
Abstract
Abscisic acid (ABA)-, stress-, and ripening-induced (ASR) proteins are involved in abiotic stress responses. However, the exact molecular mechanism underlying their function remains unclear. In this study, we report that MaASR expression was induced by drought stress and MaASR overexpression in Arabidopsis strongly enhanced drought stress tolerance. Physiological analyses indicated that transgenic lines had higher plant survival rates, seed germination rates, and leaf proline content and lower water loss rates (WLR) and malondialdehyde (MDA) content. MaASR-overexpressing lines also showed smaller leaves and reduced sensitivity to ABA. Further, microarray and chromatin immunoprecipitation-based sequencing (ChIP-seq) analysis revealed that MaASR participates in regulating photosynthesis, respiration, carbohydrate and phytohormone metabolism, and signal transduction to confer plants with enhanced drought stress tolerance. Direct interactions of MaASR with promoters for the hexose transporter and Rho GTPase-activating protein (RhoGAP) genes were confirmed by electrophoresis mobility shift array (EMSA) analysis. Our results indicate that MaASR acts as a crucial regulator of photosynthesis, respiration, carbohydrate and phytohormone metabolism, and signal transduction to mediate drought stress tolerance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Abscisic acid (ABA)-, stress-, and ripening-induced (ASR) proteins widely distribute in higher plants, whereas there is no evidence for the presence of these genes in Arabidopsis (Carrari et al. 2004). ASR proteins have been shown to possess transactivation activities and chaperone-like activity (Çakir et al. 2003; Kalifa et al. 2004; Konrad and Bar-Zvi 2008; Dai et al. 2011; Hsu et al. 2011; Ricardi et al. 2012; Hu et al. 2013). Functional analysis revealed that ASRs were involved in plant development, fruit ripening, senescence, and response to various abiotic stresses (Huang et al. 2000; Chen et al. 2011; Yang et al. 2005; Hu et al. 2013; Liu et al. 2013; Arenhart et al. 2013a, b). Recently, more important lines of evidence showed that tomato ASR1 can target with cell wall synthesis and aquaporin genes under osmotic conditions (Ricardi et al. 2014). Whether other mechanisms are involved in ASRs conferring drought stress tolerance is to be elucidated.
Several orthologous and paralogous ASR genes have been found to be transcriptionally regulated by ABA and sugar signaling (Amitai-Zeigersona et al. 1995; Çakir et al. 2003; Kalifa et al. 2004; Liu et al. 2010; Chen et al. 2011; Henry et al. 2011; Virlouvet et al. 2011; Joo et al. 2013). Additionally, overexpression of some ASR genes resulted in reduced sensitivity to ABA and changed transcription levels of ABA/stress-responsive genes (Yang et al. 2005; Shkolnik and Bar-Zvi 2008). Furthermore, a model for grape ASR gene (VvMSA) transcriptional regulation at the convergence of glucose and ABA signaling cascades through HEXOKINASE1 and SnRK1 has recently been proposed (Saumonneau et al. 2012). Also, ASR is not only involved in glucose signaling but also provides a novel link between hormone (ABA or GA) and sugar signaling in photosynthetically active leaves (Dominguez et al. 2013). Together, these molecular studies suggest that ASR could play a regulatory role between primary carbon metabolism and hormone signaling. However, it remains unclear whether these biological processes are involved in ASR enhancement of tolerance to drought stress.
Banana (Musa acuminata L.), one of the most popular fresh fruits enjoyed worldwide, is especially sensitive to water stress due to its shallow roots and a permanent green canopy (Sreedharan et al. 2013). A better understanding of the mechanisms employed by banana plants to tolerate water stress will be helpful for increasing crop production and quality of this economically valuable fruit. In the present study, overexpression of MaASR in Arabidopsis could significantly improve plants’ tolerance to drought stress. Microarray and chromatin immunoprecipitation-based sequencing (ChIP-seq) demonstrated that multiple drought stress response pathways are affected by MaASR to confer increased tolerance to drought stress.
Materials and methods
Plant materials and treatments
Young banana seedlings (M. acuminata L. AAA group cv. Brazilian) were obtained from the banana tissue culture center of the Institute of Banana and Plantain (Danzhou, Hainan). For drought stress, five-leaf stage banana seedlings were grown in soil without watering, and the soil moisture content was determined with an instrument according to manual instructions (TZS-1, TOP, Zhejiang, China). The osmotic potential of banana leaves was measured by using a Dewpoint PotentiaMeter according to the manufacturer’s instruction (WP4C, Decagon, USA). The roots and leaves were collected to analyze the MaASR expression level when banana seedlings underwent drought treatment.
All Arabidopsis seeds were sterilized in 75 % (v/v) ethanol for 10 min and then germinated on one half Murashige and Skoog (MS) medium or in soil directly after treatment to break dormancy (i.e., maintaining seeds for 2 days at 4 °C in the dark). Plants were grown at 22 °C under fluorescent light (16 h light/8 h dark cycles). The wild-type (WT) ecotype is Columbia (Col-0). The ABA-deficient (gin1-3) and ABA-insensitive (abi4-1) mutants were obtained from the Arabidopsis Biological Resource Center (Ohio State University, Columbus, OH). For drought tolerance assay, 54-week-old adult plants of each strain were analyzed. Water was withheld for 14 days, and the survival rate of plants was subsequently scored after resumption of watering for 3 days. For dehydration treatment, 14-day-old Arabidopsis seedlings of each sample were transferred to filter paper to induce dehydration. The top of the filter papers were covered by Petri dishes to prevent moisture loss from evaporation, and then the filter papers were placed into the light incubator. The temperature was controlled at 22 °C, the light intensity was 150μmol m−2 s−1, and the incubator relative humidity was 70 %. After 0 or 2 h of treatment, the whole Arabidopsis seedlings were sampled to extract RNA for further microarray analysis. For germination assay, approximately 150 seeds were analyzed in a germination assay using one half MS medium containing different concentrations of ABA or mannitol. The rate of seed germination was scored as the percentage of seeds with radicles of more than 2 mm after 5 days of incubation. To test the germination under normal conditions, the germination of fresh seeds was scored every indicated interval after they were placed at room temperature.
Identification of full-length complementary DNA encoding banana MaASR
Based on the complementary DNA (cDNA) fragment previously identified by suppression subtractive hybridization (SSH) and microarray, a cDNA fragment encoding ASR gene showed upregulation at the early stage of postharvest banana ripening (Xu et al. 2007). The full-length cDNA of MaASR was obtained by using the rapid amplification of cDNA ends (RACE) with single-stranded cDNAs from banana fruit 2 days after harvest as a template. For 5′ RACE, the forward primer was 5′-catctcgccgaggtgctccttgtgc-3′, and the reverse primer was 5′-ccttgcctcaacaacacgatc-3′ (provided in the SMART PCR cDNA Synthesis Kit as SMART CDS primer IIA). For 3′ RACE, the forward primer was 5′-cagcggtggcggttggcagcggaggc-3′, and the reverse primer was 5′-ctccgagatctggacgagc-3′ (provided in the SMART PCR cDNA Synthesis Kit as SMART CDS primer IIA). The amplified products of the 5′ and 3′ cDNA ends were inserted into the pGEM-T easy vector (Promega, Madison, WI, USA). Based on the 5′ and 3′ end sequences of the cDNA, a pair of specific primers was designed (5′-tcggccattacggccgggga-3′ and 5′-cttatttttaagggtttttgatac-3′) to allow amplification of the entire open reading frame (ORF). The sequence was analyzed by BLAST (http://ncbi.nlm.nih.gov/blast).
Plasmid construction and transgenic plants
The MaASR ORF driven by a 35S promoter was isolated following digestion with BamHI and SacI and inserted into the pBI121 vector. The constructs were transferred into Agrobacterium strain GV3101. Arabidopsis transformation was performed using the floral dip-mediated infiltration method (Clough and Bent 1998). T1 transgenic plants were selected and identified using one half MS medium containing 50 mg/L kanamycin. The homozygous T3 transgenic lines L14 and L38 with kanamycin resistance were used for functional analysis.
Measurement of water loss rate and the contents of malondialdehyde and proline
For water loss rate (WLR) analysis, 30 fully expanded leaves were obtained from 4-week-old Arabidopsis plants. We placed the plants with the abaxial side up on open Petri dishes on the laboratory bench and weighed them at different times to analyze the rate of water loss. Four-week-old Arabidopsis seedlings were grown in soil and treated with or without watering for 14 days. Leaves from both WT and MaASR-overexpressing plants were used for measurement of malondialdehyde (MDA) and proline levels. MDA content was analyzed following the method by Carmak and Horst (1991). Proline content was measured according to Bates et al. (1973).
RNA isolation and gene expression analysis
Total RNA was isolated using the TRIzol (Invitrogen, Gaithersburg, MD, USA) one-step method, concentrated by isopropanol precipitation, and further purified using the NucleoSpin® RNA clean-up kit (MACHEREY-NAGEL, Germany). The RNA quality and concentration was monitored using formaldehyde-denatured agarose gels and a spectrophotometer. Acceptable values were defined as follows: A260/A280 ratio between 1.8 and 2.2 and rRNA ratio (28S/18S) >0.9. All RNA samples prepared were suitable for subsequent microarray and quantitative real-time polymerase chain reaction (qRT-PCR) analyses.
Gene expression was determined according to the methods described by Livak and Schmittgen (2001). Gene-specific primers used in this study are listed in Table S1. The primers excluded the highly conserved protein domains and had high efficiency and specificity, which was determined by the MxProTM qRT-PCR software (Stratagene, CA, USA) and sequencing analysis. Before proceeding with the actual experiments, a series of template and primer dilutions was tested to determine the optimal concentrations for maximal target amplification. Amplification efficiencies for the internal control and target genes were between 0.92 and 1.01.
Microarray experiments
For gene expression analysis, a 29k Arabidopsis Genome Array (CapitalBio Corp.) having 29,110 probe (70-mer) sets representing 28,964 Arabidopsis transcripts was used. Arabidopsis genes were from the Arabidopsis thaliana Genome Oligo Set Version 3.0 (Operon). Internal and external controls were printed on amino silane glass slides using a SmartArray microarrayer (CapitalBio Corp.). Labeling and hybridization were performed according to the procedure described by Patterson et al. (2006). Three biological replicates of each sample were used to test hybridization reproducibility and quality.
Microarray data analyses
Arrays were scanned with a LuxScan TM-scanner confocal laser scanner (CapitalBio Corp.) and the resulting images were analyzed with LuxScanTM3.0 software (CapitalBio Corp.). cDNA spots were screened and identified using the methods described by Ouyang et al. (2007). A spatial- and intensity-dependent (LOWESS) normalization method was employed, and normalized ratio data were then log transformed (Yang et al. 2002). Differentially expressed genes were identified using a t test, and multiple test corrections were performed using false discovery rate (FDR). Genes with FDR <0.05 and a fold change ≥2 were identified as differentially expressed genes.
Chromatin immunoprecipitation-based sequencing
Two-week-old homozygous MaASR-overexpressing L14 plants after a 2-h dehydration treatment were used for ChIP assays according to the procedure described by Kaufmann et al. (2010). The specificity of the MaASR monoclonal antibody used for ChIP assay was confirmed by Western blot analysis using purified MaASR protein. The optimal sonication conditions for ChIP of A. thaliana seedlings were eight rounds of 20 s pulses at 65 % power, with a 4-min interval between each pulse, which yielded 400–800 bp DNA fragments for subsequent experiments. Five ChIP experiments were performed to get the accumulative total DNA (7.45 ng) for the following sequence analysis. ChIP sequencing was performed by BGI after DNA purification.
ChIP-seq data analysis
Sequencing reads from the results of MaASR ChIP-seq were mapped to genome release of Arabidopsis using SOAP software and allowing for up to two mismatched nucleotides. Only uniquely mapped reads were used for the subsequent analysis. Model-based Analysis of ChIP-Seq (MACS) software was used to identify peaks representing enriched binding sites, which were classified according to the following criteria: (1) if a binding site is located in the gene body region, it will have a more detailed classification according to its location in the gene body (i.e., 5′-untranslated region, exon, intron, or 3′-untranslated region); (2) if a binding site is localized in the 2000-bp region upstream of the gene transcription start site, it is classified as a binding site in the promoter region; (3) if a binding site is assigned to more than one gene by criterion (1) or (2), it is regarded as a binding site for each of these genes; and (4) the binding sites that were not selected by the above three criteria were defined as binding sites in intergenic regions.
Electrophoresis mobility shift array
His-tagged MaASR protein was expressed in Escherichia coli and purified with nickel-nitrilotriacetic acid agarose. Electrophoresis mobility shift array (EMSA) was performed using biotin-labeled probes that were made with a Biotin 3′ End DNA Labeling Kit (89818, Pierce, USA) and the Lightshift Chemiluminescent EMSA kit (20148, Pierce, USA). Double-stranded DNA was needed in EMSA, so the probes were labeled separately and annealed before use. The probes were designed according to the peak sequence derived from the ChIP-seq. The sequence of the biotin-labeled and cold probes of the hexose transporter is 5′-GAACAAATAGAACAAAAGCA-3′, and the sequences of biotin-labeled and cold probes of Rho GTPase-activating protein (RhoGAP) were amplified by PCR using the primers F: 5′-AAAGCTCTTTCGATGATATTGG-3′ and R: 5′-TCACGGACT GTGACGGTAAT-3′. The MaASR protein and biotin-labeled probe/cold probe were incubated together in the binding reactions at room temperature for 20 min, and the entire reaction mixture was then analyzed by native polyacrylamide gel electrophoresis (PAGE) on a 6 % gel.
Northern and Western blotting
Northern blotting was performed with 3 mg total RNA extracted from 200 mg of 14-day-old Arabidopsis seedlings using an RNA extraction kit (Qiagen, Germany). Total RNA was separated on 1.2 % agarose formaldehyde gels and transferred to nylon membranes (PerkinElmer). cDNA probes were used in the F: CCGAGGAGAAGCACCACCAC and R: GCCACCGCTGCAGCGATCTCCTC and labeled using a random primer labeling system (Cat. 1093657, Roche, Mannheim, Germany). Hybridization for Northern blotting was performed according to the manufacturer’s instructions (Roche, Mannheim, Germany).
Plant tissues were frozen in liquid nitrogen and then ground into fine powder. Proteins were extracted, separated on SDS-PAGE gels, and then transferred to nitro-cellulose membranes. Western blotting was performed using MaASR specific antibodies (Abmart, Shanghai, China) diluted 1:2000. Solutions used in this experiment were prepared according to the appendix of TaKaRa product instructions.
Results
Cloning and expression analysis of MaASR
The full-length cDNA, designated as MaASR (GenBank Accession No.: AY628102), was obtained using the RACE method. MaASR cDNA consists of 683 bp with a 429-bp ORF. As with other ASR genes, MaASR encodes a small N-terminal sequence that contains a stretch of six His residues and a longer C-terminal region that has an ABA/WDS domain, two Ala-rich regions, and a putative nuclear targeting signal (Fig. S1). Phylogenetic analysis showed that MaASR is close to banana ASRs, OsASR, ZmASR2, and TaASR1 in the monocot group (Fig. S2). These results suggested that MaASR is a member of the ASR family in banana.
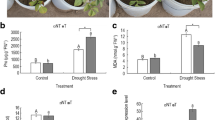
To investigate the response of MaASR to drought stress, banana seedlings were subjected to water-deficient treatment. qRT-PCR detection of MaASR expression levels in the roots and leaves showed induction when the soil water content was maintained from 45 to 30 % and the osmotic potential of banana leaves decreased between −2.7 and −3.4 MPa (Fig. 1a, b).

Leaf osmotic potential and expression patterns of MaASR in banana after drought treatment. a Osmotic potential of banana leaves was measured when soil water content reached 85, 60, 45, and 30 % respectively. b MaASR expression in leaves and roots was detected when soil water content reached 85, 60, 45, and 30 % respectively. Vertical bars indicate ±SE of three independent experiments
MaASR overexpression in Arabidopsis reduces leaf area
Two transgenic lines with different MaASR expression from the T3 generation, 35S::MaASR-14 and 35S::MaASR-38 (L14 and L38, respectively), were selected and their phenotypes were observed. The leaf areas of both L14 and L38 were significantly smaller (0.52- and 0.59-fold, respectively) than that of WT plants, and the leaf edges were curled to the adaxial surface (Fig. 2a, b). Northern and Western blots showed that MaASR expression levels were higher in L14 compared to L38 (Fig. 2c, d). In comparison with L38, the higher MaASR expression levels and smaller leaf area in L14 indicated that the phenotype of transgenic lines was relative to the expression levels of MaASR.

MaASR overexpression in Arabidopsis. a Phenotype of leaves detached from the same position and developmental stage of 4-week-old WT and transgenic lines. b Phenotype of whole seedlings from the same developmental stage of 4-week-old WT and transgenic lines. c Northern blot analysis of MaASR expression in L14 and L38. d Western blot assay of MaASR expression levels in transgenic lines L14 and L38
MaASR overexpression increases plant tolerance to drought stress
To characterize the performance of MaASR transgenic lines under drought stress, 4-week-old plants were exposed to water withholding. After 14 days of withholding water, almost all of the WT leaves yellowed and withered, but most L14 and L38 leaves were still green (Fig. 3a). Statistical analysis showed that, after withholding watering for 14 days and following resumption of watering for 3 days, the survival rate of WT plants was 14.7 %, while those of L14 and L38 plants were 83.3 and 74.0 %, respectively (Fig. 3c). Both abi4-1 and gin1-3 mutants were reported to enhance osmotic and salt stress tolerance during the seed germination stage (Yang et al. 2005). Thus, these mutants were used to evaluate the increased tolerance to osmotic stress of transgenic lines. After exposure to mannitol from 100 to 300 mM concentrations, the transgenic lines exhibited similar germination rate of seeds to the two mutants, which is significantly higher than the WT (Fig. 3b). These results showed that MaASR overexpression can significantly enhance resistance to drought and osmotic stresses.

Drought tolerance of WT and MaASR transgenic lines. a The photographs of adult WT, L14, and L38 seedlings after water withholding for 14 days. b Germination rates of WT, abi4-1, gin1-3, L14, and L38 under different concentrations of mannitol for 5 days. The survival rates (c), water loss rate (d), and the contents of proline (e) and MDA (f) in WT and transgenic lines under normal conditions and drought treatment. Vertical bars indicate ±SE of three independent experiments. Asterisks indicate a significant difference between the WT and the two transgenic lines (*p < 0.05; **p < 0.01; n = 3)
After being detached, the WLR of the leaves increased and, 350 min later, was 35.8 % for WT but only 26.5 and 27.3 % for L38 and L14, respectively (Fig. 3d). The proline content in the leaves of L14 and L38 was higher than WT after 14 days of drought treatment (Fig. 3e). At the same time, levels of MDA were measured, which indicated that MDA content in the leaves of L14 and L38 was lower than that of WT plants (Fig. 3f). Moreover, the increased tolerance and improved physiological indices in L14 and L38 were approximately relative to their expression levels in transgenic Arabidopsis. These results suggested that transgenic Arabidopsis overexpressing the MaASR gene were more tolerant to drought stress than the WT plants.
MaASR overexpression reduces ABA sensitivity of seeds
To test the ABA sensitivity in transgenic lines, fresh seeds of L14, L38, and WT were grown on one half MS medium to monitor the germination. The transgenic seeds germinated after 12 h of incubation, but for WT seeds, germination occurred 2 h later (Fig. 4a). In addition, the L14 and L38 germination rate was consistently higher than that of the WT throughout the seed germination process, although germination of all seed types was complete after 48 h of light photoperiod culture (Fig. 4a). Furthermore, transgenic lines also displayed reduced sensitivity to exogenous ABA. L14 and L38 seeds germinated and produced green cotyledons, while no green cotyledons were observed for WT seeds after 10 days of ABA treatment (Fig. 4c). Twenty days later, WT seeds produced only two cotyledons, whereas MaASR seeds had generated two to four true leaves (Fig. 4d).

Germination of WT, abi4-1, gin1-3, L14, and L38 under normal conditions and ABA treatment. Seeds germinated on one half MS for 2 days (a) or one half MS supplied with various concentrations of ABA for 5 days (b) and the germination rate was calculated. Seeds germinated on one half MS or one half MS containing 1 μM ABA and grown for 10 days (c) or 20 days (b) to detect the growth status, and then the photos were taken. Vertical bars indicate ±SE of three independent experiments
The seed germination rate of abi4-1 was unaffected by increasing ABA concentration, whereas the germination of WT and gin1-3 mutant seeds was significantly inhibited by 5 μM ABA (Fig. 4b). With increasing ABA concentrations, the germination rate of L14 and L38 seeds was gradually inhibited and was eventually lower than that of abi4-1, but higher than that of the WT and gin1-3 mutant with the rate for L14 being slightly higher than that of L38 (Fig. 4b). These results showed that MaASR overexpression can moderately enhance insensitivity to exogenous ABA.
Gene expression changes caused by MaASR overexpression
Microarray analysis was used to determine the transcript levels of downstream genes affected by MaASR overexpression. Hybridization was conducted using the 29k Arabidopsis Genome Array (CapitalBio Corp). Microarray experiments using a synthetic oligonucleotide chip demonstrated a high degree of reproducibility among the three sets of independent experiments based on the cluster data of differentially expressed gene expression profiles and a scatter plot of the gene expression profile (Fig. S3). Under normal conditions, 747 genes were found to have at least a 2-fold change in expression between L14 and WT (14 vs WT), among which 559 genes were upregulated and 188 genes were downregulated in L14 relative to WT (Tables S2 and S3). Following a 2-h dehydration treatment, 661 genes showing more than a 2-fold expression change were identified in L14 compared to WT (Y vs W), with 260 and 401 genes having enhanced and reduced expression levels, respectively (Tables S4 and S5).
Due to the prominent role of phytohormones in regulating abiotic stress responses, expression levels of 12 genes involved in ABA-, ethylene-, and auxin-metabolic/transductional genes were confirmed by qRT-PCR analysis (Fig. S4), which showed that the transcript levels of the tested genes closely correlated with the changes detected by microarray analysis (10/12; 83.3 %). When Arabidopsis seedlings were subjected to a 6-h dehydration treatment, the expression level patterns of eight genes (8/12; 66.6 %) were similar to that seen for a 2-h dehydration treatment.
Functional classification of MaASR-regulated crucial pathways
Categorization of the differentially expressed genes in the expression profile microarray was performed according to their cellular location, biological roles, and biological processes using the Gene Ontology (GO) database. The results indicated that these genes are distributed throughout the cells and participate in multiple biological processes (Figs. S5 and S6). These crucial pathways were subjected to further detailed analysis using the microarray data as basis (Tables S6 and S7).
Drought-responsive genes
In 14 vs WT, 25 drought-responsive genes, mainly those encoding protective proteins (LEA14, ERD7, ERD10, ERD15) and various transcription factors (AZF2, DREB2A, ANAC019, STZ, bHLH, ATHB-12, WRKY33, AP2, ATMYC2) showed upregulation; four drought-responsive genes showed downregulation, including PIP1B, LTP3, LTP4, and AP2. In Y vs W, 11 drought-responsive genes showed upregulation, mainly genes for protective proteins LEA14, ERD10, LTP2, and transcription factor genes ABI5 and C3HC4-type RING finger; four drought-responsive genes showed downregulation, including the bHLH transcription factor gene. These results suggested that MaASR functions in regulating the drought response process, primarily by affecting transcription of protective proteins and transcription factors.
Photosynthesis-related genes
In 14 vs WT, six and three genes involved in photosynthesis showed upregulation and downregulation, respectively. In contrast, a large number of genes (36) involved in photosynthetic electron transport in photosystem I, light-harvesting photosystem I/II, light reaction, photoperiodism, phototransduction, nonphotochemical quenching, photoinhibition, and photosystem repair showed downregulation, while only four genes showed upregulation in Y vs W. These results suggest that downregulation of photosynthetic metabolism may occur in MaASR-overexpressing plants under water-deficient conditions.
Respiration-related genes
In 14 vs WT, seven respiration-related genes showed upregulation and no genes showed downregulation. In contrast, for Y vs W, two genes involved in respiration showed upregulation and seven genes showed downregulation, mainly including respiratory electron transport chain genes such as ATP binding/ATPase, cytochrome c oxidase, and NADH dehydrogenase genes.
Carbohydrate metabolism- and response-associated genes
In 14 vs WT, 24 genes involved in carbohydrate metabolism and response showed upregulation and 13 genes showed downregulation. In Y vs W, 23 genes showed upregulation and 25 genes showed downregulation. The affected genes are mainly involved in carbohydrate biosynthesis, metabolic processes, catabolism, transport processes, cellular responses to sugar stimulus, and sugar-mediated signaling pathways.
Amino acid metabolism-related genes
Compared to WT, for genes involved in amino acid metabolism in L14, nine genes showed upregulation and six genes showed downregulation. In Y vs W, three genes showed upregulation and 10 genes showed downregulation. Genes showing expression changes are involved in metabolism, biosynthesis, and degradation of 17 different amino acids.
Phytohormone metabolism- and signaling transduction-associated genes
In 14 vs WT, 53 genes that participate in phytohormone metabolism and signal transduction showed upregulation and eight genes showed downregulation, while for Y vs W, 23 genes showed upregulation and 46 genes showed downregulation. These genes are involved in metabolism, biosynthesis, degradation and catabolic processes, as well as response and signaling pathways for several phytohormones, including ABA, ethylene (ET), auxin, cytokinin, jasmonic acid (JA), salicylic acid (SA), brassinosteroid (BR), and gibberellin (GA).
Genomewide identification of in vivo MaASR binding sites
To determine the global in vivo binding sites of MaASR, we performed ChIP-seq analysis using the transgenic line L14 that was subjected to dehydration treatment for 2 h. The specificity of MaASR monoclonal antibody used for ChIP analysis was confirmed by Western blot analysis (Fig. S7). A total of 1.7 million reads (49 bp per read) were obtained from the DNA libraries, and of these, 253 peaks were identified as in vivo MaASR binding sites (p value < 1e-05) as shown by MACS. There are a total of 410 MaASR binding sites in the Arabidopsis genome. A more detailed analysis revealed that among all these sites, 46.3 % (i.e., 190 loci) could be assigned to particular Arabidopsis Genome Initiative loci (from −2000 bp of the transcription start site to the 3′ untranslated region), whereas 53.7 % (i.e., 220 loci) of the binding sites lie in intergenic regions (Fig. 5a, Table 1). Of all binding sites in the genes, 62.1 % are located in gene promoter regions, while the other 37.9 % are located in genic regions, including 5′ untranslated regions, exons, introns, and 3′ untranslated regions (Fig. 5a, Table 1). These binding sites were assigned to a total of 193 genes that were identified in this study as potential MaASR direct target genes (Table S8), and of these, 59 had functional annotation (Table S12). Among these 59 MaASR direct targets, the binding sites of 38 genes are located in the gene promoter regions (Table S12) and five of these genes showed upregulation, while five genes showed downregulation in Y vs WT (Fig. 5b, Table S9).

MaASR binding site distribution in the Arabidopsis genome (a) and genes directly targeted by MaASR identified by comparing ChIP-seq and microarray (Y vs W) data (b)
Previous investigations showed specific binding of VvMSA to sugar-responsive element SURE1 sequences (AATAGAAAA) and sucrose box 3 (AAAATCA---TAA) in the VvHT1 promoter region (Çakir et al. 2003). In addition, tomato ASR1 abrogates the response to ABA and glucose in Arabidopsis by competing with ABI4 for CE1 (CACCG) (Shkolnik and Bar-Zvi 2008). However, a small number of the identified MaASR directly targeted genes contain the sugar-responsive element SURE1 (4/193; 2.0 %), sucrose box 3 (6/193; 3.1 %), or CE1 (18/193; 9.3 %) (Table S10). These results indicate that there are other potential elements that bind to MaASR.
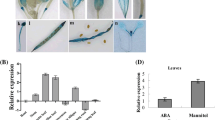
Notably, MaASR can directly target the promoter region of Arabidopsis hexose transporter gene (At5g61520), and the transcript levels of the hexose transporter gene showed upregulation in Y vs W (Fig. S8, Table 2, Table S9). The interaction of purified His-tagged MaASR with a 20-bp target (GAACAAATAGAACAAAAGCA) (repeated three times for a total of 60 bp) was evaluated by EMSA. DNA/protein complexes were observed in all assays performed with the His-tagged protein MaASR. The same fragment used as an unlabeled probe competed successfully with the labeled fragment, suggesting a specific interaction between the MaASR protein and the hexose transporter promoter in vitro (Fig. 6a). Interestingly, this MaASR target contains an element (AATAGAACA) that is similar to the SURE1 sequence (AATAGAAAA). Additionally, the promoter region of the RhoGAP that has no sugar-responsive elements, and CE1 was also found to interact with MaASR and the transcript levels of RhoGAP showed upregulation (Fig. S8, Table 2, Table S9). This interaction was also confirmed by EMSA analysis, which showed that MaASR targets a 433-bp fragment of RhoGAP (Fig. 6b).

MaASR protein binds to promoter regions of hexose transporter (a) and RhoGAP (b). a Lane 1, biotin-labeled DNA probe corresponding to a 20-bp (repeated three times for a total of 60 bp) fragment of the hexose transporter gene promoter; lane 2, DNA/protein complexes formed by the interaction between MaASR and the biotin-labeled DNA fragment of the hexose transporter gene promoter; lane 3, competition assays with the unlabeled fragment of the hexose transporter gene promoter. b Lane 1, biotin-labeled DNA probe corresponding to a 433-bp fragment of the RhoGAP promoter; lane 2, DNA/protein complexes formed by the interaction between MaASR and the biotin-labeled DNA fragment of the RhoGAP promoter; lane 3, competition assays with an unlabeled 433-bp fragment of the RhoGAP promoter
Gene ontology analysis of MaASR direct target genes
GO analysis showed that MaASR binds to a wide range of genes involved in multiple biological processes (Fig. 7, Table S11). For example, genes involved in metabolism, localization, cellular processes, and response to stimuli are highly enriched in MaASR direct target genes. Additionally, genes involved in binding, catalytic activity, enzyme regulator activity, and transporter activity are highly enriched among these MaASR-regulated genes.

GO enrichment analysis of MaASR binding target genes. The horizontal axis represents the GO term. The left vertical axis represents the proportion of genes that are related to GO categories, and the right vertical axis represents the number of genes that are associated with the GO categories
Our microarray data revealed some crucial pathways that are regulated by MaASR. Genes involved in these pathways were also enriched among MaASR directly regulated genes based on ChIP-seq analysis. Of the genes directly regulated by MaASR, 1, 20, 4, 6, and 6 genes are involved in photosynthesis, respiration, carbohydrate metabolism, phytohormone signaling transduction, and transcription factors, respectively (Table 2).
Discussion
MaASR function in photosynthesis regulation
Photosynthesis, together with cell growth, is among the primary processes that are affected by drought (Chaves 1991). Acclimation responses under drought include those that are related to growth inhibition or leaf shedding, which will help maintain a plant’s water status and therefore plant carbon assimilation (Chaves et al. 2009). We observed that MaASR-overexpressing plants have smaller leaf areas and reduced WLR, implying a role for MaASR in regulating photosynthesis (Figs. 2 and 3).
Further, a large number of genes involved in photosynthetic electron transport and light harvesting were downregulated in MaASR-overexpressing plants compared to WT plants under dehydration conditions (Table S7). Although no direct interaction of these genes with MaASR was found from our ChIP-seq data, MaASR can target the promoter of ribulose-bisphosphate carboxylase, which is similar to the ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (96 % identical as determined by BLASTp) (Table 2). Decreased activity and/or quantities of Rubisco have been observed in several different C3 and C4 species exposed to water deficit (Carmo-Silva et al. 2010). The interaction of MaASR with the ribulose-bisphosphate carboxylase promoter may contribute to the downregulation of photosynthetic electron transport and light-harvesting genes in MaASR-overexpressing plants under dehydration conditions. These results indicated that the direct influence on photosynthesis by MaASR that resulted in decreased reducing power dissipation may be a strategy by which ASRs confer drought tolerance.
MaASR function in regulating respiration
Studies on the effect of drought on respiratory pathways have produced ambiguous results, as demonstrated by the increase in respiration seen in wheat (Bartoli et al. 2005), the lack of effect in soybeans (Ribas-Carbo et al. 2005), and decreased respiration in beans and peppers (Gonzalez-Meler et al. 1997). Thus, how plant species-specific factors and/or experimental conditions affect in vivo respiratory pathways under drought stress awaits further examination. In the present study, MaASR can directly bind to a large number of respiratory electron transport chain genes and resulted in downregulation of NADH-ubiquinone oxidoreductase and cytochrome c oxidase gene expression (Table 2, Table S9). Furthermore, compared to 14 vs WT, more genes involved in respiratory electron transport showed downregulation, implying an inhibitory effect of MaASR on respiration under dehydration conditions (Tables S6 and S7).
MaASR regulates carbohydrate metabolism
Changes in cellular metabolites are thought to be associated with a protective cellular function or maintenance of the structure of cellular components under drought stress (Taji et al. 2002; Bartels and Sunkar 2005). We found that some genes related to polysaccharide, starch, glucan, maltose, galactose, hexose, inositol, and glucose metabolism showed altered transcription levels in MaASR-overexpressing plants as compared to WT under dehydration conditions (Table S7). Additionally, a gene encoding a sugar transporter family protein (At1g08890) exhibited high network connectivity, implying its key roles in sugar metabolism (Fig. S9). As such, changes in these cellular metabolites caused by MaASR overexpression may play an important role in conferring tolerance to drought stress by protecting cellular functions or preserving the structure of cellular components. In agreement with this observation, some genes encoding cellular protecting proteins such as LEA14, ERD10, and LTP2 showed upregulation in transgenic plants compared to WT under dehydration conditions (Table S7). Tobacco transgenic lines with reduced levels of ASR protein also showed impaired glucose metabolism levels and altered signaling pathway activity (Dominguez et al. 2013). Previously, a grape ASR protein was reported to bind to the sugar-responsive elements of the VvHT1 promoter to mediate sugar and ABA signaling (Çakir et al. 2003). Here, we found that MaASR not only interacted with the promoter of hexose transporter gene, but also with exostosin family protein and PYK10-BINDING PROTEIN 1 (PBP1) genes (Table 2). These interactions resulted in the upregulation of hexose transporter gene and downregulation of the exostosin gene under dehydration conditions (Table S9). All of these genes are involved in sugar metabolism (Voxeur et al. 2012; Knoch et al. 2013; Nagano et al. 2005). Collectively, we found that carbohydrate metabolism processes may be involved in drought tolerance conferred by MaASR.
MaASR regulates phytohormone metabolism and signal transduction
The phytohormone ABA plays a key role in plant adaptation to adverse environmental conditions. Continuous synthesis, transport, and degradation dynamically maintain ABA levels in plant cells. The expression levels of the ABA-biosynthetic gene ABA1 were downregulated, while the ABA degradation gene CYP707A3 showed upregulation in transgenic lines as compared to WT under dehydration conditions (Table S7). Additionally, a larger number of ABA-responsive genes showed up/downregulation in L14 than did WT under dehydration conditions (Table S7). Thus, MaASR overexpression appears to affect cellular ABA levels and transduction.
Increasing evidence also suggests that controlling ET biosynthesis and regulating ET pathway activation are important for mediating plant developmental processes and stress responses (Yu et al. 2001; Zhao and Schaller 2004; Achard et al. 2006; Cao et al. 2006). However, the relationship between ET signaling and ASR is unknown. Here, we observed increased transcription levels of genes crucial for ethylene biosynthesis, ACS6 and ACO1, ET receptor genes ETR2 and ERS2, and a large number of ET-responsive genes in MaASR-overexpressing plants under normal conditions (Table S6). The effect of ET during germination is known to be opposite that of ABA (Ghassemian et al. 2000). Thus, enhanced ET biosynthesis and transduction may contribute to inhibited ABA signaling, which leads to decreased sensitivity of MaASR-overexpressing lines to ABA. Moreover, MaASR can target the promoter of CONSTITUTIVE DISEASE RESISTANCE 1 (CDR1), which is involved in the ET response (Hall et al. 2012), to upregulate its expression under dehydration conditions (Table 2, Table S9). Collectively, these results suggest that MaASR may be a regulator of ET signaling.
In conclusion, this study demonstrated that MaASR confers tolerance to drought stress by regulating multiple biological processes, including photosynthesis, respiration, metabolism of carbohydrates and phytohormones as well as signal transduction (Fig. 8). This evidence will aid in clarifying the physiological and molecular mechanism of ASR gene function in response to drought stress.

MaASR directly regulated crucial pathways under dehydration conditions. Genes presented in boxes indicate that MaASR can directly bind to the promoter region of target genes. Genes up/downregulated by MaASR in microarray analysis are indicated by the arrow direction
References
Achard P, Cheng H, De Grauwe L, Decat J, Schoutteten H, Moritz T, Van Der Straeten D, Peng J, Harberd NP (2006) Integration of plant responses to environmentally activated phytohormonal signals. Sci 311:91–93
Amitai-Zeigersona H, Scolnik P, Bar-Zvi D (1995) Tomato Asrl mRNA and protein are transiently expressed following salt stress, osmotic stress and treatment with abscisic acid. Plant Sci 110:205–213
Arenhart RA, Bai Y, Valter de Oliveira LF, Bucker Neto L, Schunemann M, Maraschin Fdos S, Mariath J, Silverio A, Sachetto-Martins G, Margis R, Wang ZY, Margis-Pinheiro M (2013a) New insights into aluminum tolerance in rice: the ASR5 protein binds the STAR1 promoter and other aluminum-responsive genes. Mol Plant 7:709–721
Arenhart RA, Lima JC, Pedron M, Carvalho FE, Silveira JA, Rosa SB, Caverzan A, Andrade CM, Schünemann M, Margis R, Margis-Pinheiro M (2013b) Involvement of ASR genes in aluminium tolerance mechanisms in rice. Plant Cell Environ 36:52–67
Bartels D, Sunkar R (2005) Drought and salt tolerance in plants. Crit Rev Plant Sci 24:23–58
Bartoli CG, Gomez F, Gergoff G, Guiamet JJ, Puntarulo S (2005) Up-regulation of the mitochondrial alternative oxidase pathway enhances photosynthetic electron transport under drought conditions. J Exp Bot 56:1269–1276
Bates LS, Waldren RP, Teare ID (1973) Rapid determination of free proline for water stress studies. Plant Soil 39:205–208
Çakir B, Agasse A, Gaillard C, Saumonneau A, Delrot S, Atanassova R (2003) A grape ASR protein involved in sugar and abscisic acid signaling. Plant Cell 15:2165–2180
Cao WH, Liu J, Zhou QY, Cao YR, Zheng SF, Du BX, Zhang JS, Chen SY (2006) Expression of tobacco ethylene receptor NTHK1 alters plant responses to salt stress. Plant Cell Environ 29:1210–1219
Carmak I, Horst JH (1991) Effects of aluminum on lipid peroxidation, superoxide dismutase, catalase, and peroxidase activities in root tips of soybean (Glycine max). Physiol Plant 83:463–468
Carmo-Silva AE, Keys AJ, Andralojc PJ, Powers SJ, Arrabaça MC, Parry MA (2010) Rubisco activities, properties, and regulation in three different C4 grasses under drought. J Exp Bot 61:2355–2366
Carrari F, Fernie AR, Iusem ND (2004) Heard it through the grapevine? ABA and sugar cross-talk: the ASR story. Trends Plant Sci 9:57–59
Chaves MM (1991) Effects of water deficits on carbon assimilation. J Exp Bot 42:1–16
Chaves MM, Flexas J, Pinheiro C (2009) Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann Bot 103:551–560
Chen JY, Liu DJ, Jiang YM, Zhao ML, Shan W, Kuang JF, Lu WJ (2011) Molecular characterization of a strawberry FaASR gene in relation to fruit ripening. PLoS One 6:e24649
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Dai JR, Liu B, Feng DR, Liu HY, He YM, Qi KB, Wang HB, Wang JF (2011) MpAsr encodes an intrinsically unstructured protein and enhances osmotic tolerance in transgenic Arabidopsis. Plant Cell Rep 30:1219–1230
Dominguez PG, Frankel N, Mazuch J, Balbo I, Iusem N, Fernie AR, Carrari F (2013) ASR1 mediates glucose-hormone cross talk by affecting sugar trafficking in tobacco plants. Plant Physiol 161:1486–1500
Ghassemian M, Nambara E, Cutler S, Kawaide Y, Kamiya Y, McCourt P (2000) Regulation of abscisic acid signaling by the ethylene response pathway in Arabidopsis. Plant Cell 12:1117–1126
Gonzalez-Meler MA, Matamala R, Penuelas J (1997) Effects of prolonged drought stress and nitrogen deficiency on the respiratory O2 uptake of bean and pepper leaves. Photosynthetica 34:505–512
Hall BP, Shakeel SN, Amir M, Ul Haq N, Qu X, Schaller GE (2012) Histidine kinase activity of the ethylene receptor ETR1 facilitates the ethylene response in Arabidopsis. Plant Physiol 159:682–695
Henry IM, Carpentier SC, Pampurova S, Van Hoylandt A, Panis B, Swennen R, Remy S (2011) Structure and regulation of the Asr gene family in banana. Planta 234:785–798
Hsu YF, Yu SC, Yang CY, Wang CS (2011) Lily ASR protein-conferred cold and freezing resistance in Arabidopsis. Plant Physiol Biochem 49:937–945
Hu W, Huang C, Deng X, Zhou S, Chen L, Li Y, Wang C, Ma Z, Yuan Q, Wang Y, Cai R, Liang X, Yang G, He G (2013) TaASR1, a transcription factor gene in wheat, confers drought stress tolerance in transgenic tobacco. Plant Cell Environ 36:1449–1464
Huang JC, Lin SM, Wang CS (2000) A pollen-specific and desiccation-associated transcript in Lilium longiflorum during development and stress. Plant Cell Physiol 41:477–485
Joo J, Lee YH, Kim YK, Nahm BH, Song SI (2013) Abiotic stress responsive rice ASR1 and ASR3 exhibit different tissue-dependent sugar and hormone-sensitivities. Mol Cells 35:421–435
Kalifa Y, Gilad A, Konrad Z, Zaccai M, Scolnik PA, Bar-Zvi D (2004) The water- and salt-stress-regulated Asr1 (abscisic acid stress ripening) gene encodes a zinc-dependent DNA-binding protein. Biochem J 381:373–378
Kaufmann K, Muino JM, Osteras M, Farinelli L, Krajewski P, Angenent GC (2010) Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat Protoc 5:457–472
Knoch E, Dilokpimol A, Tryfona T, Poulsen CP, Xiong G, Harholt J, Petersen BL, Ulvskov P, Hadi MZ, Kotake T, Tsumuraya Y, Pauly M, Dupree P, Geshi N (2013) A β-glucuronosyltransferase from Arabidopsis thaliana involved in biosynthesis of type II arabinogalactan has a role in cell elongation during seedling growth. Plant J 76:1016–1029
Konrad Z, Bar-Zvi D (2008) Synergism between the chaperone-like activity of the stress regulated ASR1 protein and the osmolyte glycine-betaine. Planta 227:1213–1219
Liu HY, Dai JR, Feng DR, Liu B, Wang HB, Wang JF (2010) Characterization of a novel plantain Asr gene, MpAsr, that is regulated in response to infection of Fusarium oxysporum f. sp. cubense and abiotic stresses. J Integr Plant Biol 52:315–323
Liu J, Jia C, Dong F, Wang J, Zhang J, Xu Y, Xu B, Jin Z (2013) Isolation of an abscisic acid senescence and ripening inducible gene from litchi and functional characterization under water stress. Planta 237:1025–1036
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25:402–408
Nagano AJ, Matsushima R, Hara-Nishimura I (2005) Activation of an ER-body-localized beta-glucosidase via a cytosolic binding partner in damaged tissues of Arabidopsis thaliana. Plant Cell Physiol 46:1140–1148
Ouyang B, Yang T, Li H, Zhang L, Zhang Y, Zhang J, Fei Z, Ye Z (2007) Identification of early salt stress response genes in tomato root by suppression subtractive hybridization and microarray analysis. J Exp Bot 58:507–520
Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, Collins PJ, Chu TM, Bao W, Fang H, Kawasaki ES, Hager J, Tikhonova IR, Walker SJ, Zhang L, Hurban P, de Longueville F, Fuscoe JC, Tong W, Shi L, Wolfinger RD (2006) Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat Biotechnol 24:1140–1150
Ribas-Carbo M, Taylor NL, Giles L, Busquets S, Finnegan PM, Day DA, Lambers H, Medrano H, Berry JA, Flexas J (2005) Effects of water stress on respiration in soybean leaves. Plant Physiol 139:466–473
Ricardi MM, Guaimas FF, González RM, Burrieza HP, López-Fernández MP, Jares-Erijman EA, Estévez JM, Iusem ND (2012) Nuclear import and dimerization of tomato ASR1, a water stress-inducible protein exclusive to plants. PLoS One 7:e41008
Ricardi MM, González RM, Zhong S, Domínguez PG, Duffy T, Turjanski PG, Salgado Salter JD, Alleva K, Carrari F, Giovannoni JJ, Estévez JM, Iusem ND (2014) Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor. BMC Plant Biol. doi:10.1186/1471-2229-14-29
Saumonneau A, Laloi M, Lallemand M, Rabot A, Atanassova R (2012) Dissection of the transcriptional regulation of grape ASR and response to glucose and abscisic acid. J Exp Bot 63:1495–1510
Shkolnik D, Bar-Zvi D (2008) Tomato ASR1 abrogates the response to abscisic acid and glucose in Arabidopsis by competing with ABI4 for DNA binding. Plant Biotechnol J 6:368–378
Sreedharan S, Shekhawat UK, Ganapathi TR (2013) Transgenic banana plants overexpressing a native plasma membrane aquaporin MusaPIP1;2 display high tolerance levels to different abiotic stresses. Plant Biotechnol J 11:942–952
Taji T, Ohsumi C, Iuchi S, Seki M, Kasuga M, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K (2002) Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J 29:417–426
Virlouvet L, Jacquemot MP, Gerentes D, Corti H, Bouton S, Gilard F, Valot B, Trouverie J, Tcherkez G, Falque M, Damerval C, Rogowsky P, Perez P, Noctor G, Zivy M, Coursol S (2011) The ZmASR1 protein influences branched-chain amino acid biosynthesis and maintains kernel yield in maize under water-limited conditions. Plant Physiol 157:917–936
Voxeur A, André A, Breton C, Lerouge P (2012) Identification of putative rhamnogalacturonan-II specific glycosyltransferases in Arabidopsis using a combination of bioinformatics approaches. PLoS One 7:e51129
Xu BY, Su W, Liu JH, Wang JB, Jin ZQ (2007) Differentially expressed cDNAs at the early stage of banana ripening identified by suppression subtractive hybridization and cDNA microarray. Planta 226:529–539
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15
Yang CY, Chen YC, Jauh GY, Wang CS (2005) A lily ASR protein involves abscisic acid signaling and confers drought and salt resistance in Arabidopsis. Plant Physiol 139:836–846
Yu XM, Griffith M, Wiseman SB (2001) Ethylene induces antifreeze activity in winter rye leaves. Plant Physiol 126:1232–1240
Zhao XC, Schaller GE (2004) Effect of salt and osmotic stress upon expression of the ethylene receptor ETR1 in Arabidopsis thaliana. FEBS Lett 562:189–192
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31071788), the earmarked fund for Modern Agro-industry Technology Research System (CARS-32), the Ministry of Science and Technology of the People’s Republic of China (2011AA10020605), and the Major Technology Project of Hainan (ZDZX2013023-1).
Authors’ contributions
ZQJ and BYX conceived the study. LLZ, YW, RJF, YDZ, JHL, CHJ, HXM, and JBZ performed the experiments and carried out the analysis. WH designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Lili Zhang and Wei Hu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Alignment of MaASR sequence with homologues from other species: mASR1 (GU134740) from Musa acuminata subsp, mASR2 (GU134761) from Musa balbisiana, mASR4 (GU134735) from Musa banksii, TaASR1 (HQ287799) from Triticum aestivum, ZmASR2 (NP_001147703.1) from Zea mays, OsASR1 (AAX92999.1) from Oryza sativa. ABA/WDS motif was displayed in the box. Letters marked with double transverse lines showed a putative nuclear targeting signal. One His-rich region and two Ala-rich regions are underlined. Amino acid sequences alignment was performed by ClusterX 2.0. (GIF 79 kb)
Fig. S2
Phylogenetic analysis of MaASR with RcASR (XP_002524296.1) from Ricinus communis, VpASR (ABC86744.1) from Vitis pseudoreticulata, SlASR4 (AAY98026.1) from Solanum lycopersicum, ScASR4 (AAY98030.1) from Solanum corneliomuelleri, PpASR (BAA96451.1) from Pyrus pyrifolia, SpASR2 (ABY84856.1) from Solanum pimpinellifolium, SlASR2 (ABY84857.1) from Solanum lycopersicum, LpASR2 (ABS19518.1) from Lycopersicon peruvianum, NbASR (ACV52581.1) from Nicotiana benthamiana, mASR1 (GU134740) from Musa acuminata subsp. Burmannicoides, mASR2 (GU134761) from Musa balbisiana, mASR4 (GU134735) from Musa banksii, ZmASR2 (NP_001147703.1) from Zea mays, OsASR (AAX92999.1) from Oryza sativa, TaASR1 (HQ287799) from Triticum aestivum, LlASR (AAM51877.1) from Lilium longiflorum. Phylogenetic tree was constructed by using ClusterX 2.0 and Mega 4.0. (GIF 15 kb)
Fig. S3
Cluster data (A) and scatter plot (B) of differentially expressed genes identified by microarrays. (A) The red color represents upregulated genes with fold change ≥ 2, with screening criteria ratio ≥ 2; The green color represents downregulated genes with fold change ≥ 2 and screening criteria ratio ≤ 0.5. (B) X and Y axes represent the fluorescence signal intensity value of the WT and transgenic lines, respectively. Each point represents the hybridization signal of the gene in the expression profile microarray. The red color represents the genes with a screening criteria ratio ≥ 2, while the green color represents genes with a screening criteria ratio ≤ 0.5; The black color represents genes that have no significant changes with screening criteria of 0.5 < ratio < 2. (GIF 77 kb)
Fig. S4
Expression of phytohormone-metabolic/-transduction-related genes determined by qRT-PCR analysis in WT and transgenic lines. Fourteen day old Arabidopsis seedlings of each sample were transferred to filter paper for 2 h or 6 h to induce dehydration. The fold difference in mRNA was relative to that of WT under normal conditions. Vertical bars indicate ± SE of three independent measurements. (GIF 95 kb)
Fig. S5
Categorization of the differentially expressed genes identified from the Arabidopsis expression profile microarray (14 vs WT) according to position in the cellular component (A), molecular biological function (B) and biological processes (C). (GIF 82 kb)
Fig. S6
Categorization of the differentially expressed genes identified from the Arabidopsis expression profile microarray (Y vs W) according to position in the cellular component (A), molecular biological function (B) and biological processes (C). (GIF 90 kb)
Fig. S7
Analysis of the specificity of the MaASR monoclonal antibody used for chromatin immunoprecipitation (ChIP) assay. (A) SDS-PAGE analysis of expressed MaASR protein after Ni-agarose affinity chromatography. M: Protein Marker; 1: Purified protein of MaASR. (B) Western bolt analysis of the specificity of the MaASR monoclonal antibody. M: Protein marker; 1: Hybridization of MaASR monoclonal antibody with proteins expressed by vacant vector; 2: Hybridization of MaASR monoclonal antibody with purified MaASR protein. (GIF 22 kb)
Fig. S8
The expression of hexose transporter and RhoGAP genes determined by qRT-PCR analysis in WT and transgenic lines. Fourteen day old Arabidopsis seedlings of each sample were transferred to filter paper for 2 h or 6 h to induce dehydration. The fold difference in mRNA was relative to that of WT under normal conditions. Vertical bars indicate ± SE of three independent measurements. (GIF 25 kb)
Fig. S9
Network diagram (A) and connectivity (B) of differentially expressed genes in Y vs W. (A) Red represents upregulated genes and yellow represents downregulated genes. CX = mRNA co-expression between Arabidopsis genes; DC = Domain co-occurrence between Arabidopsis proteins; GN = Gene neighborhoods between Arabidopsis orthologs in bacterial genomes; LC = Literature-curated Arabidopsis protein interactions; PG = Phylogenetic profile similarity between Arabidopsis homologues. (B) The horizontal axis represents genes ID in the network diagram. The vertical axis represents gene connectivity in network diagram. (GIF 96 kb)
Table S1
qRT-PCR primers used in this study. (XLS 26 kb)
Table S2
Upregulated genes in the expression profile microarray (14 vs WT). (XLS 114 kb)
Table S3
Downregulated genes in the expression profile microarray (14 vs WT). (XLS 54 kb)
Table S4
Upregulated genes in the expression profile microarray (Y vs W). (XLS 72 kb)
Table S5
Downregulated genes in the expression profile microarray (Y vs W). (XLS 89 kb)
Table S6
Differentially expressed genes related to water deprivation, photosynthesis, respiration, carbohydrate metabolism, amino acid metabolism and phytohormone metabolism and signal transduction in the expression profile microarray (14 vs WT). (XLS 70 kb)
Table S7
Differentially expressed genes related to water deprivation, photosynthesis, respiration, carbohydrate metabolism, amino acid metabolism and phytohormone metabolism and signal transduction in the expression profile microarray (Y vs W). (XLS 79 kb)
Table S8
Summary of genes and binding sites directly targeted by MaASR. (XLS 190 kb)
Table S9
Genes directly targeted by MaASR identified by comparing ChIP-seq and microarray (Y vs W) data. (XLS 24 kb)
Table S10
Statistics of sugar-responsive elements SURE1 (AATAGAAAA), sucrose box 3 (AAAATCA---TAA) and CE1 (CACCG) among genes directly targeted by MaASR. (XLS 28 kb)
Table S11
Corresponding genes according to GO enrichment analysis of MaASR directly regulated genes. (XLS 32 kb)
Table S12
(DOC 41 kb)
Rights and permissions
About this article

Cite this article
Zhang, L., Hu, W., Wang, Y. et al. The MaASR gene as a crucial component in multiple drought stress response pathways in Arabidopsis . Funct Integr Genomics 15, 247–260 (2015). https://doi.org/10.1007/s10142-014-0415-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-014-0415-y