
Abstract
Background
Many species of red algae belonging to the phylum Rhodophyta are consumed by humans as raw materials for nutrition and medicine. As the seaweed market grows, the importance of the laver species has increased. The classification of red algal species has changed significantly, and the accuracy of this classification has improved significantly in recent years. Here, we report the complete circular genomes of the chloroplasts (cp) and mitochondria (mt) of three laver species (Neoporphyra dentata, Neoporphyra seriata, and Neopyropia yezoensis).
Objective
This study aims to assemble, annotate, and characterize the organization of the organelle genomes of three laver species, conduct comparative genomic studies, and develop molecular markers based on SNPs.
Methods
We analyzed organelle genome structures, repeat sequences, sequence divergence, gene rearrangements, and phylogenetic relationships of three laver species.
Results
The chloroplast genomes of the three species contained an average of 212 protein-coding genes (PCGs), while the mitochondrial genomes contained an average of 25 PCGs. We reconstructed the phylogenetic trees based on both chloroplast and mitochondrial genomes using 201 and 23 PCGs (in cp and mt genomes, respectively) shared in the class Bangiophyceae (and five species of Florideophyceae class used as an outgroup). In addition, 12 species-specific molecular markers were developed for qRT-PCR analysis.
Conclusions
This is the first report of Neoporphyra seriata complete organellar genomes. With the results, this study provides useful genetic information regarding taxonomic discrepancies, the reconstruction of phylogenetic trees, and the evolution of red algae. Moreover, the species-specific markers can be used as fast and easy methods to identify a target species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Humans have long consumed various seaweeds, such as green, brown, and red algae, especially in northeast Asia, including Korea, Japan, and China (Mouritsen et al. 2013; Brown et al. 2014). In addition, seaweeds can be used for various purposes such as nutrition, medicine, and food (Kandale et al. 2011; Cui et al. 2019). Although the use of seaweed in Western countries has been very limited, consumers have recently begun to consume seaweed as food. The seaweed market is growing, and marine crops have become one of the most commercially important vegetables. In Korea, several laver species (i.e., Neopyropia spp. and Neoporphyra spp.) are cultivated and exported to 114 countries worldwide, becoming the most exported agricultural and fishery products (Park 2023).
Previously, the phylum Rhodophyta was reported to have two classes: Bangiophyceae and Florideophyceae (Garbary and Gabrielson 1990; Yoon et al. 2006). As morphological and genetic research on Rhodophyta progressed, red algae were divided into seven classes (Bangiophyceae, Compsopogonophyceae, Cyanidiophyceae, Florideophyceae, Porphyridiophyceae, Rhodellophyceae, and Stylonematophyceae) (Saunders and Hommersand 2004; Yoon et al. 2006). Recently, the Bangiophyceae class and some families have been redefined (Yang et al. 2020; Necchi Jr and L Vis 2021; Park et al. 2023). The database such as NCBI or algal specific ‘AlgaeBase’ are updated by reflecting the latest research results. Unfortunately, old classifications and scientific names of some species are still used in some studies.
The most of eukaryotic cells have endosymbiotic organelles, and the organelles have their independent genomes. Mitochondria, whose primary role is to synthesize ATP, can be found in most eukaryotic cells, whereas chloroplasts, whose primary role is to carry out photosynthesis, can be found in most plants and algae. According to the endosymbiotic theory, proteobacteria and cyanobacteria evolve into mitochondria and chloroplasts, respectively. These organelle genomes are key targets for reconstructing phylogenetic relationships; in particular, the matK and rbcL genes are frequently used in chloroplast-based plant phylogenetic tree reconstruction (Gagnon et al. 2019; Costa et al. 2019). Recently, the rapid development of next-generation sequencing (NGS) technology has made it possible to exploit organellar genome sequence data. These changes have expanded the phylogenetic analysis methods. Recently, numerous sequence polymorphisms and genetic variations were identified. The family Bangiaceae contains over 170,000 species in AlgaeBase (https://www.algaebase.org/), but only a few complete organelle genome sequences have been published and registered in the NCBI GenBank despite the development of sequencing technology.
In this study, we generated the complete circular chloroplast and mitochondrial genomes of three red algal laver species, Neoporphyra dentata, Neoporphyra seriata, and Neopyropia yezoensis, using an Illumina sequencing platform. A previous study reported the circular organelle genomes of N. dentata (Choi et al. 2020, 2022), but comparative analysis using other Bangiaceae species showed missing annotations; therefore, we improved the sequences and annotations of the organelle genomes of N. dentata in this study. Here, we report the first complete organelle genome sequence of N. seriata. Our analysis focused on the organelle genome structures, phylogenetic relationships across Bangiophyceae species, and genetic diversity. Additionally, we report a strategy for designing species-specific molecular markers to identify different species using organelle genome sequences and nucleotide diversity and developed a total of 12 species-specific markers (four markers per species).
Materials and methods
Plant materials and DNA isolation
The fresh samples of two Neoporphyra species, N.dentata and N.seriata were collected from Doripo, Jeollanamdo, Republic of Korea (35°8′52.8″N, 126°20′18″E), and Hwangsan-myeon, Jeollanamdo, Republic of Korea (34°32′25.6″N, 126°25′22.5″E), respectively. The fresh sample of Neopyropia yezoensis was collected from Dangin-ri, Jeollanamdo, Republic of Korea (34°18′36.1″N, 126°36′37.1″E). Permission to collect samples was granted by the Ministry of Ocean and Fisheries, Republic of Korea. Our studies were complied with local and national regulations and following Kangwon National University (Chuncheon, Republic of Korea) and the Ministry of Oceans and Fisheries (Sejong, Republic of Korea) regulations.
Total genomic DNA was isolated using the Exgene™ Plant SV Kit (GeneAll®, Seoul, Korea) according to the manufacturer’s protocol. Paired-end sequencing of the genomic DNA was performed using the Illumina NovaSeq 6000 platform.
Organelle genomes assembly, mapping, and annotation
To construct complete circular organelle genomes, we used two assemblers (GetOrganelle and NOVOPlasty) (Dierckxsens et al. 2017; Jin et al. 2020) and Geneious Prime software for mapping as previously described (Lee et al. 2022), with some modifications. The raw reads obtained from Illumina sequencing were quality-and adapter-trimmed for GetOrganelle assembler and Geneious Prime mapping, and adapter-trimmed for NOVOPlasty as recommended by the developer using Trimmomatic (Bolger et al. 2014). The closely related species Neoporphyra haitanensis (NC_021189 for the cp genome and NC_017751 for the mt genome) was used as a reference for the assembly and mapping of the two Neoporphyra species, and Neopyropia yezoensis (NC_007932 for the cp genome and NC_017837 for the mt genome) was used as reference for the N.yezoensis in this study. Complete circular organelle genomes were constructed by comparing the results of two assemblers and Geneious Prime mapping.
The constructed organelle genomes were annotated using GeSeq (Tillich et al. 2017) and Geneious Prime using Bangiophyceae reference genomes (Table S1). tRNA genes were identified using tRNAscan-SE (Chan and Lowe 2019) v2.0.7. Maps of the cp and mt genomes were generated using OGDRAW (Greiner et al. 2019). After the complete genomes of the three species were constructed, we modified the sequence starting point and provisionally re-annotated the reference genomes (Table S1) to standardize gene names for comparative and phylogenetic studies.
Structural and comparative analyses
CodonW (http://codonw.sourceforge.net) was used to analyze codon usage for all protein-coding genes (PCGs). Simple sequence repeats (SSRs) and long repeats were detected using MISA (Beier et al. 2017) (10 > for mono-, 5 > for di- and tri-, and 3 > for tetra-, penta-, and hexanucleotides) and REPuter (Kurtz et al. 2001) (Hamming distance 3, sequence identity ≥ 90%, and minimum repeat size ≥ 30bp), respectively. The mVISTA program (Frazer et al. 2004) with Shuffle-LEGAN mode was applied to perform multiple alignments and analyze the divergences among the organelle genomes using N. haitanensis organelle genome as a reference. The progressiveMauve (Darling et al. 2004) alignment plugin tool in Geneious Prime was used to analyze gene rearrangements and synteny.
Phylogenetic analysis
Published organelle genomes of Bangiophyceae species and five Florideophyceae species used as outgroups were collected for phylogenetic analysis of the newly constructed organelle genomes in this study. The PCG sequences of 201 cp genes and 23 mt genes were aligned using MAFFT (Katoh and Standley 2013), trimmed using trimAl (Capella-Gutiérrez et al. 2009), and concatenated in PhyloSuite (Zhang et al. 2020). Phylogenetic analysis was performed based on maximum-likelihood (ML) using IQ-TREE 2 with 1000 bootstrap replicates and 1000 Sh-aLRT replicates. The best-fit model, GTR + F + I + G4, was chosen according to the Bayesian Information Criterion (BIC).
Development of molecular markers for distinguishing the three laver species
Species-specific markers were developed as previously described (Lee et al. 2022). The CDSs of the organelle genomes constructed in this study were aligned using ClustalW, and species-specific primer pairs were designed based on the polymorphic sites using Beacon Designer™ (PRIMER Biosoft, Palo Alto, CA, USA).
Results
Complete organelle genomes of three species
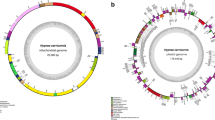
Illumina sequencing produced an average of 16 Gb of paired-end sequencing data of average 108,290 k raw reads. After trimming the low-quality raw reads, we compared the results of GetOrganelle assembly, NOVOPlasty assembly, and Geneious Prime mapping to construct the complete circular organelle genomes of the three laver species (Fig. 1). Here, we provide the complete organelle genomes of three laver species, including the first report of Neoporphyra seriata cp and mt genomes.

The chloroplast genome maps (a) and mitochondrial genome maps (b) of three laver species (Neoporphyra dentata, Neoporphyra seriata, and Neopyropia yezoensis, from left to right). The genes on the outside of the maps are transcribed in a clockwise direction, whereas the genes on the inside of the maps are transcribed in a counterclockwise direction
The average cp genome size was 193,735 bp (196,617 bp for N. dentata, 192,614 bp for N. seriata, and 191,974 bp for N. yezoensis), with a GC content of 33% (33.1% for N. dentata, 32.7% for N. seriata, and 33.1% for N. yezoensis). The cp genomes of the three species contained an average of 212 protein-coding genes (212, 211, and 213 genes in N. dentata, N. seriata and N. yezoensis, respectively). N. dentata and N. yezoensis have six rRNA genes (two sets of rrf, rrl, and rrs), whereas N. seriata has four rRNA genes (one rrs and one rrl are missing). Two tRNA genes (trnI and trnA) were located between rrs and rrl, and N. seriata also lost these two tRNA genes (37 tRNA genes in N. dentata and N. yezoensis cp genomes and 35 tRNA genes in N. seriata cp genome) (Table 1). The chloroplast genes were involved in the genetic system, photosystem, ATP synthesis, metabolism, transport, RNA genes, and unknown genes (Table S2).
The complete mt genomes of N. dentata, N. seriata, and N. yezoensis were 25,543, 32,498, and 35,863 bp in length with GC contents of 30, 31.9, and 32.4%, respectively (Fig. 1 and Table 1). The mt genomes contained 24, 25, and 26 protein-coding genes (in N. dentata, N. seriata, and N. yezoensis, respectively). All mt genomes contained 24 tRNA genes and two rRNA genes. Genes were divided into five categories (Table S2) according to their functions (oxidative phosphorylation, genetic systems, RNA genes, transport, and unknown).
Codon usage patterns
The organelle genomes of the three species were compared in terms of codon usage. Overall, 50,236 codons encoding 212 PCGs, 50,216 codons (211 PCGs), and 50,252 codons (213 PCGs) were detected in the cp genomes of N. dentata, N. seriata, and N. yezoensis, respectively. The three most commonly used codons were AUU (encoding Ile), AAA (encoding Lys), and UUA (encoding Leu2), all of which were composed of A and U. All codons with RSCU > 1 ended with A and U in the cp genomes (Fig. 2a). In the mt genomes, 6045 codons (24 PCGs), 6037 codons (25 PCGs), and 6164 codons (26 PCGs) were detected in N. dentata, N. seriata, and N. yezoensis, respectively. The codons in mt genomes showed a pattern similar to that of the cp genomes. Codons consisting of A and U accounted for the top seven frequencies (UUU, UUA, AUU, AUA, AAA, AAU, and UAU), and all codons with RSCU > 1 ended with A and U (Fig. 2b).

The RSCU (relative synonymous codon usage) of chloroplast genomes (a) and mitochondrial genomes (b)
SSR and repeat sequence analysis
In the present study, 18 (N. yezoensis) and 21 (N. dentata) SSRs were detected in the chloroplast genome (Fig. 3a). The most abundant SSRs were dinucleotide repeat units AT/AT, whereas mono- and hexanucleotide repeat units were not found. In the mitochondrial genomes, the A/T mononucleotide repeat unit was the most abundant SSR in all the three species.

Repeat analysis of organelle genomes of the three species. a The SSR analysis of organelle genomes of the three species. x-axis: repeat unit; y-axis: frequency, b Longer repeat analysis of chloroplast genomes. x-axis: repeat length; y-axis: copy number, c Longer repeat analysis of mitochondrial genomes. x-axis: repeat length; y-axis: copy number
In the cp genomes of the three species, REPuter detected five (N. dentata), 43 (N. seriata), and nine (N. yezoensis) long (> 30 bp) forward repeats and 15 (N. dentata), seven (N. seriata), and 16 (N. yezoensis) palindromic repeats (Fig. 3b). Most of the forward repeats of over 100 bp were found in the rRNA-containing region (from rrs to rrf gene) in all chloroplast genomes of the three species. The mt genomes had 2, 2, and 23 long forward repeats and 5, 3, and 10 long palindromic repeats in N. dentata, N. seriata, and N. yezoensis, respectively (Fig. 3c). Each mitochondrial genome had one long palindromic repeat of over 100 bp, and these were found in the same regions (between cob and nad6 gene) in all three species.
Analyses of genomic synteny and rearrangements
The mVISTA program was used to identify the organellar genome divergence of the three species, using the genome sequence and annotation of Neoporphyra haitanensis organellar genome as an alignment reference (Fig. 4). Gene organization was highly conserved across the cp genomes of all four species. Gene-coding regions tended to be more conserved, whereas conserved non-coding sequences (CNS) showed large variations. As illustrated in this study, N. seriata had a missing region in one set of rRNA genes, and the rRNAs were also highly conserved, except for this region. In the mt genomes, the coding regions were generally conserved, except for some orf genes. However, the mt genomes varied more than the chloroplast genome sequences.

Sequence Alignment and divergence analysis using mVISTA in chloroplast genomes (a) and mitochondrial genomes (b). Neoporphyra haitanensis cp and mt genomes were used as alignment references. UTR: untranslated region (rRNAs and tRNAs); CNS: conserved non-coding sequence
To check for gene rearrangements based on co-linear analysis of the organelle genomes, we selected 18 species, including the newly sequenced species in this study, and conducted MAUVE alignment (Fig. 5). The results showed that the organelle genomes (both cp and mt genomes) of the Bangiaceae family had no structural rearrangements.

Gene rearrangement of 18 species including three newly sequenced species in this study. a Gene rearrangement of chloroplast genomes across the species, b Gene rearrangement of mitochondrial genomes across the species. Local collinear blocks were colored to indicate syntenic regions
Phylogenetic analysis
The phylogenetic trees were reconstructed based on aligned, trimmed, and concatenated nucleotide sequences of 201 PCGs shared by 66 cp genomes (63 accessions downloaded from NCBI GenBank and three constructed cp genomes in this study) and 23 PCGs shared by 57 mt genomes (54 accessions downloaded from NCBI GenBank and three constructed mt genomes in this study), including five species of Florideophyceae, which are sister taxa of the Bangiophyceae class, as the outgroup (Fig. 6). Based on the phylogenetic analysis, the Bangiaceae family species were clearly distinguished from other families (Porphyridiaceae, Phragmonemataceae, Galdieriaceae, Cavernulicolaceae, Cyanidiaceae, and Cyanidioschyzonaceae) in both phylogenetic trees reconstructed using cp and mt genomes. Bangiaceae species were separated into five clades. Wildemania schizophylla formed a single clade, whereas two species of Bangia and two species of Porphyra formed another clade. The other three clades were clearly divided by genera (Neoporphyra, Neopyropia, and Pyropia).

The phylogenetic tree of 66 chloroplast genomes (including 63 accessions with 5 Florideophyceae species as outgroup and 3 new chloroplast genomes in this study) constructed by 201 shared PCGs (a) and the phylogenetic tree of 57 mitochondrial genomes (including 54 accessions with 5 Florideophyceae species as outgroup and 3 new mitochondrial genomes in this study) constructed by 23 shared PCGs (b)
Development of molecular markers for identifying three laver species
To validate the SNPs in genes across the three species, we aligned the complete cp genomes and compared the PCG sequences. We developed 12 species-specific markers (four markers per species) based on the SNPs of the chloroplast genes (Fig. 7) with cut-off Ct values ranging from 18 to 22 (Table S3).

Development of molecular markers based on the quantitative real-time PCR using SNPs of chloroplast genomes. Lane 1: Neopyropia yezoensis, 2: Neoporphyra seriata, 3: Neoporphyra dentata, M: DNA ladder
Discussion
In this study, the complete chloroplast and mitochondrial genomes of three laver species, Neoporphyra dentata, Neoporphyra seriata, and Neopyropia yezoensis from the family Bangiopyceae were characterized, and especially, this is the first report of the complete organelle genome sequence of N. seriata. To construct complete circular organelle genomes, NGS technology was applied and the three assembly or mapping results (two assemblies and one mapping) were compared. Two species (N. dentata and N. yezoensis) had two sets of rRNAs (rrsA and rrsB, rrlA and rrlB, and rrfA and rrfB) in their chloroplast genomes, whereas the N. seriata chloroplast genome lacked rrsB and rrlB. According to our assembly and mapping results, the two assemblers (NOVOPlasty and GetOrgenelle) showed the same results in the absence of regions including the two rRNA genes (rrsB and rrlB), while mapping to the N. haitanensis reference sequence using Geneious Prime software generated continuous but ambiguous sequences. Thus, we determined that the results of the two assemblers were correct. Loss of the region, including rrsB and rrlB didn’t occur only in N. seriata, but also in Neoporphyra perforata. Compared to other species, the two species lost about 2.9 kbp (N. seriata) and 3.3 kbp (N. perforata) of rRNA regions in similar location. Except for some orf genes (such as orf32 and orf35), the cp genomes of the three species had the same PCGs and gene orders. Owing to the loss of the rRNA region in the cp genome, N. seriata also lost two tRNA genes (trnI and trnA) located between rrsB and rrlB in other species. Except for the missing regions, the rRNA and tRNA genes were the same across the three species. The mean similarity of the whole cp genome sequences was 85.67% (88.19% between N. dentata and N. seriata, 85.14% between N. dentata and N. yezoensis, and 83.68% between N. seriata and N. yezoensis). The mt genomes showed the same PCGs and order of genes, except for some orf genes (orf72, orf88, and orf729) similar to the cp genomes. The mitochondrial genome contained the same number of rRNAs and tRNAs. Mitochondrial genes with intron sites were differentially occurred across the three species. The rnl genes of N. seriata and N. yezoensis have two intron sites, whereas the rnl gene of N. dentata has no introns. The cox1 gene in N. seriata had one intron site (and the orf729 predicted gene was encoded in the intron site), whereas the other two species had no intron site in their cox1 gene. The mean similarity of the whole mt genome sequences was 62.71% (68.09% between N. dentata and N. seriata, 56.99% between N. dentata and N. yezoensis, and 63.06% between N. seriata and N. yezoensis).
Comparative analysis of rearrangements using the Mauve program revealed that the organelle genomes of Bangiaceae family (or Bangiales order) species were more highly conserved than those of other families or orders in the Bangiophyceae class. Gene rearrangements rarely occurred in both chloroplast and mitochondrial genomes of the Bangiaceae family. The cp genome sizes of the Bangiaceae species were almost identical. In contrast, species belonging to the Porphyridiaceae family showed high variation in gene order and genome size in organelle genomes.
The classification of red algae was previously unclear. For instance, the scientific name Neoporphyra dentata has been misused as Pyropia dentata (Kim et al. 2019; Choi et al. 2020) or Porphyra dentata (Yang et al. 2022a, b). Advancements in sequencing technologies and the accumulation of genetic and phenotypic data have made the classification of red algae more accurate and precise. In the reconstructed phylogenetic trees in this study, Cyanidioschyzonaceae and Cyanidiaceae species clustered together. Recently, Park et al. suggested a new classification system for certain families of red algae (Park et al. 2023). The names of three species of the Cavernulicolaceae family registered in the NCBI have been changed (from Cyanidiales sp. SPark-2023b to Cavernulicola chilensis, from Cyanidiales sp. SPark-2023c to Gronococcus sybilensis, and from Cyanidiales sp. SPark-2023d to Sciadococcus taiwanensis). They also suggested that some Cyanidiales species should be classified under the taxonomic order Cyanidioschyzonale. Based on these suggestions, the phylogenetic tree classification in this study is clear. Although we reconstructed the phylogenetic trees based on information registered in the NCBI taxonomy (Fig. 6), we fully agree with the researcher’s suggestions. Similarly, the classification of species belonging to the Bangiaceae family is becoming more accurate. Previously, most phylogenetic trees based on nucleotide sequences were constructed using specific genes such as rbcL in the chloroplast genome or cox genes in the mitochondrial genome; however, recently, many researchers have attempted to construct phylogenetic trees using concatenated PCG sequences. We cannot determine which method provides the correct answer, but the accumulation of various data would be helpful in reconstructing more accurate phylogenetic trees. In this study, we reported the complete organelle genome sequences of N. dentata and N. yezoensis, and the first complete organelle genome sequence of N. seriata and reconstructed phylogenetic trees using the concatenated sequence of 201 shared PCGs and 23 shared PCGs in cp and mt genomes, respectively.
SSRs are frequently used to develop molecular markers; however, in this study, it was difficult to use SSRs to develop markers because of the high frequency of A and T in SSRs. In addition, codon usage analysis revealed that codons composed of A and U were used extensively. Because of the high AT content, we used SNPs in PCGs as species-specific markers and developed four markers per species for chloroplast genes (accD, atpA, atpF, and rpoC2 for N. dentata and accD, atpF, rbcL, and rpoC2 for N. seriata and N. yezoensis).
Laver species of Bangiaceae are highly valued in Korea, Japan, and China, especially in Northeast Asia. As laver gains popularity owing to its texture and flavor, the seaweed market, including laver, is growing, and the economic importance of laver species is also growing. Consequently, accurate species authentication has become important. Most species of edible algae, including laver, can be mixed during the processing stage of food materials. It is important to develop species discriminating markers to prevent such mixing or contamination for consumer’s health and rights. To accurately distinguish between species, we analyzed the organelle genomes of three laver species and developed species-specific markers. The results of this study contribute to our understanding of plant biodiversity, phylogeny, and evolution.
Data availability
The newly sequenced organellar genomes of three laver species are deposited in NCBI (OR128540.1 for N. dentata cp genome, OR123499.1 for N. dentata mt genome, OR128541.1 for N. seriata cp genome, OR123500.1 for N. seriata mt genome, OR123498.1 for N. yezoensis cp genome, OR123501.1 for N. yezoensis mt genome).
References
Beier S, Thiel T, Münch T et al (2017) MISA-web: a web server for microsatellite prediction. Bioinformatics 33:2583–2585. https://doi.org/10.1093/bioinformatics/btx198
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Brown EM, Allsopp PJ, Magee PJ et al (2014) Seaweed and human health. Nutr Rev 72:205–216. https://doi.org/10.1111/nure.12091
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. https://doi.org/10.1093/bioinformatics/btp348
Chan PP, Lowe TM (2019) tRNAscan-SE: Searching for tRNA genes in genomic sequences. Method Mol Biol 1962:1–14. https://doi.org/10.1007/978-1-4939-9173-0_1
Choi SJ, Kim Y, Shin J et al (2020) Complete chloroplast genome sequences of Pyropia dentata (Bangiales, Rhodophyta). Mitochondrial DNA B Resour 5:1785–1786. https://doi.org/10.1080/23802359.2020.1749164
Choi SJ, Kim Y, Kim DW et al (2022) The complete mitochondrial genome of Neoporphyra dentata (Bangiales, Rhodophyta). Mitochondrial DNA B Resour 7:1411–1412. https://doi.org/10.1080/23802359.2022.2102441
Costa J, Torices R, Barrett SCH (2019) Evolutionary history of the buildup and breakdown of the heterostylous syndrome in Plumbaginaceae. New Phytologist 224:1278–1289. https://doi.org/10.1111/nph.15768
Cui Y, Liu T, Wang X et al (2019) The complete chloroplast genome of Sargassum horneri and its phylogenetic analysis. Mitochondrial DNA Part B 4:3312–3313. https://doi.org/10.1080/23802359.2019.1673237
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. https://doi.org/10.1101/gr.2289704
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45:e18. https://doi.org/10.1093/nar/gkw955
Frazer KA, Pachter L, Poliakov A et al (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:W273–W279. https://doi.org/10.1093/nar/gkh458
Gagnon E, Ringelberg JJ, Bruneau A et al (2019) Global Succulent Biome phylogenetic conservatism across the pantropical Caesalpinia Group (Leguminosae). New Phytol 222:1994–2008. https://doi.org/10.1111/nph.15633
Garbary DJ, Gabrielson PW (1990) Taxonomy and evolution. In: Cole KM, Sheath RG (eds) Biology of the red algae. Cambridge University Press, Cambridge, pp 477–498
Greiner S, Lehwark P, Bock R (2019) OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res 47:W59–W64. https://doi.org/10.1093/nar/gkz238
Jin JJ, Bin YuW, Yang JB et al (2020) GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol 21:241. https://doi.org/10.1186/s13059-020-02154-5
Kandale A, Meena AK, Rao MM et al (2011) Marine algae: an introduction, food value and medicinal uses. J Pharm Res 4:219–221
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Kim JH, Choi SJ, Lee S (2019) Effects of temperature and light on photosynthesis and growth of red alga Pyropia dentata (Bangiales, Rhodophyta) in a conchocelis phase. Aquaculture 505:167–172. https://doi.org/10.1016/j.aquaculture.2019.02.058
Kurtz S, Choudhuri JV, Ohlebusch E et al (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642. https://doi.org/10.1093/nar/29.22.4633
Lee YJ, Kim YD, Uh YR et al (2022) Complete organellar genomes of six Sargassum species and development of species-specific markers. Sci Rep 12:20981. https://doi.org/10.1038/s41598-022-25443-4
Mouritsen OG, Dawczynski C, Duelund L et al (2013) On the human consumption of the red seaweed dulse (Palmaria palmata (L.) Weber & Mohr). J Appl Phycol 25:1777–1791. https://doi.org/10.1007/s10811-013-0014-7
Necchi O Jr, Vis LM (2021) Subphylum Cyanidiophytina Class Cyanidiophyceae Subphylum Proteorhodophytina Classes Compsopogonophyceae Porphyridiophyceae Rhodellophyceae, and Stylonematophyceae. In: Vis LM, Necchi O Jr (eds) Freshwater Red Algae Phylogeny Taxonomy and Biogeography. Springer International Publishing, Cham, pp 27–56
Park E-J (2023) Cross-breeding of Neopyropia spp. (Bangiales, Rhodophyta) using CAPS (Cleaved Amplified Polymorphic Sequence) markers. Korean J Fish Aquat Sci 56:124–132. https://doi.org/10.5657/KFAS.2023.0124
Park SI, Cho CH, Ciniglia C et al (2023) Revised classification of the Cyanidiophyceae based on plastid genome data with descriptions of the Cavernulicolales ord. nov. and Galdieriales ord. nov. (Rhodophyta). J Phycol. https://doi.org/10.1111/jpy.13322
Saunders GW, Hommersand MH (2004) Assessing red algal supraordinal diversity and taxonomy in the context of contemporary systematic data. Am J Bot 91:1494–1507. https://doi.org/10.3732/ajb.91.10.1494
Tillich M, Lehwark P, Pellizzer T et al (2017) GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res 45:W6–W11. https://doi.org/10.1093/nar/gkx391
Yang L-E, Deng Y-Y, Xu G-P et al (2020) Redefining Pyropia (Bangiales, Rhodophyta): Four new genera, resurrection of Porphyrella and description of Calidia pseudolobata sp. nov. From China. J Phycol 56:862–879. https://doi.org/10.1111/jpy.12992
Yang M, Ma L, Yang X et al (2022) Photosynthetic protein-based edible quality formation in various Porphyra dentata harvests determined by label-free proteomics analysis. Cells 11:1136. https://doi.org/10.3390/cells11071136
Yang M, Ma L, Yang X et al (2022) Bioinformatic Prediction and Characterization of Proteins in Porphyra dentata by Shotgun Proteomics. Front Nutr 9:924524. https://doi.org/10.3389/fnut.2022.924524
Yoon HS, Müller KM, Sheath RG et al (2006) Defining the major lineages of red algae (Rhodophyta). J Phycol 42:482–492. https://doi.org/10.1111/j.1529-8817.2006.00210.x
Zhang D, Gao F, Jakovlić I et al (2020) PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 20:348–355. https://doi.org/10.1111/1755-0998.13096
Funding
This research was supported by Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries, Korea [grant number 20210656].
Author information
Authors and Affiliations
Contributions
YJL analyzed the data; YJL and CSJ wrote the manuscript; YRU, YMK, and CMK designed and performed experiments. All authors approved the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interests.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, Y.J., Uh, Y.R., Kim, Y.M. et al. Characterization and comparative analysis of the complete organelle genomes of three red macroalgae species (Neoporphyra dentata, Neoporphyra seriata, and Neopyropia yezoensis) and development of molecular makers for their identification. Genes Genom 46, 355–365 (2024). https://doi.org/10.1007/s13258-023-01472-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-023-01472-9