
Abstract
Hypnea cervicornis J. Agardh (Gigartinales, Florideophyceae) is a commercially important carrageenan producing seaweed. Currently, there are no organellar genomes of Hypnea species available in public databases. Here, we report the complete organellar genomes of H. cervicornis using next-generation sequencing technology. The mitochondrial genome has a circular mapping organization with a total length of 25,060 bp and consists of 50 genes (24 protein-coding, 2 rRNA, and 24 tRNA). The plastid genome is also a circular molecule and is 176,446 bp in length and includes 230 genes (194 protein-coding, 3 rRNA, 30 tRNA, 1 tmRNA and 2 misc_RNA). Colinear analysis show that the organellar genomes in the Gigartinales are conserved, except for the inversion of two genes (trnY and trnR) in the mitochondrial genome and a 12.5-kb rearrangement in the plastid genome. One stem-loop structure at the intergenic regions between trnS2 and trnA, plus one short hairpin structure between cob and trnL2 are detected in the mitochondrial genome of H. cervicornis. The Ka/Ks analysis reveal that values for most of the protein-coding genes in organellar genomes of H. cervicornis are below one, reflecting the importance of those genes. Phylogenetic relationships based on shared protein-coding genes from the organellar genomes of Rhodophyta are also examined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypnea cervicornis (Cystocloniaceae, Florideophyceae), is mainly distributed in the intertidal and subtidal zones of tropical and warm temperate waters (Mshigeni and Chapman 1994; Guiry and Guiry 2021) and is a common species in China. Thalli erect, epilithic or epiphytic, reddish-pink to brownish when alive, terete, fleshy in texture, 1–4 cm long, attached to the substratum by a primary discoid holdfast and secondary holdfasts formed at the uppermost portions of branches and branchlets (Nauer et al. 2014). Due to its high κ-type carrageenan content, and analgesic, anti-inflammatory effects, H. cervicornis is used as a raw material in food, and the medical industry (Liu 2001; Bitencourt et al. 2008). It has received attention in recent years and several studies on the growth (Ribeiro et al. 2013; Chen et al. 2014), physiological structure (Miguel et al. 2014), and phylogenetic analysis (de Jesus et al. 2016; Nauer et al. 2019) have been reported.
Hypnea species show high levels of phenotypic plasticity and are difficult to identify based solely on morphological characters. Therefore, they are taxonomically challenging (Price et al. 1992; Nauer et al. 2014, 2019; de Jesus et al. 2016). Hypnea has an intricate nomenclatural history. Yamagishi and Masuda (2000) described Hypnea flexicaulis from Japan and Geraldino et al. (2006) described it from Korea, and later this name was merged into H. cervicornis by de Jesus et al. (2016). Additionally, 18S (nrDNA SSU), rbcL and cox1 were used to characterize the species Hypnea asiatica from Korea, Japan, and Taiwan of China, and conclude that Hypnea charoides should be removed from the northwestern Pacific marine flora (Geraldino et al. 2009), rbcL, cox1 and psaA were used to explore the phylogeny of the genus Hypnea (Geraldino et al. 2010), and the genetic variability within the species and the genetic lineages of Hypnea to contemporary distribution were also examined using rbcL and cox1 (Geraldino et al. 2015). These studies mentioned above were mainly based on limited sequence data. Genome data with comprehensive information could resolve more complex phylogenetic relationships (Zhang et al. 2012, 2020). It is well-known that the organellar genomes are characterized by uniparental inheritance, compact genome structure and higher copy number per cell compared to the nuclear genomes, thus they have become effective molecular tools to clarify phylogenetic relationships (Zhang et al. 2020). However, there is no report on organellar genomes of genus Hypnea.
As the next-generation sequencing and third-generation sequencing technologies become less expensive and more efficient (Heather and Chain 2016), an increasing number of organellar genomes of red algae have been sequenced and used in the study for phylogeny (Janouškovec et al. 2013; Yang et al. 2015; Li et al. 2018; Watanabe et al. 2019). For the Gigartinales, there are nine mitochondrial genomes including four from the Solieriaceae (Kappaphycus alvarezii, Kappaphycus striatus, Eucheuma denticulatum and Betaphycus gelatinus), two from the Gigartinaceae (Chondrus crispus and Sarcopeltis skottsbergii), one from the Endocladiaceae (Gloiopeltis furcata), one from the Caulacanthaceae (Caulacanthus okamurae), and one from the Phyllophoraceae (Mastocarpus papillatus), at the same time seven plastid genomes available including four from the Solieriaceae (K. alvarezii, K. striatus, E. denticulatum and B. gelatinus), one from the Gigartinaceae (C. crispus), one from the Caulacanthaceae (C. okamurae) and one from the Phyllophoraceae (M. papillatus). It's worth noting that several recent surveys have used organellar genomes to carry out the reconstructions for the Gigartinales (Leblanc et al. 1995; Janouškovec et al. 2013; Tablizo and Lluisma 2014; Sissini et al. 2016; Li et al. 2018; Liu et al. 2019; Watanabe et al. 2019; Hartnell College Genomics Group et al. 2020; Hughey et al. 2020; Zhang et al. 2020). Here, we determined the complete organellar genomes of H. cervicornis (Hypnea, Cystocloniaceae, Gigartinales) from a specimen from Guangdong Province, China and added the available organellar genomes pool of red algae. We obtained information about gene content, organization, and structure of its organellar genomes, and compared them with reported organellar genomes of species from the Gigartinales.
Materials and methods
Sample collection and DNA extraction
Samples of Hypnea cervicornis (specimen number: QD 2016030050, Fig. S1) were collected from Shanwei City, Guangdong Province, China (22°39′44″N, 115°33′58″E). They were washed carefully with sterilized seawater and then ultrahigh quality (UHQ) water to remove protozoa, epiphytes, and other attachments. After examination under the dissection microscope, these cleaned samples were frozen in liquid nitrogen (-196℃) and then kept at -80℃ in ultra-low temperature freezer prior to the subsequent research. Algal materials in this study were provided by the Culture Collection of Seaweed at the Ocean University of China.
Total DNA from one individual of H. cervicornis was prepared using a modified cetyltrimethylammonium bromide (CTAB) method (Sun et al. 2011). The completeness and concentration of total DNA were estimated using agarose-gel electrophoresis and the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA).
High-throughput sequencing and assembly
A short-insert library was constructed using the Nextera DNA Sample Preparation Kit following the manufacturer’s protocols (Illumina, USA). Paired-end reads were extracted from Illumina's HiSeq × Ten system. Approximately 9 Gb of paired-end sequence data was randomly extracted from the total sequencing output, as input into NOVOPlasty (Dierckxsens et al. 2017) to assemble the mitochondrial genome, and as input into SOAPdenovo (Luo et al. 2012) with default assembly parameters to assemble the plastid genome. The mitochondrial genome of C. crispus (GenBank accession number: NC_001677.2) and plastid genomes of C. crispus (GenBank accession number: NC_020795.1) and K. alvarezii (GenBank accession number: KU892652.1) were used as the reference sequences to determine the proportion of related contigs. Subsequently, the CodonCode Aligner (CodonCode Corporation, USA) was used to align and arrange the mitochondrion-related or plastid-related contigs into a circular structure each (Liu et al. 2019).
Annotation and colinear analysis
The protein-coding genes, ribosomal RNA (rRNA) genes, introns, and transfer messenger RNA (tmRNA) genes were annotated according to the references using Geneious R10 (Biomatters Ltd., Auckland, New Zealand; available from http://www.geneious.com/). Transfer RNA (tRNA) genes were identified by tRNAscan-SE software v.1.21 (http://lowelab.ucsc.edu/tRNAscan-SE/) using the Mito/Chloroplast model (Schattner et al. 2005). The organellar genomes map were visualized with Organellar Genome DRAW (OGDRAW) (Greiner et al. 2019).
Secondary DNA structures were predicted using RNAfold webserver (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi).
Ten available mitochondrial genomes and eight plastid genomes from species of the Gigartinales were aligned respectively at the default settings with the Progressive Mauve Genome Aligner version 2.4.0 to conduct colinear analysis (Darling et al. 2011).
Estimation of substitution rates
The calculation of Ka/Ks was an effective method to determining the selective pressure on protein-coding genes: Ka/Ks > 1, positive selection; Ka/Ks = 1, neutral selection; and Ka/Ks < 1, negative selection. To calculate the selective pressures and gene evolutionary rates of H. cervicornis, Ka/Ks was analyzed using reported organellar genomes from the Gigartinales as references. Mega 7.0 (Kumar et al. 2016) was used for sequence alignment, then KaKs_calculator 2.0 (Wang et al. 2010) was utilized to calculate Ka/Ks. The settings were as follows: genetic code table 4 (mold mitochondrial coda) and 11 (bacterial and plant plastid code), method of calculation: MYN.
Phylogenetic analysis
The phylogenetic analysis was based on the following two data sets using maximum likelihood (ML) and Bayesian inference (BI) methods: ten shared protein-coding genes (atp6, atp9, cox1, cox3, nad1, nad2, nad3, nad4, nad5, nad6) from 56 available mitochondrial genomes of Rhodophyta and 99 shared protein-coding genes from 54 available plastid genomes of Rhodophyta (Table S1). Mega 7.0 software (Kumar et al. 2016) were employed to align protein sequences. BioEdit version 7.0.9.0 (Hall 1999) was used for manual editing and trimming. The concatenated alignments were generated respectively and poorly aligned regions were removed using the Gblocks server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) (Castresana 2000). The mitochondrial genome alignment was reduced from original the 3,603 positions to 2,467 and the plastid genomes alignment from 28,525 to 19,886 positions. The best-fit model for ML was selected using ProtTest 3.4.2 (Darriba et al. 2011) based on the Akaike Information Criterion (AIC). The ML analysis was performed by RAxML (Stamatakis 2006). Bootstrap probability values were run with 1,000 replicates under the CpREV + G + I + F model. BI analysis was conducted via MrBayes v. 3. 1.2 (Huelsenbeck and Ronquist 2001) using the CpREV model. The phylogenetic analysis was performed using two independent runs with four Markov Chains Monte-Carlo for 1,000,000 generations. Output trees were sampled every 100 generations. The phylogenetic analysis was run until the average standard deviation of split frequencies was below 0.01 and the first 25% of samples were removed as burn-in. FigTree version 1.4.3 (http://tree.bio.ed.ac.uk/) was used to display the phylogenetic tree (Rambaut 2016). The protein sequences of Galdieria sulphuraria strain 074 W (mitochondrial genome: NC_024666, plastid genome: KJ700459) served as outgroups.
In order to better elucidate the systemic evolutionary relationship within the Gigartinales, we reconstructed the phylogenetic relationships of the Gigartinales using the ML and BI methods with Gracilaria chilensis as the out-group (mitochondrial genome: MF401962, plastid genome: NC_029860). The phylogenetic analysis was conducted based on two datasets: twenty shared protein-coding genes from ten mitochondrial genomes of the Gigartinales and 175 shared protein-encoding genes from eight plastid genomes. The divergence time estimation tree of Rhodophyta was inferred based on the nucleotide sequence of the shared mitochondrial genes and plastid genes, respectively. The MCMCTREE in the PAMLX (Xu and Yang 2013) software package was used based on the “indepented rates” molecular clock model and the “HKY85” nucleic acid replacement model. To better adapt to the software, Mega 7.0 was used to reconstruct the phylogenetic tree using the RAxML (Stamatakis 2006) method.
Results
Genome features
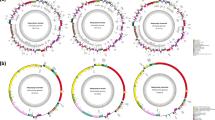
The complete organellar genomes of H. cervicornis were both assembled as single circular molecules with the lengths 25,060 bp (mitochondrial genome) and 176,446 bp (plastid genome). The average GC contents were 27.04% for the mitochondrial genome and 28.09% for the plastid genome. Its mitochondrial genome contained a set of 50 genes, including 24 protein-coding, two rRNA, and 24 tRNA genes. One group II intron positioned in the trnI gene was identified. The plastid genome of H. cervicornis encoded a total of 230 genes, including 194 protein-coding, 30 tRNA, three rRNA genes. Two misc_RNAs (ffs, rnpB) and one tmRNA gene (ssrA) were also annotated. One group II intron was located in the trnM gene. The coding proportions of the heavy and light chains of both organellar genomes were similar (Fig. 1a, b). The complete organellar genomes of H. cervicornis were submitted to GenBank under the following accession numbers: MZ682023 (mitochondrial genome) and MZ682024 (plastid genome).

Gene maps of organellar genomes of H. cervicornis (a, mitochondrial genome; b, plastid genome). Genes on the outside of the maps are transcribed in a clockwise direction, whereas those on the inside of the maps are transcribed counterclockwise
The general features of ten available mitochondrial genomes and eight plastid genomes from the Gigartinales were summarized in Table 1 and Table 2 respectively. The coding regions in the mitochondrial genome of H. cervicornis were 23,983 bp, accounting for 95.70% and 146,650 bp in its plastid genome, accounting for 83.11% of the genome.
The reported organellar genomes of the Gigartinales were compact. Four gene pairs overlap (ymf16-rps12, nad4L-rnl, rps3-rpl16, cox1-cox2) in the mitochondrial genome of H. cervicornis, with overlap lengths of 8–59 bp. There was one overlap in the mitochondrial genomes of K. striatus, K. alvarezii and G. furcata, two in E. denticulatum, three in B. gelatinus, four in C. okamurae and M. papillatus, five in S. skottsbergii, and eight in C. crispus. However, we could not find any shared gene overlaps in the mitochondrial genomes of the Gigartinales. And there were six overlaps (psbD-psbC, syfB-rnpB, trnH-ycf29, rpl24-rpl14, rpl23-rpl4, atpF-atpD) in plastid genome of H. cervicornis with overlap length of 1–95 bp. There were seven overlaps in plastid genome of C. crispus and C. okamurae, eight in M. papillatus, ten in K. striatus and K. alvarezii, eleven in B. gelatinus and E. denticulatum. Among them, three overlaps (rpl24-rpl14, rpl23-rpl4, and atpF-atpD) were conserved in all reported plastid genomes of the Gigartinales. Significantly, the rpl23-rpl4 overlapping region has also been observed in some plastid genomes of brown algae and diatom, indicating that it was highly conserved.
Protein-coding genes
A Venn diagram revealed shared and unique genes in the mitochondrial genomes of the Gigartinales (Fig. 2a). In total, 24 protein-coding genes were shared by ten mitochondrial genomes of the Gigartinales, which represented the majority of mitochondrial protein-coding gene content (100% of mitochondrial protein-coding genes of H. cervicornis, B. gelatinus, E. denticulatum, K. striatus, K. alvarezii, G. furcata and S. skottsbergii, 92.31% of M. papillatus, 82.76% of C. crispus, and 96% of C. okamurae). M. papillatus and C. crispus contained one common gene orf172, while C. crispus encoded four additional genes (orf73, orf74, orf94.1, orf105). And the mitochondrial genomes of C. okamurae and M. papillatus had one specific gene tatA.

Venn diagrams of protein-coding gene content from ten reported mitochondrial genomes (a) and eight reported plastid genomes (b) of the Gigartinales
Hypnea cervicornis encoded 194 plastid protein-coding genes. A total of 186 of those were shared by eight genomes of Gigartinales representing the majority of plastid protein-coding gene content (95.88% of plastid protein-coding genes of H. cervicornis, 91.18% of B. gelatinus, E. denticulatum, C. crispus, 92.08% of K. striatus, K. alvarezii and M. papillatus, 92.54% of C. okamurae). Several ancient genes such as glnB, chlB, chlL, and chlN were not present in the plastid genome of H. cervicornis. Shared and unique genes in the plastid genomes of Gigartinales were also revealed by Venn diagram (Fig. 2b). The plastid genomes of H. cervicornis and C. okamurae, together with four plastid genomes from the Solieriaceae (B. gelatinus, E. denticulatum, K. striatus, K. alvarezii) contained one additional gene (dfr). Two species from Kappaphycus together with M. papillatus did not encode pbsA. Three specific genes (syh, psbW, and ompR), which were found in most Florideophyceae, were only encoded by the plastid genome of M. papillatus in the Gigartinales.
The protein-coding genes utilized the mold genetic code in organellar genomes of H. cervicornis, which differed from the universal code mainly in the usage of TGA as tryptophan (Trp) in the mitochondrial genome. Of the 24 mitochondrial protein-coding genes, 23 (95.83%) initiated with ATG, which was similar with other species in the Gigartinales (Table 1). Interestingly, tatC used various alternative start codons but rarely ATG in all the reported mitochondrial genomes of Gigartinales. For example, TTG was used as the start codon for tatC in H. cervicornis, B. gelatinus, K. striatus and K. alvarezii, however it was GTT in C. crispus, ATA in S. skottsbergii, and ATC in M. papillatus. Two stop codons (TAA and TAG) were identified in the mitochondrial genome of H. cervicornis with a preference for TAA amounting to 87.50%. As in the mitochondrial genomes, the majority of the protein-coding genes (185 genes, 95.36%) in the plastid genome of H. cervicornis began with ATG. GTG was used as the alternate start codon for five plastid genes including psbT, chlI, rbcS, rps8, infC. Three plastid genes (ycf27, ycf65, trxA) used TTG as the start codon, while gene petF used ATA. Three typical stop codons TAA, TAG, and TGA were identified in plastid genome with a clear preference for TAA, which accounted for 86.60%.
Secondary structure
Mitochondrial genome of H. cervicornis was composed of two distinct transcription units with opposite transcription directions (Fig. 1a) which consistent with previously published Gigartinales. The long stem-loop (194 bp) was detected at the intergenic regions between trnS2 and trnA in its mitochondrial genome. Based on the sequence analysis, such stem-loop structures were also found in eight of nine sequenced mitochondrial genomes from the Gracilariales and only absent in S. skottsbergii (Fig. 3a). These secondary structures with complete inverted-repeat sequences located at the respective demarcation point of opposite transcriptional units and all contained A and T polymers. The number of continuous nucleotides A and T varied in different species. We could not find obvious sequence homology among these mononucleotide repeats. In addition, one short hairpin structure between cob and trnL2 was identified in mitochondrial genome of H. cervicornis. And the similar hairpin structures were also found in the other sequenced mitochondrial genomes of the Gigartinales (Fig. 3b).

A comparison of the sequences of the stem-loop structures from nine Gigartinales mitochondrial genomes (a) and hairpin structures from ten Gigartinales mitochondrial genomes (b). *Only the partial sequences of the stem -loop from C. okamurae is shown in Fig. 3a. The complete sequence is as follows: CCTACCCTATATCCCTTACCCCCTATTATACTTAAGTCATTAGTATTATATTTGTTGGAAAAAGAGAAACAGTGGGAGAGACAGTAGACCCCCCCTCTTATAGTAAACGCTTTTTTTATGTAAGGTTATTTTTTTTCAAAAGTAACG—-—A—-—CGTTACTTTTGAAAAAAAATAACCTTACATAAAAAAAGCGTTTACTATAAGAGGGGGGGTCTACTGTCTCTCCCACTGTTTCTCTTTTTCCAACAAATATAATACTAATGACTTAAGTATAATAGGGGGTAAGGGATATAGGGTAGG
Colinear analysis
Colinear analysis showed that the organellar genomes of the Gigartinales were very conserved except for some rearrangements (Fig. 4a, b). Among mitochondrial genomes of the Gigartinales, the difference in gene order could be explained by the inversion of two tRNA genes: trnY and trnR. Gene order in the mitochondrial genome of H. cervicornis (trnY-trnR) was consistent with six Gigartinales species while different from C. crispus, S. skottsbergii and M. papillatus (trnR-trnY). In the plastid genomes, the approximately 12.5 kb gene fragment from gene ycf21 to psaM of H. cervicornis and three other Gigartinales (C. crispus, M. papillatus, C. okamurae) was completely reversed to that of four Solieriaceae (B. gelatinus, E. denticulatum, K. striatus and K. alvarezii). Organellar gene order of H. cervicornis was identical with that of C. okamurae.

Whole-genome multiple alignments of ten mitochondrial genomes (a) and eight plastid genomes (b) from the Gigartinales using Mauve software
Gene substitution rates
The results of Ka/Ks calculation for 24 mitochondrial genes and 186 plastid genes in H. cervicornis compared to the other species of Gigartinales were shown in Fig. 5a and b, respectively. Most of the Ka/Ks values were less than 1, indicating negative selections of those genes during evolution. For the mitochondrial genomes, Ka/Ks values for atp9 were the lowest (1.83 × 10–11), showing very strong purifying selection pressure. The value of mitochondrial gene tatC in H. cervicornis compared to M. papillatus was equal to 1, suggesting a neutral selection occurred during evolution. For plastid genomes, psbL had the lowest substitution rates with average Ka/Ks values of 2.76 × 10–11. The Ka/Ks value of ycf86 in H. cervicornis compared to B. gelatinus was higher than 1 (1.28753), suggesting a positive selection, while the Ka/Ks value of ycf52 compared to E. denticulatum was equal to 1, indicating a neutral selection. Furthermore, there were several organellar genes such as cox1, nad1, petD, psbC, psbD, psbE, psbI, psbN and rbcL whose Ka/Ks values were far below one (< 0.01).

The Ka/Ks values of 24 protein-coding genes in the mitochondrial genomes (a) and 186 protein-coding genes in the plastid genomes (b) of H. cervicornis versus other sequenced species from the Gigartinales
Phylogenetic analysis
The ML and BI methods were used to perform the phylogenetic reconstruction. The ML tree was congruent with the Bayesian topology. Both ML and BI phylogenetic results showed that all species were clearly divided into four groups according to their classes: Florideophyceae, Bangiophyceae, Compsopogonophyceae, and Cyanidiophyceae. In the mitochondrial tree (Fig. S2a), Hypnea cervicornis and C. okamurae clustered with 86% ML bootstrap (BS) and 1.0 Bayesian posterior probability (BPP) support, in a larger clade with B. gelatinus, E. denticulatum, K. striatus, K. alvarezii (79% BS, 1.0 BPP), while C. crispus, S. skottsbergii, G. furcata and M. papillatus formed the second subclade (47% BS, 1.0 BPP). In the plastid tree (Fig. S2b), H. cervicornis together with four species of the Solieriaceae (B. gelatinus, E. denticulatum, K. striatus, K. alvarezii) and C. okamurae constituted a clade (100% BS, 1.0 BPP), while C. crispus and M. papillatus were resolved in another clade (100% BS, 1.0 BPP). Most branches were estimated with a high bootstrap support and a high posterior probability value.
The phylogenetic relationships of the Gigartinales based on shared mitochondrial genes and plastid genes showed that H. cervicornis, C. okamurae, together with four species of the Solieriaceae formed a high supported (100% BS, 1.0 BPP) sub-clade (Fig. S3a, 3b). The phylogenetic relationship of the Gigartinales based on mitochondrial genomes (Fig. S3a) was consistent with the Gigartinales clade in Fig. S2a, except that G. furcata was moderately resolved in a single clade (71% BS, 0.89 BPP). The evolutionary relationship based on plastid genomes (Fig. S3b) was identical with the Gigartinales clade of Fig. S2b.
Divergence time estimation was performed for Rhodophyta based on whole organellar genomes and provided insights on its evolutionary divergence (Fig. S4a, 4b). According to our divergence tree, most classes of Rhodophyta diversified in the timespan ranging through the Proterozoic Eon (2500–570 Mya). The class Cyanidiophyceae and Compsopogonophyceae diverged first, while the Florideophyceae differentiated relatively late showing its advanced position during the evolutionary history of Rhodophyta.
Discussion
In this study the complete organellar genomes of H. cervicornis was firstly characterized which expanded the available organellar pool of red algae. Most of the genes found in organellar genomes from the red lineage were determined in H. cervicornis. In general, the coding regions in the mitochondrial genomes for the Gigartinales species ranged from 90.19 to 95.88% (Li et al. 2018), and in plastid genomes ranged from 82.63 to 87.96% (Zhang et al. 2020). Compared with previously reported organellar genomes of the Gigartinales species, there was a high conservation in gene content. The plastid genome of H. cervicornis encoding fewer orfs showed a smaller gene content (230 genes). The missing or unique genes were mostly related to ycf or orf. The function of these genes was to encode conservative hypothetical proteins (Galperin 2001; Galperin and Koonin 2004). Therefore, it was difficult to discover their deletion effects in the organellar genomes. Additionally, the plastid genomes of H. cervicornis just like other Gigartinales species maintained fewer ancient genes compared to most red algae. The genes glnB, chlB, chlL and chlN were absent in Gigartinales, while existed in Bangiaceae (Wang et al. 2013a). The lack of these ancient genes further verified that Gigartinales species were more advanced multicellular red algae.
Among the reported organellar genomes of the Gigartinales, ATG was used as the start codon for most organellar protein-coding genes, and a few genes used GTG, TTG, ATA, ATC as start codons. GTG, which was another commonly utilized start codon, mainly existed in bacteria. In the Gigartinales, the start codon GTG was mainly in some plastid genes (especially in infC, chlI, rbcS), indicating a conserved evolution of these genes. The mitochondrial genes that used GTG as the start codon in the Gigartinales was only ymf39 in G. furcata. Analysis of the organellar genomes for algae showed that GTG as a start codon was mainly present in plastid genomes, such as Pyropia haitanensis, P. yezoensis, Ectocarpus siliculosus, Saccharina japonica, S. longissima, and so on (Le Corguillé et al. 2009; Wang et al. 2013a, 2013b; Zhang et al. 2013). TTG was known as an unusual start codon in eubacteria and archaea (Golderer et al. 1995). There were some genes using TTG as a start codon in reported organellar genomes of the Gigartinales. The same phenomenon has been found in organellar genomes of other red algae (Kim et al. 2014), which was rarely found in brown algae. This further verified that red algae were more primitive than brown algae. In addition, previous studies revealed some genes in organellar genomes used ATA as the start codon in animals (Meng et al. 2008). Here, we found that tatC in the mitochondrial genome of S. skottsbergii, plastid gene petF in H. cervicornis and the plastid ycf86 in M. papillatus used ATA as the start codon. As a nonstandard start codon, ATA has also been found in the plastid genomes of Porphyra, such as for dnaB in P. yezoensis and P. haitanensis (Wang et al. 2013a). The ATC start codon was rarely found in plants and more common in animals (Yang et al. 2019). However, tatC used ATC as the start codon in the mitochondrial genome of M. papillatus, which was very uncommon in algae. In general, the diversified start codon usage in organellar genomes indicated relaxed selection, which might be related to the transcription efficiency of each gene.
Consistent with published mitochondrial genomes of other Gigartinales, the mitochondrial genome of H. cervicornis consisted of one long stem-loop structure and one short hairpin structure. Similar secondary structures are common in the mitochondrial genomes in the Ceramiales, Gelidiales, Gracilariales, Palmariales, Halymeniales, Plocamiales, Rhodymeniales, Batrachospermales, and Bangiales (Li et al. 2018). Heretofore, no similar structures have been found in the sequenced mitochondrial genomes of brown algae and green algae. This might be one of the traits to differentiate mitochondrial genomes of red algae from the other algae. Moreover, although the specific location and nucleotide length of the secondary structures varied from different species, they were all located at their respective demarcation point of two transcriptional units. And these secondary structures were similar to the D-loop (displacement loop) structure of mammalian mitochondrial genomes (Zhang et al. 2012). The D-loop, or control region, may activate the initiation of DNA replication (Clayton 1982; Chang and Clayton 1986). Meanwhile, the polymers with low free energy tended to unlink easily and might be a recognition site of some enzymes (Zhang et al. 2012). Therefore, it could be speculated that these structures with polymers A and polymers T might play a functional role in replication, transcription, or translation.
Subsequent colinear analysis in this study indicated that ten available mitochondrial genomes and eight available plastid genomes of the Gigartinales were relatively conserved, except for two gene inversions (trnR and trnY) in the mitochondrial genomes and a gene rearrangement in the plastid genomes (a 12.5 kb gene fragment from psaM to ycf21). As previously reported, the 12.5 kb gene rearrangement in the plastid genomes of the Gigartinales could be used as an evolutionary marker for the Solieriaceae (Zhang et al. 2020). Other inversions or rearrangements have also been reported, such as the 30 kb inversion in all living vascular plants (Raubeson and Jansen 1992), two transient reversals found in all ferns (Roper et al. 2007; Gao et al. 2009), 18.5 kb from gene ycf46 to trnN detected in plastid genomes of the Gigartinales against Riquetophycus sp. (Zhang et al. 2020), and two inversions covering trnC and trnN identified in ten Sargassum species against the other two Fucales species that have been sequenced (Li et al. 2021). It has been reported that gene rearrangements, such as the reordering of genetic elements, occurred through repeat inversions (Liu et al. 2019). However, no similar inverted sequences were detected in the rearranged fragments and their adjacent related regions, and no common features were identified. Therefore, the detailed mechanisms of these inversions and rearrangements were still unclear and need research.
The calculation of Ka/Ks was of great significance for the understanding of evolutionary dynamics of protein-coding genes in closely related species (Fay and Wu 2003). Nucleotide substitutions at the gene and genome level have been well investigated in higher plants (Xu et al. 2012; Dong et al. 2013), however, their study remains limited in red algae. In this investigation, we evaluated Ka/Ks values of organellar genes from H. cervicornis versus other sequenced species from the Gigartinales. The results show that the majority of the protein-coding genes in the H. cervicornis organellar genomes had negative selection, which was largely consistent with the previous reports (Li et al. 2018). Only three genes were identified to have undergone neutral or positive selections (tatC, ycf52 and ycf86). Generally, if a gene has a low substitution rate, it indicates that purifying selective forces are maintaining a strong stability (Du et al. 2016) and the gene-related function is more conserved (Li et al. 2018). Therefore, most protein-coding genes in organellar genomes were thought to be conserved across the Gigartinales and they would play an important role in maintaining conservation of organellar genomes. The gene atp9 had the lowest substitution rate among these genes, and the same phenomenon has been observed in previous related studies (Boo et al. 2016; Li et al. 2018). The results demonstrated that the sequence stability of atp9 and high conservatism of its gene product. In this study, it was shown that mitochondrial gene cox1 and plastid gene rbcL have lower substitution rates, which might explain why these two genes have been utilized as the DNA barcoding (Hughey et al. 2001; Saunders 2005). Therefore, some genes such as nad1, petD, psbC, psbD, psbE, psbI, psbN with similar lower gene substitution according to our research was speculated that they might be potential markers for detecting molecular evolution.
The phylogenetic relationship of Rhodophyta revealed by this study divided the phylum into four clades. Due to a few low bootstrap supports or posterior probability values within the Gigartinales clade (Fig S2a, 2b), we reconstructed the phylogenetic trees of Gigartinales using more shared genes (Fig S3a, 3b) to better verify their phylogenetic relationships. The limited genomic data might lead to uncertain phylogenetic results (Zhang et al. 2020). Therefore, we could not fully resolve the phylogenetic relationships on the basis of the existing molecular data in this study. With the inclusion of more and more samples in the future, this problem will be better solved. Although some red algae fossilize (Xiao et al. 2004; Cao et al. 2018), divergence times are uncertain based on these limited samplings. Therefore, estimating divergence times with the help of molecular data is essential (Yoon et al. 2004). Here, we performed divergence time estimation for Rhodophyta based on whole organellar genomes and provided insights on its evolutionary divergence. We noticed that the divergence times based on mitochondrial genomes were correspondingly later than that based on plastid genomes. The divergence time determined here was based on mitochondrial genomes or plastid genomes; it reflected the species divergence from the perspective of mitochondrion or plastid evolution and might lead to the deviation. In conclusion, we generated a robust phylogeny to provide an evolutionary timeline for Rhodophyta diversification. It will provide the evidence to elucidate the origin and evolution of Rhodophyta.
Data availability statements
The BioProject designations for H. cervicornis are PRJNA835989. And its raw sequences in compressed FASTQ have been deposited in GenBank under the accession numbers SRR19164831 in May 2022. The assembled complete mitochondrial genome sequence was submitted to NCBI Genbank with an accession number MZ682023 and plastid genome sequence with an accession number MZ682024.
References
Bitencourt Fda S, Figueiredo JG, Mota MR, Bezerra CC, Silvestre PP, Vale MR, Nascimento KS, Sampaio AH, Nagano CS, Saker-Sampaio S, Farias WR, Cavada BS, Assreuy AM, de Alencar NM (2008) Antinociceptive and anti-inflammatory effects of a mucin-binding agglutinin isolated from the red marine alga Hypnea cervicornis. Naunyn Schmiedebergs Arch Pharmacol 377:139–148
Boo GH, Hughey JR, Miller KA, Boo SM (2016) Mitogenomes from type specimens, a genotyping tool for morphologically simple species: ten genomes of agar-producing red algae. Sci Rep 6:35337
Cao M, Bi GQ, Mao YX, Li GY, Kong FN (2018) The first plastid genome of a filamentous taxon ’Bangia’ sp. OUCPT-01 in the Bangiales. Sci Rep. 8:10688
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Chang DD, Clayton DA (1986) Identification of primary transcriptional start sites of mouse mitochondrial DNA: accurate in vitro initiation of both heavy- and light-strand transcriptions. Mol Cell Biol 6:1446–1453
Chen WZ, Zhong ZH, Jin YL, Huang ZJ (2014) Effects of light intensity, temperature and salinity on growth and biochemical constituents of Hypnea cervicornis. South China Fisheries Science 10:48–53 (in Chinese with English abstract)
Clayton DA (1982) Replication of animal mitochondrial DNA. Cell 28:693–705
Darling AE, Tritt A, Eisen JA, Facciotti MT (2011) Mauve assembly metrics. Bioinformatics 27:2756–2757
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
de Jesus PB, Nauer F, Lyra GM, Cassano V, Oliveira MC, Nunes JM, Schnadelbach AS (2016) Species-delimitation and phylogenetic analyses of some cosmopolitan species of Hypnea (Rhodophyta) reveal synonyms and misapplied names to H. cervicornis, including a new species from Brazil J Phycol 52:774–792
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45:e18
Dong WP, Xu C, Cheng T, Zhou SL (2013) Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 8:e77965
Du QW, Bi GQ, Mao YX, Sui ZH (2016) The complete chloroplast genome of Gracilariopsis lemaneiformis (Rhodophyta) gives new insight into the evolution of family Gracilariaceae. J Phycol 52:441–450
Fay JC, Wu CI (2003) Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet 4:213–235
Galperin MY (2001) Conserved “hypothetical” proteins: new hints and new puzzles. Comp Funct Genomics 2:14–18
Galperin MY, Koonin EV (2004) “Conserved hypothetical” proteins: prioritization of targets for experimental study. Nucleic Acids Res 32:5452–5463
Gao L, Yi X, Yang YX, Su YJ, Wang T (2009) Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol Biol 9:130
Geraldino PJL, Yang EC, Boo SM (2006) Morphology and molecular phylogeny of Hypnea flexicaulis (Gigartinales, Rhodophyta) from Korea. Algae 21:417–423
Geraldino PJL, Yang EC, Kim MS, Boo SM (2009) Systematics of Hypnea asiatica sp. nov. (Hypneaceae, Rhodophyta) based on morphology and nrDNA SSU, plastid rbcL, and mitochondrial cox1. Taxon 58:606–616
Geraldino PJL, Riosmena-Rodriguez R, Liao LM, Boo SM (2010) Phylogenetic relationships within the genus Hypnea (Gigartinales, Rhodophyta), with a description of Hypnea caespitosa sp. nov. J Phycol 46:336–345
Geraldino PJL, Boo GH, Boo SM (2015) Genetic variability and biogeography of the widespread red alga Hypnea flexicaulis (Gigartinales, Rhodophyta) based on rbcL and cox1 sequences. Bot Mar 58:167–174
Golderer G, Dlaska M, Grobner P, Piendl W (1995) TTG serves as an initiation codon for the ribosomal protein MvaS7 from the Archaeon Methanococcus vannielii. J Bacteriol 177:5994–5996
Greiner S, Lehwark P, Bock R (2019) Organellar Genome DRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucl Acids Res 47:W59–W64
Guiry MD, Guiry GM (2021) AlgaeBase. World-wide electronic publication, National University of Ireland, Galway. https://www.algaebase.org; searched on 10 September 2021
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hartnell College Genomics Group, Aguilar A, Ahumada TJ, Amezcua Moreno N, Bohn J et al (2020) The complete mitochondrial and plastid genomes of the invasive marine red alga Caulacanthus okamurae (Caulacanthaceae, Rhodophyta) from Moss Landing, California, USA. Mitochondrial DNA B Resour 5:2067–2069
Heather JM, Chain B (2016) The sequence of sequencers: the history of sequencing DNA. Genomics 107:1–8
Huelsenbeck JP, Ronquist F (2001) Mrbayes: bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hughey JR, Silva PC, Hommersand MH (2001) Solving taxonomic and nomenclatural problems in Pacific Gigartinaceae (Rhodophyta) using DNA from type material. J Phycol 37:1091–1109
Hughey JR, Leister GL, Gabrielson PW, Hommersand MH (2020) Sarcopeltis gen. nov. (Gigartinaceae, Rhodophyta), with S. skottsbergii comb. nov. from southern South America and S. antarctica sp. Phytotaxa 468:75–88
Janouškovec J, Liu SL, Martone PT, Carré W, Leblanc C, Collén J, Keeling PJ (2013) Evolution of red algal plastid genomes: ancient architectures, introns, horizontal gene transfer, and taxonomic utility of plastid markers. PLoS ONE 8:e59001
Kim SY, Yang EC, Boo SM, Yoon HS (2014) Complete mitochondrial genome of the marine red alga Grateloupia angusta (Halymeniales). Mitochondrial DNA 25:269–270
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Le Corguillé G, Pearson G, Valente M, Viegas C, Gschloessl B, Corre E, Bailly X, Peters AF, Jubin C, Vacherie B, Cock JM, Leblanc C (2009) Plastid genomes of two brown algae, Ectocarpus siliculosus and Fucus vesiculosus: further insights on the evolution of red-algal derived plastids. BMC Evol Biol 9:253
Leblanc C, Boyen C, Richard O, Bonnard G, Grienenberger JM, Kloareg B (1995) Complete sequence of the mitochondrial DNA of the rhodophyte Chondrus crispus (Gigartinales). Gene content and genome organization. J Mol Biol 250:484–495
Li Y, Liu N, Wang XM, Tang XM, Zhang L, Meinita MDN, Wang GL, Yin HX, Jin YM, Wang HY, Liu C, Chi S, Liu T, Zhang J (2018) Comparative genomics and systematics of Betaphycus, Eucheuma, and Kappaphycus (Solieriaceae: Rhodophyta) based on mitochondrial genome. J Appl Phycol 30:3435–3443
Li RR, Jia XL, Zhang J, Jia SG, Liu T, Qu JY, Wang XM (2021) The complete plastid genomes of seven Sargassaceae species and their phylogenetic analysis. Front Plant Sci 12:747036
Liu N, Zhang L, Tang XM, Wang XM, Meinita NMD, Wang GL, Chen WZ, Liu T (2019) Complete plastid genome of Kappaphycus alvarezii: insights of large-scale rearrangements among Florideophyceae plastid genomes. J Appl Phycol 31:3997–4005
Liu F (2001) Studies on the Mechansim of Extracting High Gelation Property Carrageenan from Hypnea. Dissertation, South China University of Technology (In Chinese with English abstract)
Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, He GZ, Chen YX, Pan Q, Liu YJ, Tang JB, Wu GX, Zhang H, Shi YJ, Liu Y, Yu C, Wang B, Lu Y, Han CL, Cheung DW, Yiu SM, Peng SL, Zhu XQ, Liu GM, Liao XK, Li YR, Yang HM, Wang J, Lam TW, Wang J (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18
Meng C, Zhang HH, Chen YC (2008) Sequencing and analysis of mitochondrial genome of Chinese Grey Wolf (Canis lupus chanco). Chin J Biochem Mol Biol 24:1170–1176 (In Chinese with English abstract)
Miguel TBAR, Schmidt EC, Bouzon ZL, Nascimento FEP, Cunha MD, Pireda SF, Nascimento KS, Nagano CS, Saker-Sampaio S, Cavada BS, Miguel EC, Sampaio AH (2014) Morphology, ultrastructure and immunocytochemistry of Hypnea cervicornis and Hypnea musciformis- (Hypneaceae, Rhodophyta) from the coastal waters of Ceará, Brazil. J Microsc Ultrastruct 2:104–116
Mshigeni KE, Chapman DJ (1994) Hypnea (Gigartinales, Rhodophyta). Biology of economic algae. SPB Academic Publishing, The Hague, pp 245–281
Nauer F, Guimarães NR, Cassano V, Yokoya NS, Oliveira MC (2014) Hypneaspecies (Gigartinales, Rhodophyta) from the southeastern coast of Brazil based on molecular studies complemented with morphological analyses, including descriptions of Hypnea edeniana sp. nov. and H. flava sp. nov. Eur J Phycol 49:550–5753
Nauer F, Jesus PB, Cassano V, Nunes JMC, Schnadelbach AS, Oliveira MC (2019) A taxonomic review of the genus Hypnea (Gigartinales, Rhodophyta) in Brazil based on DNA barcode and morphology. Braz J Bot 42:561–574
Price JH, John DM, Lawson GW (1992) Seaweeds of the western coast of tropical Africa and adjacent islands: a critical assessment. IV. Rhodophyta (Florideae) 3. Genera H-K. Bull Br Mus Nat Hist Bot 22:123–146
Rambaut A (2016) FigTree v. 1.4.3. Available: http://tree.bio.ed.ac.uk/software/figtree/
Raubeson LA, Jansen RK (1992) Chloroplast DNA evidence on the ancient evolutionary split in vascular land plants. Science 255:1697–1699
Ribeiro ALNL, Tesima KE, Souza JMC, Yokoya NS (2013) Effects of nitrogen and phosphorus availabilities on growth, pigment, and protein contents in Hypnea cervicornis J. Agardh (Gigartinales, Rhodophyta). J Appl Phycol 25:1151–1157
Roper JM, Hansen SK, Wolf PG, Karol KG, Mandoli DF, Everett KDE, Kuehl J, Boore JL (2007) The complete plastid genome sequence of Angiopteris evecta (G. Forst.) Hoffm. (Marattiaceae). Am Fern J 97:95–106
Saunders GW (2005) Applying DNA barcoding to red macroalgae: a preliminary appraisal holds promise for future applications. Philos Trans R Soc Lond B 360:1879–1888
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:W686–W689
Sissini MN, Navarrete-Fernández TM, Murray EMC, Freese JM, Gentilhomme AS, Huber SR, Mumford TF, Hughey JR (2016) Mitochondrial and plastid genome analysis of the heteromorphic red alga Mastocarpus papillatus (C. Agardh) Kützing (Phyllophoraceae, Rhodophyta) reveals two characteristic florideophyte organellar genomes. Mitochondrial DNA B Resour 1:676–677
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Sun XY, Luo D, Zhao C, Li W, Liu T (2011) DNA extraction and PCR analysis of five kinds of large seaweed under different preservation conditions. Mol Plant Breed 9:1680–1691 (in Chinese with English abstract)
Tablizo FA, Lluisma AO (2014) The mitochondrial genome of the red alga Kappaphycus striatus (“Green Sacol” variety): complete nucleotide sequence, genome structure and organization, and comparative analysis. Mar Genomics 18:155–161
Wang DP, Zhang YB, Zhang Z, Zhu J, Yu J (2010) KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom Proteom Bioinform 8:77–80
Wang L, Mao YX, Kong FN, Li GY, Ma F, Zhang BL, Sun PP, Bi GQ, Zhang FF, Xue HF, Cao M (2013a) Complete sequence and analysis of plastid genomes of two economically important red algae: Pyropia haitanensis and Pyropia yezoensis. PLoS ONE 8:e65902
Wang XL, Shao ZR, Fu WD, Yao JT, Hu QP, Duan DL (2013b) Chloroplast genome of one brown seaweed, Saccharina japonica (Laminariales, Phaeophyta): its structural features and phylogenetic analyses with other photosynthetic plastids. Mar Genomics 10:1–9
Watanabe K, Kishimoto T, Kumagai Y, Shimizu T, Uji T, Yasui H, Kishimura H (2019) Complete sequence of mitochondrial DNA of Gloiopeltis furcata (Postels and Ruprecht) J Agardh. Mitochondrial DNA B Resour 4:2543–2544
Xiao SH, Knoll AH, Yuan XL, Pueschel CM (2004) Phosphatized multicellular algae in the Neoproterozoic Doushantuo Formation, China, and the early evolution of florideophyte red algae. Am J Bot 91:214–227
Xu B, Yang ZH (2013) PAMLX: a graphical user interface for PAML. Mol Biol Evol 30:2723–2724
Xu Q, Xiong GJ, Li PB, He F, Huang Y, Wang KB, Li ZH, Hua JP (2012) Analysis of complete nucleotide sequences of 12 Gossypium chloroplast genomes: origin and evolution of allotetraploids. PLoS ONE 7:e37128
Yamagishi Y, Masuda M (2000) A taxonomic revision of Hypnea charoides-valentiae complex (Rhodophyta, Gigartinales) in Japan, with a description of Hypnea flexicaulis sp. nov. Phycol Res 48:27–35
Yang EC, Kim KM, Kim SY, Lee J, Boo GH, Lee JH, Nelson WA, Yi G, Schmidt WE, Fredericq S, Boo SM, Bhattacharya D, Yoon HS (2015) Highly conserved mitochondrial genomes among multicellular red algae of the Florideophyceae. Genome Biol Evol 7:2394–2406
Yang QC, Li X, Li Z, Pan ZH, Yang D (2019) The mitochondrial genome analysis of Trypetoptera punctulata (Diptera: Sciomyzidae). Mitochondrial DNA B Resour 4:97–98
Yoon HS, Hackett JD, Ciniglia C, Pinto G, Bhattacharya D (2004) A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol 21:809–818
Zhang L, Wang XM, Qian H, Chi S, Liu C, Lui T (2012) Complete sequences of the mitochondrial DNA of the wild Gracilariopsis lemaneiformis and two mutagenic cultivated breeds (Gracilariaceae, Rhodophyta). PLoS ONE 7:e40241
Zhang J, Wang XM, Liu C, Jin YM, Liu T (2013) The complete mitochondrial genomes of two brown algae (Laminariales, Phaeophyceae) and phylogenetic analysis within Laminaria. J Appl Phycol 25:1247–1253
Zhang J, Liu N, Meinita MDN, Wang XM, Liu T (2020) The complete plastid genomes of Betaphycus gelatinus, Eucheuma denticulatum, and Kappaphycus striatus (Solieriaceae: Rhodophyta) and their phylogenetic analysis. J Appl Phycol 32:3521–3532
Funding
This work was supported by the Natural Science Foundation of Shandong Province (Grant No. ZR2019MC049), the National Natural Science Foundation of China (Grant No. 31402300), and Agricultural Seed Project of Shandong Province and TaiShan industrial Experts Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Xia, Y., Liu, P., Liu, X. et al. Complete organellar genomes and molecular phylogeny of Hypnea cervicornis (Gigartinales, Florideophyceae) from China. J Appl Phycol 34, 2705–2717 (2022). https://doi.org/10.1007/s10811-022-02801-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-022-02801-3