
Abstract
Key message
Suitable internal control genes to normalize qPCR data from different stages of embryo development and germination were identified in two representative conifer species.
Abstract
Clonal propagation by somatic embryogenesis has a great application potentiality in conifers. Quantitative PCR (qPCR) is widely used for gene expression analysis during somatic embryogenesis and embryo germination. No single reference gene is universal, so a systematic characterization of endogenous genes for concrete conditions is fundamental for accuracy. We identified suitable internal control genes to normalize qPCR data obtained at different steps of somatic embryogenesis (embryonal mass proliferation, embryo maturation and germination) in two representative conifer species, Pinus pinaster and Picea abies. Candidate genes included endogenous genes commonly used in conifers, genes previously tested in model plants, and genes with a lower variation of the expression along embryo development according to genome-wide transcript profiling studies. Three different algorithms were used to evaluate expression stability. The geometric average of the expression values of elongation factor-1α, α-tubulin and histone 3 in P. pinaster, and elongation factor-1α, α-tubulin, adenosine kinase and CAC in P. abies were adequate for expression studies throughout somatic embryogenesis. However, improved accuracy was achieved when using other gene combinations in experiments with samples at a single developmental stage. The importance of studies selecting reference genes to use in different tissues or developmental stages within one or close species, and the instability of commonly used reference genes, is highlighted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Real-time quantitative PCR (qPCR) is increasingly used for gene expression quantification. Its main advantages are the low template inputs required due to the high sensitivity of the method, and a high resolution that allows measuring small differences in expression between samples. In addition, it is less time-consuming and cumbersome than other methods, such as northern analysis, and the cost per sample is relatively low. By contrast, its main pitfall is that measured variation can be caused not only by true biological variation but also by technical factors, resulting in non-specific variation (Bustin 2002; Nolan et al. 2006). Therefore, C t values must be normalized against the initial concentration in each sample to correct the variability of the experimental procedure. Among the several relative quantification normalization methods proposed (Thellin et al. 2009), the currently preferred one is using the expression of reference genes, because they are internal controls that are affected by all sources of variation during the experimental workflow in the same way as the genes of interest. Some authors strongly recommend using several endogenous genes in parallel (Vandesompele et al. 2002), as no single gene is universal for all experiments and frequently used reference genes are inappropriate in certain conditions (Schmittgen and Zakrajsek 2000; Dheda et al. 2004; Czechowski et al. 2005). So, prior to its use for normalization, it is important to assess the stability of the gene(s) to be used as internal controls in the experimental conditions employed (Vandesompele et al. 2009). The importance of identifying accurate endogenous genes has been widely evidenced in plants by plotting the differences in the relative expression values when using stable versus unstable reference genes (Die et al. 2010; Huis et al. 2010; Li et al. 2010; Marum et al. 2012).
Somatic embryogenesis is the process by which plant cells are induced to form somatic embryos through the manipulation of culture medium components and plant growth regulators. Clonal propagation through somatic embryogenesis has a great potential for application in conifers, but it is still difficult to achieve (von Arnold et al. 2002; von Arnold et al. 2005; Bonga et al. 2009; Humánez et al. 2012). When compared with other in vitro propagation methods, somatic embryogenesis offers several advantages in improving forest trees and gene functional studies (Harvengt 2005; Tereso et al. 2006; Lara-Chavez et al. 2012). Somatic embryos pass through similar stages of morphological development as their zygotic counterparts (Bonga et al. 2009). Zygotic embryo formation is commonly divided into three main stages in higher plants: morphogenesis, maturation and desiccation/pregermination (West and Harada 1993), which correlate with the definition of the shoot and root poles, the formation of storage organs (such as cotyledons) and embryo dormancy (Goldberg et al. 1994).
Developing embryos exhibit a dynamic genetic activity during cell differentiation, organ formation and maturation (Spencer et al. 2007), so the selection of stable references is a prior requirement in embryo gene expression qPCR studies (Gonçalves et al. 2005; Tu et al. 2007; Lin and Lai 2010). The coverage of the most representative developmental time-window regarding gene expression was considered for the selection of the biological samples included in this work. The early and mature embryo stages are the developmental points where most genes are up- or down-regulated along embryogenesis, as evidenced by microarray transcriptomic data obtained during maritime pine embryogenesis (Pinus pinaster; Simões et al. 2009 and in preparation). Also, notable expression changes in multiple pathways were found during differentiation of early embryos and development of late embryos in Norway spruce (Picea abies; Vestman et al. 2011). Additionally, extending the analysis to early germination steps may prove informative. In fact, multiple gene expression studies during embryo development extend their analysis to the germination period (von Arnold et al. 2005; Che et al. 2006; Suzuki et al. 2006; Tu et al. 2007; Aquea and Arce-Johnson 2008; Bonga et al. 2009; Humánez et al. 2012).
In a previous study (Gonçalves et al. 2005), we evaluated absolute quantification versus relative quantification with four commonly used reference genes to conclude that each of the reference genes was inadequate by itself for accurate relative quantification in the different embryonic stages, recommending the use of absolute quantification methods. Although calibration curves for absolute quantification are highly reproducible, they rely entirely on the accuracy of the standards including standard design, production, determination of the exact standard concentration and stability over long storage time, which can be problematic. Relative quantification is easier to perform and the approaches implemented in statistical algorithms such as geNorm (Vandesompele et al. 2002), NormFinder (Andersen et al. 2004) and BestKeeper (Pfaffl et al. 2004) developed for the evaluation of best-suited reference gene(s) for normalization of qPCR data in a given set of biological samples, provide robust normalization strategies.
In this study, we identified and validated the most suitable internal control genes to normalize qPCR data at different stages of somatic embryo development and germination in two representative conifer species, P. pinaster and P. abies.
Materials and methods
Somatic embryogenesis
Two embryogenic cell lines, one of P. pinaster and another of P. abies, were used in this work. The embryogenic cell lines, initiated and cryopreserved according to established protocols in P. pinaster (Marum et al. 2004; Miguel et al. 2004; Marum et al. 2009) and P. abies (von Arnold and Clapham 2008), were regrown from cryopreserved cultures. The P. pinaster thawed line was proliferated on modified Litvay medium (Litvay et al. 1985) as described by Klimaszewska et al. (2001b) with 0.72 mg/l NiCl2·6H2O, 0.125 mg/l CoCl2·6H2O, 4.4 μM N6-benzyladenine, 9 μM 2,4-dichlorophenoxyacetic acid, 0.5 g/l casein hydrolysate, 0.44 g/l glutamine, 10 g/l sucrose and 4 g/l gelrite. Maturation of somatic embryos and germination were performed following previously described procedures (Tereso et al. 2006; Marum et al. 2009). P. abies embryogenic cell line (kindly provided by S. von Arnold, Sweden) was proliferated on half-strength LP medium (von Arnold and Clapham 2008) supplemented with 4 μM benzyladenine and 9 μM 2,4-dichlorophenoxyacetic acid solidified with 3.5 g/l gelrite. Somatic embryo maturation and germination were performed as described previously (von Arnold and Clapham 2008). Tissue samples were collected from both embryogenic lines at three time-points: (1) embryogenic tissue on proliferation medium 1 week after subculture, consisting of early stage somatic embryos, i.e. T0–T2 developmental stages according to Gonçalves et al. (2005); (2) mature somatic embryos at T7 developmental stage after 3 months on maturation medium and (3) germinating embryos after 7 days on germination medium at 23 °C in the dark with visible hypocotyl elongation. Tissues were frozen in liquid nitrogen and kept at −80 °C until RNA extraction.
RNA extraction and DNAse treatment
Approximately 0.1 g of embryogenic tissue or embryos was ground to a fine powder in liquid nitrogen with mortar and pestle and then incubated at 65 °C for 10 min in the extraction buffer consisting of 0.3 M Tris HCl, 0.025 M EDTA, 2 M NaCl, CTAB 2 % (w/v), PVPP 2 % (w/v) and β-mercaptoethanol 2 % (v/v). After centrifugation, the supernatant was extracted twice with chloroform:isoamyl alcohol (24:1) followed by precipitation with isopropanol for 1 h at −80 °C and centrifugation at 4 °C. Pellet was resuspended in DEPC-treated water and total RNA was precipitated with 8 M LiCl at −20 °C. 4 μg of RNA were treated with Ambion Turbo DNA-free kit (Applied Biosystems, USA) following manufacturer indications. The RNA concentration and quality (260/280 and 260/230 nm absorbance ratios) were determined using a ND-1000 Spectrophotometer (NanoDrop Technologies Inc., USA) after RNA extraction and DNAse treatment and integrity was also checked by gel electrophoresis. Genomic DNA contaminations were discarded during qPCR by absence of amplicon when using RNA as template.
Sequence selection and primers design
A total of ten genes were selected as candidate internal controls. They were identified either from published data or a previous work (Simões et al. 2009) in which they showed a minimal relative expression variation along P. pinaster embryo development according to microarray data deposited in NCBI Gene Expression Omnibus Database (Edgar et al. 2002) and accessible through GEO Series accession number GSE32551. Primers (Table 1) were designed based on NCBI GenBank available ESTs for Pinus taeda and later checked in P. pinaster ESTs database (SustainPine DB, http://www.scbi.uma.es/sustainpine), P. abies, and other Picea spp., if available. Primers were designed with Primer 3 program (http://primer3.sourceforge.net/) to have a Tm of 58 °C, bind the orthologous genes conserved regions and amplify around 100 bp. For the validation experiments, PpRab1 (DQ372931; Gonçalves et al. 2007), E11A12 (AT1G18100; Vestman et al. 2011) and L1L (AT5G47G70; Vestman et al. 2011) primers were designed based on the NCBI public sequences, for the same PCR conditions. PpRab1 primers were 5′-TTGCT ACTGC GGTTT GCGGA-3′ and 5′-CCGCC TTTCT GTTGA AGGGG CT-3′, L1L primers were 5′-CGAAG TTGGC AGCCC TACAA-3′ and 5′-TCACA GCACA GTTCT CCCTC TCT-3′, and E11A12 primers were 5′-CCCAG CGAGC CCAAC AT-3′ and 5′-GCGGC GCCTG GAATG-3′.
qPCR
Reverse transcription was performed using Transcription High Fidelity cDNA Synthesis Kit (Roche diagnostics, USA) as indicated by manufacturer from 3 μg of total RNA and 300 ng of Primer Random Hexamers. Subsequent quantitative PCR (qPCR) was performed with Lightcycler 480 SYBR Green I Master kit (Roche diagnostics, USA) using 3 μl of cDNA and 300 nM of primers. DNA contamination was discarded by the absence of signal after qPCR amplification of RNA samples with each combination of primers. Real-time PCR reactions were performed in a Roche LightCycler 480 system (Applied Biosystems, USA) following standard cycling conditions. For the validation experiments, elongation step duration was 40 s instead of the standard 20 s. C t and fluorescence measures per cycle values were exported to Microsoft Excel tables after run. Five biological replicates of each sample and three technical replicates per biological replicate were analyzed. For each biological replicate, samples were distributed in three 96-reaction plates that were prepared together and consecutively run. One sample (calibrator) was included in all the plates to verify accuracy. The presence of only one specific peak was checked in the melting curve (dissociation curve), and qPCR product size was checked by electrophoresis in 2.5 % (w/v) agarose gels that were stained with ethidium bromide and visualized under UV light.
Statistical analysis
geNorm (Vandesompele et al. 2009), Normfinder (Andersen et al. 2004) and Bestkeeper (Pfaffl et al. 2004) methods are implemented in three Microsoft Excel Plugins. Bestkeeper directly used C t values, but normalized expression was used as input in geNorm and Normfinder. In that case, the amplification efficiency of primers was calculated for each gene and species based on the regression analysis of the PCR reactions kinetics by program LinReg 11.3 (Ruijter et al. 2009). The relative expressions were calculated using the formula Q = E Δ C t where E is the efficiency of the gene amplification and Δ C t is the difference between the sample with the lowest expression in the data set and the C t value of the sample in question.
We implemented a new approach with program Bestkeeper to estimate the ideal number of endogenous genes in each condition, in a similar way to geNorm method. The step-wise approach of geNorm was simulated in Bestkeeper by repeating the statistical analysis after consecutively discarding the gene with the lower stability value. The Bestkeeper stability value of a gene is calculated as the Pearson correlation (r) between the gene C t and the Bestkeeper index, which is the geometric mean of all the genes. The variation of the Pearson correlation values after consecutively discarding the gene with the lower stability value can be calculated. A high variation reveals a higher effect when an extra endogenous gene is incorporated in the normalization. For the validation experiments, the C t tables were imported in qBase Plus program (Hellemans et al. 2007) to be normalized against the geometric average of several endogenous genes. Next, relative expression was quantified in comparison with the average expression.
Results
Real-time qPCR amplification and accuracy
Embryogenic cell lines after 1 week of subculture on proliferation medium, mature somatic embryos and germinating embryos after hypocotyl elongation, but before light acclimation, were analysed by qPCR in two conifer species, P. pinaster and P. abies (Fig. 1). These developmental stages are commonly selected in expression analysis as representative of early and late embryogenesis (Gonçalves et al. 2005; Uddenberg et al. 2011; Vestman et al. 2011) and multiple studies also extend their analysis to the germination period (von Arnold et al. 2005; Che et al. 2006; Suzuki et al. 2006; Tu et al. 2007; Aquea and Arce-Johnson 2008; Bonga et al. 2009; Humánez et al. 2012).

Somatic embryogenesis main steps in Pinus pinaster and Picea abies showing the sampled stages selected for the analysis. From left to right, images correspond to Pinus pinaster embryogenic cell lines in proliferation, mature somatic embryos, and germinating embryos after hypocotyl elongation but before exposure to photoperiod
The tested candidate reference genes (Table 1) were chosen based on several criteria: actin (ACT), α-tubulin (ATUB), elongation factor-1α (EF1), adenosine kinase (AK) and ubiquitin (UBI) candidate genes were selected from previous qPCR studies in Pinus spp. (Bishop-Hurley et al. 2003; Gonçalves et al. 2005; Alonso et al. 2007; Aquea and Arce-Johnson 2008; Bomal et al. 2008; Ratnaparkhe et al. 2009; Liu et al. 2010; Vásquez-Robinet et al. 2010); histone 3 (HISTO3), SAND protein family (SAND) and clathrin adaptor complex subunit (CAC) are commonly used in model plants (Tu et al. 2007; Expósito-Rodríguez et al. 2008; Li et al. 2010) but to our knowledge not in P. pinaster or P. abies; two additional genes, one of them homologous to a gene encoding a heat shock protein (HEATS, accession no. EU268067) and the other to an ether reductase (REDUC, accession no. AF242490), showing a minimal relative expression variation along five P. pinaster embryo developmental stages in a microarray gene expression experiment (Simões et al. 2009 and in preparation; GEO Series accession number GSE32551). The homologous Arabidopsis loci, as well as the corresponding Gene Ontology terms, are described in Table 1.
For a given gene, average C ts from each of the five biological replicates were individually included in the statistical analysis. Pearson correlation coefficient (r) between biological replicates was 90.9 % in P. pinaster and 91.21 % in P. abies.
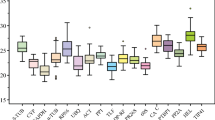
The studied reference genes displayed a wide range of expression levels according to their C t values (Fig. 2). None of the P. abies samples amplified HEATS. C ts median varied from around 20 in the case of EF1 in P. abies (19.7) and P. pinaster (20.42), to almost 40 for CAC (39.86) and ACT (39.48) in P. abies. The biggest expression differences across samples were observed in ACT, AK and CAC in P. pinaster. In general, differences among samples were lower in P. abies. Gene expression along developmental stages followed different patterns. While for some genes (e.g. ACT, ATUB and HISTO3 in P. pinaster) expression levels increased from early stage embryos to germinating embryos, for other genes (e.g. SAND and UBI in P. abies) expression decreased along the three developmental stages analysed or was highest in mature embryos (e.g. CAC, EF1 and SAND in P. pinaster).

Range of Ct values of candidate reference genes in the three tested developmental stages in Pinus pinaster (a) and Picea abies (b). The Ct values distributions are represented as standard box-plots, indicating interquartile (ITQ) range (box), median (horizontal line) and outliers within 1.5 × ITQ range (whiskers)
Amplification efficiencies were determined for each amplicon based on the kinetic model of PCR amplification using LinRegPCR program (Ruijter et al. 2009). Efficiencies were calculated from the 45 amplification plots and all were higher than 1.68 (Table 1). The SD of the efficiency values was very low, indicating comparable results among the different experiments. Most of the candidate genes had close amplification efficiency values in P. pinaster and P. abies. This evidenced a similar amplicon length and GC distribution. Even HISTO3, REDUC and ATUB genes, which showed different expression ranges for both species, exhibited close amplification efficiency values. However, ACT exhibited different amplification efficiency values in both species.
Stability analysis of candidate reference genes
Three different approaches implemented in geNorm, Bestkeeper and Normfinder programs were used to determine the expression stability of a candidate gene according to the relation with the other candidate genes. The ranking of genes according to their stability values was consistent among the different programs. When the analysis was performed at a single stage rather than across all tested time points, the ranking of the most stable genes was not identical in all cases.
In the analysis of the most stable genes across all the tested time points in P. pinaster (Fig. 3a), gene stability was higher for HISTO3, ATUB and EF1, whereas HEATS, REDUC, ACT and UBI were the less stable genes. The more relevant disagreement among rankings was found for ACT, which ranked as the fifth more stable gene in Bestkeeper, and therefore it was more stable than SAND and CAC. With Bestkeeper, there were minimal differences among the seven genes showing higher stability, which grouped separately from the other three less stable genes. In P. abies (Fig. 3b), ATUB, AK and CAC genes were clearly more stable than the remaining genes, their stability values being lower than one in geNorm and higher than 0.95 in Bestkeeper. A small gap was observed between these three genes and the remaining genes in both rankings. However, stable genes showed minimal differences in the Normfinder ranking, where CAC, EF1 and SAND had equivalent stability values. In all the rankings, ACT, REDUC, UBI and HISTO3 showed less stability.

Stability values of candidate reference genes calculated by different statistical methods. Expression stability across all time points in Pinus pinaster (a) and Picea abies (b), and of both P. pinaster and P. abies in proliferating embryogenic cell lines (c), in mature embryos (d) and germinating embryos (e) of candidate genes ordered from less stable (left) to more stable (right) according to geNorm (squares, primary axis). Normfinder (circles, primary axis) and Bestkeeper (triangles, secondary axis) rankings are also indicated
When analysing both P. pinaster and P. abies samples at identical stages of development diverse results were obtained. In the proliferating embryogenic cell lines (Fig. 3c), EF1, CAC, UBI, ATUB and REDUX were the more stable genes in the Normfinder and Bestkeeper rankings. However, ATUB and REDUX were less stable than other genes such as UBI, CAC, EF1, SAND and AK, in the geNorm ranking. On the other hand, HISTO3 and ACTIN were unstable in the geNorm and Normfinder rankings but not in the Bestkeeper. In the mature embryos (Fig. 3d) there were differences among rankings. CAC, ACTIN, UBI and ATUB were the most stable genes in Normfinder and Bestkeeper rankings. Among these, UBI, ATUB and specially ACTIN were badly ranked in the geNorm. However, the geNorm ranking was contradictory with the other two in this stage and it should be considered with caution. In the germinating embryos (Fig. 3e), UBI, CAC, ACTIN and ATUB were the most stable in both Normfinder and Bestkeeper rankings. However, ACTIN and ATUB ranked worse than other genes (such as EF1, AK and SAND) in the geNorm. HISTO3 and REDUX were unstable in the three rankings.
Ideal number of reference genes for normalization
The optimal number of endogenous genes required for accurate normalization was the same using either geNorm or Bestkeeper programs (Fig. 4). The ideal number of endogenous genes was three in P. pinaster (Fig. 4). The addition of the third gene to the two more stable genes had a clear positive effect (Pairwise variation—V, V2/3 = 0.423 and 0.216 with geNorm and Bestkeeper, respectively) while the effect was progressively smaller from the fourth gene onwards (V3/4 lower than 0.3 and 0.124 with geNorm and Bestkeeper, respectively). In P. abies, the ideal number of endogenous genes was four (Fig. 4). In both methods, a clear effect was observed by the addition of a fourth gene (V3/4 = 0.328 and 0.221) to the three more stable genes.

Pairwise variation between a n and n + 1 number of genes of stability values obtained with geNorm (a) and Bestkeeper (b), to test the effect of adding additional reference genes in the normalization of data from Pinus pinaster and Picea abies across all time points and in proliferating cell lines, mature embryos and germinating embryos
When considering developmental stages separately, the ideal number of endogenous genes during proliferation of embryogenic cells was four. V2/3 and V3/4 were very similar (0.39–0.378 in geNorm, and 0.215–0.24 in Bestkeeper) in both approaches, so the addition of a fourth gene increased the accuracy as the addition of a third. The ideal number of endogenous genes in mature embryos was three using geNorm or four using Bestkeeper, because the addition of a fourth gene has a strong effect on Bestkeeper (V2/3 = 0.111–V3/4 = 0.409) that was not observed in geNorm. Finally, the ideal number of endogenous genes was three during embryo germination. In both approaches, V2/3 is clearly higher than V3/4 (0.481–0.339 in geNorm, and 0.338–0.203 in Bestkeeper).
Validation of the more stable reference genes
Three genes were analysed during different stages of P. pinaster or P. abies embryogenesis using the more stable endogenous genes. PpRab1 gene was differentially expressed during P. pinaster somatic embryogenesis according to qPCR and semi-quantitative RT-PCR (Gonçalves et al. 2007). E11A12 and L1L genes were differentially expressed during P. abies somatic embryogenesis according to microarray analysis (Vestman et al. 2011).
Using the three endogenous genes more stable through all P. pinaster somatic embryogenesis stages (HISTO3, ATUB and EF1), PpRab1 expression in proliferating embryogenic tissue showed a fold-change of approximately four and two with respect to mature and germinating embryos, respectively (Fig. 5a). Using the four endogenous genes proposed for P. abies. (CAC, AK, ATUB and EF1), E11A12 expression was 4.4 and 11.6 times higher in germinating embryos than in embryogenic tissue and mature embryos, respectively (Fig. 5b). L1L expression was 34 and 42 times higher in embryogenic tissue in proliferation that in mature and germinating embryos (Fig. 5c).

Relative expression of genes PpRab1 (a), E11A12 (b) and L1L (c) normalized with different reference genes or combination of reference genes
In the three cases, the relative expression when using the geometric mean of the three more stable genes was different than when using only one of them. However, the variation in the relative expression of E11A12 when using three or four endogenous genes was minor compared with the difference when using only one endogenous gene. The addition of a fourth gene did not have a clear effect.
Discussion
A correct normalization is a pre-requisite for the reliability of the qPCR results (Nolan and Bustin 2004; Huggett et al. 2005). Stable endogenous genes have been identified in several plants, including trees (Cruz et al. 2009; de Almeida et al. 2010; Chen et al. 2011; Marum et al. 2012; Yan et al. 2012) and in several conditions, including somatic embryogenesis (Tu et al. 2007; Lin and Lai 2010). However, such studies have not been reported in any conifer species.
We report a selection of endogenous genes to be used as internal control in qPCR experiments during somatic embryogenesis in conifers. Plant embryogenesis includes an initial morphogenetic phase characterized by cell division and differentiation, followed by a maturation phase that involves accumulation of major storage products and preparation for seed desiccation, dormancy and germination (Meinke 1995). In this work the sampling of tissues covered both the morphogenetic and maturation phases and germination. Highly different genome-wide transcript profiles among the three sampled tissues are expected due to the major developmental changes occurring during embryogenesis and germination. In fact, we have previously found that a high number of genes are differentially expressed when P. pinaster early and mature embryo stages are compared (Simões et al. 2009). Therefore, it is expected that many genes stable at a specific period of embryogenesis are no longer stable at other developmental stages. Somatic embryogenesis is essential for functional gene analysis in conifers as this system is currently used for regenerating transformed plants in both P. pinaster (Trontin et al. 2007, 2013) and P. abies (Clapham et al. 2000; Klimaszewska et al. 2001a). Thus, it is desirable to have a set of reference genes that can be routinely used in proliferating embryogenic masses, which are the first tissues used to evaluate the success of transgene integration and expression, and in mature and germinating embryos to evaluate the stability of transgene expression. On the other hand, by analysing together P. pinaster and P. abies gene expressions, we aimed at identifying reference genes that can likely be extended to similar studies in other conifers.
Single PCR products were obtained for all candidate genes except HEATS in P. abies samples. Statistical analysis of the C t values evidenced significant differences between both species and, therefore, stability expression analysis was performed separately for each species. Biological replicates were obtained from independent somatic embryogenesis assays and the correlation between them was clear in both species. The average C ts of five biological replicates were used as input to avoid losing information, as tools for expression stability analysis do not consider the SD. Efficiency of primers was calculated by linear regression analysis using LinReg program (Ruijter et al. 2009). Most of the efficiencies were between 1.75 and 1.85, which were within the expected range (Nordgård et al. 2006; Ruijter et al. 2009). Efficiencies lower but close to 2 can be found in similar studies (Artico et al. 2010; Giménez et al. 2011; Xu et al. 2011). Only ACT showed different efficiencies between species. Efficiency is dependent on the length and nucleotide distribution in the amplicon, so it indicates differences in the ACT fragment amplified in P. pinaster and P. abies.
Three different approaches, geNorm, Bestkeeper and Normfinder, were used to determine the expression stability of a candidate gene according to the relation with the other candidate genes. The underlying principle in geNorm (Vandesompele et al. 2009) is that the expression ratios of two genes between the same two samples have to be the same, and so in a perfect scenario the ratio of these ratios would have to be 1. The pairwise variation of a candidate gene with all the other genes is calculated and the average is defined as stability value (M). The lower the M, the more stable the expression. The software ranks the candidate genes according to the M value in a set of samples by a repeated process of stepwise exclusion of the worst scoring or less stable gene. Bestkeeper (Pfaffl et al. 2004) uses directly C t values instead of normalized expression values and employs a different measure of expression stability based on the principle that more stable candidate genes should display a similar expression pattern. The program calculates the correlation (by Pearson coefficient) of all possible pairs of candidate genes and calculates their geometric mean. Finally, correlation between each gene and this geometric mean is calculated and the “stability” of a gene is expressed as the coefficient of correlation. Normfinder (Andersen et al. 2004) is based on a statistical model for describing the overall variation of the candidate reference genes and the variation between sample groups. The variance of the log-average of the expression of the candidate reference genes can be calculated and used for stability ranking. The lower the variance, the more stable the gene. Furthermore, we repeatedly used Bestkeeper to simulate a step-wise approach similar to the one in geNorm, in which the worst scoring gene is discarded before repeating the corresponding calculation step. Our results in P. pinaster or P. abies (Fig. 3a, b) showed that these algorithms result in equivalent rankings. This is evidenced by a single difference between geNorm and Normfinder regarding ACT in P. pinaster and CAC in P. abies, which was mostly equivalent to Bestkeeper, with three position shifts (SAND, AK and ACTIN) in P. pinaster and two (CAC and UBI) in P. abies. However, this is not the case when our study was done with both P. pinaster and P. abies samples at the same developmental stage. Differences among rankings are commonly found when samples are diverse enough (Klimaszewska et al. 2001a, b; Artico et al. 2010; Huis et al. 2010).
The stability values can also be used to determine the optimal number of reference genes for accurate normalization by comparison of values for a n and n + 1 number of endogenous genes. A small total variation between both conditions, represented by a small column, entails that the increase in one additional gene does not significantly increase the accuracy. This estimation is automatically made by geNorm. We used a new approach to estimate the ideal number of endogenous genes based on the comparison of the correlation values calculated by Bestkeeper for a decreasing number of endogenous genes. These new predictions showed equivalent results to those obtained with geNorm, thus evidencing that it is a valid approach. Estimating the optimal number of candidate genes is fundamental in this kind of analysis, so having at least a second method to calculate them appeared relevant. The ideal number of genes was three in most of the conditions. However, the inclusion of an additional gene(s) can be required for reliable normalization and the threshold values here proposed must not be taken as a strict cut-off. In fact, the commonly used 0.15 cut-off value when using geNorm is not good enough to discriminate our results, while a 0.2 cut-off value is.
The geometric averaging of the expression of three genes, EF1, ATUB and HISTO3, is the better combination in P. pinaster. These three genes are the better ranked with the three methods, only surpassed by AK in Bestkeeper. On the other hand, ACT, UBI, REDUC and HEATS were unstable in all the programs and should be avoided. HISTO3 and ATUB are classified in the GO Biological Process category Cellular organization and biogenesis (level 5 category), which may raise concerns about co-regulation. However, this is a very broad category that in Arabidopsis includes 4,693 genes. While the Arabidopsis homologous to HISTO3 functions in DNA binding and is involved in DNA packaging, chromatin assembly and nucleosome assembly, the Arabidopsis homologous to ATUB is constituent of the cytoskeleton and is involved in GTP catabolic processes and cytoskeleton organization. The possibility of co-regulation cannot be excluded but seems unlikely because in P. abies, where the somatic embryogenesis is expected to be regulated in a similar way as in P. pinaster, HISTO3 and ATUB are not ranked together. Furthermore, one of the advantages of using different algorithms for reference gene selection is to avoid the selection of co-regulated transcripts (Tong et al. 2009; Paim et al. 2012).
The better option in P. abies is the combination of four genes, ATUB, AK, CAC and EF1. All these genes showed stability values lower than the proposed threshold by geNorm authors (M < 1.4). The remaining genes showed a progressive decrease in stability according to all the analyses. However, the difference was not significant when three or four endogenous genes were compared for transcript quantification of two genes with different expression profiles. Only in certain conditions requiring discrimination of small differences, the addition of a fourth gene to increase of accuracy would be justified. After comparing both species, the combination of EF1 and ATUB is recommended as a generic option to consider in other conifers.
We verified if any of these genes was unstable during any specific developmental stage, which could be masked by doing simultaneously the analysis for all the stages. For example, ACT stability in P. pinaster was unclear. However, it is commonly used for qPCR in plants. Stage-specific rankings showed that ACT was unstable in proliferating embryogenic cells but stable in germinating embryos. So, it appears to be a good option for seedlings but not for developing embryos. Also, REDUX was discarded after the analysis for all the stages simultaneously, but it is a good option for studies in proliferating embryogenic cells, in agreement with the microarray results that covered early embryo stages up to embryo maturation (Simões et al. 2009).
The proposed endogenous genes were validated by quantifying the relative expression of P. pinaster PpRab1 and P. abies L1L and E11A12 genes. These were previously verified as differentially expressed in conditions similar to those here described by non-qPCR techniques. Gonçalves et al. (2007) demonstrated by semi-quantitative RT-PCR an increase of PpRab1 expression starting on mature embryos (T7 stage) towards seedlings with 3, 5, 7 and 9 days on germination (S3, S5, S7 and S9 stages). We observed that PpRab1 expression in mature embryos was half the value obtained in germinating embryos. Since our mature and germinating embryos would, respectively, correspond to T7 and S7 stages, the results here obtained are in accordance with those obtained by Gonçalves et al. (2007). Vestman et al. (2011) analysed the expression of L1L and E11A12 in P. abies somatic embryogenesis by microarray expression analysis and found that L1L expression significantly decreased from proliferation to early embryogenesis and from early embryogenesis to late embryogenesis. By contrast, E11A12 expression significantly increased among those stages. The qPCR results for quantification of L1L and E11A12 transcripts here reported are in agreement with these studies. Therefore, we can conclude that the proposed endogenous genes are reliable for obtaining accurate expression profiles in different expression ranges and samples during somatic embryogenesis in conifers.
Eventually, three conclusions highlight the importance of this kind of studies. (1) Genes stable in microarray analysis showed variations in our qPCR experiments and were unstable in some stages. The studied genes were selected among those showing a minimal variation in expression according to microarray analysis, as evidenced by the lowest standard deviation among samples, as previously done in Arabidopsis (Czechowski et al. 2005) and rice (Jain and Khurana 2009). Though a minimal variation among different samples, typical of house-keeping genes, is a good indicator of expression stability in certain conditions, this is not necessarily extensive to all other experiments, especially those covering a wider range of developmental periods. The fact that the microarray time-frame (T0–T2, T3–T4, T4B, T5–T6 and T7; Simões et al. 2009) does not exactly match the three developmental time-points used here may explain the obtained results. So, candidate genes should be identified from studies under conditions as similar as possible to the ones being used.
(ii) A reference gene stable for a species or developmental period can be unstable in a closely related species or developmental stage. Although our study showed that most of the genes had equivalent stability values in both species, there were clear exceptions such as HISTO3, which is stable in P. pinaster but the most unstable in P. abies. Additionally, REDUX was the most stable gene in proliferating embryogenic cells, but it was unstable in mature embryos. The statistical algorithms, such as the ones used in the present work, allow the quick and accurate identification of internal control that has to be validated for the specific experimental conditions before qPCR analysis. It is also important to remark that considering a fixed efficiency value is not acceptable because the ΔΔ C t method (Livak and Schmittgen 2001; Pfaffl 2001; Schmittgen and Livak 2008) is highly dependent on C ts and amplification efficiency values. Efforts have focused on improving the determination of amplification efficiency (Ramakers et al. 2003). Actually, novel quicker methods based on regression analysis of the PCR reactions kinetics after qPCR (Ruijter et al. 2009) lead to reproducible efficiency values (Nordgård et al. 2006), as also supported by our results.
And finally, (3) the commonly used actin gene (ACT) ranked as the worst scoring with some of the used algorithms. Common reference genes, such as those involved in basic cellular process, were used for many years as reference in Northern blots and RT-PCR assays. However, numerous studies have demonstrated that the expression of these genes may vary enormously depending on the tested conditions (Czechowski et al. 2005; Huggett et al. 2005).
Abbreviations
- SE:
-
Somatic embryogenesis
- qPCR:
-
Real-time quantitative PCR
- C t :
-
Cycle threshold
- ACT:
-
Actin
- ATUB:
-
α-Tubulin
- EF1:
-
Elongation factor-1α
- AK:
-
Adenosine kinase
- UBI:
-
Ubiquitin
- HISTO3:
-
Histone 3
- SAND:
-
SAND protein family
- CAC:
-
Clathrin adaptor complex subunit
- HEATS:
-
Heat shock protein
- REDUC:
-
Ether reductase protein
- V n :
-
Pairwise variation
- L1L :
-
Leafy cotyledon 1-like
- PpRab1 :
-
Pinus pinaster Rab GTPase
- CTAB:
-
Hexadecyltrimethylammonium bromide
- PVPP:
-
Polyvinylpyrrolidone
- DEPC:
-
Diethylpyrocarbonate
References
Alonso P, Cortizo M, Cantón FR, Fernández B, Rodríguez A, Centeno ML, Cánovas FM, Ordás RJ (2007) Identification of genes differentially expressed during adventitious shoot induction in Pinus pinea cotyledons by subtractive hybridization and quantitative PCR. Tree Physiol 27:1721–1730
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Aquea F, Arce-Johnson P (2008) Identification of genes expressed during early somatic embryogenesis in Pinus radiata. Plant Physiol Biochem 46:559–568
Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa M, Alves-Ferreira M (2010) Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol 10:49
Bishop-Hurley SL, Gardner RC, Walter C (2003) Isolation and molecular characterization of genes expressed during somatic embryo development in Pinus radiata. Plant Cell Tiss Organ Cult 74:267–281
Bomal C, Bedon F, Caron S, Mansfield SD, Levasseur C, Cooke JEK, Blais S, Tremblay L, Morency M-J, Pavy N et al (2008) Involvement of Pinus taeda MYB1 and MYB8 in phenylpropanoid metabolism and secondary cell wall biogenesis: a comparative in planta analysis. J Exp Bot 59:3925–3939
Bonga JM, Klimaszewska KK, Aderkas P (2009) Recalcitrance in clonal propagation, in particular of conifers. Plant Cell Tiss Organ Cult 100:241–254
Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23–39
Che P, Love TM, Frame BR, Wang K, Carriquiry AL, Howell SH (2006) Gene expression patterns during somatic embryo development and germination in maize Hi II callus cultures. Plant Mol Biol 62:1–14
Chen L, Zhong H-Y, Kuang J-F, Li J-G, Lu W-J, Chen J-Y (2011) Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta 234:377–390
Clapham D, Newton R, Sen S, Von Arnold S (2000) Transformation of Picea species. In: Jain M, Minocha SC (eds) Molecular biology of woody plants, vol 2. Springer, Heidelberg, pp 105–110
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LMG, Romano E, Grossi-de-Sá MF, Vaslin M, Alves-Ferreira M (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Mol Breeding 23:607–616
Czechowski T, Stitt M, Altmann T, Udvardi M, Scheible W (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
de Almeida MR, Ruedell CM, Ricachenevsky FK, Sperotto RA, Pasquali G, Fett-Neto AG (2010) Reference gene selection for quantitative reverse transcription-polymerase chain reaction normalization during in vitro adventitious rooting in Eucalyptus globulus Labill. BMC Mol Biol 11:73
Dheda K, Huggett J, Bustin S, Johnson MA, Rook G, Zumla A (2004) Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 37:112–119
Die J, Román B, Nadal S, González-Verdejo C (2010) Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta 232:145–153
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210
Expósito-Rodríguez M, Borges A, Borges-Pérez A, Pérez J (2008) Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol 8:131
Giménez MJ, Pistón F, Atienza SG (2011) Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta 233:163–173
Goldberg RB, de Paiva G, Yadegari R (1994) Plant embryogenesis: zygote to seed. Science 266:605–614
Gonçalves S, Cairney J, Maroco J, Oliveira M, Miguel C (2005) Evaluation of control transcripts in real-time RT-PCR expression analysis during maritime pine embryogenesis. Planta 222:556–563
Gonçalves S, Cairney J, Rodríguez MP, Cánovas F, Oliveira M, Miguel C (2007) PpRab1, a Rab GTPase from maritime pine is differentially expressed during embryogenesis. Mol Genet Genomics 278:273–282
Harvengt L (2005) Somatic embryogenesis in maritime pine (Pinus pinaster Ait.). In: Jain SM, Gupta PK (eds) Protocol of somatic embryogenesis in woody plants, vol. 77. Springer Verlag, Berlin, pp 107–120
Hellemans J, Mortier G, de Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19
Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6:279–284
Huis R, Hawkins S, Neutelings G (2010) Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol 10:71
Humánez A, Blasco M, Brisa C, Segura J, Arrillaga I (2012) Somatic embryogenesis from different tissues of Spanish populations of maritime pine. Plant Cell Tiss Organ Cult 111:373–383
Jain M, Khurana JP (2009) Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J 276:3148–3162
Klimaszewska K, Lachance D, Pelletier G (2001a) Regeneration of transgenic Picea glauca, P. mariana, and P. abies after cocultivation of embryogenic tissue with Agrobacterium tumefaciens. In Vitro Cell Dev Biol Plant 37:748–755
Klimaszewska KK, Park YS, Overton C, MacEacheron I, Bonga JM (2001b) Optimized somatic embryogenesis in Pinus strobus L. In Vitro Cell Dev Biol Plant 37:392–399
Lara-Chavez A, Egertsdotter U, Flinn BS (2012) Comparison of gene expression markers during zygotic and somatic embryogenesis in pine. In Vitro Cell Dev Biol Plant 48:341–354
Li Q-F, Sun SSM, Yuan D-Y, Yu H-X, Gu M-H, Liu Q-Q (2010) Validation of candidate reference genes for the accurate normalization of real-time quantitative RT-PCR data in rice during seed development. Plant Mol Biol Rep 28:49–57
Lin Y, Lai Z (2010) Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci 178:359–365
Litvay JD, Verma DC, Johnson MA (1985) Influence of loblolly pine (Pinus taeda L.) culture medium and its components on growth and somatic embryogenesis of the wild carrot (Daucus carota L.). Plant Cell Rep 4:325–328
Liu J-J, Zamani A, Ekramoddoullah AKM (2010) Expression profiling of a complex thaumatin-like protein family in western white pine. Planta 231:637–651
Livak K, Schmittgen T (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-[Delta][Delta] CT method. Methods 25:402–408
Marum L, Estêvão C, Oliveira MM, Amâncio S, Rodrigues L, Miguel C (2004) Recovery of cryopreserved embryogenic cultures of maritime pine: effect of cryoprotectant and suspension density. Cryo Lett 25:363–374
Marum L, Rocheta M, Maroco J, Oliveira MM, Miguel C (2009) Analysis of genetic stability at SSR loci during somatic embryogenesis in maritime pine (Pinus pinaster). Plant Cell Rep 28:673–682
Marum L, Miguel A, Ricardo CP, Miguel C (2012) Reference gene selection for quantitative real-time PCR normalization in Quercus suber. PLoS One 7:e35113
Meinke DW (1995) Molecular genetics of plant embryogenesis. Annu Rev Plant Physiol Plant Mol Biol 46:369–394
Miguel C, Gonçalves S, Tereso S, Marum L, Maroco J, Oliveira MM (2004) Somatic embryogenesis from 20 open-pollinated families of Portuguese plus trees of maritime pine. Plant Cell Tiss Org Cult 76:121–130
Nolan T, Bustin SA (2004) Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech 15:155
Nolan T, Hands RE, Bustin SA (2006) Quantification of mRNA using real-time RT-PCR. Nat Protoc 1:1559–1582
Nordgård O, Kvaløy JT, Farmen RK, Heikkilä R (2006) Error propagation in relative real-time reverse transcription polymerase chain reaction quantification models: the balance between accuracy and precision. Anal biochem 356:182–193
Paim RM, Pereira MH, Di Ponzio R, Rodrigues JO, Guarneri AA, Gontijo NF, Araújo RN (2012) Validation of reference genes for expression analysis in the salivary gland and the intestine of Rhodnius prolixus (Hemiptera, Reduviidae) under different experimental conditions by quantitative real-time PCR. BMC Res Notes 5:128
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Ramakers C, Ruijter J, Deprez R, Moorman A (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66
Ratnaparkhe S, Egertsdotter E, Flinn B (2009) Identification and characterization of a matrix metalloproteinase (Pta1-MMP) expressed during Loblolly pine (Pinus taeda) seed development, germination completion, and early seedling establishment. Planta 230:339–354
Ruijter J, Ramakers C, Hoogaars W, Karlen Y, Bakker O, van den Hoff M, Moorman A (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37(6):e45
Schmittgen T, Livak K (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108
Schmittgen TD, Zakrajsek BA (2000) Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 46:69–81
Simões M, Lorenz WW, Alba R, Gonçalves S, Maroco J, Dean J, Miguel CM (2009) Embryogenesis in maritime pine: a transcriptomics approach. In: IUFRO tree biotechnology conference, Jun 28–Jul 2, Whistler, Canada (Poster)
Spencer MWB, Grene R, Lindsey K (2007) Transcriptional profiling of the Arabidopsis embryo. Plant Physiol 143:924–940
Suzuki M, Wang HHY, McCarty DR (2006) Repression of the LEAFY COTYLEDON 1/B3 regulatory network in plant embryo development by VP1/ABSCISIC ACID INSENSITIVE 3-LIKE B3 genes. Plant Physiol 143:902–911
Tereso S, Miguel C, Zoglauer K, Valle-Piquera C (2006) Stable Agrobacterium-mediated transformation of embryogenic tissues from Pinus pinaster Portuguese genotypes. Plant Growth Regul 50:57–68
Thellin O, ElMoualij B, Heinen E, Zorzi W (2009) A decade of improvements in quantification of gene expression and internal standard selection. Biotechnol Adv 27:323–333
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol 10:71
Trontin J, Walter C, Klimaszewska K, Park Y, Lelu-Walter M (2007) Recent progress in genetic transformation of four Pinus spp. Transgenic Plant J 1:314–329
Trontin JF, Debille S, Canlet F, Harvengt L, Lelu-Walter MA, Label P, Teyssier C, Lesage-Descauses MC, Le Metté C, Miguel C et al (2013) Somatic embryogenesis as an effective regeneration support for reverse genetics in maritime pine: the Sustainpine collaborative project as a case study. In: Proceedings of the IUFRO Working Party 2.09.02, integrating vegetative propagation, biotechnology and genetic improvement for tree production and sustainable forest management
Tu L, Zhang X, Liu D, Jin S, Cao J, Zhu L, Deng F, Tan J, Zhang C (2007) Suitable internal control genes for qRT-PCR normalization in cotton fiber development and somatic embryogenesis. Chin Sci Bull 52:3110–3117
Uddenberg D, Valladares S, Abrahamsson M, Sundström JF, Sundås-Larsson A, von Arnold S (2011) Embryogenic potential and expression of embryogenesis-related genes in conifers are affected by treatment with a histone deacetylase inhibitor. Planta 234:527–539
Vandesompele J, de Preter K, Pattyn F, Poppe B, van Roy N, de Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3(7):research0034
Vandesompele J, Kubista M, Pfaffl M (2009) Reference gene validation software for improved normalization. In: Logan J, Edwards K, Saunders N (eds) Real-time PCR: current technology and applications. Caister Academic Press, London, pp 47–64
Vásquez-Robinet C, Watkinson JI, Sioson AA, Ramakrishnan N, Heath LS, Grene R (2010) Differential expression of heat shock protein genes in preconditioning for photosynthetic acclimation in water-stressed loblolly pine. Plant Physiol Biochem 48:256–264
Vestman D, Larsson E, Uddenberg D, Cairney J, Clapham D, Sundberg E, von Arnold S (2011) Important processes during differentiation and early development of somatic embryos of Norway spruce as revealed by changes in global gene expression. Tree Genet Genomes 7:347–362
von Arnold S, Clapham D (2008) Spruce embryogenesis. In: Suarez MF, Bozhkov PV (eds) Plant embryogenesis methods in molecular biology. Humana, Totowa, pp 31–47
von Arnold S, Sabala I, Bozhkov P, Dyachok J, Filonova L (2002) Developmental pathways of somatic embryogenesis. Plant Cell Tiss Organ Cult 69:233–249
von Arnold S, Bozhkov P, Clapham D, Dyachok J, Filonova L, Hogberg K-A, Ingouff M, Wiweger M (2005) Propagation of Norway spruce via somatic embryogenesis. Plant Cell Tiss Organ Cult 81:323–329
West M, Harada JJ (1993) Embryogenesis in higher plants: an overview. Plant Cell Online 5:1361–1369
Xu M, Zhang B, Su X, Zhang S, Huang M (2011) Reference gene selection for quantitative real-time polymerase chain reaction in Populus. Anal Biochem 408:337–339
Yan J, Yuan F, Long G, Qin L, Deng Z (2012) Selection of reference genes for quantitative real-time RT-PCR analysis in citrus. Mol Biol Rep 39:1831–1838
Acknowledgments
This work has been supported by Fundação para a Ciência e Tecnologia (FCT, Portugal) through projects PTDC/AGR-GPL/102877/2008, P-KBBE/AGR-GPL/0001/2009 and grant SFRH/BD/32037/2006 (MS). The authors acknowledge Prof. Sara von Arnold and Dr. David Clapham (SLU, Uppsala, Sweden) for providing the Picea embryogenic cell line used in these experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Bennett.
Rights and permissions
About this article
Cite this article
de Vega-Bartol, J.J., Santos, R.R., Simões, M. et al. Normalizing gene expression by quantitative PCR during somatic embryogenesis in two representative conifer species: Pinus pinaster and Picea abies . Plant Cell Rep 32, 715–729 (2013). https://doi.org/10.1007/s00299-013-1407-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-013-1407-4