
Abstract
A novel endoglucanase encoding gene was cloned from Alicyclobacillus vulcanalis and expressed in E. coli. The deduced amino acid sequence showed highest identity with α-l-arabinofuranosidase-like proteins from glycoside hydrolase family 51. The recombinant enzyme was purified by affinity chromatography and characterised in terms of its potential suitability for lignocellulose hydrolysis at high temperature in the production of bioethanol. The purified enzyme displayed maximum activity at 80 °C and pH 3.6–4.5. Tween 20 was found to have a beneficial effect on enzyme activity and thermal stability. When incubated in the presence of 0.1 % Tween 20, the enzyme retained full activity after 72 h at 70 °C and 78 % of original activity after 72 h at 75 °C. Maximum activity was observed on carboxymethyl cellulose, and the purified enzyme also hydrolysed lichenan, barley β-glucan and xylan. The purified enzyme decreased the viscosity of carboxymethyl cellulose when assessed at 70–85 °C and was capable of releasing reducing sugars from acid-pretreated straw at 70 and 75 °C. The results indicate the potential suitability of the enzyme for industrial application in the production of cellulosic bioethanol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellulosic bioethanol, a renewable transport fuel produced from non-food lignocellulosic material, has received increasing global attention as a sustainable alternative to fossil fuels. The widespread use of cellulosic bioethanol is highly desirable to decrease greenhouse gas emissions, reduce dependency on fossil fuels, improve energy security and meet the ambitious targets set for biofuels in most world regions (Viikari et al. 2012; Menon and Rao 2012). The production of bioethanol from lignocellulosic material involves an initial pretreatment step to disrupt the lignocellulose structure, followed by enzymatic hydrolysis of the cellulose and hemicellulose components to their constituent monomeric sugars, which are then fermented to produce bioethanol. The enzymatic hydrolysis of lignocellulose, which is composed of cellulose, hemicellulose and lignin, requires multiple enzymes with different specificities acting in synergy (Bhalla et al. 2013). One of the main enzymes involved is endoglucanase (EC 3.2.1.4) which randomly hydrolyses accessible internal β-1,4-glucosidic bonds in cellulose chains (Wang et al. 2014).
The production of cellulosic bioethanol is technically challenging due to the recalcitrance of lignocellulose and improvements in process efficiency, and economics are required to make cellulosic bioethanol more cost-competitive (Viikari et al. 2012). Thermostable lignocellulose-degrading enzymes display realistic potential to address current limitations and contribute towards process improvement (Bhalla et al. 2013; Miller and Blum 2010). Existing enzymatic hydrolysis steps employ mesophilic enzymes which are optimally active at 50–55 °C (Singhania et al. 2010) and exhibit poor stability and decreased enzyme performance due to inactivation during the hydrolysis period, requiring high enzyme dosages (Bhalla et al. 2013). The use of thermostable enzymes, which have enhanced stability and activity at high temperatures, would result in improved hydrolysis performance, decreased enzyme dosage and reduced hydrolysis time (Viikari et al. 2012; Elleuche et al. 2014). Enzymatic hydrolysis at higher temperatures would also likely promote increased substrate solubility and biomass disorganisation, improved rheological properties and lower viscosity as well as reducing the risk of microbial contamination (Bhalla et al. 2013; Viikari et al. 2012; Turner et al. 2007). In addition, enzymatic hydrolysis at high temperature is more compatible with commonly employed high temperature pretreatment methods reducing the need for costly biomass cooling after pretreatment (Elleuche et al. 2014; Karnaouri et al. 2014). Thermostable enzymes have also been reported to be more suitable for potential enzyme recovery and recycling than mesophilic enzymes due to their enhanced tolerance to temperature and ethanol (Skovgaard and Jorgensen 2013).
The use of thermostable enzymes facilitates various modifications of existing lignocellulose to bioethanol process configurations. A partial prehydrolysis or liquefaction step at high temperature has been proposed whereby thermostable cellulolytic enzyme activity is used to decrease the viscosity of the substrate prior to the hydrolysis step (Viikari et al. 2012). This could potentially address some of the problems associated with the high initial solids loading (15–20 % dry matter) required to achieve an economically feasible final ethanol concentration in the fermentation broth (>4 % w/w), which include poor mixing and mass transfer conditions due to the high viscosity (Szijarto et al. 2011a, b). Endoglucanase has been identified as the enzyme activity with greatest liquefaction ability (Szijarto et al. 2011b) with its action on cellulose resulting in a decrease in chain length and hence viscosity (Karnaouri et al. 2014). Ideally, the enzyme(s) should also contribute to substrate hydrolysis, reducing the enzyme requirement in the subsequent hydrolysis step (Szijarto et al. 2011a). Optimisation of this approach is expected to decrease total enzyme use and cost in cellulosic bioethanol production (Szijarto et al. 2011a) while facilitating high-solids loadings with associated benefits in terms of increased sugar and ethanol yields, decreased water and energy use and overall improved process efficiency (Modenbach and Nokes 2012). Thermostable enzymes are also of interest for use in ‘thermophilic SSF’, whereby hydrolysis and fermentation are undertaken in a single reactor at high temperature using fermentative thermophiles to achieve more cost-effective processing (Bhalla et al. 2013).
Bioprospecting for novel thermostable lignocellulose-degrading enzymes suitable for application in bioethanol production is therefore of growing interest. Members of the genus Alicyclobacillus are heterotrophic, Gram-positive, thermoacidophilic bacteria and inhabit mostly acidic geothermal environments (Simbahan et al. 2004). Alicyclobacillus species are a source of thermostable glycosyl hydrolases, and some have been reported to produce lignocellulose-degrading enzymes. These include a family 9 endoglucanase (CelA) (Eckert et al. 2002), a family 51 endoglucanase (CelB) (Eckert and Schneider 2003) and a cellulase (CelG) (Morana et al. 2008) from Alicyclobacillus acidocaldarius, and an acidic β-1,4-glucanase (CelA4) (Bai et al. 2010a, b) and a family 9 β-1,3(4)-glucanase (Agl9A) (Bai et al. 2010c) from Alicyclobacillus sp. A4, with temperature optima ranging from 55 to 80 °C. This paper describes the cloning, expression, purification and characterisation of a thermostable endoglucanase from Alicyclobacillus vulcanalis (DSM 16176), which was originally isolated from an acidic geothermal pool in Mojave Desert, CA, and grows optimally at 55 °C and pH 4 (Simbahan et al. 2004). The purified enzyme was assessed in terms of its potential application in the production of cellulosic bioethanol. This is the first known report of the characterisation of a lignocellulose-degrading enzyme from this strain.
Materials and methods
Materials and strains
All chemicals used were obtained from Sigma-Aldrich Ireland Limited or Fisher Scientific Ireland Ltd, unless otherwise stated. Carboxymethyl cellulose (CMC) 4M, barley β-glucan, CM-curdlan and cello-oligosaccharides were obtained from Megazyme, Ireland. Restriction enzymes were from Roche Diagnostics Ltd., UK. T4 DNA Ligase and AcTEV Protease were from Invitrogen. A. vulcanalis (DSM 16176) was obtained from DSMZ German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany, and was routinely grown at 55 °C and pH 4. E. coli DH5α, used for gene cloning and recombinant protein production, was routinely grown at 37 °C in Luria-Bertani medium.
Cloning and expression of A. vulcanalis endoglucanase
A. vulcanalis genomic DNA was prepared using QIAGEN DNeasy Blood and Tissue Kit. The gene encoding endoglucanase was amplified by PCR using TaKaRa Ex Taq with the following primers: CelP_For GTATCTAGAATGAGTGTCCACAGCGCGGCCACG and CelP_Rev GGGAAGCTTTCAGCTGATCACGAAGTTGACCAC. The primers were designed using Alicyclobacillus sp. nucleotide sequence (GenBank DJ359535.1) and incorporated recognition sites for XbaI and HindIII (underlined). The PCR product was purified (QIAGEN QIAquick PCR Purification Kit), digested with XbaI and HindIII and ligated into the corresponding sites of the vector pProEX HTb using T4 DNA ligase. The ligation mixture was transformed into E. coli DH5α competent cells by the CaCl2 method (Sambrook and Russell 2001). Positive clones were identified by restriction enzyme analysis and small-scale expression studies and submitted to Eurofins Genomics for sequencing. Database searches were undertaken using the National Center for Biotechnology Education BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi ). The deduced amino acid sequence was analysed using EXPASY tools (http://www.expasy.org/). Sequence alignments were undertaken using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). A phylogenetic tree was constructed using the neighbour-joining method with 1000 replicates in MEGA 6 (http://www.megasoftware.net/) based on the amino acid sequences of A. vulcanalis endoglucanase and 31 representative glycoside hydrolase family 51 (GH51) members from bacteria. Optimisation of expression conditions to achieve maximum enzyme yield was undertaken with respect to media (Luria-Bertani, Terrific broth), isopropyl-β-d-thio-galactopyranoside (IPTG) concentration (0.01–0.2 mM), temperature (18, 25, and 37 °C) and time (4–24 h). For optimum expression, an overnight culture grown at 37 °C and 250 rpm in Luria-Bertani medium with 100 μg/ml ampicillin was diluted 1 in 100 in the same medium and grown to an optical density of 0.6 at 600 nm. IPTG was added to a final concentration of 0.01 mM, and the culture was incubated at 18 °C, 250 rpm for 20 h. Cells were harvested by centrifugation (4500g, 10 min, 4 °C).
Purification of recombinant endoglucanase
Cells were disrupted by sonication in 50 mM Tris–HCl, pH 7, and the cell debris was removed by centrifugation (12,000g, 20 min, 4 °C). Purification was undertaken by immobilized metal ion affinity chromatography (IMAC) using Chelating Sepharose Fast Flow charged with Ni2+ (GE Healthcare). After washing with 20 mM sodium phosphate, 500 mM NaCl, pH 7, bound protein was removed by a stepwise decrease in pH with the protein of interest eluted using 20 mM sodium phosphate, 500 mM NaCl, pH 4.55. For removal of the polyhistidine affinity tag, pooled activity was incubated with AcTEV Protease at 4 °C according to the supplier’s instructions, and the recombinant cellulase was purified from the cleavage reaction by IMAC chromatography. Endoglucanase activity was determined as outlined below. Protein concentration was determined by the method of Bradford (1976). Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using a 10 % gel, as described by Laemmli (1970). Protein staining was carried out using EZBlue Gel Staining Reagent. For zymogram analysis, samples were heated at 80 °C for 5 min prior to running on a 10 % gel containing 0.1 % CMC. After electrophoresis, the gel was washed (2 × 15 min) in 4:1 50 mM sodium acetate buffer, pH 4: isopropyl alcohol followed by washing (2 × 15 min) in 50 mM sodium acetate buffer, pH 4. After incubation at 80 °C, pH 4 for 60 min, bands of substrate hydrolysis were observed by staining with 0.1 % (w/v) Congo Red for 30 min, followed by destaining with 1 M NaCl.
Endoglucanase assay
Endoglucanase activity was determined by measuring the amount of reducing sugars released from 1 % (w/v) CMC in 0.5 M sodium acetate buffer, pH 4, using the dinitrosalicyclic acid (DNS) method of Miller (1959). The enzyme assay was undertaken at 80 °C and pH 4, unless otherwise stated. One unit of enzyme activity was defined as the amount of enzyme that catalyses the production of 1 μmol reducing sugar (as glucose equivalents) per minute under the assay conditions.
Biochemical characterisation of the purified recombinant enzyme
The effect of temperature on enzyme activity was determined by measuring activity on CMC, as outlined above, at 50–94 °C. The effect of pH on enzyme activity was determined by measuring activity at pH 2–5.5 at 80 °C using the following buffers: KCl-HCl (pH 2), glycine-HCl (pH 2.45–3.15) and sodium acetate-acetic acid (pH 3.6–5.5). The effect of various additives on enzyme activity was determined by preparing the enzyme in a final concentration of 10 mM CaCl2, 10 mM MgCl2, 2 mM EDTA, 0.1 % Tween 20 or 0.5 % Triton X-100 and immediately measuring the enzyme activity of each additive-containing enzyme sample as outlined above. Enzyme activity was expressed as a percentage of the activity of a similarly prepared enzyme sample containing distilled water instead of additive. Enzyme thermal stability was assessed by measuring residual endo-β-glucanase activity after heating at 70, 75 or 80 °C for the indicated time period in 50 mM sodium acetate-acetic acid buffer, 0.1 % Tween 20, pH 4. Substrate specificity was determined by measuring the release of reducing sugars from the following substrates (1 % (w/v)) in 0.5 M sodium acetate buffer, pH 4 at 80 °C: CMC, lichenan (Cetraria islandica), barley β-glucan, beechwood xylan, birchwood xylan, laminarin (Laminaria digitata), starch, CM-curdlan, avicel and filter paper. Determination of 1,4-β-d-xylosidase, β-d-glucosidase, α-d-galactosidase and α-l-arabinofuranosidase activities was undertaken by measuring the release of p-nitrophenol from p-nitrophenyl β-d-xylopyranoside, p-nitrophenyl β-d-1,4-glucopyranoside, 4-nitrophenyl-α-d-galactopyranoside and 4-nitrophenyl-α-l-arabinofuranoside, respectively, at 80 °C and pH 4. One unit of enzyme activity is defined as the amount of enzyme that releases 1 μmol p-nitrophenol per minute under the assay conditions. Kinetic constants were measured at 80 °C, pH 4, using 1–12 mg/ml CMC as substrate. GraphPad Prism Version 6.05 (GraphPad Software, Inc.) was used to determine K m and V max from the data generated.
Analysis of hydrolysis products by thin-layer chromatography
Enzyme reactions were undertaken at 80 °C and contained an equal volume of enzyme (2.45 units/ml) and substrate (1 % CMC or 8.25 mg/ml cello-oligosaccharide in 0.5 M sodium acetate buffer, pH 4). At the indicated time points, a 5 μl aliquot of the reaction mixture was spotted onto a silica gel plate and separated by migration in butanol/ethanol/water (5:3:2). Hydrolysis products were visualised by spraying with sulphuric acid/ethanol (1:9) and heating in an oven at 100 °C.
Viscometric assay
Viscometric assay of enzyme activity was undertaken at 70–85 °C using 1 % (w/v) CMC as substrate, based on the methods of Bathgate (1979) and McCleary (2001) with some modifications. The viscosity of the enzyme reaction mixture was measured over a 10-min period using a miniature U-tube viscometer. Enzyme activity was calculated in inverse reciprocal viscosity units (IRVU), where 1 IRVU is the increase in reciprocal viscosity per hour per milliliter of enzyme under the assay conditions.
Hydrolysis of pretreated straw
Ten percent (w/v) straw was pretreated in 0.75 % (w/v) sulphuric acid at 121 °C for 15 min and washed extensively with distilled water prior to enzymatic hydrolysis. For hydrolysis, the straw was incubated with the purified enzyme (27 units/g straw) in 50 mM sodium acetate buffer, 0.1 % Tween 20, pH 4 at 70 or 75 °C. The amount of reducing sugars released after 3 h was quantified using the DNS method of Miller (1959). A control hydrolysis reaction was also undertaken using distilled water instead of enzyme.
Nucleotide sequence accession number
The nucleotide sequence of A. vulcanalis endoglucanase was deposited in the EMBL database under accession number LN626709.
Results
Sequence analysis
The cloned DNA fragment showed the highest nucleotide sequence identity (79 %) to A. acidocaldarius celB gene for cellulase precursor (AJ551527.1) and was predicted to encode a protein of 936 amino acids with a deduced molecular mass of 98,466 Da and a calculated pI of 4.30. The deduced amino acid sequence showed the highest identities of 81 and 77 % with alpha-l-arabinofuranosidase-like proteins from A. acidocaldarius subsp. acidocaldarius DSM 446 (ACV57112.1) (identical to CelB) and A. acidocaldarius subsp. acidocaldarius Tc-4-1 (AEJ42043.1), respectively. Both proteins belong to GH51, members of which cleave the glycosidic bond with a retention of the anomeric configuration. The deduced amino acid sequence showed 42 % identity with Alicyclobacillus sp. A4 β-1,4-glucanase (ADI82825.1), CelA4, characterised by Bai et al. (2010a, 2010b) which is also a member of GH51. Additional matches included several α-l-arabinofuranosidases, which catalyse the hydrolysis of α-1,2 and α-1,3 l-arabinofuranosidic bonds in hemicelluloses. Sequence analysis indicates that A. vulcanalis endoglucanase does not contain a carbohydrate-binding domain. Sequence alignment with family 51 glycoside hydrolases is shown in supplementary material (Fig. S1). The deduced amino acid sequence of A. vulcanalis endoglucanase (Thr199 to Pro679) shows 88 % identity with the central catalytic domain (Thr223 to Pro702) identified in CelB by sequence comparison with GH51 members (Eckert and Schneider 2003). Conserved amino acids include the proposed acid/base catalyst (Glu366) and catalytic nucleophile residue (Glu510) of CelB. Phylogenetic tree analysis (Fig. 1) shows that A. vulcanalis endoglucanase forms a distinct cluster with the only four enzymes characterised as endoglucanases in GH family 51 and of these is most closely related with A. acidocaldarius subsp. acidocaldarius DSM 446 (ACV57112.1) (CelB).

Phylogenetic tree showing the relationship between the amino acid sequences of A. vulcanalis endoglucanase, GH51 members with endoglucanase activity and representative GH51 members with α-l-arabinofuranosidase activity
Cloning and expression
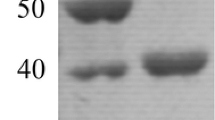
The DNA fragment amplified from A. vulcanalis by PCR was cloned into pProEX HTb and expressed in E. coli with a N-terminal His-tag. Upon optimisation of expression conditions, maximum enzyme yield was observed following induction with 0.01 mM IPTG at 18 °C for 20 h. Under these conditions, endoglucanase activity of 13.3 units/mg protein was detected in the cell lysate while no endoglucanase activity was detected in the cell lysate from a transformant containing empty pProEX HTb vector. The recombinant His-tagged endoglucanase was purified to apparent homogeneity by a single IMAC chromatography step with a purification factor of 35 and a percentage yield of 43 %. The specific activity of the purified enzyme was 469.2 units/mg. Upon SDS-PAGE analysis, the purified enzyme migrated as a single band of molecular mass similar to the calculated value (Fig. 2a). The endoglucanase activity of the purified enzyme was confirmed by zymogram analysis, with a band of CMC hydrolysis observed corresponding to the position of the purified enzyme (Fig. 2b).

a SDS-PAGE analysis of purification: M, protein molecular weight marker, masses indicated alongside; lane 1, crude enzyme preparation; lane 2, purified enzyme. b Zymogram analysis of purified enzyme on a CMC-containing gel: lane 1, stained for protein; lane 2, stained for endoglucanase activity using Congo Red
Effect of temperature, pH and additives
The purified enzyme displays maximum activity at 80 °C and exhibits over 80 % of its maximum activity in the temperature range 70–85 °C, with 40 % maximum activity observed at 90 °C (Fig. 3). The purified enzyme exhibited maximum activity at pH 3.6–4.5, with 90 % maximum activity observed at pH 3.15 (Fig. 4). Enzyme solutions containing 10 mM CaCl2 or 2 mM EDTA displayed 97 and 98 % activity, respectively, relative to the control enzyme solution without additives while 10 mM MgCl2 had a slight inhibitory effect on enzyme activity with 86 % activity observed relative to the control (Table 1). Inclusion of 0.1 % Tween 20 or 0.5 % Triton X-100 in the enzyme solution had a significant beneficial effect on enzyme activity with 124 and 126 % activity observed, respectively, relative to the control enzyme without additive. Inclusion of 0.1 % Tween 20 or Triton X-100 in the enzyme solution was also found to have a positive effect on enzyme thermal stability. Residual activity determined after incubating the enzyme at 70 °C for 2 h was 70 % of original activity in comparison with 103 and 105 % of original activity upon inclusion of 0.1 % Tween 20 and 0.1 % Triton X-100, respectively. Due to the observed beneficial effect on enzyme activity and thermal stability, Tween 20 was added to the purified enzyme at a final concentration of 0.1 % for all further thermal stability studies. Under these conditions, the enzyme was found to be extremely thermostable at 70 °C, retaining full activity after 72 h while 78 % of original activity was observed after 72 h at 75 °C (Fig. 5). At 80 °C, residual enzyme activity determined after 30, 60 and 120 min was 46, 23 and 6 %, respectively.

Temperature versus activity profile of the purified recombinant endoglucanase. Data expressed as a percentage of maximum activity. Each value represents mean ± SD (n = 3)

pH versus activity profile of the purified recombinant endoglucanase. Data expressed as a percentage of maximum activity. Each value represents mean ± SD (n = 3)

Thermal stability of the purified recombinant endoglucanase at 70 and 75 °C in 50 mM sodium acetate buffer, 0.1 % Tween 20, pH 4. Data expressed as percentage activity remaining after heating at the indicated conditions, relative to the activity of an unheated enzyme sample. Error bars represent mean ± SD (n = 3)
Substrate specificity and kinetic parameters
Maximum enzyme activity was observed on CMC with 94 and 92 % of maximum activity observed on lichenan and barley β-glucan, respectively. The purified enzyme also displayed activity on beechwood xylan and birchwood xylan (34 and 31 % of activity on CMC, respectively). No activity was detected on the other substrates tested, under the conditions used. Upon determination of kinetic parameters using CMC as substrate, the purified enzyme had an apparent K m of 4.358 mg/ml and a V max of 0.29 μmol/min.
Analysis of hydrolysis products
The products produced upon hydrolysis of CMC and cello-oligosaccharides by the purified enzyme were analysed by thin-layer chromatography (Fig. 6). Upon incubation under optimum enzyme conditions (80 °C, pH 4), no detectable activity was observed towards cellobiose while a small amount of cellobiose was observed upon hydrolysis of cellotriose. The enzyme hydrolysed cellotetraose and cellopentose producing cellobiose and cellotriose. No glucose was detected in any of the reactions. Analysis of the hydrolysis of CMC by A. vulcanalis endoglucanase (Fig. 6b) shows the production of a mixture of cellobiose, cellotriose, cellotetraose and cellopentose after 2 h.

Analysis of hydrolysis products by thin-layer chromatography. a Hydrolysis products observed upon incubation (80 °C, pH 4, 16 h) of purified endo-β-glucanase with individual cello-oligosaccharides (lane 1, cellobiose; lane 2, cellotriose; lane 3, cellotetraose; lane 4, cellopentose). b Hydrolysis products observed upon incubation (80 °C, pH 4) of purified endo-β-glucanase with CMC for 2 h (lane 1). S oligosaccharide standards, G glucose, C2 cellobiose, C3 cellotriose, C4 cellotetraose, C5 cellopentose
Viscometric assay and hydrolysis of straw
The endoglucanase activity of the purified enzyme was confirmed by viscometric assay using CMC as substrate. Upon incubation of the enzyme with CMC at 70, 75, 80 or 85 °C, the viscosity of the reaction mixture decreased rapidly while that of the control reaction without enzyme remained constant. When the results were expressed in inverse reciprocal viscosity units, maximum enzyme activity was observed at 80 °C with 84 % maximum activity observed at 85 °C. The ability of the purified enzyme to hydrolyse pretreated straw lignocellulose at elevated temperature was assessed at 70 and 75 °C. After 3 h, the amount of reducing sugars released by the enzyme at 70 and 75 °C was 88.55 ± 2.96 and 94.58 ± 10.10 μmol/g straw, respectively, relative to 57.01 ± 4.68 and 57.29 ± 1.49 μmol/g straw for the corresponding control samples without enzyme.
Discussion
Thermostable lignocellulose-degrading enzymes have been identified as a means of achieving improved process efficiency and economics in the production of cellulosic bioethanol. In this study, a novel endoglucanase from A. vulcanalis was cloned, expressed, purified and characterised in terms of its potential suitability for this application. Key features of this A. vulcanalis endoglucanase which warrant attention and distinguish it from previously described mesophilic endoglucanases include its higher temperature optimum (80 vs 50–55 °C), superior thermal stability over extended periods at high temperature, enhanced activity and stability in the presence of Tween and hence greater suitability for high temperature lignocellulose hydrolysis.
The purified enzyme exhibits maximum activity at 80 °C and on the basis of its temperature profile (Fig. 3), is more suitable for lignocellulose degradation at high temperature than the commerical enzymes currently available which are generally used at 50 °C (Singhania et al. 2010). The high temperature optimum is similar to that of CelB endoglucanase from A. acidocaldarius (Eckert and Schneider 2003) and higher than that of the other Alicyclobacillus endoglucanases characterised to date which range from 55 to 70 °C (Eckert et al. 2002; Morana et al. 2008; Bai et al. 2010a, b). The purified enzyme displays optimum activity at pH 3.6–4.5, and the relatively broad pH profile observed (Fig. 4) is likely to offer flexibility in terms of hydrolysis pH which would be advantageous in terms of industrial application. The optimum pH is similar to that of CelB and CelG from A. acidocaldarius, both of which display maximum activity at pH 4 but differs from the two glucanases characterised from Alicyclobacillus sp. A4, which are optimally active at pH 2.6 and pH 5.8. The thermoacidophilic nature of the purified enzyme is in line with the environmental conditions from which A. vulcanalis was isolated (78 °C, pH 1.7) (Simbahan et al. 2004).
The effect of Ca2+ and Mg2+ ions on A. vulcanalis endoglucanase activity (Table 1) is in contrast to A. acidocaldarius CelB which displays optimal activity in the presence of these ions (Eckert and Schneider 2003). The significant beneficial effect of Tween 20 and Triton X-100 on enzyme activity may be attributed to the effect of surfactant binding on enzyme secondary and tertiary structure or flexibility or alternatively to changes in the enzyme reaction environment due to presence of the surfactant (Rubingh 1996). These additives were also found to have a beneficial effect on enzyme thermal stability with the enzyme retaining full activity after 72 h at 70 °C and 78 % of original activity after 72 h at 75 °C (Fig. 5) in the presence of 0.1 % Tween 20. Such surfactants have previously been reported to have a stabilising effect on other enzymes (Kaar and Holtzapple 1998; Yoon and Robyt 2005). For bioethanol production, thermal stability over extended periods is important with enzymatic hydrolysis times of up to 72 h reported. In terms of hydrolysis temperature, the enzyme is therefore suitable for short-term hydrolysis at 80 °C or long-term hydrolysis at up to 75 °C. The thermal stability of the purified enzyme is far superior to that of three commercial enzyme products assessed by Pribowo et al. 2012. Upon incubation at 40-–75 °C for 24 h, the three commercial products retained more than 85 % of their activity on CMC at temperatures up to 50 °C, but activity decreased significantly at higher temperatures with little or no activity detected after incubation at 70–75 °C for 24 h. Similarly, Viikari et al. (2007) reported the rapid inactivation of commercial enzymes during hydrolysis of lignocellulose at temperatures above 60 °C with no hydrolysis observed after 24 h at 60 °C or 48 h at 55 °C.
The observed thermal stability of the purified enzyme (Fig. 5) in conjunction with its temperature profile (Fig. 3) strongly indicate potential suitability for high-temperature enzymatic hydrolysis at 70 or 75 °C with associated advantages as outlined earlier. Addition of surfactants, including Tween 20 and Triton X-100, during enzymatic hydrolysis of lignocellulose has been reported to improve the conversion of cellulose to soluble sugars, thereby reducing the amount of enzyme required (Eriksson et al. 2002; Kristensen et al. 2007). This has been attributed to reduced unproductive adsorption of enzymes to lignin, changes in the nature of the substrate, effects on enzyme-substrate interactions and/or reduced enzyme denaturation (Eriksson et al. 2002; Kristensen et al. 2007). Of the surfactants and additives tested to improve cellulose hydrolysis, the non-ionic surfactant Tween has frequently been reported to perform the best (Van Dyk and Pletschke 2012) and is also more environmentally favourable for large-scale application than Triton (Eriksson et al. 2002). Enhanced enzyme activity and thermal stability in the presence of Tween is therefore highly beneficial in terms of the industrial application of A. vulcanalis endoglucanase in lignocellulose hydrolysis.
While maximum activity was observed on carboxymethyl cellulose, the purified enzyme also hydrolysed lichenan, barley β-glucan and xylan. Activity on lichenan and barley β-glucan, both of which contain β-1,3 and β-1,4 linkages, and inability to hydrolyse laminarin, which consists primarily of β-1,3 linked d-glucose with some β-1,6 linkages, indicates specificity for β-1,4 linkages. Enzymes with activity on both CMC and xylan have previously been reported, for example a cellulase from Trichoderma viride, and may be advantageous in terms of reducing enzyme requirements for overall lignocellulose hydrolysis (Van Dyk and Pletschke 2012). Despite the described sequence similarity to GH51 α-l-arabinofuranosidase-like proteins, the purified enzyme showed no activity on 4-nitrophenyl-α-l-arabinofuranoside. While known activities of GH51 include endoglucanase, endo-β-1,4-xylanase, β-xylosidase and α-l-arabinofuranosidase, members are mostly α-l-arabinofuranosidases. Known exceptions are CelB from A. acidocaldarius (ACV57112.1) (cellulase/xylanase), CelA4 from Alicyclobacillus sp. A4 (ADI82825.1) (β-1,4-glucanase), Cel51A from Fibrobacter succinogenes (AAC45377.1) (endoglucanase), an endo-β-1,4-glucanase from an uncultured bacterium (CAF22222.1) and α-l-arabinofuranosidase/β-xylosidase from Arabidopsis thaliana (CAZy; http://www.cazy.org). The purified enzyme therefore likely belongs to a minority group of GH51 proteins not exhibiting α-l-arabinofuranosidase activity. This is further corroborated by phylogenetic analysis (Fig. 1) with A. vulcanalis endoglucanase part of a group with the four GH51 endoglucanases known to date that is clearly separate from the representative GH51 α-l-arabinofuranosidases analysed. The observed substrate specificity of A. vulcanalis endoglucanase is broadly similar to that of the Alicyclobacillus endoglucanases CelB (Eckert and Schneider 2003) and CelA4 (Bai et al. 2010b), both of which are reported to hydrolyse CMC and oat spelt xylan with maximum activity observed on barley β-glucan in the case of CelA4. In contrast, CelG from A. acidocaldarius (Morana et al. 2008) is specific for CMC with no activity observed on xylan. The K m value of the purified A. vulcanalis endoglucanase is lower than those of the thermostable endoglucanase Thcel9A from Thermobifida halotolerans (12.02 mg/ml) (Zhang et al. 2011) and two thermostable endoglucanases from Aspergillus fumigatus Z5 (37.88 and 51.82 mg/ml) (Liu et al. 2011), indicating higher substrate affinity. The production of a small amount of cellobiose upon hydrolysis of cellotriose indicates that the enzyme may display low activity on cellotriose. Cellobiose and cellotriose were observed upon hydrolysis of cellotetraose and cellopentose while no glucose was detected. Eckert and Schneider (2003) attributed the production of the cellotriose intermediate and no detectable glucose upon hydrolysis of cellotetraose by CelB to a possible transglycosidase activity of the enzyme under the conditions used and degradation of the larger transglycosylation products. Analysis of the hydrolysis of CMC by A. vulcanalis endoglucanase indicates an endo- mode of action.
Upon viscometric assay, the purified enzyme was capable of decreasing the viscosity of CMC at high temperature confirming endoglucanase activity and also indicating potential suitability for use in a prehydrolysis/liquefaction step in the production of bioethanol with associated benefits as outlined earlier. The temperature optimum determined using the viscometric assay is similar to that determined by quantification of reducing sugars using DNS. The release of reducing sugars observed upon incubation of the purified enzyme with pretreated straw at 70 and 75 °C show that the enzyme is capable of hydrolysing straw lignocellulose at high temperature indicating potential suitability for use in high-temperature enzymatic hydrolysis in the production of bioethanol. While efficient prehydrolysis/liquefaction can be achieved using a single endoglucanase, additional lignocellulose-degrading enzymes which work in synergy with endoglucanase are required for efficient lignocellulose degradation in the subsequent enzymatic hydrolysis step. Therefore, further assessment of enzyme suitability for this step would necessitate the use of a thermostable enzyme cocktail containing purified A. vulcanalis endoglucanase together with additional thermostable synergistically acting lignocellulosic enzyme activities. The recommended operating temperatures (50–55 °C) and poor thermal stability of currently available commercial enzyme cocktails render them unsuitable for such studies. Reported bioprospecting efforts focused on identifying novel thermostable lignocellulose-degrading enzymes (Bhalla et al. 2013) are likely to continue to yield suitable enzymes which will facilitate the development of thermostable enzyme cocktails for this application. Identification of the sequence encoding A. vulcanalis endoglucanase activity and functional expression of the protein in E. coli, as reported in this study, facilitates overexpression of this novel protein for further application-relevant assessment as well as potential protein engineering studies.
The biochemical characteristics of A. vulcanalis endoglucanase, in particular its activity and stability at high temperature as well as increased activity and thermal stability in the presence of Tween, a known enhancer of cellulose hydrolysis, strongly indicate its potential suitability for use in the production of cellulosic bioethanol. While further studies are necessary to confirm industrial applicability, potential uses may include the prehydrolysis/liquefaction of pretreated lignocellulose at high temperature or inclusion in a thermostable enzyme cocktail for high-temperature enzymatic hydrolysis.
References
Bai YG, Wang JS, Zhang ZF, Shi PJ, Luo HY, Huang HQ, Luo CL, Yao B (2010a) Expression of an extremely acidic β-1,4-glucanase from thermoacidophilic Alicyclobacillus sp. A4 in Pichia pastoris is improved by truncating the gene sequence. Microb Cell Fact 93(3). doi: 10.1186/1475-2859-9-33
Bai Y, Wang J, Zhang Z, Shi P, Luo H, Huang H, Feng Y, Yao B (2010b) Extremely acidic beta-1,4-glucanase, CelA4, from thermoacidophilic Alicyclobacillus sp. A4 with high protease resistance and potential as a pig feed additive. J Agric Food Chem 58(3):1970–1975. doi:10.1021/jf9035595
Bai YG, Wang JS, Zhang ZF, Shi PJ, Luo HY, Huang HQ, Luo CL, Yao B (2010c) A novel family 9 beta-1,3(4)-glucanase from thermoacidophilic Alicyclobacillus sp. A4 with potential applications in the brewing industry. Appl Microbiol Biotechnol 87(1):251–259. doi:10.1007/s00253-010-2452-3
Bathgate G (1979) The institute of brewing analysis committee the determination of endo-β-glucanase activity in malt. J Inst Brew 85:92–94. doi:10.1002/j.2050-0416.1979.tb06833.x
Bhalla A, Bansal N, Kumar S, Bischoff KM, Sani RK (2013) Improved lignocellulose conversion to biofuels with thermophilic bacteria and thermostable enzymes. Bioresour Technol 128:751–759. doi:10.1016/j.biortech.2012.10.145
Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein dye-binding. Anal Biochem 72(1–2):248–254. doi:10.1016/0003-2697(76)90527-3
Eckert K, Schneider E (2003) A thermoacidophilic endoglucanase (CelB) from Alicyclobacillus acidocaldarius displays high sequence similarity to arabinofuranosidases belonging to family 51 of glycoside hydrolases. Eur J Biochem 270(17):3593–3602. doi:10.1046/j.1432-1033.2003.03744.x
Eckert K, Zielinski F, Lo Leggio L, Schneider E (2002) Gene cloning, sequencing, and characterization of a family 9 endoglucanase (CelA) with an unusual pattern of activity from the thermoacidophile Alicyclobacillus acidocaldarius ATCC27009. Appl Microbiol Biotechnol 60(4):428–436. doi:10.1007/s00253-002-1131-4
Elleuche S, Schroder C, Sahm K, Antranikian G (2014) Extremozymes-biocatalysts with unique properties from extremophilic microorganisms. Curr Opin Biotechnol 29:116–123. doi:10.1016/j.copbio.2014.04.003
Eriksson T, Borjesson J, Tjerneld F (2002) Mechanism of surfactant effect in enzymatic hydrolysis of lignocellulose. Enzyme Microb Technol 31(3):353–364. doi:10.1016/s0141-0229(02)00134-5
Kaar WE, Holtzapple MT (1998) Benefits from Tween during enzymic hydrolysis of corn stover. Biotechnol Bioeng 59(4):419–427. doi:10.1002/(SICI)1097-0290(19980820)59:4<419::AID-BIT4>3.3.CO;2-9
Karnaouri AC, Topakas E, Christakopoulos P (2014) Cloning, expression, and characterization of a thermostable GH7 endoglucanase from Myceliophthora thermophila capable of high-consistency enzymatic liquefaction. Appl Microbiol Biotechnol 98(1):231–242. doi:10.1007/s00253-013-4895-9
Kristensen JB, Borjesson J, Bruun MH, Tjerneld F, Jorgensen H (2007) Use of surface active additives in enzymatic hydrolysis of wheat straw lignocellulose. Enzyme Microb Technol 40(4):888–895. doi:10.1016/j.enzmictec.2006.07.014
Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227(5259):680–685. doi:10.1038/227680a0
Liu D, Zhang R, Yang X, Xu Y, Tang Z, Tian W, Shen Q (2011) Expression, purification and characterization of two thermostable endoglucanases cloned from a lignocellulosic decomposing fungi Aspergillus fumigatus Z5 isolated from compost. Protein Expression Purif 79(2):176–186. doi:10.1016/j.pep.2011.06.008
McCleary B (2001) Analysis of Feed Enzymes. In: Bedford M, Partridge G (eds) Enzymes in farm animal nutrition. CABI Publishing, Oxon, pp 85–107
Menon V, Rao M (2012) Trends in bioconversion of lignocellulose: biofuels, platform chemicals & biorefinery concept. Prog Energy Combust Sci 38(4):522–550. doi:10.1016/j.pecs.2012.02.002
Miller GL (1959) Use of dinitrosalicyclic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428. doi:10.1021/ac60147a030
Miller PS, Blum PH (2010) Extremophile-inspired strategies for enzymatic biomass saccharification. Environ Technol 31(8–9):1005–1015. doi:10.1080/09593330903536113
Modenbach AA, Nokes SE (2012) The use of high-solids loadings in biomass pretreatment—a review. Biotechnol Bioeng 109(6):1430–1442. doi:10.1002/bit.24464
Morana A, Esposito A, Maurelli L, Ruggiero G, Ionata E, Rossi M, La Cara F (2008) A novel thermoacidophilic cellulase from Alicyclobacillus acidocaldarius. Protein Pept Lett 15(9):1017–1021. doi:10.2174/092986608785849209
Pribowo A, Arantes V, Saddler JN (2012) The adsorption and enzyme activity profiles of specific Trichoderma reesei cellulase/xylanase components when hydrolyzing steam pretreated corn stover. Enzyme Microb Technol 50(3):195–203. doi:10.1016/j.enzmictec.2011.12.004
Rubingh DN (1996) The influence of surfactants on enzyme activity. Curr Opin Colloid Interface Sci 1(5):598–603
Sambrook J, Russell DW (2001) Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory Press, New York
Simbahan J, Drijber R, Blum P (2004) Alicyclobacillus vulcanalis sp nov., a thermophilic, acidophilic bacterium isolated from Coso Hot Springs, California, USA. Int J Syst Evol Microbiol 54:1703–1707. doi:10.1099/ijs. 0.03012-0
Singhania RR, Sukumaran RK, Patel AK, Larroche C, Pandey A (2010) Advancement and comparative profiles in the production technologies using solid-state and submerged fermentation for microbial cellulases. Enzyme Microb Technol 46(7):541–549. doi:10.1016/j.enzmictec.2010.03.010
Skovgaard PA, Jorgensen H (2013) Influence of high temperature and ethanol on thermostable lignocellulolytic enzymes. J Ind Microbiol Biotechnol 40(5):447–456. doi:10.1007/s10295-013-1248-8
Szijarto N, Horan E, Zhang J, Puranen T, Siika-aho M, Viikari L (2011a) Thermostable endoglucanases in the liquefaction of hydrothermally pretreated wheat straw. Biotechnol Biofuels 4(2). doi: 10.1186/1754-6834-4-2
Szijarto N, Siika-aho M, Sontag-Strohm T, Viikari L (2011b) Liquefaction of hydrothermally pretreated wheat straw at high-solids content by purified Trichoderma enzymes. Bioresour Technol 102(2):1968–1974. doi:10.1016/j.biortech.2010.09.012
Turner P, Mamo G, Karlsson EN (2007) Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb Cell Fact 6. doi: 910.1186/1475-2859-6-9
Van Dyk JS, Pletschke BI (2012) A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes—factors affecting enzymes, conversion and synergy. Biotechnol Adv 30(6):1458–1480. doi:10.1016/j.biotechadv.2012.03.002
Viikari L, Alapuranen M, Puranen T, Vehmaanpera J, Siika-Aho M (2007) Thermostable enzymes in lignocellulose hydrolysis. In: Olsson L (ed) Biofuels, Vol 108. Springer Berlin Heidelberg, pp 121–145
Viikari L, Vehmaanpera J, Koivula A (2012) Lignocellulosic ethanol: from science to industry. Biomass Bioenergy 46:13–24. doi:10.1016/j.biombioe.2012.05.008
Wang K, Luo H, Bai Y, Shi P, Huang H, Xue X, Yao B (2014) A thermophilic endo-1,4-beta-glucanase from Talaromyces emersonii CBS394.64 with broad substrate specificity and great application potentials. Appl Microbiol Biotechnol 98(16):7051–7060. doi:10.1007/s00253-014-5680-0
Yoon SH, Robyt JF (2005) Activation and stabilization of 10 starch-degrading enzymes by Triton X-100, polyethylene glycols, and polyvinyl alcohols. Enzyme Microb Technol 37(5):556–562. doi:10.1016/j.enzmictec.2005.04.002
Zhang F, Chen JJ, Ren WZ, Nie GX, Ming H, Tang SK, Li WJ (2011) Cloning, expression and characterization of an alkaline thermostable GH9 endoglucanase from Thermobifida halotolerans YIM 90462(T). Bioresour Technol 102(21):10143–10146. doi:10.1016/j.biortech.2011.08.019
Acknowledgments
This work was funded as part of the Science, Technology, Research and Innovation for the Environment (STRIVE) Programme 2007–2013. The programme is financed by the Irish Government under the National Development Plan 2007–2013, and it is administered on behalf of the DEHLG by the Environmental Protection Agency (EPA) which has the statutory function of coordinating and promoting environmental research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 36 kb)
Rights and permissions
About this article

Cite this article
Boyce, A., Walsh, G. Characterisation of a novel thermostable endoglucanase from Alicyclobacillus vulcanalis of potential application in bioethanol production. Appl Microbiol Biotechnol 99, 7515–7525 (2015). https://doi.org/10.1007/s00253-015-6474-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6474-8