
Abstract
De novo assembly of reads produced by next-generation sequencing (NGS) technologies offers a rapid approach to obtain expressed gene sequences for non-model organisms. Senna (Cassia angustifolia Vahl.) is a drought-tolerant annual undershrub of Caesalpiniaceae, a subfamily of Fabaceae. There are insufficient transcriptomic and genomic data in public databases for understanding the molecular mechanism underlying the drought tolerance of senna. Therefore, the main purpose of this study was to know the transcriptome profile of senna, with special reference to drought stress. RNA from two different stages of leaf development was extracted and sequenced separately using the Illumina technology. A total of 200 million reads were generated, and a de novo assembly of processed reads in the pooled transcriptome using Trinity yielded 43,413 transcripts which were further annotated using NCBI BLAST with “green plant database (txid 33090),” Swiss Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), and Gene Ontology (GO). Out of the total transcripts, 42,280 (95.0 %) were annotated by BLASTX against the green plant database of NCBI. Senna transcriptome showed the highest similarity to Glycine max (41 %), followed by Phaseolus vulgaris (16 %), Cicer arietinum (15 %), and Medicago trancatula (5 %). The highest number of GO terms were enriched for the molecular functions category; of these “catalytic activity” (GO: 0003824) (25.10 %) and “binding activity” (GO: 0005488) (20.10 %) were most abundantly represented. We used InterProscan to see protein similarity at domain level; a total of 33,256 transcripts were annotated against the Pfam domains. The transcripts were assigned with various KEGG pathways. Coding DNA sequences (CDS) encoding various drought stress-regulated pathways such as signaling factors, protein-modifying/degrading enzymes, biosynthesis of phytohormone, phytohormone signaling, osmotically active compounds, free radical scavengers, chlorophyll metabolism, leaf cuticular wax, polyamines, and protective proteins were identified through BLASTX search. The lucine-rich repeat kinase family was the most abundantly found group of protein kinases. Orphan, bHLH, and bZIP family TFs were the most abundantly found in senna. Six genes encoding MYC2 transcription factor, 9-cis-epoxycarotenoid dioxygenase (NCED), l -ascorbate peroxidase (APX), aminocyclopropane carboxylate oxidase (ACO), abscisic acid 8′-hydroxylase (ABA), and WRKY transcription factor were confirmed through reverse transcriptase-PCR (RT-PCR) and Sanger sequencing for the first time in senna. The potential drought stress-related transcripts identified in this study provide a good start for further investigation into the drought adaptation in senna. Additionally, our transcriptome sequences are the valuable resource for accelerated genomics-assisted genetic improvement programs and facilitate manipulation of biochemical pathways for developing drought-tolerant genotypes of crop plants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plants are exposed to numerous stresses, abiotic and biotic. Both of these stresses have substantial impacts on plant growth and productivity. Abiotic stress caused by several factors including supra-optimal (high temperature) or sub-optimal (low temperature), excess water or water deficient, increased salt levels, increased chemicals, increased light incidence, and increased levels of pollutants. Often, stress conditions are compounded in the field due to occurrence of more than one stress at the same time. Among the many abiotic stresses, drought stress is one of the most important environmental stresses affecting the productivity of most crop plants. About 26 % of world’s arable land is affected by drought stress (Blum 1988). The effects of drought on crop plants are complex, variable, and accentuated by a number of interacting factors. Drought delays plant development and affects morphology, as well as physiological processes such as photosynthesis, respiration, and translocation of assimilates (Do et al. 2013). Hence, it is important to understand the effects of drought stress on plants and identify crops tolerant to stressful environments in order to increase crop productivity and to mitigate food crisis without expending cultivated lands.
Plant species have developed diverse strategies to adapt and thrive in all kinds of climates. Drought avoidance and drought tolerance are the strategies by which a crop can minimize the loss in yield under drought stress. Drought avoidance can be achieved through morphological changes in plants, such as decreased stomatal conductance, reduced leaf area, and extensive root systems (Levitt 1980; Budak et al. 2013; Rama Reddy et al. 2014). Drought tolerance is achieved by physiological and molecular mechanisms, including osmotic adjustment, antioxidant, and scavenger compounds (Bartels and Sunkars 2005). These strategies are supported by rich and complex metabolic and gene networks that enable the plant to synthesize a wide range of compounds (Yamaguchi-Shinozaki and Shinozaki 2006; Shanker et al. 2014). Plant responses to drought stress involve interactions and crosstalk between many molecular pathways (Kantar et al. 2011). High-throughput screening techniques such as transcriptome sequencing have been used to study the adaptability of plants to drought (Ergen and Budak 2009; Raney et al. 2014; Thumma et al. 2012; Bowman et al. 2013; do Amara et al. 2016). This has led to identification of many genes related to drought stress (Zhang et al. 2013; Zhou et al. 2012; Akpinar et al. 2013; Dong et al. 2014). However, few natural allelic variants have been cloned for drought-related traits, so transcriptome analysis, quantitative trait loci (QTL), and other trait isolation methods are much essential to improve methodology for exploring drought tolerance which is a complex trait. Elucidation of the complex molecular mechanisms underlying drought tolerance in crops will accelerate the development of new varieties with enhanced drought tolerance.
The Caesalpiniaceae, a subfamily under Fabaceae, is a large family with several economically important species produceing economically important products, such as drugs (Cassia angustifolia, Cassia tora, Cassia italica, Saraca asoca), sour preparation (Tamarindus indica), dyes (Caesalpina sappan), timber (Cassia fistula), and ornamentals (Bauhinia purpuria, Delonix regia, Poinciana pulchrrima). Senna (Cassia angustifolia) (2n = 28) is a drought-tolerant annual undershrub of Caesalpiniaceae (Ayoub 1977; Khalid et al. 2012). It can survive arid environments by maintaining its water content under severe stress conditions. It is one of the important medicinal crops in the world and included in the pharmacopeia of USA, Germany, UK, India, and many other countries mainly for its cathartic properties (Lemli 1986; Folkard 1995). Medicinal properties are due to presence of sennosides in the leaves and pods (Hammouda et al. 2005). Senna is native of Yemen and Hardramaunt province of Saudi Arabia (Abulafatih 1987; Ghazanfar and Al-Sabahi 1993). The species grows in arid areas of Sudan and Egypt (Hammouda et al. 2005) and cultivated commercially in arid parts of Rajasthan, Gujarat, and Tamil Nadu in India.
Senna is a plant suitable to study the genes related to drought tolerance ultimately leading to enhancement of drought tolerance in legume crops through genetic manipulation. Physiological, developmental, and morphological characteristics collectively confer drought stress tolerance in senna (Ratnayaka and Kincaid 2005). These include maintenance of high carbon gain and water efficiency over a wide range of stress levels, drought deciduousness with re-allocation of resources to apical leaves, plasticity of stomatal and trichome numbers on a given leaflet surface, densely deposited epicuticular wax, isobilateral leaf anatomy with large bundle sheath extensions, and paraheliotropic leaflet movement. Also, production of various drought stress-related proteins and enzymes probably aids in the tolerance to drought stress in senna (Ratnayaka and Kincaid 2005). Abiotic stress-related enzymes like glutathione reductase, catalase, superoxide dismutase, and osmoprotectant proline production were enhanced in response to drought stress (Khammari et al. 2012), salt stress (Agarwal and Pandey 2004), and increased heavy metals (Qureshi et al. 2007) in senna.
Abiotic stress-tolerant plants are a potential source of genes for further research and breeding of stress-tolerant plants. Senna is considered as an excellent model plant to study plant adaptation to abiotic stress and have a deeper understanding of genetic control and strategies for adaptation to dry environment in Fabaceae. However, this objective is difficult to achieve due to lack of genomic sequence information of senna. Fortunately, availability of next-generation sequencing (NGS) has made sequencing affordable and reliable and has led to availability of transcriptomes of non-model organisms as well. RNA-Seq is a method of choice for study of drought adaptation and biological features in non-model plants (Hirayama and Shinozaki 2010; Van Eck et al. 2014; Singh et al. 2016). RNA-seq has been used to study abiotic stress response genes in many plants such as parsley (Li et al. 2014a, b), common bean (Hiz et al. 2014), chrysanthemum (Xu et al. 2013), tall fescue (Hu et al. 2014), and grapevine (Rocheta et al. 2014).
In the present study, leaf transcriptome of senna was sequenced by next-generation sequencing (NGS) technology to implicate specific pathways putatively associated with drought stress tolerance. This study for the first time provides a whole dataset of genes expressed in senna along with the glimpse of biochemical pathways in this uniquely drought-tolerant member of Fabaceae and thus will provide a base for understanding the molecular mechanism underlying drought stress tolerance.
Material and methods
Total RNA isolation, library construction, and deep sequencing
Developing leaf samples were collected at flowering stage from Cassia angustifolia (var. Sona) plants grown in the experimental farm at the ICAR-Directorate of Medicinal and Aromatic Plants Research (ICAR-DMAPR), Anand, Gujarat, India, in the year 2013. RNA from the each sample was isolated using RaFlex Total RNA isolation kit (Merck Millipore, MA, USA) by using the standard protocol described by the manufacturer. The yield and quality of RNA were determined using a Nanodrop-8000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). For RNA library construction and deep sequencing, equal quantity of RNA from young and mature leaves was used. The samples were sequenced using Illumina Miseq platform (Illumina®, San Diego, CA, USA).
Raw data processing
Next-generation sequencing using illumina Miseq generated raw data in FASTQ format. Quality of the raw data was validated based on Phred quality score (Q). This raw data was filtered and trimmed for low-quality score reads; adaptor and primer sequences were removed and reads less than 40 bp was removed using Trimmomatic v0.30 (Bolger et al. 2014). Sequencing data with Phred quality score Q ≥20 was further used assembly of raw data.
De novo assembly
High-quality data obtained after filtration from young and mature leaf samples were pooled to serve as a representative transcriptome and were provided as an input for transcriptome assembly using a Trinity RNA-Sequence assembler (Version 2013) (Grabherr et al. 2011) on optimized parameters and K-mer size set to 25. CLC Genomics workbench (CLC Bio, Boston, MA 02108 USA) was used to validate the assembled transcript contigs by mapping high-quality reads back to the assembled transcript contigs. To identify the coding DNA sequences (CDS) from assembled transcript contigs, an online tool ORF-Predictor (Min et al. 2005) (http://proteomics.ysu.edu/tools/OrfPredictor.html) with the default parameters was used.
Annotation
All the predicted CDS were functionally annotated by aligning to green plant database (txid 33090) of NCBI using basic local alignment search tool (BLASTX) (Altschul et al. 1990) with an E value threshold of 1e−06. Functions of predicted CDS were classified and each CDS was provided with the ontology of defined terms using gene ontology (GO) assignment and mapping. GO terms for all the BLASTX functionally annotated CDS was retrieved using GO mapping. CDS were categorized into 45 functional groups by WEGO analysis which involved sketching a WEGO plot based on GO hits. To retrieve GO terms for annotated CDS, the GO mapping used defined criteria. This included use of (i) BLASTX result accession IDs to retrieve gene names or symbols, (ii) UniProt IDs, and (iii) direct search in the dbxref table of the GO database. Gene names or symbols thus identified were then searched against the species-specific entries of the gene-product tables in GO database. To retrieve UniProt IDs, Protein Information Resource (PIR) was used. PIR includes protein sequence database (PSD), UniProt, SwissProt, TrEMBL, RefSeq, GenPept, and PDB databases. Using GO analysis, all the annotated nodes comprising GO functional groups were specified.
All predicted CDS were annotated against protein database so as to assign putative function of the transcriptome after translation into protein. CDS were searched against the non-redundant protein sequences available in the Uni-ProtKB/SwisProt database using BLASTX with an E value threshold of 1e−06. The CDS were categorized in 24 functional clusters of phylogenetically widespread domain families of proteins by comparing the CDS to Clusters of Orthologus Groups (COG) protein database. For higher-level groupings of related protein families, also known as clans and the identification of domains that occurs within proteins, transcripts were compared against Pfam database. For the identification of transcription factors, predicted CDS were searched against all the transcription factor and protein kinases at Plant transcription factor database (http://planttfdb.cbi.pku.edu.cn/) using BLASTX with an E value cut-off of <1e−05 (Jin et al. 2014).
KEGG mapping
KEGG automatic annotation server (KAAS) (Moriya et al. 2007) was used for ortholog assignment and mapping of CDS to metabolic pathways. BLASTX with threshold bit-score value of 60 (default) was used to search all CDS against the KEGG database. The CDS mapped in 24 different functional KASS pathway categories represented different enzymes involved in different metabolic pathways. KAAS (version 1.6) (http://www.genome.jp/tools/kaas/) with default parameters was used to perform the KEGG orthology (KO) assignment reconstructions. KASS carry out functional annotation of genes by BLAST comparison of CDS against the manually curated KEGG genes database. Thus, CDS were annotated by KEGG Orthology (KO) assignments and also were mapped to various KEGG metabolic pathways.
Mining genes involved in drought stress response
To identify putative CDS/transcripts involved in drought stress response, the literature was searched for functional genes, transcription factors and enzymes involved directly or indirectly in drought stress regulation. Orthologs of such genes were identified in the annotated transcripts and CDS of senna using various databases.
RT-PCR and Sanger sequencing
Sequences of CDS of six genes known to be involved in drought stress tolerance were used to design gene specific primers so as to assess the quality as well as precession of assembly and annotation. cDNA was used as a template for amplification using these primers. Total RNA was extracted from leaf using plant RNA-isolation kit (GeNei, Bangalore, India) according to manufacturer’s protocol. To check the quality of RNA, 8 μl of isolated RNA sample was loaded after adding 2 μl of 5× formaldehyde RNA loading dye (Thermo Scientific) and heat denaturation at 65 °C for 10 min, was loaded on 1 % denaturing agarose gel prepared in 1X MOPS buffer and formaldehyde. Gel electrophoresis was carried out using 1X MOPS buffer prepared in DEPC treated water. First strand cDNA was synthesized using M-MuLV RT-PCR kit (GeNei, Bangalore, India) with oligo-dT primer and 4 μl of isolated RNA as template according to manufacturer’s protocol. Gene specific primers were designed using transcriptome sequence was used to amplify six drought related genes using first strand cDNA as template in a 50 μl system. Touchdown PCR using conditions of 95 °C for 5 min, 95 °C for 15 s, 61 °C for 20 s (−1 °C/cycle), 72 °C for 1 min 30 s, 95 °C for 10 s, 54 °C for 20 s, 72 °C for 30 s repeated for 30 cycles and final extension 72 °C for 20 min was carried out. Amplified product was loaded on 1.5 % agarose gel. Band of amplified product was extracted from gel using Gel extraction kit (GeNei, Bangalore, India) according to manufacturer’s protocol. Extracted amplified products were sequenced using Sanger sequencing (Eurofins Scientific, India). The sequence obtained was compared to predicted CDS sequence. Sequence was BLAST searched against existing sequence on NCBI to confirm the identity of amplified sequence. Sequences were deposited in GeneBank of NCBI.
Results and discussion
Sequencing and assembly
We sequenced two cDNA library from two different stages of leaf development using Illumina Mi-Seq platform. Sequencing data included 2,20,26,329 raw reads containing 6,34,59,92,202 nucleotide bases (Table 1). The raw paired-end sequencing data in FASTQ format was deposited in the National Center for Biotechnology Information (NCBI) BioProject database (as Short Read Archive) under accession number PRJNA273534. A pooled data set was created by combining the reads from two libraries and subjected to de novo assembly. The transcriptome shotgun assembly project was deposited at DDBJ/EMBL/GenBank under the accession GEEB00000000. The version described in this paper is the first version, GEEB01000000. The assembly using Trinity yielded 43,413 non-redundant transcript contigs after filtering out those shorter than 200 bases. The details of the pooled transcriptome are provided in Table 1. The total transcript length was 72,058,285 bases (72.0 Mb), with average transcript length of 1659 bases. The GC content of transcripts was 41.96 %, which was marginally lower than AT content of 58.04 %. The size distribution of transcripts ranged from more than 1000 bp to 3500 bp and above (Table 1), wherein the maximum number of transcripts i.e., 21,192 transcripts had size in the range of 1000–1499 followed by 12,950 transcripts, size of which was in the range of 1500–1999 bp (Figure S1). Maximum contig length was 8715 bp with an average contig length of 1659 bp (Table 1). The N50 of assembly was 1697 bp which is slightly higher than that reported in senna (Rama Reddy et al. 2015) and is higher than most of the recently published plant transcriptome assemblies like Phaseolus vulgaris L. (1449 bp) (Hiz et al. 2014), Salvia hispanica L. (1338 bp) (Sreedhar et al. 2015), Raphanus sativus (773 bp) (Wu et al. 2015), Gentiana rigescens (1384 bp) (Zhang et al. 2015a, b, c), Camelia sinensis (521 bp) (Wang et al. 2016) and Codonopsis pilosula (1243 bp) (Gao et al. 2015) indicating good transcriptome assembly. In medicinal plant Cassia obtusifolia, the transcriptome sequencing of seed using Illumina and its assembly yielded 40,102 unigenes with average length of 681 bases (Liu et al. 2014). The assembly was validated by mapping high quality reads back to the assembled transcript contigs using CLC Bio Genomics workbench, 82.72 % of reads, leaf library was mapped to the transcript thereby suggesting that the assembly was highly valid. However, all but 17.28 % of reads from the leaf transcriptome library were not mapped to transcript contigs, which might be due to the presence of certain very low expressing transcripts, the reads for which might either be partially assembled or left out completely during the assembly process. This might lead to small portion of reads that are not used in the transcript contigs assembly. Identification of coding DNA sequences (CDS) for assembled transcript contigs lead to prediction of a total of 43,413 CDS from the total transcripts. The maximum CDS length was found to be 8691 bp, whereas minimum CDS length was 201 bp (Table 1). The size distribution CDS ranged from 200 bp to 1000 bp and above, wherein, the maximum number of CDS were in range of 1000 and above bp (19,312 CDS) which was followed by 4079 CDS and 4065 CDS in range of 800–899 and 900–900 respectively (Figure S2).
Functional annotation
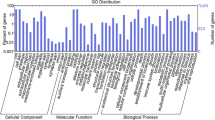
We first annotated the assembled senna unique transcripts through homologous search against green plant database (txid 33090) of NCBI using BLASTX search and threshold E-value as 1e−06. A total of 42,280 CDS (97.39 %) which had significant BLAST hits. Based on BLASTX annotation, top hit species with senna transcriptome showed highest similarity to Glycine max (41 %), followed by Phaseolus vulgaris (16 %), Cicer arietinum (15 %), and Medicago trancatula (5 %) (Fig. 1). Gene ontology (GO) was used for classification of CDS into functional categories by Blast2GO analysis. Function of predicted CDS were classified and each CDS were provided with ontology of defined terms. GO terms were enriched for 26,326 CDS and were grouped into three main domains: Biological process, Molecular function and Cellular component (Table 2 and Fig. 2). For the annotated CDS, a total of 55,318 GO terms were enriched using GO assignment. The number of enriched terms were more than that total number of CDS which is due to overlapping i.e., multiple CDS assigned to one GO term and the single CDS can have multiple GO terms. In biological processes category, 20,776 CDS were enriched, while 21,763 CDs were enriched in molecular functions category and 12,779 CDS were enriched in cellular components category (Table 2). CDS were categorized into 47 functional groups by WEGO analysis which involved making of WEGO plots based on GO hits (Fig. 2). In the biological process category, highest number of CDS were enriched in metabolic process (GO:0008152) (21.77 %) group, followed by cellular process (GO:0009987) (20.12 %) group. In the molecular function category, “catalytic activity” (GO: 0003824) (25.10 %) and “binding activity” (GO: 0005488) (20.10 %) were most abundantly represented. This indicates that the diverse metabolic processes are active in the C. angustifolia leaf, and a variety of metabolites synthesized. Under the cellular component category, the highest number of CDS were associated with “cell” (GO: 0005623) (26.10 %) and “cell part” (GO: 0044464) (21.10 %). Extremely low percentage of genes were classified in terms of “protein tag” (GO: 0031386), “locomotion” (GO: 0040011), “metallochaperone” (GO: 0016530) and “viral reproduction” (GO: 0016032).

BLASTX top hit species distribution of transcript contigs in th leaf transcriptome of senna

GO Classification. GO terms were derived based on the similarity search within leaf CDS in the transcriptome of Cassia angustofolia
We searched CDS against the non-redundent protein sequences available in the Uni-ProtKB/SwisProt database using BLASTX with E-value threshold of 1e−06 and their putative protein functions were predicted. A total of 33,256 (76.6 %) CDS showed significant hits thus indicated overall gene conservation across species. In addition, many CDS were annotated as unknown, hypothetical and expressed proteins as the CDS showed homology to uncharacterised proteins.
A total of 17,966 CDS were categorized in 24 functional clusters of phylogenetically wide spread domain families of proteins by comparing the CDS to the Clusters of Orthologous Groups (COG) protein database (Fig. 3). The highest number of CDS were represented under “General Functional Prediction only [R]” followed by “Secondary Structure [O]” and “Carbohydrate metabolism and transport [G].” The lowest number of CDS was represented under “Cell motility [N]” and “Cytoskeleton [Y].”

Clusters of orthologous groups (COG) functional classification of CDS predicted in the senna transcriptome
InterProscan was used to see protein similarity at domain level. Transcripts were annotated against Pfam domains and 37,872 transcripts were annotated (Fig. 4). The pentatricopeptide repeat (PPR) (PF01535.15) domain represented the most (4880 transcripts) which was followed by PPR_3 (PF13812.1) (4526 transcripts), PPR_2 (PF13041.1) (4293 transcripts), and PPR_1 (PF12854.2) (3985 transcripts). Other domains frequently represented in the leaf library include LRR_1 (PF00560.28) (3705 transcripts), LRR_6 (PF13516.1) (3469 transcripts) TPR_14 (PF13428.1) (3382 transcripts), LRR_7 (PF13504.1) (3307 transcripts), WD40 (PF00400.27) (3203 transcripts), and LRR_4 (PF12799.2) (2952 transcripts) in the transcripts indicating strong signal transduction mechanisms.

Top 10 Pfam domains represented in InterProScan transcript annotations of the Cassia angustifolia leaf transcriptome

Transcript annotated to different protein kinases family in the transcriptome of senna
KEGG pathway mapping
KEGG automatic annotation server (KAAS) was used for ortholog assignment and mapping of CDS to metabolic pathways. BLASTX with threshold bit-score value of 60 (default) was used to search all CDS against the KEGG database. The CDS mapped in 24 different functional KASS pathway categories represented different enzymes involved in metabolic pathways. A total of 8250 CDS were enriched in functional KASS pathway categories (Table 3). All CDS were assigned to 191 KEGG pathways (Table S1). The highest number of CDS was represented under Translation (846) and Carbohydrate metabolism (744) pathway category indicating that many active metabolic processes occurred in senna leaves, while the least number of CDS was represented under “signal molecules and interaction” and membrane transport. Under environmental adaptation category, 201 CDS were represented.
Genes encoding enzymes involved in drought stress response
Drought stress triggers a wide variety of plant responses, including alterations in gene expression, the accumulation of secondary metabolites or osmotically active compounds, and the synthesis of specific proteins and others (Ramchandra Reddy et al. 2004; Ergen et al. 2009). The ecotypic expression or suppression of regulatory genes could potentially activate multiple mechanisms of drought tolerance. Signaling factors, protein-modifying/degrading enzymes, biosynthesis of phytohormones (abscisic acid, ethylene, jasmonate, salicylic acid, brassinosteroid, gibberellic acid, and nitric oxide), phytohormone signaling (ABA, abscisic acid, ethylene, jasmonate, salicylic acid, brassinosteroid, gibberellic acid, and auxin), biosynthesis of osmotically active compounds (proline, glycin betaine, trehalose, sorbitol, mannitol, and galactinol), synthesis of free radical scavengers (catalase, peroxidase, antioxidant enymes, glutathionine, ascorbic acid, and phytochelatin), chlorophyll biosynthesis and degradation, leaf cuticular wax biosynthesis, polyamine biosynthesis, protective proteins (heat shock proteins, LEA proteins, chaprones, osmatin, and aquaporin), and others have been exploited for engineering drought tolerance in plants (Hu and Xiong 2014; Umezawa et al. 2006).
Drought signaling factors in senna
Plants have developed complex and efficient signal transduction networks to cope with the continual challenges of an unfavorable environment especially during drought stress. Protein kinases, transcription factors, and others such as cyclin-dependent protein kinase, ATPase/hydrogen-translocating pyrophosphatase (AVP1), and ABA catabolism (Chl-NADP-ME) are major signaling factors involved in drought stress signaling (Akpinar et al. 2012).
Protein kinases
Protein kinases play essential roles in developmental and environmental signal transduction in plants (Rodriguez et al. 2010; Liu et al. 2016). Protein kinases activate transcription factors and drought-responsive proteins through post-transcriptional modification, thus important candidates for improving drought tolerance. Mitogen-activated protein kinase (MAPK) cascades, calcineurin B-like protein-interacting protein kinase (CIPK), calcium-dependent protein kinase (CDPK or CPK), receptor-like kinases, and others have roles in drought stress signaling and regulation pathways. Genes encoding various PK have been identified in plants as they are involved in stress signaling (Long et al. 2014; Yang et al. 2008; Wei et al. 2014; Zhao et al. 2013). In the present study, BLASTX search of senna transcripts against protein kinase family database (http://bioinfo.bti.cornell.edu/tool/itak/) resulted in 2060 PKs which were classified into 41 known PK families based the kinase domins (Table 4 , Fig. 5). The most abundant group of protein kinases was leucine-rich repeat kinase family (346; 17 %), receptor like cytoplasmic kinase family (316; 15 %), domain of unknown function 26 (DUF26) kinase (126; 6 %), CDC2 like kinase family (114; 6 %), GmPK6/AtMRK1 family (109; 5 %), and IRE/NPH/PI dependent/S6 kinase (104; 5 %) in senna. Genes from these families have been reported to play significant roles in plant responses to drought stress in plants (Hu and Xiong 2014; Sun et al. 2015a, b). For example, there were CDS detected for MAPK cascades (74) and CDPK (84), receptor-like kinases (42) involved in drought signaling forms important candidates for improvement of drought tolerance (Hu and Xiong 2014). Other PKs were present in lower number.
Transcription factors
Transcription factors (TFs) are the important upstream regulatory proteins and play critical roles in various plant developmental processes and plant responses to abiotic and biotic stresses. Genes encoding various TF family members were identified as involved in regulating drought tolerance and are promising in improving the drought tolerance in plants (Ciftci-Yilmaz and Mittler 2008; Fang et al. 2008; Lucas et al. 2011a, b; Sun et al. 2016). The BLASTX search of senna transcripts against Plant Transcription Factor database resulted in 10,833 transcription factors which were classified into 78 known transcription factor families based on their DNA binding domains. The largest group of transcription factors included was the Orphan family (263, 2.40 %), followed by bHLH (235, 2.10 %), C3H (193, 1.78 %), and C2C2 Family (190, 1.75 %) (Table 4). Genes from these families have been reported to play significant roles in plant responses to drought stress in plants (Hu and Xiong 2014). NAM-ATAF-CUC2 (NAC) is a plant-specific TF family with a highly conserved DNA-binding domain, and many genes belonging to this family are responsive to drought stress (Fang et al. 2008). There were 183 CDS encoding NAC TF family (Table 4). Over expression of SNAC1 a NAC family TF, in rice and wheat showed improved drought resistance (Nakashima et al. 2012). Similarly, CDS for TF families such as MYB-related (Xiong et al. 2014), MYB (Su et al. 2014), MYC (Abe et al. 1997), C2H2 type zinc Finger (Shi et al. 2014), bZIP (Zhong et al. 2015), HB (Jain et al. 2008), WRKY (Ren et al. 2010; Van Eck et al. 2014), AP2-EREBP (Liu et al. 1998; Wu et al. 2016),CCAAT/NF-Y (Li et al. 2008), HSF (Li et al. 2014a, 2014b), CAMTA (Pandey et al. 2013), and zf-HD (Tran et al. 2007) were identified which have been reported to play important roles in plant responses to drought stress, and were also highly abundant in our transcriptome dataset. CDS encoding other signaling factors such as Cyclin-dependent protein kinase (95), ATPase/hydrogen-translocating pyrophosphatase (AVP1) (5), and ABA catabolism (Chl-NADP-ME) (65) were also identified in the senna leaf transcriptome.
Protein-modifying/degrading enzymes
Protein modifications and degrading enzymes play important roles in ABA signaling, hence used for engineering drought tolerance in crop plants. The cytosolic enzymes viz., E3 ubiquitin-protein ligase, ubiquitin-conjugating enzyme E2, and ubiquitin-activating enzyme E1 play an important role in degradation of ubiquitinated proteins, are demonstrated to function as a positive regulator of ABA-dependent response to drought stress (Ryu et al. 2010). Genes encoding various protein-modifying/degrading enzymes have been identified in plants (Gao et al. 2011; Kuzuoglu-Ozturk et al. 2012; Song et al. 2016) and were use in improving drought tolerance in plants (Ning et al. 2011; Park et al. 2010). In the present study, CDS encoding for E3 ubiquitin-protein ligase (609), ubiquitin-conjugating enzyme E2 (57), and ubiquitin-activating enzyme E1 (14) were identified through BLASTX search which forms important resource to understand the drought adaptation in senna (Table 4). Farnesylation is a posttranslational modification in which a farnesyl group is added to inactive target proteins so that they are targeted to membranes. Suppressing the rice farnesyltransferase SQS by RNA interference greatly enhanced drought resistance (Manavalan et al. 2012). There were nine CDS encoding farnesyltransferase/squalene synthase in senna which may be exploited for improving drought tolerance in crop plants. Ski-Interacting protein/SNW domain-containing protein and ribosome-inactivating proteins are involved in drought signaling (Lim et al. 2010; Jiang et al. 2012). There were six CDS encoding Ski-Interacting protein/SNW domain-containing protein in senna which form additional candidates for developing drought-tolerant transgenic plants.
Biosynthesis of phytohormone
Plant hormones play a major role in abiotic stress response in plants (Khan et al. 2012) and regulate developmental processes and signaling networks under abiotic stress. In plants, accumulation of abscisic acid (ABA) plays an important role in drought stress signaling and transduction pathways, mediating many responses. Plants perceive and respond adaptively to drought stress controlled mainly by the phytohormone abscisic acid (ABA). Other plant hormones involved in drought stress response include ethylene, jasomnate and methyl jasmonate, salicylic acid, brasinosteroids, gibberlic acid, and nitric oxide.
ABA biosynthesis
ABA is synthesized and accumulated in guard cells where it triggers stomatal closer under drought (Schroeder et al. 2001). ABA is synthesized from the C40 carotenoid precursor beta-carotene in plants (Cutler and Krochko 1999). In senna, 38 CDS encoding eight enzymes involved in ABA biosynthesis, i.e., beta-carotene 3-hydroxylase, beta-ring hydroxylase, zeaxanthin epoxidase, violaxanthin de-epoxidase, 9-cis-epoxycarotenoid dioxygenase, xanthoxin dehydrogenase, abscisic-aldehyde oxidase, and abscisic acid 8′-hydroxylase were identified (Table 5 and Fig. 6). Beta-carotene 3-hydroxylase (BCH) has been shown to be critical for drought tolerance and oxidative stress (Du et al. 2010). There were the CDS encoding BCH in senna. Recently, a key ABA biosynthesis gene ABA8H encoding abscisic acid 8′-hydroxylase plays a critical role in regulating ABA levels during seed imbibition and dehydration stress (Saito et al. 2004; Kushiro et al. 2004; Xu et al. 2014a, b). There were seven CDS encoding abscisic acid 8′-hydroxylase in senna, which form important resource for improvement of drought tolerance.

ABA biosynthetic pathway in senna (numbers in brackets represent number of CDS)
Ethylene biosynthesis
Under drought conditions, ethylene causes leaf abscission and consequently reduced water loss (Zhu et al. 2011). Several studies in model plants have evaluated the importance of ethylene hormone in crosstalk signaling with different metabolic pathways, in addition to responses to biotic stresses (Arraes et al. 2015; Shang et al. 2014; Kantar et al. 2011). Ethylene is derived from the amino acid methionine provided by the Yang cycle (Roje 2006) in which the precursor S-adenosylmethionine (AdoMet or SAM) is synthesized from ATP and methionine by S-adenosylmethionine synthetase (SAMS; EC 2.5.1.6). AdoMet is then converted into 1-aminocyclopropane-1-carboxylic acid (ACC) and 5-methylthioadenosine (MTA) by the enzyme 1-aminocyclopropane-1-carboxylase synthase (ACS, EC 4.4.1.14). MTA is recycled through a series of Yang cycle reactions back to methionine (Argueso et al. 2007). In the present study, 49 CDS encoding for three enzymes involved in ethylene biosynthesis namely S-adenosylmethionine synthetase (8), 1-aminocyclopropane-1-carboxylate synthase (ACS) (2), and aminocyclopropanecarboxylate oxidase (39) were identified (Table 5) thus providing important candidates for engineering drought tolerance in crops.
Jasmonate and methyl-jasmonate biosynthesis
Plant responses to abiotic stresses particularly drought stress are orchestrated locally and systemically by signaling molecules known as the jasmonates (JAs), a class of polyunsaturated fatty acid-derived phytohormones (Turner et al. 2002). The biosynthesis of JA initiates in chloroplasts, involving the release of α-linolenic acid (α-LeA, 18:3 or 18:2) from the lipid membrane by phospholipases (PLDs). In this study, from the leaf transcriptome of senna, 71 CDS encoding for 10 enzymes involved in jasmonate and methyl-jasmonate biosynthesis, i.e., phospholipase A2 (3), lipoxygenase (24), hydroperoxide dehydratase (5), allene oxide cyclase (4), 12-oxophytodienoic acid reductase (4), OPC-8:0 CoA ligase (3), acyl-CoA oxidase (9), enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase (13), acetyl-CoA acyltransferase (4), and jasmonate O-methyltransferase (2) were identified (Table 5) which will form on invaluable resource to understand the JA pathway which in turn lead to practical biotechnological applications in plants.
Salicylic acid biosynthesis
Salicylic acid (SA) is a seven carbon-containing, naturally occurring phenolic compound and endogenously synthesized signaling molecule in plants (Wang et al. 2015). The most common pathway in plants for SA synthesis is phenylalanine pathway; however, SA biosynthesis may also be accomplished by isochorismate pathway (Kawano et al. 2004; Mustafa et al. 2009). SA is produced after a series of chemical reactions catalyzed by many enzymes. There were 21 CDS identified encoding for three enzymes involved in salicylic acid biosynthesis namely isochorismate synthase (9), phenylalanine ammonia-lyase (7), and trans-cinnamate 4-monooxygenase (5) in our dataset (Table 5). We did not find CDS for benzoic acid 2-hydroxylase involved in catalyzing the biosynthesis of salicylic acid from benzoic acid. This might be due to the presence of very low levels of transcripts in the sample or possibility of being a key enzyme, and thus, its expression might be tightly regulated and thus absence of the transcript. Similarly, the CDS encoding enzymes involved in biosynthesis of brassinosteroids, gibbarlic acid, and nitric oxide were identified (Table 5) in the leaf transcriptome of senna.
Plant hormone signaling in senna
Plant hormones regulate plant responses under biotic and abiotic stresses through specific signaling networks (Kohli et al. 2013). Plant hormones such as abscisic acid, ethylene, jasmonic acid, salicyclic acid, brassinosetroids, gibbarlic acid, and auxin were studied for their role in abiotic stress responses (Peleg and Blumwald 2011; Santner et al. 2009). Putative plant hormone signaling genes detected in senna during the drought response are listed in Table 6. Abscisic acid (ABA), acts as an endogenous messenger in the regulation of the plant’s water status (Swamy and Smith 1999). ABA-dependent stress signaling involves many enzymes. The soluble PYR/PYL/RCAR receptors function at the apex of a negative regulatory pathway to directly regulate PP2C phosphatases, which in turn directly regulate SnRK2 kinases (Cutler et al. 2010). A maximum number of CDS (253) encoding for protein phosphatase 2C PP2C, a negative regulator of ABA response in plants, were detected in senna. Similarly, CDS for abscisic acid receptor PYR/PYL family (11), serine/threonine-protein kinase SRK2 SNRK2 (25), and ABA responsive element binding factor ABF (21) were also detected in the leaf transcriptome of senna which forms candidates for understanding and improvement of drought stress in plants. Ethylene, a gaseous plant hormone, regulates growth and development as well as responses to biotic and abiotic stresses in plants (Bleecker and Kende 2000). Over the last few decades, key elements involved in ethylene signal transduction have been identified in plants (Shakeel et al. 2013). Almost all of the ethylene-signaling homologous members was detected in the C. agustifolia transcriptome. Serine/threonine-protein kinase CTR1 was well-known for mediating stress responses and development in plants (Xu et al. 2014a). JAs also regulate such diverse processes as pollen maturation and wound responses in Arabidopsis. Several core factors in the JA signaling pathway are listed in Table 6. MYC transcription factors and Coronatine-insensitive protein 1 (COI1) are key regulator of genes involved in wound- and methyl jasmonate-induced secondary metabolism, defense, and hormone interactions (Devoto et al. 2005 and Zhang et al. 2015a, b, c). In the salicyclic acid signaling pathway, NPR1 (non-specific disease resistance 1) is a key regulator in SA-dependent defense signaling (Boatwright and Pajerowska-Mukhtar 2013). Similarly, WRKY and TGA play major roles as transcriptional regulators in the SA pathway. We detected three NPR1-related proteins and 22 TGAs in C. angustifolia, which might function in the SA signaling pathway in C. angustifolia. Similarly, brassinosteroids (BRs) are growth-promoting steroid hormones that regulate diverse physiological processes in plants. The BR signal is transduced by a receptor kinase-mediated signal transduction pathway, which is distinct from animal steroid-signaling systems. CDS for majority of the BR signaling homologous members were detected in the C. angustifolia transcriptome. We detected 14 gibberellin receptor GIBBERELLIN INSENSITIVE DWARF1 (GID1), nine DELLA growth inhibitors (DELLAs), two F-box proteins (GID2), and 13 phytochrome-interacting factor 3, which play important roles in GA signaling pathways (Daviere and Achard 2013; Richards et al. 2001) in senna. Auxin regulates transcription by rapidly modulating levels of Aux/IAA proteins throughout development. Auxin binds to TIR1, the F-box subunit of the ubiquitin ligase complex SCF (TIR1), and stabilizes the interaction between TIR1 and Aux/IAA substrates (Mockaitis and Estelle 2008). The auxin response factor (ARF) family contains transcription factors that bind to auxin-responsive elements (AREs) in the promoters of primary auxin-responsive genes (Sun et al. 2015a, b; Wang et al. 2010). All of the major auxin signaling factors are found in the senna transcriptome (Table 6) which will enrich our knowledge on molecular basis of phytohormone signaling in plants during drought stress.
Osmolytes in drought stress response
Plants respond to stress by production of low molecular weight, non-toxic solutes which are known as osmolytes or osmoprotectants. Osmolytes stabilize cell proteins and structures under stress. The major osmolytes accumulated by plants under abiotic stress include proline, glycine betaine (GB), trehalose, sorbitol and mannitol and galactinol. Plants accumulate proline which functions as an osmolyte to stabilize cell proteins and structures under drought stress. Proline is considered as a scavenger of free radicals, an energy sink, and a stress-regulated signal (Seki et al. 2007). Tissue-specific proline synthesis and catabolism have been found promoting growth and maintain a higher NADP/NADPH ratio at lower water potential (Sharma et al. 2011; Zhang et al. 2015a, b, c). In the present study, there were 17 CDS encoding for two enzymes involved in proline biosynthesis of which 14 were encoding delta-1-pyrroline-5-carboxylate synthetase (P5CS) and three encoding pyrroline-5-carboxylate reductase (P5CR) enzymes (Table 6 and Fig. 7). Similarly, for the biosynthesis of glycine betaine, four CDS encoding for two enzymes namely choline monooxygenase (one CDS) and betaine-aldehyde dehydrogenase (three CDS) were identified. In addition, we also found unique transcripts in the trehalose, sorbitol, and mannitol and galactinol biosynthesis in senna. The accumulation of these osmolytes could be critical to improve stress tolerance especially osmotic stress of senna.

Proline biosynthetic pathway in senna (numbers in brackets represent number of CDS)
Analysis of genes for free radical scavengers in senna
Plants, when subjected to drought stress, increase the production of reactive oxygen species (ROS) such as superoxide radical, hydrogen peroxide, and hydroxyl radical. To protect cells and subcellular systems from the effect of these ROS, plants produce free radical scavengers (D’Autréaux and Toledano 2007; Kumari et al. 2015). These include enzymes such as superoxide dismutase (SOD), catalase (CAT) and peroxidase (APX), and non-enzyme compounds such as ascorbic acid, phytochelatin, and glutathione. In the present study, ten CDS encoding for superoxide dismutase (SOD), two CDS encoding for catalase (CAT), and 29 CDS encoding for peroxidase (APX) were identified. Glutathione reductase (GR, EC 1.6.4.2) and tripeptide glutathione (GSH, γ-glutamyl-cysteinyl-glycine) are two major components of the ascorbate-glutathione (AsA-GSH) pathway which play a significant role in protecting cells against ROS and its reaction products-accrued potential anomalies (Gill et al. 2013). There were CDS encoding for glutathione S-transferase (48), glutathione reductase (9), glutathione peroxidase (5), glutamate cysteine ligase (2), and glutathione synthase (2) in the transcriptome of senna which are involved in the biosynthesis of glutathione, a ubiquitous intracellular peptide with diverse functions (Table 6). l-Ascorbic acid (vitamin C) is a major antioxidant in plants and plays a significant role in mitigation of excessive cellular reactive oxygen species activities caused by a number of abiotic stresses (Venkatesh and Park 2014). There were 75 CDS encoding for eight enzymes involved in ascorbic acid biosynthesis which were identified namely GDP-d-mannose 3′, 5′-epimerase, GDP-l-galactose phosphorylase, inositol-phosphate phosphatase/l-galactose 1-phosphate phosphatase, l-galactose dehydrogenase, l-galactono-1,4-lactone dehydrogenase, l-ascorbate peroxidase, monodehydroascorbate reductase, and l-ascorbate oxidase. We also found nine CDS encoding for phytochelatin synthase also known as glutathione gamma-glutamylcysteinyltransferase which is responsible for phytochelatin synthesis in senna which suggests drought stress may trigger the complex antioxidant network, and finely tuned ROS accumulation to facilitate appropriate signaling functions (Munné-Bosch et al. 2013).
Chlorophyll metabolism
Delayed leaf senescence or stay green is an important drought adaptation in crop plants (Rama Reddy et al. 2014). The stay-green trait reflects impaired or delayed chlorophyll catabolism (Thomas and Ougham 2014). Plants are engineered to overproduce chlorophyll—for example by overexpression of the gene encoding chlorophyllide a oxygenase (Kusaba et al. 2013). In chlorophyll metabolism, there are 15 enzymes catalyzing chlorophyll biosynthesis and five enzymes catalyzing its degradation and the genes coding these enzymes have been cloned from this model plant (Beale 2005; Hortensteiner 2006). We searched the orthologs of these genes in the leaf transcriptome of senna and found CDS for all the enzymes except ferredoxin:protochlorophyllide reductase involved in the chlorophyll biosynthesis. Similarly, out of five enzymes, CDS were identified for four enzymes viz., chlorophyll(ide) b reductase (11), chlorophyllase (6), Mg-dechelatase, pheophorbide a oxygenase (7), red chlorophyll catabolite reductase (16) involved in chlorophyll degradation (Table 7 and Fig. 8) and form important candidates to understand chlorophyll metabolism in senna.

Chlorophyll metabolism pathway under stress in senna (numbers in brackets indicate number of CDS encoding the enzyme in the pathway
Gene involved in cuticular wax biosynthesis
Cuticular wax covers outer organs of plants and functions as the outermost barrier against non-stomatal water loss and UV light. Cuticular waxes are composed of very-long-chain fatty acids (VLCFAs) and their derivatives, such as aldehydes, alkanes, esters, and primary and secondary alcohols. Many genes involved in cuticular wax biosynthesis and export have been characterized by forward and reverse genetic approaches in plants (Hooker et al. 2002; Kim et al. 2013) and their role in conferring water stress tolerance has been reported (Zhou et al. 2015; Krugman et al. 2010). There were 51 CDS encoding for three enzymes of leaf cuticular wax biosynthesis namely fatty acyl-CoA reductase (6), aldehyde decarbonylase (6), and diacylglycerol O-acyltransferase (39) which were identified which form important candidates for improvement of drought tolerance in senna (Table 7).
Genes involved in biosynthesis of polyamines
Polyamines (PAs) (putrescine, spermidine, and spermine) are a group of phytohormone-like aliphatic amine natural compounds with aliphatic nitrogen structure involved in cell growth and development, and respond to stress tolerance to various environmental factors (Gill and Tuteja 2010). We explored the gene encoding the enzymes in the production of polyamines and found that there were 64 CDS encoding for eight enzymes, arginine dacarboxylase (2), agmatine deiminase (2), S-adenosylmethionine decarboxylase (8), spermidine synthase (15), spermine oxidase (2), polyamine oxidase (25), and thermospermine synthase (8) regulating the production of PAs in senna (Table 8). However, none were found for agmatinase and N-carbamoylputrescine amidase may due to presence very low levels of transcripts in the transcriptome or possibility of being a key enzyme and thus its expression might be tightly regulated.
Protective proteins and transporters
Protective proteins (PP) include heat shock proteins (HSPs), late embryogenesis abundant proteins (LEA), chaperones, osmatin and aquaporins produced in response to drought stress in plants. HSPs are the family of proteins produced in response to stress (Song et al. 2014; Leng et al. 2015). HSPS protect cells from injury and facilitate recovery and survival after a return to normal growth conditions. HSPs function as chaperones involved in protein folding, assembly, translocation, and degradation, and also help to stabilize proteins and membranes (Boston et al. 1996; Lucas et al. 2011a, b). The well-characterized HSPs belong to the HSP70 family. We explored the senna transcriptome and found 79 CDS putatively encoding for heat shock protein. However, the role of these HSPs needs further study. We also identified many CDS encoding protective proteins such as LEA proteins (6), cheperones (236), osmatin (1), and aquaporins (34) and transporter such as Na(+)/H(+) antiporter (17) and auxin efflux carrier (18) (Table 8). Constitutive overexpression of these candidates genes could improve defense against drought stress in plants.
Validation of transcriptome
Since transcript assembly needs to be validated and as our interest was in drought stress metabolism, we choose to validate using six genes involved in drought stress (Table 9). The transcripts of all the six abiotic stress genes, viz., transcription factor MYC2, 9-cis-epoxycarotenoid dioxygenase, l-ascorbate peroxidase, aminocyclopropanecarboxylate oxidase, (+)-abscisic acid 8′-hydroxylase, and WRKY transcription factor were confirmed by reverse transcription polymerase chain reaction (RT-PCR), as was observed in an agarose gel. The amplicon sizes matched with the expected size of the gene based on assembled transcripts (Fig. 9). Transcription factor MYC2, 9-cis-epoxycarotenoid dioxygenase, l-ascorbate peroxidase, aminocyclopropanecarboxylate oxidase, (+)-abscisic acid 8′-hydroxylase, and WRKY transcription factor PCR products were further Sanger sequenced for confirmation, giving ∼100 % identity with the assembled transcriptome sequences (File S1). This is the first time that these genes have been identified in Cassia angustifolia. The sequences have been deposited at NCBI as mRNA sequences.

Detection and validation of different drought-related genes in Cassia angustifolia. cDNA was used as template to amplify drought stress-related genes. The amplified fragments were analyzed by 1.5 % agarose gel electrophoresis. DNA ladder containing 14 bands (from down 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1200, 1500, 2000, 3000; O’gene ruler 100 bp plus DNA ladder SM1153) was used. Lane L: DNA ladder; lane 1: WrkyIDCa01 (∼1200 bp); lane 2: MycIDCa02 (∼1500 bp); lane 3: NcedIDCa02 (∼1300 bp); lane 4: Aba8hIDCa02 (∼1100 bp); lane 5: ApxIDCa01 (∼1400 bp); lane 6: AccoIDCa01 (∼600 bp)
Conclusions
In this study, we performed large-scale transcriptome sequencing of senna, an important drought-tolerant herb cultivated in arid and semi-arid tropics. Insufficient transcriptomic and genomic data in public databases has limited our understanding of the molecular mechanism underlying the drought stress tolerance of senna. More than 200 million reads were generated and assembled into 43,413 unique transcripts which were further extensively annotated by comparing sequences with different databases. Coding DNA sequences (CDS) encoding various drought stress-regulated pathways such as signaling factors, protein-modifying/degrading enzymes, biosynthesis of phytohormone, phytohormone signaling, osmotically active compounds, free radical scavengers, chlorophyll metabolism, leaf cuticular wax, polyamines, and protective proteins were identified through BLASTX search. Six genes (transcription factor MYC2, 9-cis-epoxycarotenoid dioxygenase, l-ascorbate peroxidase, aminocyclopropane carboxylate oxidase, (+)-abscisic acid 8′-hydroxylase, and WRKY transcription factor) encoding enzymes involved in drought stress regulation were confirmed through RT-PCR and Sanger sequencing for the first time in senna. The potential drought stress-related transcripts identified in this study provide a good start for further investigation into the drought adaptation in senna. Additionally, our transcriptome sequences can be a valuable resource for accelerated genomics-assisted genetic improvement programs and facilitate a better understanding and more effective manipulation of biochemical pathways for developing drought-tolerant crop plants.
References
Abe H, Yamaguchi-Shinozaki K, Urao T, Iwasaki T, Hosokawa D, Shinozaki K (1997) Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 9(10):1859–1868
Abulafatih HA (1987) Medicinal plants of Southern Arabia. Econ Bot 41:354–360
Agarwal S, Pandey V (2004) Antioxidant enzyme responses to NaCl stress in Cassia angustifolia. Biol Plantarum 48(4):555–560
Akpinar BA, Avsar B, Lucas SJ, Budak H (2012) Plant abiotic stress signalling. Plant Signal Behav 7(11):1450–1455
Akpinar BA, Lucas SJ, Budak H (2013) Genomics approaches for crop improvement against abiotic stress. Sci World J 15:361921
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Argueso CT, Hansen M, Kieber JJ (2007) Regulation of ethylene biosynthesis. J Plant Growth Regul 26(2):92–105
Arraes FB, Beneventi MA, Lisei de Sa ME, Paixao JF, Albuquerque EV, Marin SR, Purgatto E, Nepomuceno AL, Grossi-de-Sa MF (2015) Implications of ethylene biosynthesis and signaling in soybean drought stress tolerance. BMC Plant Biol 15:213
Ayoub AT (1977) Some primary features of salt tolerance in senna (Cassia angastifolia). J Exp Bot 28:484–492
Bartels D, Sunkar R (2005) Drought and salt tolerance in plants. Cr Rev Plant Sci 24(1):23–58
Beale SI (2005) Green genes gleaned. Trends Plant Sci 10(7):309–312
Bleecker AB, Kende H (2000) Ethylene: a gaseous signal molecule in plants. Annu Rev Cell Dev Biol 16:1–18
Blum A (1988) Plant breeding for stress environments. CRC Press, Inc., Boca Raton
Boatwright JL, Pajerowska-Mukhtar K (2013) Salicylic acid: an old hormone up to new tricks. Mol Plant Pathol 14(6):623–634
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120
Boston RS, Viitanen PV, Vierling E (1996) Molecular chaperones and protein folding in plants. In: Filipowicz W, Hohn T (eds) Post-transcriptional control of gene expression in plants. Springer, Dordrecht, pp 191–222
Bowman MJ, Park W, Bauer PJ, Udall JA, Page JT, Raney J, Scheffler BE, Jones DC, Campbell BT (2013) RNA-Seq transcriptome profiling of upland cotton (Gossypium hirsutum L.) root tissue under water-deficit stress. PLoS One 8(12):e82634
Budak H, Kantar M, Kurtoglu KY (2013) Drought tolerance in modern and wild wheat. Sci World J 15:548246
Ciftci-Yilmaz S, Mittler R (2008) The zinc finger network of plants. Cell Mol Life Sci 65(7–8):1150–1160
Cutler AJ, Krochko JE (1999) Formation and breakdown of ABA. Trends Plant Sci 4(12):472–478
Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR (2010) Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol 61:651–679
D’Autréaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8(10):813–824
Daviere JM, Achard P (2013) Gibberellin signaling in plants. Development 140(6):1147–1151
Devoto A, Ellis C, Magusin A, Chang HS, Chilcott C, Zhu T, Turner JG (2005) Expression profiling reveals COI1 to be a key regulator of genes involved in wound- and methyl jasmonate-induced secondary metabolism, defence, and hormone interactions. Plant Mol Biol 58(4):497–513
do Amaral MN, Arge LW, Benitez LC, Danielowski R, Silveira SF, Farias DD, de Oliveira AC, da Maia LC, Braga EJ (2016) Comparative transcriptomics of rice plants under cold, iron, and salt stresses. Funct Integr Genom 16(5):567–579
Do PT, Degenkolbe T, Erban A, Heyer AG, Kopka J, Köhl KI, Hincha DK, Zuther E (2013) Dissecting rice polyamine metabolism under controlled long-term drought stress. PLoS One 8(4):e60325
Dong Y, Fan G, Deng M, Xu E, Zhao Z (2014) Genome-wide expression profiling of the transcriptomes of four Paulownia tomentosa accessions in response to drought. Genomics 104(4):295–305
Du H, Wang N, Cui F, Li X, Xiao J, Xiong L (2010) Characterization of the beta-carotene hydroxylase gene DSM2 conferring drought and oxidative stress resistance by increasing xanthophylls and abscisic acid synthesis in rice. Plant Physiol 154(3):1304–1318
Ergen NZ, Budak H (2009) Sequencing over 13 000 expressed sequence tags from six subtractive cDNA libraries of wild and modern wheats following slow drought stress. Plant Cell Environ 32(3):220–236
Ergen NZ, Thimmapuram, Bohnert J, Hans J, Budak H (2009) Transcriptome pathways unique to dehydration tolerant relatives of modern wheat. Funct Integr Genomics 9(3):377–396
Fang Y, You J, Xie K, Xie W, Xiong L (2008) Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol Genet Genomics 280(6):547–563
Folkard C (1995) Encyclopedia of herbs and their uses. Herb Society of America, Dorling Kindersley Publishing Inc., New York
Gao T, Wu Y, Zhang Y, Liu L, Ning Y, Wang D, Tong H, Chen S, Chu C, Xie Q (2011) OsSDIR1 overexpression greatly improves drought tolerance in transgenic rice. Plant Mol Biol 76(1–2):145–156
Gao JP, Wang D, Cao LY, Sun HF (2015) Transcriptome sequencing of Codonopsis pilosula and identification of candidate genes involved in polysaccharide biosynthesis. PLoS One 10(2):e0117342
Ghazanfar SA, Al-Sabahi AA (1993) Medicinal plants of northern and central Oman (Arabia). Econ Bot 41:89–98
Gill SS, Tuteja N (2010) Polyamines and abiotic stress tolerance in plants. Plant Signal Behav 5(1):26–33
Gill SS, Anjum NA, Hasanuzzaman M, Gill R, Trivedi DK, Ahmad I, Pereira E, Tuteja N (2013) Glutathione and glutathione reductase: a boon in disguise for plant abiotic stress defense operations. Plant Physiol Biochem 70:204–212
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29:644–652
Hammouda FM, Ismail SI, Abdel-Azim NS, Shams KA (2005) A guide to medicinal plants in North Africa. In: Batanouny KH (eds) IUCN Centre for Mediterranean Cooperation, Malaga, Andalusia, Spain, pp 217–218
Hirayama T, Shinozaki K (2010) Research on plant abiotic stress responses in the postgenome era: past, present and future. Plant J 61(6):1041–1052
Hiz MC, Canher B, Niron H, Turet M (2014) Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS One 9(3):e92598
Hooker TS, Millar AA, Kunst L (2002) Significance of the expression of the CER6 condensing enzyme for cuticular wax production in Arabidopsis. Plant Physiol 129(4):1568–1580
Hortensteiner S (2006) Chlorophyll degradation during senescence. Annu Rev Plant Biol 57:55–77
Hu H, Xiong L (2014) Genetic engineering and breeding of drought-resistant crops. Annu Rev Plant Biol 65:715–741
Hu T, Sun X, Zhang X, Nevo E, Fu J (2014) An RNA sequencing transcriptome analysis of the high-temperature stressed tall fescue reveals novel insights into plant thermotolerance. BMC Genomics 15:1147
Jiang SY, Bhalla R, Ramamoorthy R, Luan HF, Venkatesh PN, Cai M, Ramachandran S (2012) Over-expression of OSRIP18 increases drought and salt tolerance in transgenic rice plants. Transgenic Res 21(4):785–795
Jin JP, Zhang H, Kong L, Gao G, Luo JC (2014) PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res 42(D1):1182–1187
Kantar M, Lucas SJ, Budak H (2011) Drought stress: molecular genetics and genomics approaches. Adv Bot Res 57:445–493
Kawano T, Furuichi T, Muso S (2004) Controlled free salicylic acid levels and corresponding signaling mechanisms in plants. Plant Biotechnol 21:319–335
Khalid H, Abdalla WE, Abdelgadir H, Opatz T, Effert T (2012) Gems from traditional north-African medicine: medicinal and aromatic plants from Sudan. Nat Prod Bioprospect 2(3):92–103
Khammari I, Galavi M, Ghanbari A, Solouki M, Poorchaman MRA (2012) The effect of drought stress and nitrogen levels on antioxidant enzymes, proline and yield of Indian Senna (Cassia angustifolia L.). J Med Plants Res 6(11):2125–2130
Khan NA, Nazar R, Iqbal N, Anjum NA (2012) Phytohormones and abiotic stress tolerance in plants. Springer, Berlin
Kim J, Jung JH, Lee SB, Go YS, Kim HJ, Cahoon R, Markham JE, Cahoon EB, Suh MC (2013) Arabidopsis 3-ketoacyl-coenzyme a synthase9 is involved in the synthesis of tetracosanoic acids as precursors of cuticular waxes, suberins, sphingolipids, and phospholipids. Plant Physiol 162(2):567–580
Kohli A, Sreenivasulu N, Lakshmanan P, Kuman PP (2013) The phytohormone crosstalk paradigm takes center stage in understanding how plants respond to abiotic stresses. Plant Cell Rep 32(7):945–957
Krugman T, Chagué V, Peleg Z, Balzergue S, Just J, Korol AB, Nevo E, Saranga Y, Chalhoub B, Fahima T (2010) Multilevel regulation and signalling processes associated with adaptation to terminal drought in wild emmer wheat. Funct Integr Genomics 10(2):167–186
Kumari S, Joshi R, Singh K, Roy S, Tripathi AK, Singh P, Singla-Pareek SL, Pareek A (2015) Expression of a cyclophilin OsCyp2-P isolated from a salt-tolerant landrace of rice in tobacco alleviates stress via ion homeostasis and limiting ROS accumulation. Funct Integr Genomics 15(4):395–412
Kusaba M, Tanaka A, Tanaka R (2013) Stay-green plants: what do they tell us about the molecular mechanism of leaf senescence. Photosynth Res 117(1–3):221–234
Kushiro T, Okamoto M, Nakabayashi K, Yamagishi K, Kitamura S, Asami T, Hirai N, Koshiba T, Kamiya Y, Nambara E (2004) The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: key enzymes in ABA catabolism. EMBO J 23(7):1647–1656
Kuzuoglu-Ozturk D, Cebeci Yalcinkaya O, Akpinar BA, Mitou G, Korkmaz G, Gozuacik D, Budak H (2012) Autophagy-related gene, TdAtg8, in wild emmer wheat plays a role in drought and osmotic stress response. Planta 236(4):1081–1092
Lemli J (1986) The chemistry of senna. Fitoterapia 57:33–40
Leng X, Mu Q, Wang X, Li X, Zhu X, Shangguan L, Fang J (2015) Transporters, chaperones, and P-type ATPases controlling grapevine copper homeostasis. Funct Integr Genomics 15(6):673–684
Levitt J (1980) Responses of plants to environmental stress: chilling, freezing and high temperature stresses, vol 1, 2nd edn. Academic, New York
Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and post-transcriptionally to promote drought resistance. Plant Cell 20(8):2238–2251
Li MY, Tan HW, Wang F, Jiang Q, Xu ZS, Tian C, Xiong AS (2014) De novo transcriptome sequence assembly and identification of AP2/ERF transcription factor related to abiotic stress in parsley (Petroselinum crispum). PLoS One 9(9):e108977
Li PS, Yu TF, He GH, Chen M, Zhou YB, Chai SC, Xu ZS, Ma YZ (2014) Genome-wide analysis of the Hsf family in soybean and functional identification of GmHsf-34 involvement in drought and heat stresses. BMC Genom 15:1009
Lim GH, Zhang X, Chung MS, Lee DJ, Woo YM, Cheong HS, Kim CS (2010) A putative novel transcription factor, AtSKIP, is involved in abscisic acid signalling and confers salt and osmotic tolerance in Arabidopsis. New Phytol 185(1):103–113
Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi-Shinozaki K, Shinozaki K (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought- and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell 10(8):1391–1406
Liu Z, Song T, Zhu Q, Wang W, Zhou J, Liao H (2014) De novo assembly and analysis of Cassia obtusifolia seed transcriptome to identify genes involved in the biosynthesis of active metabolites. Biosci Biotechnol Biochem 78(5):791–799
Liu H, Che Z, Zeng X, Zhou X, Sitoe HM, Wang H, Yu D (2016) Genome-wide analysis of calcium-dependent protein kinases and their expression patterns in response to herbivore and wounding stresses in soybean. Funct Integr Genom 16(5):481–493
Long L, Gao W, Xu L, Liu M, Luo X, He X, Yang X, Zhang X, Zhu L (2014) GbMPK3, a mitogen-activated protein kinase from cotton, enhances drought and oxidative stress tolerance in tobacco. Plant Cell Tiss Organ Cult 116:153–162
Lucas S, Dogan E, Budak H (2011a) TMPIT1 from wild emmer wheat: first characterisation of a stress-inducible integral membrane protein. Gene 483(1):22–28
Lucas S, Durmaz E, Akpınar BA, Budak H (2011b) The drought response displayed by a DRE-binding protein from Tritium dicoccoides. Plant Physiol Biochem 49(3):346–351
Manavalan LP, Chen X, Clarke J, Salmeron J, Nguyen HT (2012) RNAi-mediated disruption of squalene synthase improves drought tolerance and yield in rice. J Exp Bot 63(1):163–175
Min XJ, Butler G, Storms R, Tsang A (2005) OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. Web Server Issue W677–W680. (http://bioinformatics.ysu.edu/tools/OrfPredictor.html)
Mockaitis K, Estelle M (2008) Auxin receptors and plant development: a new signaling paradigm. Annu Rev Cell Dev Biol 24:55–80
Moriya Y, Itoh M, Okuda S, Yoshizawa A, Kanehisa M (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182–W185
Munné-Bosch S, Queval G, Foyer CH (2013) The impact of global change factors on redox signaling underpinning stress tolerance. Plant Physiol 161(1):5–19
Mustafa NR, Kim HK, Choi YH, Erkelens C, Lefeber AW, Spijksma G, van der Heijden R, Verpoorte R (2009) Biosynthesis of salicylic acid in fungus elicited Catharanthus roseus cells. Phytochemistry 70(4):532–539
Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) NAC transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819(2):97–103
Ning Y, Jantasuriyarat C, Zhao Q, Zhang H, Chen S, Liu J, Liu L, Tang S, Park CH, Wang X, Liu X, Dai L, Xie Q, Wang GL (2011) The SINA E3 ligase OsDIS1 negatively regulates drought response in rice. Plant Physiol 157(1):242–255
Pandey N, Ranjan A, Pant P, Tripathi RK, Ateek F, Pandey HP, Patre UV, Sawant SV (2013) CAMTA 1 regulates drought responses in Arabidopsis thaliana. BMC Genomics 14:216
Park GG, Park JJ, Yoon J, Yu SN, An G (2010) A RING finger E3 ligase gene, Oryza sativa delayed seed germination 1 (OsDSG1), controls seed germination and stress responses in rice. Plant Mol Biol 74(4–5):467–478
Peleg Z, Blumwald E (2011) Hormone balance and abiotic stress tolerance in crop plants. Curr Opin Plant Biol 14(3):290–295
Qureshi MI, Abdin MZ, Qadir S, Iqbal M (2007) Lead-induced oxidative stress and metabolic alterations in Cassia angustifolia Vahl. Biol Plantarum 51(1):121–128
Rama Reddy NR, Ragimasalawada M, Sabbavarapu MM, Nadoor S, Patil JV (2014) Detection and validation of stay-green QTL in post-rainy sorghum involving widely adapted cultivar, M35-1 and a popular stay-green genotype B35. BMC Genom 18(15):909
Rama Reddy NR, Mehta RH, Soni PH, Makasana J, Gajbhiye NA, Ponnuchamy M, Kumar J (2015) Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS One 10(6):e0129422
Ramchandra Reddy A, Chaitanya KV, Vivekanandan M (2004) Drought induced responses of photosynthesis and antioxidant metabolism in higher plants. J Plant Physiol 161(11):1189–1202
Raney JA, Reynolds DJ, Elzinga DB, Page J, Udall JA, Jellen EN, Bonfacio A, Fairbanks DJ, Maughan PJ (2014) Transcriptome analysis of drought induced stress in Chenopodium quinoa. Am J Plant Sci 5(3):338–357
Ratnayaka HH, Kincaid D (2005) Gas exchange and leaf ultrastructure of tinnevelly senna, cassia angustifolia, under drought and nitrogen stress. Crop Sci 45(3):840–847
Ren X, Chen Z, Liu Y, Zhang H, Zhang M, Liu Q, Hong X, Zhu J, Gong Z (2010) ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J 63(3):417–429
Richards DE, King KE, Ait-Ali T, Harberd NP (2001) How gibberellin regulates plant growth and development: a molecular genetic analysis of gibberellins signaling. Annu Rev Plant Physiol Plant Mol Biol 52:67–88
Rocheta M, Becker JD, Coito JL, Carvalho L, Amâncio S (2014) Heat and water stress induce unique transcriptional signatures of heat-shock proteins and transcription factors in grapevine. Funct Integr Genomics 14(1):135–148
Rodriguez MC, Petersen M, Mundy J (2010) Mitogen-activated protein kinase signaling in plants. Annu Rev Plant Biol 61:621–649
Roje S (2006) S-adenosyl-L-methionine: beyond the universal methyl group donor. Phytochemistry 67(15):1686–1698
Ryu MY, Cho SK, Kim WT (2010) The Arabidopsis C3H2C3-type RING E3 ubiquitin ligase AtAIRP1 is a positive regulator of an abscisic acid-dependent response to drought stress. Plant Physiol 154(4):1983–1997
Saito S, Hirai N, Matsumoto C, Ohigashi H, Ohta D, Sakata K, Mizutani M (2004) Arabidopsis CYP707As encode (+)-abscisic acid 8′-hydroxylase, a key enzyme in the oxidative catabolism of abscisic acid. Plant Physiol 134(4):1439–1449
Santner A, Calderon-Villalobos LI, Estelle M (2009) Plant hormones are versatile chemical regulators of plant growth. Nat Chem Biol 5(5):301–307
Schroeder JI, Allen GJ, Hugouvieux V, Kwak JM, Waner D (2001) Guard cell signal transduction. Annu Rev Plant Physiol Plant Mol Biol 52:627–658
Seki M, Umezawa T, Urano K, Shinozaki K (2007) Regulatory metabolic networks in drought stress responses. Curr Opin Plant Biol 10(3):296–302
Shakeel SN, Wang X, Binder BM, Schaller GE (2013) Mechanisms of signal transduction by ethylene: overlapping and non-overlapping signalling roles in a receptor family. AoB Plants 5:plt010
Shang J, Song P, Ma B, Qi X, Zeng Q, Xiang Z, He N (2014) Identification of the mulberry genes involved in ethylene biosynthesis and signaling pathways and the expression of MaERF-B2-1 and MaERF-B2-2 in the response to flooding stress. Funct Integr Genomics 14(4):767–777
Shanker AK, Maheswari M, Yadav SK, Desai S, Bhanu D, Attal NB, Venkateswarlu B (2014) Drought stress responses in crops. Funct Integr Genomics 14(1):11–22
Sharma S, Villamor JG, Verslues PE (2011) Essential role of tissue-specific proline synthesis and catabolism in growth and redox balance at low water potential. Plant Physiol 157(1):292–304
Shi H, Wang X, Ye T, Chen F, Deng J, Yang P, Zhang Y, Chan Z (2014) The cysteine2/histidine2-type transcription factor ZINC FINGER OF ARABIDOPSIS THALIANA6 modulates biotic and abiotic stress responses by activating salicylic acid-related genes and C-REPEAT-BINDING FACTOR Genes in Arabidopsis. Plant Physiol 165(3):1367–1379
Singh R, Kumar R, Mahato AK, Paliwal R, Singh AK, Kumar S, Marla SS, Kumar A, Singh NK (2016) De novo transcriptome sequencing facilitates genomic resource generation in Tinospora cordifolia. Funct Integr Genomics 16(5):581–591
Song A, Zhu X, Chen F, Gao H, Jiang J, Chen S (2014) A chrysanthemum heat shock protein confers tolerance to abiotic stress. Int J Mol Sci 15(3):5063–5078
Song L, Jiang L, Chen Y, Shu Y, Bai Y, Guo C (2016) Deep-sequencing transcriptome analysis of field-grown Medicago sativa L. crown buds acclimated to freezing stress. Funct Integr Genom 16(5):495–511
Sreedhar RV, Kumari P, Rupwate SD, Rajasekharan R, Srinivasan M (2015) Exploring triacylglycerol biosynthetic pathway in developing seeds of Chia (Salvia hispanica L.): a transcriptomic approach. PLoS One 10(4):e0123580
Su LT, Li JW, Liu DQ, Zhai Y, Zhang HJ, Li XW, Zhang QL, Wang Y, Wang QY (2014) A novel MYB transcription factor, GmMYBJ1, from soybean confers drought and cold tolerance in Arabidopsis thaliana. Gene 538(1):46–55
Sun R, Wang K, Guo T, Jones DC, Cobb J, Zhang B, Wang Q (2015a) Genome-wide identification of auxin response factor (ARF) genes and its tissue-specific prominent expression in Gossypium raimondii. Funct Integr Genomics 15(4):481–493
Sun W, Chen H, Wang J, Sun HW, Yang SK, Sang YL, Lu XB, Xu XH (2015b) Expression analysis of genes encoding mitogen-activated protein kinases in maize provides a keylink between abiotic stress signaling and plant reproduction. Funct Integr Genomics 15(1):107–120
Sun X, Xie Z, Zhang C, Mu Q, Wu W, Wang B, Fang J (2016) A characterization of grapevine of GRAS domain transcription factor gene family. Funct Integr Genomics 16(4):347–363
Swamy PM, Smith BN (1999) Role of abscisic acid in plant stress tolerance. Curr Sci 76:1220–1227
Thomas H, Ougham H (2014) The stay-green trait. J Exp Bot 65(14):3889–3900
Thumma BR, Sharma N, Southerton SG (2012) Transcriptome sequencing of Eucalyptus camaldulensis seedlings subjected to water stress reveals functional single nucleotide polymorphisms and genes under selection. BMC Genomics 13:364
Tran LS, Nakashima K, Sakuma Y, Osakabe Y, Qin F, Simpson SD, Maruyama K, Fujita Y, Shinozaki K, Yamaguchi-Shinozaki K (2007) Co-expression of the stress-inducible zinc finger homeodomain ZFHD1 and NAC transcription factors enhances expression of the ERD1 gene in Arabidopsis. Plant J 49(1):46–63
Turner JG, Ellis C, Devoto A (2002) The jasmonate signal pathway. Plant Cell 14(Suppl):S153–S164
Umezawa T, Fujita M, Fujita Y, Yamaguchi-Shinozaki K, Shinozaki K (2006) Engineering drought tolerance in plants: discovering and tailoring genes to unlock the future. Curr Opin Biotechnol 17(2):113–122
Van Eck L, Davidson RM, Wu S, Zhao BY, Botha AM, Leach JE, Lapitan NL (2014) The transcriptional network of WRKY53 in cereals links oxidative responses to biotic and abiotic stress inputs. Funct Integr Genomics 14(2):351–362
Venkatesh J, Park SW (2014) Role of L-ascorbate in alleviating abiotic stresses in crop plants. Botan Stud 55:38
Wang S, Bai Y, Shen C, Wu Y, Zhang S, Jiang D, Guilfoyle TJ, Chen M, Qi Y (2010) Auxin-related gene families in abiotic stress response in Sorghum bicolor. Funct Integr Genomics 10(4):533–546
Wang Z, Jia C, Li J, Huang S, Xu B, Jin Z (2015) Activation of salicylic acid metabolism and signal transduction can enhance resistance to Fusarium wilt in banana (Musa acuminata L. AAA group, cv. Cavendish). Funct Integr Genomics 15(1):47–62
Wang YN, Tang L, Hou Y, Wang P, Yang H, Wei CL (2016) Differential transcriptome analysis of leaves of tea plant (Camellia sinensis) provides comprehensive insights into the defense responses to Ectropis oblique attack using RNA-Seq. Funct Integr Genomics 16(4):383–398
Wei S, Hu W, Deng X, Zhang Y, Liu X, Zhao X, Luo Q, Jin Z, Li Y, Zhou S, Sun T, Wang L, Yang G, He G (2014) A rice calcium-dependent protein kinase OsCPK9 positively regulates drought stress tolerance and spikelet fertility. BMC Plant Biol 14:133
Wu G, Zhang L, Yin Y, Wu J, Yu L, Zhou Y, Li M (2015) Sequencing, de novo assembly and comparative analysis of Raphanus sativus transcriptome. Front Plant Sci 6:198
Wu ZJ, Li XH, Liu ZW, Li H, Wang YX, Zhuang J (2016) Transcriptome-based discovery of AP2/ERF transcription factors related to temperature stress in tea plant (Camellia sinensis). Funct Integr Genomics 15(6):741–752
Xiong H, Li J, Liu P, Duan J, Zhao Y, Guo X, Li Y, Zhang H, Ali J, Li Z (2014) Overexpression of OsMYB48-1, a novel MYB-related transcription factor, enhances drought and salinity tolerance in rice. PLoS One 9(3):e92913
Xu Y, Gao S, Yang Y, Huang M, Cheng L, Wei Q, Fei Z, Gao J, Hong B (2013) Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. BMC Genomics 14:662
Xu A, Zhang W, Wen CK (2014a) Enhancing ctr1-10 ethylene response2 is a novel allele involved in constitutive triple-response 1-mediated ethylene receptor signaling in Arabidopsis. BMC Plant Biol 14:48
Xu DB, Gao SQ, Ma YZ, Xu ZS, Zhao CP, Tang YM, Xy L, Li LC, Chen YF, Chen M (2014b) ABI-like transcription factor gene TaABL1 from wheat improves multiple abiotic stress tolerances in transgenic plants. Funct Integr Genomics 14(4):717–730
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular response and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yang W, Kong Z, Omo-Ikerodah E, Xu W, Li Q, Xue Y (2008) Calcineurin B-like interacting protein kinase OsCIPK23 functions in pollination and drought stress responses in rice (Oryza sativa L.). J Genet Genomics 35(9):531–543, S1-2
Zhang J, Mao Z, Chong K (2013) A global profiling of uncapped mRNAs under cold stress reveals specific decay patterns and endonucleolytic cleavages in Brachypodium distachyon. Genome Biol 14(8):R92
Zhang F, Yao J, Ke J, Zhang L, Lam VQ, Xin XF, Zhou XE, Chen J, Brunzelle J, Griffin PR, Zhou M, Xu HE, Melcher K, He SY (2015a) Structural basis of JAZ repression of MYC transcription factors in jasmonate signalling. Nature 525(7568):269–273
Zhang L, Hu W, Wang Y, Feng R, Zhang Y, Liu J, Jia C, Miao H, Zhang J, Xu B, Jin Z (2015b) The MaASR gene as a crucial component in multiple drought stress response pathways in Arabidopsis. Funct Integr Genomics 15(2):247–260
Zhang X, Allan AC, Li C, Wang Y, Yao Q (2015c) De novo assembly and characterization of the transcriptome of the Chinese medicinal herb, Gentiana rigescens. Int J Mol Sci 16(5):11550–11573
Zhao J, Gao Y, Zhang Z, Chen T, Guo W, Zhang T (2013) A receptor-like kinase gene (GbRLK) from Gossypium barbadense enhances salinity and drought-stress tolerance in Arabidopsis. BMC Plant Biol 13:110
Zhong L, Chen D, Min D, Li W, Xu Z, Zhou Y, Li L, Chen M, Ma Y (2015) AtTGA4, a bZIP transcription factor, confers drought resistance by enhancing nitrate transport and assimilation in Arabidopsis thaliana. Biochem Biophys Res Commun 457(3):433–439
Zhou Y, Gao F, Liu R, Feng J, Li H (2012) De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus. BMC Genomics 13:266
Zhou X, Li L, Xiang J, Gao G, Xu F, Liu A, Zhang X, Peng Y, Chen X, Wan X (2015) OsGL1-3 is involved in cuticular wax biosynthesis and tolerance to water deficit in rice. PLoS One 10(1):e116676
Zhu H, Dardick CD, Beers EP, Callanhan AM, Xia R, Yuan R (2011) Transcriptomics of shading-induced and NAA-induced abscission in apple (Malus domestica) reveals a shared pathway involving reduced photosynthesis, alterations in carbohydrate transport and signaling and hormone crosstalk. BMC Plant Biol 11:138
Acknowledgments
The authors gratefully acknowledge the Department of Science and Technology (DST), Government of India (GOI), for supporting this work under the Fast Track Scheme for Young Scientist under grant number SB/FT/LS-329/2012 and the project entitled “Transcriptome analysis of senna (Cassia angustifolia Vahl.) to identify potential genes involved in the biosynthesis of sennosides” and the Director, ICAR-Directorate of Medicinal and Aromatic Plants Research, Boriavi, Anand, Gujarat, India, and Indian Council of Agricultural research (ICAR), New Delhi, India for the facilities to undertake the study.
Authors’ contributions
Conceived and designed the experiments: NRRR. Performed the experiments: NRRR RHM. Analyzed the data: NRRR MP JK. Contributed reagents/materials/analysis tools: NRRR MP JK. Wrote the paper: RHM NRRR MP JK
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Availability of supporting data
The datasets supporting the results of this article are available at the National Center for Biotechnology Information (NCBI) BioProject database (Short Read Archive) under accession number PRJNA273534 and the Transcriptome Shotgun Assembly (TSA) at DDBJ/EMBL/GenBank under the accession GEEB00000000. The version described in this paper is the first version, GEEB01000000.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
Transcript size (bp) distribution (DOC 61 kb)
Figure S2
Size distribution of CDS (DOC 67 kb)
Figure S3
Sequence alignment to show conformity between transcriptome assembly (trans) and Sanger sequencing (seq) of the in vitro amplified PCR product of drought stress regulated genes in senna (DOCX 30 kb)
Table S1
KEGG categories of CDS in the transcriptome of leaf of C. angustifolia (PDF 3957 kb)
Rights and permissions
About this article

Cite this article
Mehta, R.H., Ponnuchamy, M., Kumar, J. et al. Exploring drought stress-regulated genes in senna (Cassia angustifolia Vahl.): a transcriptomic approach. Funct Integr Genomics 17, 1–25 (2017). https://doi.org/10.1007/s10142-016-0523-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-016-0523-y