducing sugars released was quantified using DNS.
Results
Cloning and sequence analysis
The DNA fragment cloned from S. shibatae was predicted to encode a protein of 311 amino acids with a deduced molecular mass of 35,128 Da and a theoretical pI of 4.98. The predicted amino acid sequence shows 96–97% identity to several endoglucanase/endoglucanase precursor sequences from Sulfolobus islandicus (Accession numbers ADX81754.1, WP_014513759.1, WP_012718693.1, WP_012710983.1, WP_016732182.1, WP_048050150.1, ACR41545.1, PVU76659.1, WP_012715810.1) and 96% and 95% identity, respectively to the glycoside hydrolase family 12 protein from S. solfataricus 98/2 (ACX92839.1) and endoglucanase from S. solfataricus (WP_063492797.1). The predicted amino acid sequence showed 96% and 94% identity to the endoglucanase precursors SSO1949 (GenBank AAK42142.1) and SSO1354 (WP_010923347.1) from S. solfataricus. Lower homology (55 and 50% identity) was observed with two glycoside hydrolase family 12 proteins from DSM 17230 Ignisphaera aggregans (ADM27292.1 and ADM27702.1) while additional matches (31–36% identity) included endo-glucanases from thermophilic bacteria and archaea of the genera Thermococcus, Pyrococcus, Thermoproteus, Caldivirga, Thermotoga and Dictyoglomus. Weak homology (28% sequence identity) was observed between S. shibatae endoglucanase and SSO2534 endoglucanase from S solfataricus P2 (AAK42663.1) upon blastx alignment. Analysis of the amino acid sequence of S. shibatae endoglucanase, revealed a conserved glycosyl hydrolase family 12 (GH12) domain (residues 158–310). This indicates that the enzyme can be classified as a member of GH family 12, members of which cleave the glycosidic bond via a retaining mechanism with glutamic acid residues acting as nucleophile and proton donors. Sequence alignment with family 12 glycoside hydrolases from S. solfataricus P2 is shown in Fig. 1. Conserved amino acids include the catalytic residues identified in SSO1354 (Glu211 and Glu308), SSO2534 (Glu193 and Glu299) and SSO1949 (Glu213 and Glu310). Sequence analysis indicates that S. shibatae endoglucanase does not contain a carbohydrate-binding domain. Upon analysis for putative N-glycosylation sites (Asn-Xaa-Ser/Thr sequons) using NetNGlyc, 12 potential N-glycosylation sites were identified of which 4 (at positions 66, 87, 104 and 294) were predicted by the program as very likely to be glycosylated.

Amino acid sequence alignment of S. shibatae endo-1,4-β-glucanase (CZS63539.1) and family 12 glycoside hydrolases from Sulfolobus solfataricus P2: SSO1949 endoglucanase precursor (AAK42142.1), SSO1354 endoglucanase (WP_010923347.1) and SSO2534 endo 1,4 β-glucanase (AAK42663.1). (*) Indicates positions which have a fully conserved residue, (:) and (.) indicate conservation between groups of strongly similar properties and weakly similar properties, respectively. The shaded regions represent catalytic residues identified in SSO1354 (Glu211 and Glu308), SSO2534 (Glu193 and Glu299) and SSO1949 (Glu213 and Glu310)
Expression and purification of S. shibatae endo-1,4-β-glucanase
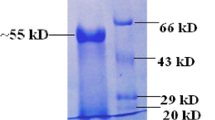
The DNA fragment amplified from S. shibatae was cloned into the vector pProEX HTb and functionally expressed in E. coli Rosetta-gami™ B(DE3). Upon induction in Terrific broth, as outlined above, endoglucanase activity of 0.19 units/mg protein was detected in the cell lysate from sonication. No endoglucanase activity was detected in a similarly prepared cell lysate from E. coli Rosetta-gami™ B(DE3) containing empty pProEX HTb vector. The recombinant enzyme was purified by a combination of heat denaturation and immobilized metal ion affinity chromatography with a purification factor of 2535 and a percentage yield of 14%. The purified enzyme had a specific activity of 482 units/mg and upon SDS-PAGE analysis the molecular mass of the His-tagged enzyme reflected the mass of 35,128 Da predicted from the amino sequence of the enzyme (Fig. 2a). Zymogram analysis (Fig. 2b) confirmed the endoglucanase activity of the purified enzyme with a band of CMC hydrolysis observed in the Congo Red stained gel at a molecular mass consistent with that of the purified enzyme.

SDS-PAGE analysis of S. shibatae endoglucanase expressed in E. coli. a Analysis of purification: M, protein molecular weight marker (Invitrogen Mark12), masses indicated alongside; lane 1, crude enzyme preparation; lane 2, purified enzyme; b Zymogram analysis of purified enzyme on a CMC-containing gel: lane 1, stained for endoglucanase activity using Congo Red
Biochemical properties of the purified recombinant enzyme
The purified enzyme has an optimum temperature of 95–100 °C and displays 50% or more of its maximum activity in the temperature range of 85–105 °C (Fig. 3). The enzyme has a broad pH profile, displaying over 91% of its maximum activity in the pH range 3–5 with 83% and 80% of maximum activity observed at pH 2.7 and 5.4, respectively (Fig. 4). Upon assessment of enzyme thermal stability, full activity was retained after incubation at 75, 80 or 85 °C for 1 h, while 98%, 90% and 84% of original activity was detected after 2 h at 75, 80 and 85 °C, respectively (Fig. 5). The enzyme was less thermostable at 90 °C, retaining 30%, 8% and 4% of original activity after 30 min, 1 h and 2 h, respectively. Inclusion of various metal ions and reagents in the standard enzyme reaction had little or no effect on enzyme activity under the conditions used (Table 1) with the exception of ZnSO4 which had a slight inhibitory effect (87% activity relative to the control) and 0.1% Triton X-100 and 0.2% PEG, both of which had a beneficial effect on enzyme activity (133% and 119%, respectively, relative to the control enzyme).

Effect of temperature on the activity of the purified recombinant endoglucanase. Data expressed as a percentage of maximum activity. Each value represents mean ± SD (n = 3)

Effect of pH on the activity of the purified recombinant endoglucanase. Data expressed as a percentage of maximum activity. Each value represents mean ± SD (n = 3)

Thermal stability of the purified recombinant endoglucanase. Data expressed as percentage activity remaining after heating the enzyme in 50 mM sodium acetate buffer (pH 4) at the indicated conditions, relative to the activity of an unheated sample. Each value represents mean ± SD (n = 3)
Substrate specificity and kinetic parameters
Of the substrates tested, the purified enzyme displays maximum activity on barley β-glucan and lichenan (100%), with 52% of maximum activity observed on CMC. Activity was also observed on beechwood and birchwood xylan (29% and 19% of activity on barley β-glucan, respectively). Under the conditions used, no activity was observed on the other substrates tested. Upon determination of kinetic constants with barley β-glucan as substrate, the purified enzyme had an apparent Km of 1.772 mg/ml and a Vmax of 0.03 µmol/min.
Analysis of hydrolysis products
Analysis of the reaction products produced upon hydrolysis of cellooligosaccharides, CMC and barley β-glucan by the purified enzyme was undertaken by thin-layer chromatography. The enzyme did not hydrolyse cellobiose or cellotriose under the conditions used but cellotetraose and cellopentose were hydrolysed producing cellobiose and cellotriose (Fig. 6a). Hydrolysis of CMC and β-glucan for 2 h resulted in the production of cellobiose, cellotriose, cellotetraose and cellopentose (Fig. 6b). Glucose was not detected in any of the reactions.

Thin-layer chromatography analysis of hydrolysis products. a Hydrolysis products observed upon reaction of the purified endoglucanase with cellobiose (lane 1), cellotriose (lane 2), cellotetraose (lane 3) and cellopentose (lane 4). b Hydrolysis products observed upon reaction of the purified endoglucanase with CMC (lane 1) and barley β-glucan (lane 2). All reactions were undertaken at 95 °C, pH 4 for 2 h. S: oligosaccharide standards, G: glucose, C2: cellobiose, C3: cellotriose, C4: cellotetraose, C5: cellopentose
Viscometric assay and hydrolysis of straw
The endoglucanase activity of the enzyme was confirmed by comparing the reduction of substrate viscosity and the release of reducing sugars during reaction of the purified enzyme with CMC and barley β-glucan. Upon co-incubation of the enzyme and substrate, a rapid decrease in substrate viscosity was observed immediately, corresponding to a 65% and 71% reduction in viscosity after 12 min and 14.6 min for barley β-glucan and CMC, respectively, relative to the viscosities of similarly treated samples containing buffer instead of enzyme (Fig. 7). This rapid decrease in viscosity coincided with a slow increase in the concentration of reducing ends produced which increased gradually as the reactions proceeded, indicating endoglucanase activity. The ability of the crude enzyme to hydrolyse pretreated straw lignocellulose was assessed at 75, 80 and 85 °C. After 5 h, the amount of reducing sugars released was approximately twofold the amount released in the corresponding control samples without enzyme at each temperature.

Reduction in viscosity and production of reducing ends during reaction of the purified recombinant enzyme with CMC and barley β-glucan
Discussion
Thermally adapted enzymes produced by thermophiles are valuable resources in the development of a bio-based economy owing to their ability to tolerate the high temperatures and extreme conditions of many industrial processes [7]. In this study, a novel thermophilic endoglucanase was cloned from the thermophilic archaea S. shibatae, expressed in E. coli, purified and characterised. Recombinant enzyme production offers several advantages over production from the native source including the potential for producing larger quantities of the enzyme in a well-characterised host organism at a more industrially favourable mesophilic growth temperature [5]. In addition, recombinant production allows the attachment of affinity tag amino acid sequences to the enzyme facilitating affinity-based protein purification [5] and also facilitates potential improvement of the enzyme application-relevant properties by protein engineering [2].
The temperature and pH optima of the purified enzyme (95–100 °C (Fig. 3) and pH 3–5 (Fig. 4)) indicate adaptation to the environmental conditions from which S. shibatae was isolated and reflect the extreme cultivation conditions of the strain (75 °C and pH 3–3.5). Despite displaying high amino acid sequence similarity to the previously characterised S. solfataricus endoglucanases SSO1949 and SSO1354, the temperature optimum of the S. shibatae endoglucanase is significantly higher than that reported for S. solfataricus SSO1949 endoglucanase expressed in E. coli, which displays optimum activity at 80 °C with less than 45% and 20% of maximum activity observed at 95 and 100 °C, respectively [25]. The optimum temperature of S. shibatae endoglucanase is also slightly higher than that reported for S. solfataricus SSO1354 endoglucanase expressed in S. solfataricus [26] and in tobacco [27] (90 °C) or produced natively (95 °C) [28]. The pH profile of S. shibatae endoglucanase differs significantly from that reported for S. solfataricus SSO1949 endoglucanase which exhibits a bell-shaped pH profile with an optimum of approximately pH 1.8 and low activity at pH 3 and higher [25]. The pH optima reported for SSO1354 endoglucanase (pH 3.5 [28], pH 4 [26] and pH 4.5 [27]) are within the optimum pH range observed for S. shibatae endoglucanase but the later exhibits a higher proportion of its maximum activity in the pH range 2.7–3 than that reported for SSO1354 produced natively [28] or in tobacco [27] while comparative data is not available for SSO1354 expressed in S. solfataricus [26]. In addition to SSO1354 and SSO1949, the deduced amino acid sequence of S. shibatae endoglucanase showed high identity with several endoglucanase sequences from Sulfolobus islandicus strains. No S. islandicus strains are currently available from culture collections [29] and as there are no known reports on the production and characterisation of endoglucanase activity from S. islandicus, comparison of biochemical properties is not possible. Endoglucanases displaying optimal activity within the range 95–106 °C have been reported from Pyrococcus furiosus, Pyrococcus horikoshii and Thermotoga neapolitana [2].
The thermal stability of S. shibatae endoglucanase facilitated its partial purification by heat denaturation at 90 °C which induced denaturation and precipitation of native E. coli proteins while recombinant endoglucanase activity (72% yield) remained in the supernatant. The thermal stability of the purified S. shibatae endoglucanase (Fig. 5) is greater than that reported for commercial lignocellulosic enzymes, for example, Viikari et al. [15] reported rapid inactivation of commercial preparations during the first 2 h of hydrolysis at temperatures above 60 °C while Skovgaard and Jorgensen [30] reported a 50% reduction in activity after 15 min and 5 min at 60 °C and 65 °C, respectively. The purified enzyme also exhibits greater thermal stability than some previously reported thermophilic endoglucanases, including CelA and CelB from Alicyclobacillus acidocaldarius, with CelA having a half-life of 30 min at 75 °C [31] and CelB retaining 60% activity after 1 h at 80 °C [32] as well as Caldibacillus cellulovorans endoglucanase with half-lives of 32 min and 2 min at 80 °C and 85 °C, respectively [33], AcCEl12B endoglucanase from Acidothermus cellulolyticus 11B with a half-life of 12 min at 75 °C [34] and TeEgl5A endoglucanase from Talaromyces emersonii CBS394.64, which retained 50% and 30% activity after 1 h incubation at 80 and 85 °C, respectively [35]. The purified enzyme also exhibits greater thermal stability at 80 °C than two recently reported thermostable endoglucanases from Chaetomium thermophilum which retained 61.3% and 65.6% of original activity after 1 h at 80 °C [36]. Pyrococcus endoglucanases expressed in E. coli exhibit greater thermostability, with Pyrococcus horikoshii endoglucanase retaining 80% residual activity after 3 h at 97 °C [37] and Pyrococcus furiosus endoglucanase with a half-life of 40 h at 95 °C [38].
The thermal stability of purified S. shibatae endoglucanase at 80 °C is similar to that of S. solfataricus SSO1949 (expressed in E. coli), with no significant decrease in activity observed after 2 h [25] while the enzyme is less thermostable than S. solfataricus SSO1354 (expressed in S. solfataricus) which has a reported half-life of 180 min at 90 °C [26]. The poorer thermal stability of the S. shibatae endoglucanase, which has 12 potential N-glycosylation sites, can potentially be attributed to its production as a non-glycosylated protein in E. coli. Girfoglio et al. [26] expressed SSO1354 in S. solfataricus and E. coli and reported marked instability and lower specific activity in the case of the latter which was attributed to the lack of N-glycosylation which is found in extracellular proteins produced in Sulfolobus. The role of glycosylation in improving enzyme thermal stability has previously been reported, for example Candida guilliermondii invertase displayed 76–92% lower thermostability at 50–70 °C upon deglycosylation [39], deglycosylated β-glucuronidase from P. purpurogenum was inactivated after 2 h at 65 °C while the glycosylated enzyme retained 80% activity [40] and deglycosylation of A. niger phytase reduced thermostability by 34% [41]. The glycan moieties reportedly confer stability by decreasing flexibility and preventing aggregation at high temperatures likely via steric hindrance [40, 42].
The addition of non-ionic surfactants including Tween and Triton and the polymer PEG has been reported to improve the enzymatic hydrolysis of various lignocellulosic substrates by reducing adsorption of enzymes to lignin [43,44,45]. Addition of PEG4000 has also been reported to reduce liquefaction time, which is beneficial in terms of process energy demand [45]. The enhanced activity of S. shibatae endoglucanase observed in the presence of Tween, Triton and PEG (Table 1) is therefore beneficial in the context of potential application of the enzyme in the bioethanol industry. The reduced activity observed in the presence of Zn2+ (Table 1) has also been reported for other endo-glucanases [18, 46, 47]. The ability of the purified S. shibatae enzyme to hydrolyse CMC in addition to barley β-glucan and lichenan, both of which contain β-1,3 and β-1,4 linkages and its inability to hydrolyse laminarin, which consists mainly of β-1,3 linked d-glucose with some β-1,6 linkages, indicates that the enzyme hydrolyses β-1,4 linkages and based on the observed substrate specificity can be described as an endo-1,4-β-d-glucanase (EC 3.2.1.4). Glycoside hydrolase family 12 contains several other endo-1,4-β-d-glucanases. Activity was also observed on xylan which is composed of β-1,4-linked d-xylose units. The observed substrate specificity of the purified enzyme is broadly similar to that reported for SSO1354 which displays higher activity on lichenan than CMC and xylan, lacks exoglucanase activity but unlike S. shibatae endoglucanase also displays activity on curdlan [26]. The substrate specificity of the purified enzyme differs significantly from that of SSO1949 which hydrolyses CMC but shows no activity on lichenan or xylan under the assay conditions used [25]. The hydrolysis products observed upon TLC analysis (Fig. 6) indicate an endo- mode of action and are similar to those reported for SSO1354 and SSO1949 [25, 26]. The endoglucanase activity of the purified enzyme was further confirmed by the rapid decrease in substrate viscosity observed at the beginning of the enzyme reaction (Fig. 7). The Km value determined on barley β-glucan (1.77 mg/ml) is similar to that reported for thermostable HiCel6C β-1,4-glucanase from Humicola insolens (1.29 mg/ml) [48] and lower than those reported for GH family 12 endoglucanase Cel12A from Gleophyllum trabeum (3.2 mg/ml) [49] and three thermostable endo-β-glucanases from Talaromyces emersonii (9.1–28.6 mg/ml) [50], indicating higher substrate affinity.
The biochemical properties of S. shibatae endoglucanase strongly indicate suitability for industrial applications at high temperature. The release of reducing sugars observed upon incubation of the enzyme with pretreated straw at 75–85 °C indicate that the enzyme can hydrolyse this industrially-relevant substrate at high temperature. On the basis of its high temperature optimum (Fig. 3) and enhanced thermal stability (Fig. 5), the enzyme is more suitable for high temperature enzymatic hydrolysis of lignocellulose than currently used commercial enzymes and may be of interest for proposed high temperature liquefaction approaches prior to enzymatic hydrolysis. The observed substrate specificity is compatible with the enzymatic requirements for bioethanol production and brewing. The observed activity on xylan is favourable for bioethanol production where saccharification of both cellulose and xylan is desirable to provide glucose and xylose for fermentation while the observed activity on β-1,3–1,4-glucans would be suitable for the brewing industry where degradation of barley 1,3–1,4-β-glucans is required [17, 18]. The endo-acting activity of the purified enzyme and ability to rapidly decrease the viscosity of CMC and barley β-glucan at high temperature (Fig. 7) further underlines its potential suitability to reduce mash viscosity in brewing and to decrease the viscosity of pretreated lignocellulose with associated benefits for bioethanol production as outlined above. The broad pH profile of the purified enzyme (Fig. 4) allows flexibility in terms of operating pH, which would be favourable in terms of industrial application.
Conclusions
The identification of thermophilic enzymes exhibiting enhanced stability and activity at high temperature is desirable for the development of a biobased economy [2, 7]. Based on its thermophilic nature and biochemical properties, this previously unreported endoglucanase from S. shibatae may be of use in high temperature industrial processes such as in the prehydrolysis/liquefaction of pretreated lignocellulose, as a component of thermostable enzyme cocktails for enzymatic hydrolysis during the production of bioethanol or for glucan degradation during the brewing process. The purified enzyme differs in terms of its application-relevant properties, including the effect of temperature and pH on enzyme activity and substrate specificity, from previously characterised S. solfataricus endoglucanases. Identification of the sequence encoding this activity facilitates its overexpression in alternative hosts for further application-relevant assessment studies and industrial-scale production. In the case of the latter, for ease of enzyme recovery it would be desirable to use an expression system, such as B. subtilis or selected species of Aspergillus, capable of secreting the recombinant enzyme. This would eliminate the need for cell disruption which is required to release the intracellular enzyme upon expression in E. coli and which adds to the cost and complexity of industrial-scale production. Furthermore, expression in a host system also capable of performing glycosylation may improve the thermal stability of the enzyme as discussed above. With characterisation studies to date on Sulfolobus endoglucanases limited to those produced by S. solfataricus, this study provides characterisation data on a thermophilic endoglucanase from a distinct Sulfolobus species, extending our knowledge of the potential of this genus to produce enzymes of industrial interest.
References
Raddadi N, Cherif A, Daffonchio D, Neifar M, Fava F (2015) Biotechnological applications of extremophiles, extremozymes and extremolytes. Appl Microbiol Biotechnol 99(19):7907–7913. https://doi.org/10.1007/s00253-015-6874-9
Akram F, Haq IU, Imran W, Mukhtar H (2018) Insight perspectives of thermostable endoglucanases for bioethanol production: a review. Renewable Energy 122:225–238. https://doi.org/10.1016/j.renene.2018.01.095
Haq I, Akram F (2017) Enhanced production of a recombinant multidomain thermostable GH9 processive endo-1,4-β-glucanase (CenC) from Ruminiclostridium thermocellum in a mesophilic host through various cultivation and induction strategies. Appl Biochem Biotechnol 183(1):171–188. https://doi.org/10.1007/s12010-017-2437-0
Sharma A, Tewari R, Rana SS, Soni R, Soni SK (2016) Cellulases: classification, methods of determination and industrial applications. Appl Biochem Biotechnol 179(8):1346–1380. https://doi.org/10.1007/s12010-016-2070-3
Bhalla A, Bansal N, Kumar S, Bischoff KM, Sani RK (2013) Improved lignocellulose conversion to biofuels with thermophilic bacteria and thermostable enzymes. Bioresour Technol 128:751–759. https://doi.org/10.1016/j.biortech.2012.10.145
Dos Santos LV, De Barros Grassi MC, Gallardo JCM, Pirolla RAS, Calderón LL, De Carvalho-Netto OV, Parreiras LS, Camargo ELO, Drezza AL, Missawa SK, Teixeira GS, Lunardi I, Bressiani J, Pereira GAG (2016) Second-generation ethanol: the need is becoming a reality. Ind Biotechnol 12(1):40–57. https://doi.org/10.1089/ind.2015.0017
Kuhad RC, Deswal D, Sharma S, Bhattacharya A, Jain KK, Kaur A, Pletschke BI, Singh A, Karp M (2016) Revisiting cellulase production and redefining current strategies based on major challenges. Renewable Sustain Energy Rev 55:249–272. https://doi.org/10.1016/j.rser.2015.10.132
Irfan M, Tayyab A, Hasan F, Khan S, Badshah M, Shah AA (2017) Production and characterization of organic solvent-tolerant cellulase from Bacillus amyloliquefaciens AK9 isolated from hot spring. Appl Biochem Biotechnol 182(4):1390–1402. https://doi.org/10.1007/s12010-017-2405-8
Jain KK, Kumar S, Deswal D, Kuhad RC (2017) Improved production of thermostable cellulase from Thermoascus aurantiacus RCKK by fermentation bioprocessing and its application in the hydrolysis of office waste paper, algal pulp, and biologically treated wheat straw. Appl Biochem Biotechnol 181(2):784–800. https://doi.org/10.1007/s12010-016-2249-7
Kazeem MO, Shah UKM, Baharuddin AS, AbdulRahman NA (2017) Prospecting agro-waste cocktail: supplementation for cellulase production by a newly isolated thermophilic B-licheniformis 2D55. Appl Biochem Biotechnol 182(4):1318–1340. https://doi.org/10.1007/s12010-017-2401-z
Elleuche S, Schroder C, Sahm K, Antranikian G (2014) Extremozymes-biocatalysts with unique properties from extremophilic microorganisms. Curr Opin Biotechnol 29:116–123. https://doi.org/10.1016/j.copbio.2014.04.003
Guerriero G, Hausman J-F, Strauss J, Ertan H, Siddiqui KS (2015) Destructuring plant biomass: focus on fungal and extremophilic cell wall hydrolases. Plant Sci 234:180–193. https://doi.org/10.1016/j.plantsci.2015.02.010
Kallioinen A, Puranen T, Siika-Aho M (2014) Mixtures of thermostable enzymes show high performance in biomass saccharification. Appl Biochem Biotechnol 173(5):1038–1056. https://doi.org/10.1007/s12010-014-0893-3
Szijarto N, Horan E, Zhang J, Puranen T, Siika-aho M, Viikari L (2011) Thermostable endoglucanases in the liquefaction of hydrothermally pretreated wheat straw. Biotechnol Biofuels 4:2
Viikari L, Vehmaanpera J, Koivula A (2012) Lignocellulosic ethanol: from science to industry. Biomass Bioenerg 46:13–24. https://doi.org/10.1016/j.biombioe.2012.05.008
Miller PS, Blum PH (2010) Extremophile-inspired strategies for enzymatic biomass saccharification. Environ Technol 31(8–9):1005–1015. https://doi.org/10.1080/09593330903536113
Bai Y, Wang J, Zhang Z, Shi P, Luo H, Huang H, Luo C, Yao B (2010) A novel family 9 beta-1,3(4)-glucanase from thermoacidophilic Alicyclobacillus sp A4 with potential applications in the brewing industry. Appl Microbiol Biotechnol 87(1):251–259. https://doi.org/10.1007/s00253-010-2452-3
Wang J, Niu C, Liu X, Chen X, Li Q (2014) Characterization of a new 1,3–1,4-β-glucanase gene from Bacillus tequilensis CGX5-1. Appl Biochem Biotechnol 173(3):826–837. https://doi.org/10.1007/s12010-014-0900-8
Grogan D, Palm P, Zillig W (1990) Isolate-B12, which harbors a virus-like element, represents a new species of the archaebacterial genus Sulfolobus, Sulfolobus shibatae, sp-nov. Arch Microbiol 154(6):594–599. https://doi.org/10.1007/BF00248842
Sambrook J, Russell DW (2001) Molecular cloning. A laboratory manual, 3 edn. Cold Spring Harbor Laboratory Press, New York
Bradford MM (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem 72(1–2):248–254. https://doi.org/10.1006/abio.1976.9999
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685. https://doi.org/10.1038/227680a0
Boyce A, Walsh G (2015) Characterisation of a novel thermostable endoglucanase from Alicyclobacillus vulcanalis of potential application in bioethanol production. Appl Microbiol Biotechnol 99(18):7515–7525. https://doi.org/10.1007/s00253-015-6474-8
Miller GL (1959) Use of dinitrosalicyclic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428. https://doi.org/10.1021/ac60147a030
Huang YW, Krauss G, Cottaz S, Driguez H, Lipps G (2005) A highly acid-stable and thermostable endo-beta-glucanase from the thermoacidophilic archaeon Sulfoloblus solfataricus. Biochem J 385:581–588. https://doi.org/10.1042/bj20041388
Girfoglio M, Rossi M, Cannio R (2012) Cellulose degradation by Sulfolobus solfataricus requires a cell-anchored endo-beta-1-4-glucanase. J Bacteriol 194(18):5091–5100. https://doi.org/10.1128/jb.00672-12
Klose H, Roeder J, Girfoglio M, Fischer R, Commandeur U (2012) Hyperthermophilic endoglucanase for in planta lignocellulose conversion. Biotechnol Biofuels. https://doi.org/10.1186/1754-6834-5-63
Maurelli L, Giovane A, Esposito A, Moracci M, Fiume I, Rossi M, Morana A (2008) Evidence that the xylanase activity from Sulfolobus solfataricus O alpha is encoded by the endoglucanase precursor gene (sso1354) and characterization of the associated cellulase activity. Extremophiles 12(5):689–700. https://doi.org/10.1007/s00792-008-0175-5
Zeldes BM, Keller MW, Loder AJ, Straub CT, Adams MWW, Kelly RM (2015) Extremely thermophilic microorganisms as metabolic engineering platforms for production of fuels and industrial chemicals. Front Microbiol. https://doi.org/10.3389/fmicb.2015.01209
Skovgaard PA, Jorgensen H (2013) Influence of high temperature and ethanol on thermostable lignocellulolytic enzymes. J Ind Microbiol Biotechnol 40(5):447–456. https://doi.org/10.1007/s10295-013-1248-8
Eckert K, Zielinski F, Lo Leggio L, Schneider E (2002) Gene cloning, sequencing, and characterization of a family 9 endoglucanase (CeIA) with an unusual pattern of activity from the thermoacidophile Alicyclobacillus acidocaldarius ATCC27009. Appl Microbiol Biotechnol 60(4):428–436. https://doi.org/10.1007/s00253-002-1131-4
Eckert K, Schneider E (2003) A thermoacidophilic endoglucanase (CelB) from Alicyclobacillus acidocaldarius displays high sequence similarity to arabinofuranosidases belonging to family 51 of glycoside hydrolases. Eur J Biochem 270(17):3593–3602. https://doi.org/10.1046/j.1432-1033.2003.03744.x
Huang XP, Monk C (2004) Purification and characterization of a cellulase (CMCase) from a newly isolated thermophilic aerobic bacterium Caldibacillus cellulovorans gen. nov., sp nov. World J Microbiol Biotechnol 20(1):85–92. https://doi.org/10.1023/B:WIBI.0000013316.12730.e7
Wang J, Gao G, Li Y, Yang L, Liang Y, Jin H, Han W, Feng Y, Zhang Z (2015) Cloning, Expression, and characterization of a thermophilic endoglucanase, AcCel12B from Acidothermus cellulolyticus 11B. Int J Mol Sci 16(10):25080–25095. https://doi.org/10.3390/ijms161025080
Wang K, Luo H, Bai Y, Shi P, Huang H, Xue X, Yao B (2014) A thermophilic endo-1,4-beta-glucanase from Talaromyces emersonii CBS394.64 with broad substrate specificity and great application potentials. Appl Microbiol Biotechnol 98(16):7051–7060. https://doi.org/10.1007/s00253-014-5680-0
Hua C, Li W, Han W, Wang Q, Bi P, Han C, Zhu L (2018) Characterization of a novel thermostable GH7 endoglucanase from Chaetomium thermophilum capable of xylan hydrolysis. Int J Biol Macromol 117:342–349. https://doi.org/10.1016/j.ijbiomac.2018.05.189
Ando S, Ishida H, Kosugi Y, Ishikawa K (2002) Hyperthermostable endoglucanase from Pyrococcus horikoshii. Appl Environ Microbiol 68(1):430–433. https://doi.org/10.1128/AEM.68.1.430-433.2002
Bauer MW, Driskill LE, Callen W, Snead MA, Mathur EJ, Kelly RM (1999) An endoglucanase, eg1A, from the hyperthermophilic archaeon Pyrococcus furiosus hydrolyzes β-1,4 bonds in mixed-linkage (1→3),(1→4)-β-D-glucans and cellulose. J Bacteriol 181(1):284–290
Plascencia-Espinosa M, Santiago-Hernández A, Pavón-Orozco P, Vallejo-Becerra V, Trejo-Estrada S, Sosa-Peinado A, Benitez-Cardoza CG, Hidalgo-Lara ME (2014) Effect of deglycosylation on the properties of thermophilic invertase purified from the yeast Candida guilliermondii MpIIIa. Process Biochem 49(9):1480–1487. https://doi.org/10.1016/j.procbio.2014.05.022
Zou S, Huang S, Kaleem I, Li C (2013) N-glycosylation enhances functional and structural stability of recombinant β-glucuronidase expressed in Pichia pastoris. J Biotechnol 164(1):75–81. https://doi.org/10.1016/j.jbiotec.2012.12.015
Han YM, Lei XG (1999) Role of glycosylation in the functional expression of an Aspergillus niger phytase (phyA) in Pichia pastoris. Arch Biochem Biophys 364(1):83–90. https://doi.org/10.1006/abbi.1999.1115
Hoiberg-Nielsen R, Fuglsang CC, Arleth L, Westh P (2006) Interrelation ships of glycosylation and aggregation kinetics for Peniophora lycii phytase. Biochemistry 45(15):5057–5066. https://doi.org/10.1021/bi0522955
Méndez Arias J, de Oliveira Moraes A, Modesto LFA, de Castro AM, Pereira N (2017) Addition of surfactants and non-hydrolytic proteins and their influence on enzymatic hydrolysis of pretreated sugarcane bagasse. Appl Biochem Biotechnol 181(2):593–603. https://doi.org/10.1007/s12010-016-2234-1
Parnthong J, Kungsanant S, Chavadej S (2018) The influence of nonionic surfactant adsorption on enzymatic hydrolysis of oil palm fruit bunch. Appl Biochem Biotechnol 1–14. https://doi.org/10.1007/s12010-018-2783-6
Rocha-Martín J, Martinez-Bernal C, Pérez-Cobas Y, Reyes-Sosa FM, García BD (2017) Additives enhancing enzymatic hydrolysis of lignocellulosic biomass. Bioresour Technol 244:48–56. https://doi.org/10.1016/j.biortech.2017.06.132
Deep K, Poddar A, Das SK (2016) Cloning, overexpression, and characterization of halostable, solvent-tolerant novel β-endoglucanase from a marine bacterium Photobacterium panuliri LBS5T (DSM 27646T). Appl Biochem Biotechnol 178(4):695–709. https://doi.org/10.1007/s12010-015-1903-9
Huang XM, Li QQ, Chen XL, Fan JX, Xu XH, Sun XD, Li DY, Zhao HX (2017) Expression and characteristics of an endoglucanase from Trichoderma atroviride (TaEGII) in Saccharomyces cerevisiae. Appl Biochem Biotechnol 182(3):1158–1170. https://doi.org/10.1007/s12010-016-2389-9
Xu X, Li J, Zhang W, Huang H, Shi P, Luo H, Liu B, Zhang Y, Zhang Z, Fan Y, Yao B (2015) A neutral thermostable beta-1,4-glucanase from Humicola insolens Y1 with potential for applications in various industries. PLoS ONE. https://doi.org/10.1371/journal.pone.0124925
Miotto LS, de Rezende CA, Bernardes A, Serpa VI, Tsang A, Polikarpov I (2014) The characterization of the endoglucanase Cel12A from Gloeophyllum trabeum reveals an enzyme highly active on beta-glucan. PLoS ONE. https://doi.org/10.1371/journal.pone.0108393
McCarthy T, Hanniffy O, Savage AV, Tuohy MG (2003) Catalytic properties and mode of action of three endo-beta-glucanases from Talaromyces emersonii on soluble beta-1,4- and beta-1,3–1,4-linked glucans. Int J Biol Macromol 33(1–3):141–148. https://doi.org/10.1016/s0141-8130(03)00080-1
Acknowledgements
This work was supported by Science Foundation Ireland/Enterprise Ireland Technology Innovation Development Award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Boyce, A., Walsh, G. Expression and characterisation of a thermophilic endo-1,4-β-glucanase from Sulfolobus shibatae of potential industrial application. Mol Biol Rep 45, 2201–2211 (2018). https://doi.org/10.1007/s11033-018-4381-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4381-7