
Abstract
Two complete mitochondrial genome sequences for Laminaria longissima (=Saccharina longissima) and Laminaria hyperborea are reported in this study. They had circular mapping organization with slight difference in size (37,628 and 37,976 bp, respectively) and contained almost the same set of mitochondrial genes, including the genes for three rRNAs (23S, 16S, and 5S), 25 tRNAs, 35 known mitochondrial proteins, and three to four Open Reading Frame genes (ORFs). Both mitochondrial genomes exhibited typical gene content and organization of Laminaria mtDNAs except for the existence of ORF157 genes being located between rRNA large subunit gene 5 (rpl5) and ORF129-139 in L. hyperborea as found in that of Laminaria digitata. The phylogenetic analysis based on mitochondrial genomes supported the hypothesis of the split of the genus Laminaria, and the result of this study provided important information on the molecular evolution of Laminaria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Laminarialian seaweeds (Laminariales, Phaeophyceae) are the most abundant algae in coastal waters of cold-temperate regions from Arctic to Antarctic. Laminaria is not only an ecologically important genus but also a main marine resource. It comprises a number of economically important species used as food whose extracts are of great economical value in medical products (Chapman 1952; Ohno and Crithchley 1998).
Species classification in the genus Laminaria has been mainly based on morphological and ecological features of the sporophytes, such as the shape of holdfast or blade base, and the geographical distribution (Yoshida 1998). Since phenotype character is highly variable in response to different environmental conditions and the geographical distribution of some species is not clear, classification based on those criteria is now being challenged by molecular systematics. Genes from three genetic compartments (nucleus, plastid, and mitochondrion) have recently been utilized for phylogenetic reconstruction and have also provided useful information for taxonomic study.
As a consequence of recent molecular researches, the genus Laminaria is suggested to be polyphyletic (Yoon et al. 2001), and a separation into the two genera (Laminaria Lamouroux and a resurrected Saccharina Stackhouse) are proposed (Lane et al. 2006). Nevertheless, the conclusion drawn above is only based on limited sequence data, which might not exhaust the phylogenetic relationship of the genus Laminaria, and further comprehensive probe is still urgently needed.
Mitochondrial genes are considered to be a valuable alternative to nuclear genes, for the origin of mitochondria being probably concomitant with that of eukaryotic cells (Martin and Müller 1998; Vellai et al. 1998), thus they facilitate the investigation of the molecular evolutionary relationship. Moreover, the complete mitochondrial genome sequences provide adequate information especially for the reliable resolution of the phylogenetic relationships of closely related species. Therefore, we decided to reevaluate the phylogeny of Laminaria using the whole mitochondrial genome. To date, eight complete mtDNA sequences from Laminaria species have been reported (Oudot-Le Secq et al. 2002; Yotsukura et al. 2009), while there was no information of the mitochondrial genomes from L. longissima (=S. longissima) and Laminaria hyperborea. According to previous reports, L. longissima (=S. longissima) was the important natural species of resurrecting Stackhouse’s genus Saccharina, and the genus Laminaria contains the type of the genus, L. hyperborea, besides Laminaria digitata. In this study, we determined and characterized the complete mitochondrial sequences of L. longissima (=S. longissima) and L. hyperborea, and reconstruct the phylogenetic relationships within the genus Laminaria together with all datasets of mitochondrial genomes available.
Material and methods
Laminaria japonica (=Saccharina japonica) individuals were collected from Hokkaido of Japan and L. hyperborea individuals were collected from Germany. They were all provided by the Culture Collection of Seaweed in the Ocean University of China.
Genomic DNA was extracted following the improved CTAB method (Guillemaut and Drouard 1992).The mitochondrial genome sequences were amplified from the total DNA extracts using the polymerase chain reaction (PCR). Primers for PCR were designed with the reference to the nucleotide alignment of mtDNAs from L. japonica (=S. japonica) and L. digitata. Subsequent sequencing was accomplished by the strategy of primer walking method. Primers for the interfaces obstructing the whole mitochondrial genome assembly were designed from the adjacent flanking sequenced fragments. All PCR reactions (25 μL) contained 0.4 mM of each primer, 2.5 U TaKaRa Taq (Takara), 2.5 μL 10× PCR buffer, 2 mM deoxyribonucleotide triphosphate mixture, and 1 μL of total DNA (5–30 ng). The reaction profile went as follows: initial denaturation at 95 °C for 3 min, followed by 30 cycles of denaturation at 95 °C for 45 s, annealing temperature depending on primer sequences for 45 s and elongation step at 72 °C for a time depending on the expected size of the amplified fragment, and a final elongation of 15 min at 72 °C.
The target fragments of PCR amplification were checked on 1.0 % agarose gels, and subsequently purified using the Wizard SV Gel and PCR Clean-up System (Promega) following the manufacturer’s instructions. Sequencing reactions were conducted on an ABI 3730 automated sequencer (Applied Biosystems).
Sequence alignment and genome analysis
All sequences, for both strands, were edited and aligned employing the DNAStar (DNASTAR, Inc., Madison, USA), and then assemblies and sequence quality were verified by manual operation. Gene content was determined by BLAST similarity searches (Altschul et al. 1997) against the non-redundant database of National Center for Biotechnology Information. Transfer RNA genes were identified with tRNAscan-SE 1.21 software with default parameters (http://lowelab.ucsc.edu/tRNAscan-SE/; Lowe and Eddy 1997). The ribosomal RNA genes and protein-encoding genes were annotated by DOGMA (Wyman et al. 2004) and BLAST. Gene maps of mitochondrial genomes were generated using software OGDRAW (Lohse et al. 2007).
Phylogenetic analysis
Together with L. longissima (=S. longissima) and L. hyperborea, all currently available complete mitochondrial genome data of Laminaria in GenBank (Table 1) were used in the phylogenetic analysis, using Ectocarpus siliculosus as an out-group. Phylogenetic relationships of Laminaria were conducted with two datasets, the concatenation of all shared mitochondrial protein-encoding genes as well as the concatenation of two rRNA genes (rnl, rns), respectively. Two sets of DNA sequences were performed with Clustal W computer program (Thompson et al. 1994) using the default settings. Base composition and pairwise comparison were examined using MEGA version 4.0 (Kumar et al. 2008).
Bayesian phylogenetic analyses were conducted in both datasets with MrBayes 3.0 (Ronquist and Huelsenbeck 2003). We set three partitions (first, second, and third codon positions) in the protein-encoding dataset and two partitions (rnl and rns) in rRNA-encoding dataset, assuming that functional constraints on sequence evolution were more similar within codon positions across genes than across codon positions within genes. MrBayes phylogenetic parameters of the best-fit model of individual partitions were selected by AIC in MrModeltest 3.7 (Posada and Crandall 1998). Bayesian inference using Markov chain Monte Carlo methods was performed within each partition analysis as well as within each individual sequence, each including one cold Markov chain and three incrementally heated chains, which ran simultaneously for 1,000,000 generations with trees sampled every 100 generations. The “unlink” option was employed to unlink model parameters across character partitions while all partitioned analyses accommodated among-partition rate variation by using the “prset ratepr = variable” option. Models used in partitioned Bayesian analyses were listed in Table 2. In addition, maximum likelihood (ML) trees, which based on protein-encoding dataset and rRNA-encoding dataset, were also conducted using Kimura 2-parameter model with 1,000 bootstrap replicates via MEGA5.05 (Tamura et al. 2011).
Results
Gene composition and overall organization
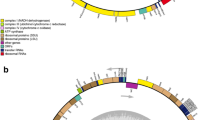
The complete mtDNAs of L. longissima (=S. longissima) and L. hyperborea exhibited circular mapping organizations with 37,628 bp (S. longissima) and 37,976 bp (L. hyperborea) in length, respectively (Fig. 1). The complete mtDNA sequences were deposited in GenBank under the following accession number: L. longissima (=S. longissima), JN099684 and L. hyperborea, JN099683.

Gene map of mitochondrial genomes of S. longissima (a) and L. hyperborea (b). Genes shown on the outside of the map are transcribed in a clockwise direction, whereas those on the inside of the map are transcribed counterclockwise
Different characteristics of the mitochondrial genomes, such as the A + T content for different categories of sequences, the total number of gene overlaps, and the size of protein-coding gene were summarized in Table 3. All these values were in the same range as those of previously reported Laminaria species. The AT content varied considerably among different regions of the mtDNAs in the two species, exhibiting a high AT richness with overall values estimated to be 64.7 and 64.8 %, respectively. Cumulative GC-skew and AT-skew analysis of the mtDNAs reflected a slight bias toward G and T in nucleotide composition on H-strand.
The gene contents and encoding order of Laminaria mtDNAs were highly conserved except for the insertion of ORF157 gene in the mtDNA of L. hyperborea and L. digitata. More than 60 putative coding regions were identified in each mtDNAs (Table 4), including three ribosomal RNA genes (rrn), 25 transfer RNA genes (trn), 17 ribosomal protein genes (11 small subunit genes (rps) and six large subunit genes (rpl)), ten NADH dehydrogenease genes (nad), one apocytochrome b gene (cob), three cytochrome oxidase genes (cox), F0-ATPase genes (atp), one Sec-independent protein translocation pathway gene (tat), and three or four open reading frame genes (ORF). Notably, none of the two new mtDNAs contained introns, which was consistent with the observation in other reported Laminaria mtDNAs.
Genes
The protein-encoding region in the mitochondrial genome of L. longissima (=S. longissima) was 29,007 bp in length, accounting for 77.1 % of the whole genome, while L. hyperborea included 29,454 bp for encoding proteins which accounted for 77.6 % of its mtDNA. In the two Laminaria mtDNAs, six protein genes were oriented to the L-strand, while other genes were encoded on the H-strand. One gene cluster, composed of rps8, rpl6, rps2, and rps4 (genes encoding the ribosomal proteins) with the initiation codon ATG and termination codon (TGA or TAA), was exactly conserved both in the two mtDNAs described here and in the other Laminaria species. The overlapping bases between adjacent genes were the following: rps8 and rpl6 sharing “ATGA”, rpl6 overlapping an “A” with rps2, and rps2 sharing an “A” with rps4 in order.
By protein sequence alignments, the universal genetic code exhibited an excellent suitability in all these Laminaria mtDNAs. All the protein-coding genes used ATG as start codons, except for ORF157 in L. digitata and L. hyperborea using GTG as the initiation codon. All of the three stop codons (TAA, TAG, and TGA) were employed with an obvious preference to TAA amounting to 68.42 % in S. longissima and 74.36 % in L. hyperborea.
At the first and second codon positions under more functional constrain, the base compositions were slightly AT rich, with values of about 57 and 63 %, respectively. The protein-encoding genes in the mitochondrial genomes showed a strong tendency toward A and T at the third codon position (76 %), which might be an implication of the phenomenon of AT rich (65.7 %) in protein-encoding genes from Laminaria mtDNA.
Three ribosomal RNA genes including 23S rRNA gene (rnl), 16S rRNA gene (rns), and 5S rRNA gene (rrn5) were encoded in the Laminaria mtDNAs. The size of rnl gene in L. longissima (=S. longissima) (2,745 bp) was identical to those in L. japonica (=S. japonica), Laminaria angustata (=Saccharina angustata), Laminaria diabolica (=Saccharina diabolica), Laminaria longipdalis (=Saccharina longipdalis), Laminaria ochotensis (=Saccharina ochotensis), Laminaria religiosa (=Saccharina religiosa), and Laminaria coriacea (=Saccharina coriacea), but 2 bp longer than its counterparts in L. hyperborea and L. digitata. Attracting attention, the same case above could be observed in rrn5, whereas, the rns genes of all Laminaria mtDNAs were the same in length.
The rns genes of L. longissima (=S. longissima) and L. hyperborea mitochondrial genome were separated from the rrn5 by four bases. The 4-bp spacer between rns and rrn5 was AGGG in L. longissima (=S. longissima) mitochondrial genome, as in those of L. japonica (=S. japonica), L. angustata (=S. angustata), L. diabolica (=S. diabolica), L. longipdalis (=S. longipdalis), L. ochotensis (=S. ochotensis), and L. religiosa (=S. religiosa), but AGAG in L. hyperborea, L. coriacea (=S. coriacea), and L. digitata.
The L. longissima (=S. longissima) and L. hyperborea mtDNAs encoded 25 tRNA genes on the H-strand respectively, of which two separate genes were determined for methionine that were initiator (trnM(cau)f) and elongator (trnM(cau)e). The 25 tRNA genes interspersed among the mitochondrial genome, ranging from 71 to 88 bp in size. Moreover, all tRNA sequences were potential to form standard cloverleaf secondary structures.
Phylogenetic relationships within Laminaria based on mitochondrial genes
The combined protein-encoding genes of 24,511 bp shared by all ten Laminaria algae as well as the combined rRNA genes datasets of 4,314 bp were utilized to reconstruct the Laminaria phylogeny. Bayesian and ML analysis based upon protein-encoding genes and rRNA genes (Fig. 2) exhibited similar topologies with robust statistical support at most of the nodes. Both analysis supported the polyphyletic origin of the genus Laminaria, and the lineage was divided into a clade formed by L. digitata and L. hyperborea and a monophyletic clade of the remaining eight Laminaria species (designated as Saccharina). For the clade of eight species, L. coriacea (=S. coriacea) was the basal species, and L. angustata (=S. angustata) subsequently derived, showing a sister relationship with the remaining six highly clustered species (S. diabolica, S. japonica, S. longipdalis, S. ochotensis, S. religiosa, and S. longissima).

Phylogenetic trees of Laminaria constructed based on concatenated nucleotide sequences of 30 mtDNA protein-coding genes (a Topology derived from Bayesian analysis; b Topology derived from maximum likelihood analysis)
Discussion
The mitochondrial genomes of L. longissima (=S. longissima) and L. hyperborea exhibited the canonical features commonly found in the other Laminaria mitochondrial genomes. The main difference observed in L. hyperborea and L. digitata mtDNAs was the presence of ORF157, which was not detected in other Laminaria species. The ORF157 in L. digitata was considered being similar to a fragment of T7-phage type RNA polymerase gene of Pylaiella littoralis (Oudot-Le Secq et al. 2002). It will be interesting to investigate more species to determine if this gene was lost after the divergence of L. hyperborea and L. digitata, or was lost earlier but acquired secondarily by L. hyperborea and L. digitata.
Another notable feature of Laminaria mtDNAs was that the ORF157 gene has the unusual start codon GTG instead of ATG. As an alternative start codons, GTG were commonly used in the mitochondrial genetic code of different animal or green-algal groups (e.g., invertebrates, molds, protozoans, and coelenterates), in the plant plastidial code, and in the bacterial code (Elzanowski and Ostell 2000). No post-transcriptional RNA editing mechanism occurred in algal mitochondrion, and it was reasonable to assume that start codon GTG remains unedited in RNA transcripts.
It is well known that all codons work as an integral unity in the process of protein translation, but the trn set employed for amino acid transportation in mtDNAs is not sufficient. As a result, the lacking tRNAs have to be imported from the cytosol to mitochondria for the accomplishment of encoding proteins, in accordance with the case in many organisms (Schneider and Mare´chal-Drouard 2000). According to the current research, we speculated that this case might also implement in Laminaria.
Classification of algae has been a long-standing controversial issue, especially for the genus Laminaria. Taxonomic category can be established by light-harvesting pigment system in higher units, while no reliable standard was available for lower units. As far as it went, simple structure of algae with few measurable properties showed no advantages in taxonomy, and insufficient support and bad performance in algae-related molecular biology research were proved to be the main reason. So, it was impossible to draw a comprehensive conclusion in phylogenetic construction with limited evidence.
Short mitochondrial gene fragments can only resolve phylogenetic relationships of higher taxonomies to a limited extent (Stepien and Kocher 1997). Phylogenetic analysis on the basis of all shared mitochondrial protein-encoding genes and two rRNA genes in this study supported previous researches based on single and double-gene studies (Yoon et al. 2001; Lane et al. 2006), although the investigations involved different affiliations. Yoon et al. (2001) found that species in the genus Laminaria fell in clearly separated clades: L. digitata, L. hyperborea, Laminaria setchellii, and Laminaria sinclairii formed the “Laminaria clade”, whereas L. japonica, L. religiosa, L. diabolica, L. longipdalis, L. longissima, and L. saccharina belonged to the “Hedophyllum clade”. Further, Lane et al. (2006) suggested retaining the genus Laminaria, containing the type of the genus L. digitata, and the other Laminaria species classified into the new genus Saccharina were: L. angustata, Laminaria cichorioides, L. coriacea, Laminaria dentigera, L. diabolica, Laminaria groenlandica, L. japonica, Laminaria longicruris, L. longipdalis, L. longissima, L. ochotensis, L. religiosa and Laminaria yendoana.
The phylogenetic relationships of Laminaria species in our study were in accordance with previous study on morphological characteristics as well as their geographic distribution. The species in the two clades detected in our study involved two kinds of diverse morphologies: L. digitata and L. hyperborea endowed with digitate blade while all the species in the other clade had simple blade. Corresponding to the morphological characteristics, species with digitate blade are found along the coast of the Atlantic Ocean, while simple blade species were mainly distributed along the coast of the Pacific Ocean. Thus, Laminaria species from different distributions had not only different morphological characteristics but also significant differentiation at the molecular level. Further investigations not only on molecular phylogeny of all members within the genus but also on the cytology and molecular biology will be necessary to test and refine the above hypothesis.
Reference
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Chapman VJ (1952) Seaweeds and their uses, 2nd edn. Methuen, London, p 304
Elzanowski AA, Ostell J (2000) The genetic codes. NCBI web site. http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
Guillemaut P, Drouard LM (1992) Isolation of plant DNA: a fast inexpensive and reliable method. Plant Mol Bio Rep 10:60–65
Lane CE, Mayes C, Druehl LD, Saunders GW (2006) A multi-gene molecular investigation of the kelp (Laminariales, Phaeophyceae) supports substantial reorganization. J Phycol 42:493–512
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Lohse M, Drechsel O, Bock R (2007) Organellar Genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52:267–274
Martin WF, Müller M (1998) The hydrogen hypothesis for the first eukaryote. Nature 392:37–41
Ohno M, Critchley AT (1998) Seaweed resources of the world. Japan International Cooperative Agency, Yokosuka, p 431
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Ronquist F, Huelsenbeck JP (2003) Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Schneider A, Maréchal-Drouard L (2000) Mitochondrial tRNA import: are there distinct mechanisms? Trends Cell Biol 10:509–513
Oudot-Le Secq M-P, Klogreg B, Loiseaux-De Goer S (2002) The mitochondrial genome of the brown alga Laminaria digitata: a comparative analysis. Eur J Phycol 37:163–172
Kumar S, Dudley J, Nei M, Tamura K (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9:299–306
Stepien CA, Kocher TD (1997) Molecules and morphology in studies of fish evolution. Molecular Systematics of Fishes 1-11
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. doi:10.1093/molbev/msr121
Thompson JD, Higgins DJ, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 22:425–430
Vellai T, Takacs K, Vida G (1998) A new aspect to the origin and evolution of eukaryotes. J Mol Evol 46:499–507
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Yoon HS, Lee JL, Boo SM, Bhattacharya D (2001) Phylogeny of Alariaceae, Laminariaceae, and Lessoniaceae (Phaeophyceae) based on plastid-encoded RuBisCo spacer and nuclear-encoded ITS sequence comparisons. Mol Phylogen Evol 21:231–243
Yoshida T (1998) Marine algae of Japan. Uchida Rokakuho, Tokyo, pp 1-1222 (in Japanese)
Yotsukura N, Shimizu T, Katayama T, Druehl LD (2009) Mitochondrial DNA sequence variation of four Saccharina species (Laminariales, Phaeophyceae) growing in Japan. J Appl Phycol 22:243–251
Acknowledgments
We thank the anonymous reviewers for their constructive comments. This work was supported by the National Public Benefit Research Foundation of China (200805075, 200903030), the Natural Science Foundation Project of China (40606035), and Fund projects of National Agricultural Transformation (2010 GB23600666).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 129 kb)
Rights and permissions
About this article
Cite this article
Zhang, J., Wang, X., Liu, C. et al. The complete mitochondrial genomes of two brown algae (Laminariales, Phaeophyceae) and phylogenetic analysis within Laminaria . J Appl Phycol 25, 1247–1253 (2013). https://doi.org/10.1007/s10811-012-9915-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-012-9915-0