
Abstract
Purpose
In this review, we aimed to present and discuss the available preclinical and epidemiological evidences regarding the modulation of cancer cell proliferation by β-adrenoceptors (β-AR), with a specific focus on the putative effects of β-blockers according to their pharmacological properties.
Methods
A comprehensive review of the published literature was conducted, and the evidences concerning the involvement of β-AR in cancer as well as the possible role of β-blockers were selected and discussed.
Results
The majority of reviewed studies show that: (1) All the cancer types express both β1- and β2-AR, with the exception of neuroblastoma only seeming to express β2-AR; (2) adrenergic agonists are able to increase proliferation of several types of cancers; (3) the proliferative effect seems to be mediated by both β1- and β2-AR; (4) binding to β-AR results in a cAMP transient flux which activates two major downstream effector systems: protein kinase A and EPAC and (5) β-blockers might be putative adjuvants for cancer treatment.
Conclusions
Overall, the reviewed studies show strong evidences that β-AR activation, through several intracellular mechanisms, modulate tumor cell proliferation suggesting β-blockers can be a feasible therapeutic approach to antagonize β-adrenergic response or have a protective effect per se. This review highlight the need for intensifying the research not only on the molecular mechanisms underlying the β-adrenergic influence in cancer, but also on the implications of biased agonism of β-blockers as potential antitumor agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer figures among the leading causes of death worldwide, accounting for 8.2 million deaths and 14 million new cases in 2012 (Ferlay et al. 2015). The burden of cancer is increasing in economically developed countries, and the number of new cases is expected to rise by about 70 % over the next 20 years, as a result of population aging and growth as well as of the adoption of cancer-associated lifestyle choices (Torre et al. 2015).
Over the last three decades, clinical and epidemiological studies have identified psychosocial factors including stress, chronic depression and lack of social support as risk factors for cancer progression (Moreno-Smith 2010; Spiegel 1994; Spiegel and Giese-Davis 2003). A meta-analysis by Chida et al. (2008) showed that stress-related psychosocial factors are associated with higher cancer incidence even in healthy populations (Chida et al. 2008). Others reported that stressful life experiences are related to poor cancer survival and higher mortality, despite not affecting incidence (Chida et al. 2008).
Stress response is a key mechanism for the constant adaptation to changes in social and physical environments (Goldstein 2003). Multicellular organisms cope with stress through the activation of two main systems, the hypothalamic–pituitary–adrenal axis and the sympathoadrenomedullary (SAM) system, and the release of cortisol and the catecholamines (CAs), adrenaline (AD) and noradrenaline (NA), respectively. The effects of CA are mediated through interactions with α- and β-adrenoceptors (AR) (Guimaraes and Moura 2001).
A growing number of studies suggest that stress-related persistent stimulus can result in CA overproduction, which might impact cancer prognosis and mortality (Tang et al. 2013). Over the last years, chronic stress effects on cancer progression have been focused on tumor cell proliferation, resistance to apoptosis, invasion, metastasis, angiogenesis, stroma cells microenvironment and cellular immune responses (Cole and Sood 2012; Moreno-Smith 2010). Studies addressing the link between stress-activated pathways and cancer progression suggested that CA, besides affecting the antitumor immune response (Marino and Cosentino 2013), may also display direct tumor-promoting effects in, but not limited to, breast, ovary, colorectal, esophagus, lung, prostate, nasopharynx, melanoma, leukemia, hemangioendothelium and angiosarcoma (Tang et al. 2013).
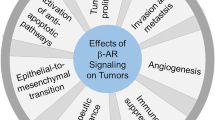
Among therapeutic drugs acting on β-AR, β-AR antagonists, commonly known as β-blockers, are widely used to treat cardiac ailments, such as hypertension and arrhythmia and other ailments. Recently, antitumor effects involving the inhibition of multiple pro-survival pathways in tumor cells have been demonstrated for many of these drugs (Baker et al. 2011). Growing epidemiological evidences have revealed strong correlations between both progression-free and long-term survival and β-blockers usage in cancer patients (Eng et al. 2014). A meta-analysis assessing 12 studies using β-blockers in cancer patients showed a positive association with overall and disease-free survival (Choi et al. 2014). Overall, the impact on survival was more pronounced for patients submitted to surgery, meaning that the perioperative period might be an opportunity to arrest tumor progression or to promote its eradication (Choi et al. 2014). Tumor cells may express β-AR, which are associated with multiple intracellular signal transduction pathways involved in cellular replication, inflammation, angiogenesis, apoptosis/anoikis, cell motility and trafficking, activation of tumor-associated viruses, DNA damage repair, cellular immune response and epithelial–mesenchymal transition (Cole and Sood 2012; Eng et al. 2014; Lutgendorf et al. 2010; Marino and Cosentino 2013). Although the involvement of β-adrenergic signaling in the progression of malignant diseases has been increasingly recognized, the underlying detailed cellular mechanisms remain elusive so far (Tang et al. 2013).
The aim of this review is to present and discuss available preclinical and clinical evidence regarding β-AR-mediated regulation of cancer cell proliferation, a crucial step in cancer development and progression, with a specific focus on the pharmacological properties and on the possible effects of β-blockers.
Physiology and pharmacology of β-AR
NA is a neurotransmitter in the central and peripheral nervous systems. AD, synthesized from NA through demethylation, is produced by chromaffin cells in the adrenal medulla and released in the bloodstream upon stimulation by the sympathetic nervous system. In the central nervous system, NA is involved in attention, arousal and vigilance, while in peripheral tissues, NA is the main transmitter of sympathetic postganglionic fibers.
NA and AD act on 7-transmembrane, G-protein-coupled receptors named “adrenoceptors” (AR), which control blood pressure, heart rate and force, airway reactivity, glucose metabolism and many central nervous system functions. AR include α1, α2 and β-AR types, each further divided into three subtypes. In particular, β-AR are expressed in heart (β1 and a few β2, mediating contraction), in smooth muscle (β2, inducing relaxation) and in skeletal muscle (β2, inducing hypertrophy). β2-AR are possibly expressed in all normal human cell types. Usually, β1-AR are located close to sympathetic terminals and are targeted mainly by NA released from nerves, while β2-AR are often extrajunctional receptors and may be preferentially acted upon by circulating NA and AD. β3-AR are mainly expressed in adipose tissue, where they control lipolysis, and in skeletal muscle, where they contribute to thermogenesis. Extensive information about physiology and pharmacology of AR can be found in Perez et al. (2016) (Dianne Perez).
AR agonists and antagonists are used as therapeutics for several indications, including cardiovascular disease, asthma, benign prostatic hypertrophy and glaucoma. In particular, β-blockers are used in cardiac arrhythmias, in the secondary prevention of myocardial infarction, and as second choice antihypertensives (reviewed in López-Sendón et al. 2004).
β-AR influence proliferation of several cancer cell lines
In 1989, Schuller and Cole (1989) provided the first evidence that β-AR activation promotes the proliferation of lung adenocarcinoma cells. Indeed, they demonstrated that isoprenaline (ISO), a synthetic agonist, was able to increase the proliferation of these cells and that β-AR blockade with propranolol (PRO) reverted this effect (Schuller and Cole 1989). Afterward, several in vitro and in vivo studies have shown that both CA promote cell proliferation in different types of cancer (Bernabé et al. 2011; Lin et al. 2013; Liu et al. 2008; Wong et al. 2011).
Stimulation of β-AR change intracellular cAMP levels which, in turn, can affect cell proliferation, differentiation and quiescence (Perez-Sayans et al. 2010). However, low receptor density might indicate deficient production of cAMP, resulting in downregulation of cell growth and differentiation (Cole and Sood 2012). In vitro studies have shown that exogenous cAMP can inhibit or stimulate cell proliferation depending on the cell type, the oncogene controlling growth or the amount of cAMP (Perez-Sayans et al. 2010). A large number of studies have also suggested that hormones that stimulate Gαs-coupled receptors and cAMP/PKA activity, such as CA, regulate a diverse array of cellular processes in cancer cell biology. In a widely range of cancer cell lines, these mediators lead to the activation of signals and proteases that are active drivers of tumor behavior such as PI3 K/Akt, Ras-ERK1/2, AP-1, Stat3, NF-kB, and CREB, and increased expression of VEGF, IL-6, IL-8 and metalloproteases (MMP) (McCarty 2014).
Tables 1 and 2, respectively, summarize the variety of cancer cell lines from different types of tumors expressing β-AR, as the effect of adrenergic agonists upon cellular proliferation.
A study using the esophageal squamous cell carcinoma cell line, HKESC-1, demonstrated that AD, via β1/β2-AR/ERK/COX-2 signaling pathways, stimulates cellular proliferation, an effect abolished by the selective blockade of both β1- and β2-AR (Liu et al. 2008). Nonetheless, the authors did not advance the mechanisms by which β-AR activation leads to ERK1/2 phosphorylation and cyclooxygenase-2 (COX-2) induction. In the same cell line, they also showed that AD was able to increase protein levels of the cell cycle regulators, CDK-4, CDK-6, cyclin D1 and cyclin E2, an effect mainly reversed by β2-AR blockade (Liu et al. 2008). The same authors showed that β-AR are functionally activated by EGF (epidermal growth factor) which increases HKESC-1 cellular proliferation via up-regulation of PKA. They also suggest that EGF can indirectly affect proliferation through the increase in TH (tyrosine hydroxylase) expression and subsequently AD production, which then will increase proliferation after binding to β-AR. Interesting enough, the blockade of β-AR with ATE and ICI almost completely reversed the proliferative effect of EGF (Liu et al. 2008).
Evidences have shown that there is an overexpression of β2-AR in human gastric cancer tissues (Shan et al. 2014). In a study using the gastric cancer cell lines, BGC-823 and SGC-7901, ISO-enhanced cellular proliferation (Liao et al. 2010). PRO was able to decrease cell proliferation in a concentration-dependent manner by reducing NF-κB DNA binding activity and concomitantly inhibiting the expression of COX-2, MMP-2/9 and VEGF at both mRNA and protein levels (Liao et al. 2010).
Oral squamous carcinoma cell proliferation also seems to be affected by adrenergic activation. A retrospective clinical study showed that β2-AR expression is a favorable prognostic factor for oral squamous carcinoma patients and could be a target for new antineoplastic pharmacological strategies (Bravo-Calderon et al. 2011). Bernabé et al. (2011) showed that NA induces the proliferation of different oral squamous carcinoma cell lines (SCC-9, SCC-15 and SCC-25) through activation of both β1- and β2-AR, an action inhibited by PRO (Bernabé et al. 2011). In another study, NA was a potent mitogen for TCa8113 and ACC cell lines, an affect again abolished by PRO (Shang et al. 2009). In these cells, only β2-AR were expressed and, given their correlation with age, tumor size, clinical stage and with cervical lymph node metastasis, they putatively related with tumor development and clinical outcomes (Shang et al. 2009).
Zhang and colleagues (Zhang et al. 2010) demonstrated that the proliferation increase in the pancreatic cancer cells, MIA PaCa-2 and BxPC-3, probably occurred through the activation of β2-AR, given that both PRO and ICI were significantly more effective than metoprolol (MET), a β1-AR selective antagonist (Zhang et al. 2010). Furthermore, they showed that β2-AR blockade suppressed proliferation by inhibition of both cAMP/PKA and Ras, which regulate activation of the MAPK pathway and transcription factors, such as NFκB, AP-1 and CREB, as well as expression of its target genes, MMP-9, MMP-2 and VEGF. However, the β1-adrenergic antagonists suppressed invasion by solely inhibiting the cAMP/PKA pathway, suggesting these drugs as novel preventive and therapeutic approaches for pancreatic cancer (Zhang et al. 2010).
A wide variety of studies have assessed the effect of stress hormones, and other adrenergic agonists, upon colon tumor biology. In fact, in this context, colon cancer seems to be the most well-studied tumor. In 2005, Wu et al. (2005) demonstrated that the activation of β-AR by ISO and NA results in an increase in colon cancer cellular proliferation accompanied by the up-regulation of arachidonic acid (AA) cascade. Wong et al. (2011) also showed that AD was able to increase HT-29 cell proliferation probably by binding to both β1- and β2-AR (Wu et al. 2005). Results from our group were also consistent with these previous reports, by showing that stress hormones and ISO were able to increase HT-29 colon cancer cell proliferation, most likely through the involvement of both β-AR (Coelho et al. 2015). Lin et al. (2013) demonstrated that AD, NA and ISO enhanced cell proliferation through β-AR-dependent pathways in three human colon cancer cell lines (HT-29, SW116 and LS174T). These findings were similar to an in vivo study where a chronic restraint stress model was used to show the effect of stress upon tumor growth (Lin et al. 2013). All together, these results strongly support the role of stress hormones in the promotion of colon cancer cell proliferation through β-AR activation.
A study of Yang et al. (2009) in melanoma tumor cell lines showed that NA can stimulate the aggressive potential of C8161, 1174MEL and Me18105 cells, not only via the promotion of cellular proliferation, but also by evolving the release of pro-angiogenic factors, such as VEGF, IL-8 and IL-6. Besides the solid data about the role of β1- and β2-AR on melanoma progression, recent works have shown that β3-AR as well play a key role in this tumor. In 2014, Dal Monte et al. (2014) using B16F10 cells, a murine melanoma cell line, demonstrated that β3-AR modulate melanoma cell proliferation and survival through nitric oxide signaling. The iNOS-produced NO acted as a downstream effector of β3-AR proving that the beneficial effects of β3-AR blockade on cell proliferation and apoptosis were functionally linked to reduced iNOS expression and NO production (Dal Monte et al. 2014). Studies by Chiarugi P. and colleagues (Calvani et al. 2015; Moretti et al. 2013) reported that β3-AR expression in human melanoma is correlated with tumor aggressiveness, being up-regulated in malignant and advanced lesions when compared with melanocytic lesions. Moreover, they also showed that NA, through β3-AR, stimulates the activation of cancer-associated macrophages, the recruitment of monocytes as well as their polarization into M2 macrophages (pro-tumorigenic type), and sustains the secretion of pro-inflammatory cytokines (Calvani et al. 2015; Moretti et al. 2013). β3-AR are also involved in the recruitment of bone marrow-derived precursors to tumor cells and promote their differentiation into mature cancer-associated fibroblast and endothelial cells, sustaining tumor inflammation, angiogenesis and ultimately promoting melanoma malignancy (Calvani et al. 2015). These findings suggest that β3-AR exert an extensive influence on the tumor microenvironment and open new and promising perspectives for the role of β3-AR in cancer biology.
The paradoxical nature of AR action in breast cancer cells was reviewed by Luthy et al. (2009). In breast cancer cells, the proliferative effect of adrenergic drugs seems to be dependent on the cellular experimental model and the activated AR-subtype (Luthy et al. 2009). Cakir et al. (2002) investigated the adrenergic influence upon breast cancer cell proliferation in six different cell lines (estrogen-responsive and non-responsive). These authors showed that ISO was able to enhance proliferation of two estrogen non-responsive cell lines (MDA-MB-435 and MDA-MB-453), but did not affect any of the estrogen-responsive cell lines (MCF-7, ZR-75, MDA-MB-361). Interestingly enough, PRO was able to significantly inhibit cell proliferation, regardless the estrogen-responsiveness. In addition, ATE and ICI also inhibited the proliferation of all the above mentioned cancer cell lines, with ICI having the greater effect. These authors also suggested that AA cascade is directly triggered by β-AR activation, as MD-MB-435 cells after exposure to ISO released high levels of AA. Shi et al. (2011) showed that ISO, in a concentration-dependent manner, markedly increased the proliferation of the breast cancer cells, MCF-7. In this study, the overexpression of Her2 increased AD release from these cells, through the activation of ERK by phosphorylation, resulting in the up-regulation of β2-AR expression. A positive feedback loop is afterward established when, after stimulation of these receptors with different agonists (including AD), there is an increase in Her2 expression (Shi et al. 2011). Pérez Piñero et al. (2012) clearly showed that AD significantly enhanced proliferation of the human cell lines, IBH-4, IBH-6 and MDA-MB-231. Nevertheless, AD seems to increase or decrease breast cancer cell proliferation, depending on its binding to α2 or β-AR, respectively (Pérez Piñero et al. 2012). In addition, PRO did not completely abolish AD-induced proliferation, also suggesting the involvement of α-AR. These authors also showed that both ISO and salbutamol (SALB, β2-AR agonist) repressed cell proliferation, probably by the inhibition of ERK1/2 phosphorylation mediated by PKA, but not by EPAC. Madden et al. (2011) concluded that β-AR activation with ISO and terbutaline (TER, β2-AR agonist) did not alter MDA-MB-231 breast cancer cell proliferation. However, these authors reported that in cells with high β-AR density, stimulation of these receptors regulates VEGF production through the classical β/AR/cAMP/PKA pathway. Gargiulo et al. (2014) showed that both AD and ISO are able to decrease cell proliferation of the non-tumorigenic cell lines, MCF-10A and HBL-100, mainly via β-AR, and to increase proliferation of the tumor cell lines, MCF-7 and MDA-231, through α2-AR.
The majority of studies performed in lung cancer mention that the nicotine-derived nitrosamine NNK induces its development in experimental in vitro and in vivo animal models, thereby indicating a direct causative association between smoking and lung cancer incidence (Al-Wadei et al. 2012b; Hoffmann et al. 1991; Schuller 2013; Schuller et al. 1990). Interestingly enough, NNK seems to act as a high affinity agonist for both β1- and β2-AR leading to the development and progression of lung cancer through the activation of AA cascade and related cellular events (Schuller et al. 1999). Al-Wadei et al. (2012a) showed that AD increased NCI-H322 and NCI-H441 lung cancer cell proliferation, and PRO was able to revert this effect. Indeed, proliferation of these cells seems to be regulated by both nicotinic and β-AR (Al-Wadei et al. 2012b; Hoffmann et al. 1991; Schuller 2013; Schuller et al. 1990). The activation of nicotinic receptors by nicotine increases NA production, that, by interacting with β-AR, ultimately leads to cellular proliferation through p-ERK and p-CREB overexpression (Al-Wadei et al. 2012a). Once smoking is the most lethal risk factor associated with lung cancer cell proliferation, current literature has shed light upon the multiple molecular mechanisms by which components of tobacco smoke can initiate tumor development, induce cell cycle progression and proliferation in multiple cancer types (Al-Wadei et al. 2012b; Hoffmann et al. 1991; Schuller 2013; Schuller et al. 1990, 1999).
In 2011, Zang et al. (2011) showed that β2-AR activation by ISO-enhanced proliferation of the prostatic cancer cells, LNCaP and PC3, and that PRO reverted this effect. Furthermore, the scaffold protein β-arrestin2 was found to be involved in both β2-AR-mediated activation of ERK1/2 and proliferation increase in LNCaP cells overexpressing this protein (LNCaP-βArr2). Besides, the activation of β2-AR in these cells leads to the formation of the complex β-arrestin2/c-Src, an effect that disappears with the inhibition of c-Src (Zhang et al. 2011).
Overall, the above studies show that adrenergic agonists mainly through β-AR are able to increase cancer cell proliferation. However, a few studies showed that these drugs have antiproliferative effects upon some cancer types. For instance, NA decreases proliferation of pancreatic (Zhang et al. 2010) and melanoma cancer cells (Yang et al. 2009) and ISO of breast cancer cells (Pérez Piñero et al. 2012). In this type of cancer, proliferation increase seems to be mainly mediated by α2-AR (Pérez Piñero et al. 2012). On the other hand, the finding by our (Coelho et al. 2015) and other groups (Cakir et al. 2002; Coelho et al. 2015; Liao et al. 2010; Shang et al. 2009; Wang et al. 2012; Wong et al. 2011; Wu et al. 2005; Zhang et al. 2010) that β-blockers per se are able to decrease cellular proliferation suggests that they act as inverse agonists.
β-AR signaling on tumor microenvironment
Despite the clear importance of the cell proliferation on cancer progression, it is well established that tumor cells do not proliferate and progress as isolated entities. During carcinogenesis, the acquisition of malignant traits is influenced by the surrounding microenvironment (Hanahan and Weinberg 2011). Activation of β-AR through physiological or pharmacological stimuli induces a pro-metastatic gene expression signature in the tumor microenvironment (Cole and Sood 2012). These alterations remodel the primary tumor architecture and increase the possibility of cell dissemination through several ways, including: recruitment of macrophages into the tumor microenvironment (Lamkin et al. 2016), influences on immune response (Eng et al. 2014) and remodeling of blood vessels (Chakroborty et al. 2009) and lymph vessels (Le et al. 2016). Cancer-related molecular pathways can be influenced not only by β-AR expressed on tumor cells (which is the focus of the present review) but also by activation of β-AR expressed on other cell types present in the tumor microenvironment (Cole et al. 2015). For instance, macrophages play a key role in mediating inflammation, modulating the tumor microenvironment and promoting metastasis (Pimentel et al. 2012), and β-AR signaling strongly enhances macrophage recruitment into tumor parenchyma through at least two different mechanisms: by stimulating production of chemotactic factors from tumor cells and by promoting myelopoietic development of monocyte precursors in the bone marrow and spleen (Armaiz-Pena et al. 2015; Scanzano and Cosentino 2015). In addition, in macrophages, the expression of several genes related to tumor progression such as TGF-β, VEGF, IL-6, MMP9 and PTGS2 is substantially increased in response to β-AR stimulation, suggesting a shift toward an immunosuppressive M2-like myeloid phenotype (Lamkin et al. 2016; Sloan et al. 2010). β-AR signaling is also involved in the reduction in lymphocyte proliferation, decrease in NK cell cytotoxicity and reduction in T cell response to mitogen stimulation, which strongly contribute to cancer progression (Inbar et al. 2011; Marino and Cosentino 2013).
Several authors also showed that AD and NA may up-regulate the expression of important pro-angiogenic factors such as VEGF, interleukin 6 (IL-6), IL-8, MMP-2 and MMP-9 in several types of cancer cells through β-AR signaling (Chakroborty et al. 2009; Cole et al. 2015; Moretti et al. 2013; Thaker et al. 2006; Yang et al. 2009). These factors are crucial catalysts of angiogenesis, which is essential in the support of tumor growth and metastasis. Recently, Le et al. (2016) demonstrated that the activation of β-AR promotes tumor dissemination also by increasing the density of intratumoral lymph vessels in a process mediated by PGE2 and VEGFC.
Another interesting point is in the tumor microenvironment immune cells themselves may also be a source of CAs, thus possibly contributing to trigger local β-AR-dependent mechanisms involved in tumor progression. Indeed, results from non-tumor models show that the synthesis of CAs occurs in both macrophages (Flierl et al. 2007; Nguyen 2011) and T lymphocytes (Cosentino et al. 2000, 2015). No evidences of CA synthesis by these cells have been shown so far within tumor microenvironment, and the possible contribution of immune cells-derived CAs to tumor progression and the antitumor immune response deserves consideration.
β-blockers: a novel class of antitumor agents?
β-blockers, one of the most currently widely prescribed classes of drugs, represent a heterogeneous group of agents with distinct pharmacological properties (Baker et al. 2011; Poirier and Tobe 2014). These drugs differ in β-AR specificity, intrinsic sympathomimetic activity, vasodilatory effects and ability to cross the blood–brain barrier (Frishman and Saunders 2011). β-blockers are usually divided into three categories according to their β1/β2-AR selectivity. Non-selective β-blockers show equal antagonistic activity at both β1- and β2-AR; selective β-blockers display higher affinity for β1-AR. In addition, α/β-AR blocking agents differ from the previous categories as they are also antagonists at α-AR (Table 3).
Given the high expression of β-AR in tumor cells, and the tight relationship between stress response and cancer progression, a significant amount of epidemiological studies have emerged to clarify the association between β-blockers use and mortality (Monami et al. 2013; Thiele et al. 2015; Watkins et al. 2015). The majority of these studies showed that oncological patients, treated with β-blockers for other clinical conditions, present lower mortality rates comparing to their counterparts (Diaz et al. 2012; Johannesdottir et al. 2013b). Since β-adrenergic signaling can modulate multiple intracellular pathways underlying tumor progression and metastasis, β-blockers may be highly desirable for therapeutic intervention (Ji et al. 2012; Vaklavas et al. 2011).
Grytli et al. (2014) investigated the potential association between β-blockers use and prostate cancer-specific mortality. These authors observed a reduction in prostate cancer mortality among patients with high-risk or metastatic disease taking β-blockers, regardless the clinical characteristics at diagnosis and the use of statins or acetylsalicylic acid.
Wang et al. (2012, 2013) tested the hypothesis of β-blockers use for reducing the rates of disease progression and improving overall survival in locally advanced non-small cell lung cancer (NSCLC). This study concluded that the incidental use of β-blockers in patients with NSCLC was associated with improved distant metastasis-free survival, disease-free survival and overall survival, but not with locoregional progression-free survival, after radiotherapy. These findings were in accordance with those of previous preclinical studies, suggesting that β-blockers have specific effects on the metastatic cascade (Armaiz-Pena et al. 2009; Moreno-Smith 2010; Schuller 2010; Thaker et al. 2007).
A recent large population-based cohort study aimed to investigate whether the use of β-blockers increase survival in patients with malignant melanoma (Lemeshow et al. 2011). This study showed an association between β-blockers use and reduced mortality risk in patients diagnosed with malignant melanoma, a finding supporting the hypothesis that CA affects melanoma progression and that β-blockers may have unrecognized therapeutic applications (Colucci and Moretti 2016; Lemeshow et al. 2011).
In a cohort of patients with epithelial ovarian cancer, the potential correlation between β-blockers use and survival in woman with advanced stage disease was investigated (Diaz et al. 2012). An association between β-blockers use and progression-free and overall survival was identified, suggesting that these drugs may improve clinical outcomes in advanced epithelial ovarian carcinoma (Diaz et al. 2012). Nonetheless, another population-based studies cohort examined whether ß-blockers affect mortality, following ovarian cancer diagnosis and found no association (Heitz et al. 2013; Johannesdottir et al. 2013a).
Powe et al. (2010) hypothesized whether patients started on, and maintained with, antihypertensive β-blocker therapy, prior to a breast cancer diagnosis, would show reduced distant metastasis compared to both non-hypertensive breast cancer patients and those treated with other antihypertensive drugs. These authors evaluated 466 women with invasive breast cancer and found that those taking β-blockers showed a significant reduction in tumor recurrence and longer disease-free interval. Furthermore, this study reported 57 % reduced risk of metastasis and 71 % reduction in breast cancer mortality after 10 years. Barron et al. (2011) evaluated 5801 women with stages I–IV breast cancer and matched those taking PRO or ATE to those not taking β-blockers. They found that PRO users were significantly less likely to present metastatic disease and had significantly lower cumulative probability of breast cancer-specific mortality, compared with nonusers. Surprisingly, there was no difference between ATE users and nonusers. These data suggest improved outcomes in patients with breast cancer under non-selective β-blockers therapy. In a very recent meta-analysis, Childers et al. (2015) showed, for the first time, that the use of β-blockers significantly reduced the risk of breast cancer death (Childers et al. 2015).
Recently, Choi et al. (2014) published a very interesting meta-analysis about the association between β-blockers and survival of cancer patients. Twelve studies published between 1993 and 2013 were included in this meta-analysis, that showed that β-blockers use was associated with prolonged survival, especially in patients with early-stage cancer who had been primarily submitted to surgery. The authors argued that β-blockers can be considered a standard approach for adjuvant therapy in various types of cancer.
The observation that β-blockers exert multiple anticancer effects and improve survival opens a novel way of research about the role of β-adrenergic signaling in cancer. Nevertheless, the majority of studies do not distinguish the affinity of the β-blockers more often associated with positive outcomes, and the signaling pathways implicated in these responses remain poorly understood.
Indeed, biased agonism may be relevant for β-blockers therapeutic use in cancer, since distinct signaling through several pathways is considered to have specific functional consequences (Galandrin and Bouvier 2006; Rajagopal et al. 2010). In fact, β-blockers are not solely antagonists for the G-protein pathways, but may independently regulate more than one pathway, behaving as partial agonists, inverse agonists or pure antagonists in each pathway (Galandrin and Bouvier 2006; Rajagopal et al. 2010). Inverse agonists means that rather than just occupying the binding site and blocking the action of the agonists, they are associated with conformations of the receptor that turn off signaling leading to a suppression of basal receptor activity, while partial agonists can block one effector pathway but stimulate one or more alternative pathways (Baker et al. 2011; Evans et al. 2010; Luttrell et al. 2015). Actually, among β-blockers described in Table 3, the most part of them are already described as inverse agonists or partial agonists (Galandrin and Bouvier 2006; Grazia Perrone and Scilimati 2010). For example, ICI-118,551 and propranolol, which act as inverse agonists on the β2-AR toward the adenylyl cyclase signaling pathway, were shown to be partial agonists when tested on the extracellular signal-regulated kinase (ERK) activity which display a great complexity in terms of biased agonism (Fig. 1). In this context, compounds can be agonist for the two pathways, inverse agonist for the two pathways or have opposite efficacies on each of the pathways. (Azzi et al. 2003; Baker et al. 2003; Galandrin and Bouvier 2006).

Schematic representation of biased signaling of propranolol on β2-AR. Propranolol acts on β2-AR as a partial agonist by β-arrestin signaling activating MAPK pathway and as inverse agonist on the canonical Gs pathway decreasing the levels of cAMP. In the latter case is suggested the existence of two pathways downstream from cAMP: one involving a cAMP-dependent activation of MAPK signaling and another involving the PKA activation. Both pathways seem to increase the phosphorylation of transcription factors such as CREB. These evidences show that drugs acting on a unique receptor can have different efficacies depending on the signaling pathway activated, which add a new level of complexity in the study of β-blockers effects not only in cancer, in which these drugs have been gaining momentum, but also in other diseases. PRO—propranolol; cAMP—3′–5′-cyclic adenosine monophosphate; CREB—cAMP response element transcription factor; PKA—protein kinase A; MAPK—mitogen-activated protein kinase; β2-AR—β2-adrenoceptor
In addition, several studies, aiming to characterize the properties of clinically relevant β-blockers at β1- and β2-AR level, have shown that these drugs have divergent effects on GαS- and β-arrestin-mediated signaling (Evans et al. 2010). For instance, β-blockers per se are able to diminish the proliferation of oral, gastric, pancreatic, colon and breast cancer cell lines (Cakir et al. 2002; Coelho et al. 2015; Liao et al. 2010; Shang et al. 2009; Wong et al. 2011; Wu et al. 2005; Zhang et al. 2010). However, the intracellular ramifications of these findings and the implications of either GαS or β-arrestin-mediated signaling via β-AR in general, and specifically in cancer, are mostly unknown.
Various authors have suggested the use of β-blockers as chemotherapy adjuvants (Ji et al. 2012; Nagaraja et al. 2013). In fact, several clinical trials are currently studying the effect of β-blockers and chemotherapy agents in lung, breast and ovarian cancers (Nagaraja et al. 2013). In addition, future prospective trials are needed to confirm the retrospective findings and establish whether the timing and extent of β-blocker use impact cancer survival outcomes.
On the other hand, knowledge about the expression of β-AR according to tumor type could help to fit therapy, maximizing benefits and decreasing side effects (Nagaraja et al. 2013).
The majority of studies reviewed here show that: (1) All the cancer types express both β1- and β2-AR, with the possible exception of neuroblastoma which only seems to express β2-AR; (2) adrenergic agonists are able to increase proliferation of several types of cancers; (3) the proliferative effect seems to be mediated by both β1- and β2-AR and (4) binding to β-AR results in a cAMP transient flux which activates two major downstream effector systems: protein kinase A (PKA) and EPAC. More recently, signaling mediated by β-arrestin is known to have distinct functional and physiological consequences from that mediated by G-proteins. “In vitro” studies exploring the underlying mechanisms involved in cellular response to CA through β-AR are lacking.
Concluding remarks
The association between stress and cancer has been described and well-studied over time, and evidences seem to support that chronic stress increases cancer progression (Cole and Sood 2012; Tang et al. 2013). In the last years, many clinical and epidemiological studies were performed in order to clarify this association (Chida et al. 2008; Lutgendorf et al. 2010; Tang et al. 2013). In the present review, we discuss the role of β-adrenergic signaling on proliferation of several human cancer types. Overall, the scrutinized studies display strong evidence that adrenergic agonists, through multiple intracellular mechanisms, enhance tumor cell proliferation and that β-blockers can be used to reverse this effect.
Unraveling the β-signaling pathways involved in cancer cell proliferation eventually will repurpose β-blockers currently in use as novel adjuvants drugs for cancer. Some open issues that should be addressed by future research are: (1) evaluation of β-AR expression on human tumor tissues which may be an useful tool to select which patients can benefit with β-blockers treatment; (2) to characterize the β-blockers more often associated with positive outcomes and which are more likely to benefit cancer patients; (3) To characterize the properties of clinically relevant β-blockers in terms of their action on Gαs and β-arrestin-mediated signaling, given that their implications in vivo are mostly unknown.
Abbreviations
- SAM:
-
Sympathoadrenomedullary
- AD:
-
Adrenaline
- NA:
-
Noradrenaline
- ISO:
-
Isoprenaline
- PRO:
-
Propranolol
- CAs:
-
Catecholamines
- ATE:
-
Atenolol
- ICI:
-
ICI-118,551
- MET:
-
Metoprolol
- NEB:
-
Nebivolol
- LAB:
-
Labetalol
- BUT:
-
Butoxamine
- SALB:
-
Salbutamol
- BIS:
-
Bisoprolol
- CAR:
-
Carvedilol
- TERB:
-
Terbutraline
- MMP:
-
Matrix metalloproteinase
- VEGF:
-
Vascular endothelial growth factor
- PKA:
-
Protein kinase A
- cAMP:
-
Cyclic adenosine monophosphate
- ERK:
-
Extracellular signal-regulated kinase
- NFκB:
-
Nuclear factor κB
- AP-1:
-
Activator protein 1
- CREB:
-
CAMP response element binding protein
- AA:
-
Arachidonic acid
- GPCR:
-
G-protein-coupled receptor
- EGF:
-
Epidermal growth factor
References
Al-Wadei HA, Al-Wadei MH, Schuller HM (2012a) Cooperative regulation of non-small cell lung carcinoma by nicotinic and β-adrenergic receptors: a novel target for intervention PloS One 7:e29915. doi:10.1371/journal.pone.0029915
Al-Wadei HA, Ullah MF, Al-Wadei MH (2012b) Intercepting neoplastic progression in lung malignancies via the β adrenergic (β-AR) pathway: implications for anti-cancer drug targets. Pharmacol Res 66:33–40. doi:10.1016/j.phrs.2012.03.014
Armaiz-Pena GN, Lutgendorf SK, Cole SW, Sood AK (2009) Neuroendocrine modulation of cancer progression. Brain Behav Immun 23:10–15. doi:10.1016/j.bbi.2008.06.007
Armaiz-Pena GN et al (2015) Adrenergic regulation of monocyte chemotactic protein 1 leads to enhanced macrophage recruitment and ovarian carcinoma growth. Oncotarget 6:4266–4273. doi:10.18632/oncotarget.2887
Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G (2003) Β-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA 100:11406–11411. doi:10.1073/pnas.1936664100
Baker JG, Hall IP, Hill SJ (2003) Influence of agonist efficacy and receptor phosphorylation on antagonist affinity measurements: differences between second messenger and reporter gene responses. Mol Pharmacol 64:679–688. doi:10.1124/mol.64.3.679
Baker JG, Hill SJ, Summers RJ (2011) Evolution of β-blockers: from anti-anginal drugs to ligand-directed signalling. Trends Pharmacol Sci 32:227–234. doi:10.1016/j.tips.2011.02.010
Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K (2011) Β blockers and breast cancer mortality: a population- based study. J Clin Oncol 29:2635–2644. doi:10.1200/jco.2010.33.5422
Bernabé DG, Tamae AC, Biasoli ER, Oliveira SH (2011) Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells. Brain Behav Immun 25:574–583
Bravo-Calderon DM, Oliveira DT, Marana AN, Nonogaki S, Carvalho AL, Kowalski LP (2011) Prognostic significance of β-2 adrenergic receptor in oral squamous cell carcinoma. Cancer Biomark 10:51–59. doi:10.3233/cbm-2012-0228
Cakir Y, Plummer HK 3rd, Tithof PK, Schuller HM (2002) Β-adrenergic and arachidonic acid-mediated growth regulation of human breast cancer cell lines. Int J Oncol 21:153–157
Calvani M et al (2015) Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 6:4615–4632. doi:10.18632/oncotarget.2652
Chakroborty D, Sarkar C, Basu B, Dasgupta PS, Basu S (2009) Catecholamines regulate tumor angiogenesis. Cancer Res 69:3727–3730. doi:10.1158/0008-5472.can-08-4289
Chida Y, Hamer M, Wardle J, Steptoe A (2008) Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat Clin Pract Oncol 5:466–475. doi:10.1038/ncponc1134
Childers WK, Hollenbeak CS, Cheriyath P (2015) β-Blockers reduce breast cancer recurrence and breast cancer death: a meta-analysis. Clin Breast Cancer 15:426–431. doi:10.1016/j.clbc.2015.07.001
Choi CH et al (2014) Meta-analysis of the effects of β blocker on survival time in cancer patients. J Cancer Res Clin Oncol 140:1179–1188. doi:10.1007/s00432-014-1658-7
Coelho M, Moz M, Correia G, Teixeira A, Medeiros R, Ribeiro L (2015) Antiproliferative effects of β-blockers on human colorectal cancer cells. Oncol Rep 33:2513–2520. doi:10.3892/or.2015.3874
Cole SW, Sood AK (2012) Molecular pathways: β-adrenergic signaling in cancer. Cancer Res 18:1201–1206. doi:10.1158/1078-0432.ccr-11-0641
Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK (2015) Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 15:563–572. doi:10.1038/nrc3978
Colucci R, Moretti S (2016) The role of stress and β-adrenergic system in melanoma: current knowledge and possible therapeutic options. J Cancer Res Clin Oncol 142:1021–1029. doi:10.1007/s00432-015-2078-z
Cosentino M et al (2000) HPLC-ED measurement of endogenous catecholamines in human immune cells and hematopoietic cell lines. Life Sci 68:283–295
Cosentino M, Marino F, Maestroni GJ (2015) Sympathoadrenergic modulation of hematopoiesis: a review of available evidence and of therapeutic perspectives. Front Cell Neurosci 9:302. doi:10.3389/fncel.2015.00302
Dal Monte M, Fornaciari I, Nicchia GP, Svelto M, Casini G, Bagnoli P (2014) β3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn Schmiedebergs Arch Pharmaco 387:533–543. doi:10.1007/s00210-014-0969-1
Diaz ES, Karlan BY, Li AJ (2012) Impact of β blockers on epithelial ovarian cancer survival. Gynecol Oncol 127:375–378. doi:10.1016/j.ygyno.2012.07.102
Eng JW, Kokolus KM, Reed CB, Hylander BL, Ma WW, Repasky EA (2014) A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol Immunother 63:1115–1128. doi:10.1007/s00262-014-1617-9
Evans BA, Sato M, Sarwar M, Hutchinson DS, Summers RJ (2010) Ligand-directed signalling at β-adrenoceptors. Br J Pharmacol 159:1022–1038. doi:10.1111/j.1476-5381.2009.00602.x
Ferlay J et al (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386. doi:10.1002/ijc.29210
Flierl MA et al (2007) Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 449:721–725. doi:10.1038/nature06185
Frishman WH, Saunders E (2011) β-Adrenergic blockers. J Clin Hypertens (Greenwich) 13:649–653. doi:10.1111/j.1751-7176.2011.00515.x
Galandrin S, Bouvier M (2006) Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol 70:1575–1584. doi:10.1124/mol.106.026716
Gargiulo L et al (2014) Differential β2-adrenergic receptor expression defines the phenotype of non-tumorigenic and malignant human breast cell lines. Oncotarget 5:10058–10069. doi:10.18632/oncotarget.2460
Goldstein DS (2003) Catecholamines and stress. Endocr Regul 37:69–80
Grazia Perrone M, Scilimati A (2010) β(3)-Adrenoceptor agonists and (antagonists as) inverse agonists history, perspective, constitutive activity, and stereospecific binding. Methods Enzymol 484:197–230. doi:10.1016/b978-0-12-381298-8.00011-3
Grytli HH, Fagerland MW, Fossa SD, Tasken KA (2014) Association between use of β-blockers and prostate cancer-specific survival: a cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol 65:635–641. doi:10.1016/j.eururo.2013.01.007
Guimaraes S, Moura D (2001) Vascular adrenoceptors: an update. Pharmacol Rev 53:319–356. doi:10.3892/or_00001032
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. doi:10.1016/j.cell.2011.02.013
Heitz F et al (2013) Impact of β blocker medication in patients with platinum sensitive recurrent ovarian cancer-a combined analysis of 2 prospective multicenter trials by the AGO study group NCIC-CTG and EORTC-GCG. Gynecol Oncol 129:463–466. doi:10.1016/j.ygyno.2013.03.007
Hoffmann D, Rivenson A, Chung FL, Hecht SS (1991) Nicotine-derived N-nitrosamines (TSNA) and their relevance in tobacco carcinogenesis. Crit Rev Toxicol 21:305–311. doi:10.3109/10408449109017917
Inbar S, Neeman E, Avraham R, Benish M, Rosenne E, Ben-Eliyahu S (2011) Do stress responses promote leukemia progression? an animal study suggesting a role for epinephrine and prostaglandin-E(2) through reduced NK activity. PLoS One. doi:10.1371/journal.pone.0019246
Ji Y, Chen S, Xiao X, Zheng S, Li K (2012) β-blockers: a novel class of antitumor agents. Onco Targets Ther 5:391–401. doi:10.2147/ott.s38403
Johannesdottir SA, Schmidt M, Phillips G, Glaser R, Yang EV, Blumenfeld M, Lemeshow S (2013a) Use of β-blockers and mortality following ovarian cancer diagnosis: a population-based cohort study. BMC Cancer 13:85. doi:10.1186/1471-2407-13-85
Johannesdottir SA, Schmidt M, Phillips G, Glaser R, Yang EV, Blumenfeld M, Lemeshow S (2013b) Use of ss-blockers and mortality following ovarian cancer diagnosis: a population-based cohort study. BMC Cancer 13:85. doi:10.1186/1471-2407-13-85
Lamkin DM et al (2016) β-Adrenergic-stimulated macrophages: comprehensive localization in the M1-M2 spectrum. Brain Behav Immun 57:338–346. doi:10.1016/j.bbi.2016.07.162
Le CP et al (2016) Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nature Commun 7:10634. doi:10.1038/ncomms10634
Lemeshow S et al (2011) β-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev 20:2273–2279. doi:10.1158/1055-9965.epi-11-0249
Liao X, Che X, Zhao W, Zhang D, Bi T, Wang G (2010) The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor κB signaling. Oncol Rep 24:1669–1676. doi:10.3892/or_00001032
Lin X, Luo K, Lv Z, Huang J (2012) β-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepatogastroenterology 59:584–588
Lin Q, Wang F, Yang R, Zheng X, Gao H, Zhang P (2013) Effect of chronic restraint stress on human colorectal carcinoma growth in mice. PLoS One 8:e61435. doi:10.1371/journal.pone.0061435
Liu X, Wu WK, Yu L, Li ZJ, Sung JJ, Zhang ST, Cho CH (2008) Epidermal growth factor-induced esophageal cancer cell proliferation requires transactivation of β-adrenoceptors. J Pharmacol Exp Ther 326:69–75. doi:10.1124/jpet.107.134528
López-Sendón J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, Tendera M, Waagstein F, Kjekshus J, Lechat P, Torp-Pedersen C (2004) Expert consensus document on angiotensin converting enzyme inhibitors in cardiovascular disease. The task force on ACE-inhibitors of the European Society of Cardiology. Eur Heart J 25:1454–1470. doi:10.1016/j.ehj.2004.06.003
Lutgendorf SK et al (2003) Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Cancer Res 9:4514–4521
Lutgendorf SK, Sood AK, Antoni MH (2010) Host factors and cancer progression: biobehavioral signaling pathways and interventions. J Clin Oncol 28:4094–4099. doi:10.1200/jco.2009.26.9357
Luthy IA, Bruzzone A, Pinero CP, Castillo LF, Chiesa IJ, Vazquez SM, Sarappa MG (2009) Adrenoceptors: non conventional target for breast cancer? Curr Med Chem 16:1850–1862. doi:10.2174/092986709788186048
Luttrell LM, Maudsley S, Bohn LM (2015) Fulfilling the promise of “biased” G protein-coupled receptor agonism. Mol Pharmacol 88:579–588. doi:10.1124/mol.115.099630
Madden KS, Szpunar MJ, Brown EB (2011) β-Adrenergic receptors (β-AR) regulate VEGF and IL-6 production by divergent pathways in high β-AR-expressing breast cancer cell lines. Breast Cancer Res Treat 130:747–758. doi:10.1007/s10549-011-1348-y
Marino F, Cosentino M (2013) Adrenergic modulation of immune cells: an update. Amino Acids 45:55–71. doi:10.1007/s00726-011-1186-6
McCarty MF (2014) A role for cAMP-driven transactivation of EGFR in cancer aggressiveness—therapeutic implications. Med Hypotheses 83:142–147. doi:10.1016/j.mehy.2014.05.009
Monami M et al (2013) Further data on β-blockers and cancer risk: observational study and meta-analysis of randomized clinical trials. Curr Med Res Opin 29:369–378. doi:10.1185/03007995.2013.772505
Moreno-Smith M (2010) Impact of stress on cancer metastasis. Future Oncol 6:1863–1881. doi:10.2217/fon.10.142
Moretti S et al (2013) β-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab Invest 93:279–290. doi:10.1038/labinvest.2012.175
Nagaraja AS, Sadaoui NC, Lutgendorf SK, Ramondetta LM, Sood AK (2013) β-blockers: a new role in cancer chemotherapy? Expert Opin Investig Drugs 22:1359–1363. doi:10.1517/13543784.2013.825250
Nguyen KD (2011) Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480:104–108. doi:10.1038/nature10653
Penn RB, Frielle T, McCullough JR, Aberg G, Benovic JL (1996) Comparison of R-, S-, and RS-albuterol interaction with human β 1- and β 2-adrenergic receptors. Clin Rev Allergy Immunol 14:37–45. doi:10.1007/bf02772201
Perez D, Hébert T, Cotecchia S, Doze VA, Graham RM, Bylund DB, Altosaar K, Devost D, Gora S, Goupil E, Kan S, Machkalyan G, Michel MC, Sleno R, Summers R, Zylbergold P, Balaji P, Bond RA, Eikenburg DC, Hieble JP, Minneman KP, Sergio P, Hills R (2016) Adrenoceptors, introduction. Last modified on 10/08/2015. IUPHAR/BPS Guide Pharmacol. http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4. Accessed 30 May 2016
Pérez Piñero C, Bruzzone A, Sarappa MG, Castillo LF, Lüthy IA (2012) Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br J Pharmacol 166:721–736. doi:10.1111/j.1476-5381.2011.01791.x
Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Diz PG, Gandara Rey JM, Garcia-Garcia A (2010) β-adrenergic receptors in cancer: therapeutic implications. Oncol Res 19:45–54. doi:10.3727/096504010X12828372551867
Pimentel MA, Chai MG, Le CP, Cole SW, Sloan EK (2012) Sympathetic nervous system regulation of metastasis. In: Rahul Jandial and Kent Hunter (eds) Metastatic cancer: integrated organ system and biological approach, pp 1–11
Poirier L, Tobe SW (2014) Contemporary use of β-blockers: clinical relevance of subclassification. Can J Cardiol 30:S9–S15. doi:10.1016/j.cjca.2013.12.001
Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, Entschladen F (2010) β-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 1:628–638. doi:10.18632/oncotarget.101009
Rajagopal S, Rajagopal K, Lefkowitz RJ (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9:373–386. doi:10.1038/nrd3024
Ramberg H et al (2008) Hormonal regulation of β2-adrenergic receptor level in prostate cancer. Prostate 68:1133–1142. doi:10.1002/pros.20778
Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, Bos JL (2003) Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the β 2-adrenergic receptor. J Cell Biol 160:487–493. doi:10.1083/jcb.200209105
Scanzano A, Cosentino M (2015) Adrenergic regulation of innate immunity: a review. Front Pharmacol. doi:10.3389/fphar.2015.00171
Schuller HM (2010) β-adrenergic signaling, a novel target for cancer therapy? Oncotarget 1:466–469. doi:10.18632/oncotarget.101102
Schuller HM (2013) Effects of tobacco constituents and psychological stress on the β-adrenergic regulation of non-small cell lung cancer and pancreatic cancer: implications for intervention. Cancer Biomark 13:133–144. doi:10.3233/cbm-130323
Schuller HM, Cole B (1989) Regulation of cell proliferation by β-adrenergic receptors in a human lung adenocarcinoma cell line. Carcinogenesis 10:1753–1755. doi:10.1093/carcin/10.9.1753
Schuller HM, Witschi HP, Nylen E, Joshi PA, Correa E, Becker KL (1990) Pathobiology of lung tumors induced in hamsters by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the modulating effect of hyperoxia. Cancer Res 50:1960–1965
Schuller HM, Tithof PK, Williams M, Plummer H 3rd (1999) The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a β-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via β-adrenergic receptor-mediated release of arachidonic acid. Cancer Res 59:4510–4515
Shan T, Cui X, Li W, Lin W, Li Y, Chen X, Wu T (2014) Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci 105:847–856. doi:10.1111/cas.12438
Shang ZJ, Liu K, de Liang F (2009) Expression of β2-adrenergic receptor in oral squamous cell carcinoma. J Oral Pathol Med 38:371–376. doi:10.1111/j.1600-0714.2008.00691.x
Shen SG, Zhang D, Hu HT, Li JH, Wang Z, Ma QY (2008) Effects of alpha-adrenoreceptor antagonists on apoptosis and proliferation of pancreatic cancer cells in vitro. World J Gastroenterol 14:2358–2363. doi:10.3748/wjg.14.2358
Shi M et al (2011) The β2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res Treat 125:351–362. doi:10.1007/s10549-010-0822-2
Shi M et al (2013) Catecholamine-induced β2-adrenergic receptor activation mediates desensitization of gastric cancer cells to trastuzumab by upregulating MUC4 expression. J Immunol (Baltimore, MD: 1950) 190:5600–5608. doi:10.4049/jimmunol.1202364
Sloan EK et al (2010) The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res 70:7042–7052. doi:10.1158/0008-5472.can-10-0522
Spiegel D (1994) Health caring. Psychosocial support for patients with cancer. Cancer 74:1453–1457
Spiegel D, Giese-Davis J (2003) Depression and cancer: mechanisms and disease progression. Biol Psychiatry 54:269–282
Tang J, Li Z, Lu L, Cho CH (2013) β-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol 23:533–542. doi:10.1016/j.semcancer.2013.08.009
Thaker PH et al (2006) Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 12:939–944. doi:10.1038/nm1447
Thaker PH, Lutgendorf SK, Sood AK (2007) The neuroendocrine impact of chronic stress on cancer. Cell Cycle (Georgetown, TX) 6:430–433. doi:10.4161/cc.6.4.3829
Thiele M, Albillos A, Abazi R, Wiest R, Gluud LL, Krag A (2015) Non-selective β-blockers may reduce risk of hepatocellular carcinoma: a meta-analysis of randomized. Liver Int 35:2009–2016. doi:10.1111/liv.12782
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65:87–108. doi:10.3322/caac.21262
Vaklavas C, Chatzizisis YS, Tsimberidou AM (2011) Common cardiovascular medications in cancer therapeutics. Pharmacol Ther 130:177–190. doi:10.1016/j.pharmthera.2011.01.009
Wang L, Liu H, Chen X, Zhang M, Xie K, Ma Q (2012) Immune sculpting of norepinephrine on MHC-I, B7-1, IDO and B7-H1 expression and regulation of proliferation and invasion in pancreatic carcinoma cells. PLoS One 7:e45491. doi:10.1371/journal.pone.0045491
Wang HM et al (2013) Improved survival outcomes with the incidental use of β-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol 24:1312–1319. doi:10.1093/annonc/mds616
Watkins JL et al (2015) Clinical impact of selective and nonselective β-blockers on survival in patients with ovarian cancer. Cancer 121:3444–3451. doi:10.1002/cncr.29392
Westfall TCWaDP (2011) Adrenergic agonists and antagonists. In: Brunton LL (ed) Goodman & Gilman’s the pharmacological basis of therapeutics, 12th edn. The McGraw-Hill
Wilson JM, Lorimer E, Tyburski MD, Williams CL (2015) β-Adrenergic receptors suppress Rap1B prenylation and promote the metastatic phenotype in breast cancer cells. Cancer Biol Ther 16:1364–1374. doi:10.1080/15384047.2015.1070988
Wolter JK et al (2014) Anti-tumor activity of the β-adrenergic receptor antagonist propranolol in neuroblastoma. Oncotarget 5:161–172. doi:10.18632/oncotarget.1083
Wong HP et al (2011) Effects of adrenaline in human colon adenocarcinoma HT-29 cells. Life Sci 88:1108–1112. doi:10.1016/j.lfs.2011.04.007
Wu WK et al (2005) 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone from cigarette smoke stimulates colon cancer growth via β-adrenoceptors. Cancer Res 65:5272–5277. doi:10.1158/0008-5472.can-05-0205
Yang EV et al (2006) Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res 66:10357–10364. doi:10.1158/0008-5472.can-06-2496
Yang EV et al (2009) Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun 23:267–275. doi:10.1016/j.bbi.2008.10.005
Zhang D, Ma QY, Hu HT, Zhang M (2010) β2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFkappaB and AP-1. Cancer Biol Ther 10:19–29. doi:10.4161/cbt.10.1.11944
Zhang P, He X, Tan J, Zhou X, Zou L (2011) β-Arrestin2 mediates β-2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncol Rep 26:1471–1477. doi:10.3892/or.2011.1417
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Author Marisa Coelho declares that she has no conflict of interest. Author Cátia Soares-Silva declares that she has no conflict of interest. Author Daniela Brandão declares that she has no conflict of interest. Author Franca Marino declares that she has no conflict of interest. Author Marco Cosentino declares that he has no conflict of interest. Author Laura Ribeiro declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Coelho, M., Soares-Silva, C., Brandão, D. et al. β-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. J Cancer Res Clin Oncol 143, 275–291 (2017). https://doi.org/10.1007/s00432-016-2278-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-016-2278-1