
Abstract
Comparative transcriptomics are useful to determine the role of orthologous genes among Triticeae species. Thus they constitute an interesting tool to improve the use of wild relatives for crop breeding. Reverse transcription quantitative real-time PCR (qPCR) is the most accurate measure of gene expression but efficient normalization is required. The choice and optimal number of reference genes must be experimentally determined and the primers optimized for cross-species amplification. Our goal was to test the utility of wheat-reference genes for qPCR normalization when species carrying the following genomes (A, B, D, R, H v and H ch) are compared either simultaneously or in smaller subsets of samples. Wheat/barley/rye consensus primers outperformed wheat-specific ones which indicate that consensus primers should be considered for data normalization in comparative transcriptomics. All genes tested were stable but their ranking in terms of stability differed among subsets of samples. CDC (cell division control protein, AAA-superfamily of ATPases, Ta54227) and RLI (68 kDa protein HP68 similar to Arabidopsis thaliana RNase L inhibitor protein, Ta2776) were always among the three most stable genes. The optimal number of reference genes varied between 2 and 3 depending on the subset of samples and the method used (geNorm vs. coefficient of determination between sequential normalization factors). In any case a maximum number of three reference genes would provide adequate normalization independent of the subset of samples considered. This work constitutes a substantial advance towards comparative transcriptomics using qPCR since it provides useful primers/reference genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Triticeae tribe includes the cereals wheat (Triticum spp.), rye (Secale cereale L.), barley (Hordeum vulgare L.), the modern cereal triticale, plus above 350 species (Barkworth and Bothmer 2009). The main species of this tribe dominate agricultural landscapes in temperate areas. However, the progressive narrowing of their genetic base (Tanksley and McCouch 1997; Warburton et al. 2006) has promoted a renewed interest in wild relatives such as Aegilops tauschii, donor of the D genome of common wheat (van Ginkel and Ogbonnaya 2007) or wild barley such as Hordeum chilense (Atienza et al. 2007b; Martin et al. 2008, 2009, 2010) or Hordeum spontaneum in barley (Matus et al. 2003; Inostroza et al. 2009) as well as in new species such as triticale or ×Tritordeum (Villegas et al. 2010). An efficient use of these genetic resources in breeding requires both the precise introduction of the genes of interest and the determination of their effect in the crop genetic background. The limited inputs dedicated to wild relatives compared to major crops hamper their use in crop breeding. Comparative genomic studies in cereals have lead to the generation of genomic tools useful for gene isolation and genetic studies in these species and, in particular, in those of the Triticeae lacking a physical map or genome sequences (Feuillet and Salse 2009). For instance, the development of conserved orthologous sequences (COS) markers will help to identify candidate genes in one species from the knowledge obtained in other (Bolot et al. 2009). Similarly, comparative transcriptomics are experiencing a growing interest for a wide range of studies including abiotic stress response (Campoli et al. 2009), regulation of developmental processes (Gil-Humanes et al. 2009) or differential regulation between polyploid species (He et al. 2003; Poole et al. 2007; Salentijn et al. 2009).
In this context, microarrays designed for one species are being used to determine expression divergence among species (Becher et al. 2004; Weber et al. 2004; Chain et al. 2008). However, sequence mismatches could cause bias when expression divergence of two species are compared using an array designed for only one of them (Chain et al. 2008). As a consequence these studies should validate their results using microarray-independent approaches such as quantitative real-time PCR (qPCR). In addition to this, qPCR would also be useful to test candidate genes in minor crops or wild relatives where no microarrays are available.
Gene expression analyses methods such as qPCR or microarray require efficient normalization approaches to be informative (Bustin 2002; Quackenbush 2002). The most common way to normalize qPCR data is to use appropriate internal reference genes which need to be expressed at a constant level in all samples tested. However, qPCR data are normalized poorly in many molecular analyses, especially in plant science, since genes validated as references are not usually used (Gutierrez et al. 2008; Guenin et al. 2009). For instance, most gene expression studies in wheat are normalized using reference genes not previously validated (for a detailed review see Paolacci et al. 2009).
The situation has improved in the last years since reference genes have been defined for a wide range of plant species including wheat (Paolacci et al. 2009), soybean (Hu et al. 2009), coffee (Cruz et al. 2009), Brachypodium distachyon (Hong et al. 2008), perennial ryegrass (Lee et al. 2010), faba bean (Gutierrez et al. 2010) or peach (Tong et al. 2009) among others.
Although comparative transcriptomics between grasses use reference genes for data normalization (Campoli et al. 2009; Gil-Humanes et al. 2009), these genes are selected on the basis of their good performance in a single species such as wheat. However, the reference genes should be stably expressed across species. Besides, the existence of mismatches between the primers and the target sequences must be avoided since it may result in detrimental effects on the quantification of the target genes (Bru et al. 2008; Ghedira et al. 2009). Consequently, the establishment of a valid reference set for comparing selected species in the Triticeae is a crucial pre-requisite to obtain meaningful expression data.
A set of reference genes has been recently proposed for common wheat (Paolacci et al. 2009). Among them, the gene for cell division control protein, AAA-superfamily of ATPases (CDC, Ta54227), ADP-ribosylation factor (ADP-RF, Ta2291) and RNase L inhibitor-like protein (RLI, Ta2776) were more effective than other reference genes used to normalize gene expression in wheat (Paolacci et al. 2009).
The aim of this work is to establish the basis for comparative expression profiling among selected Triticeae species by validating the stability of reference genes suitable for common wheat transcriptomics studies. Besides, we aim to determine the relative performance of consensus primers versus wheat-specific primers for qPCR normalization.
Materials and methods
Sample collection
A total of 11 accessions were considered in this work. Plants of ten different species belonging to the Triticeae tribe were used (Table 1). Two genotypes of H. chilense Roem. et Schultz. were included since they are representatives of two main subgroups within the species. Seeds (all obtained from GermPlasm Bank of the IAS-CSIC, Cordoba, Spain) were germinated in water-saturated filter paper and kept in the dark at 4°C for 1 week to synchronize germination. After this seeds were maintained at 25°C for 1 day in the dark. Seedlings were transplanted to pods and grown at 22/16°C day/night with 12/12 h light/darkness. The most important items of the MIQE checklist (Bustin et al. 2009) were considered as described in a practical approach to RT-qPCR experiments (Taylor et al. 2010). For each accession, five different plants were cultivated. The second fully expanded leaf of three plants was harvested in the middle of the light period, frozen in liquid nitrogen and stored at −80°C until RNA extraction. Two biological replications were arranged for each accession following the procedure described above.
Total RNA and genomic DNA isolation
Total RNA was isolated from every leaf sample using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacture’s instructions (33 samples/experiment). For eliminating DNA contamination 50 μL of 8 M LiCL2 was added to the washed pellet diluted in 150 μL of RNAse free milliQ water, mixed by inversion and incubated overnight at −20°C. After 30 min of centrifugation at 20,000g upper phases were discarded and the pellets were washed with 75% ethanol prior to their dilution in 40 μL of RNAse-free milliQ water.
RNA integrity was verified on 2% agarose gel electrophoresis and ethidium bromide staining. RNA quantity and purity was determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Walthman, MA, USA). All RNA samples were adjusted to the same concentration for subsequent reverse-transcription reactions.
Leaf tissue of individual plants was harvested, frozen in liquid nitrogen, and stored at −80°C until DNA extraction. Genomic DNA was extracted according to Murray and Thompson (1980) with minor modifications.
Selection of candidate reference gene and primer design
Genes coding for actin (Unigene accession Ta1868), translation elongation factor 1α subunit (Unigene accession Ta659, Tef-1α), the cell division control protein, AAA-superfamily of ATPases (Unigene accession Ta54227, CDC), the ADP-ribosylation factor (Unigene accessions Ta45379/Ta2291, ADP-RF) and the RNase L inhibitor-like protein (Unigene accession Ta2776, RLI) were selected as candidate reference genes in this study. A set of wheat-specific primers (Uauy et al. 2006; Paolacci et al. 2009; A. Distelfeld, University of California at Davis, USA, personal communication) (Table 2), as well as consensus primers designed for cross-specific amplification of CDC, ADP-RF and RLI in wheat, barley and rye, was used for qPCR analysis (Table 2).
All the sequences of the Unigenes Ta54227 (CDC), Ta2291 (ADP-RF) and Ta2776 (RLI) were downloaded and assembled using CAP3 (Huang and Madan 1999) to obtain their respective contig sequence. These contigs were exploited to find homeologous barley and rye genes in the GenBank database using BLAST search (Supplementary Tables S1–S3). Sequences from the different species were aligned using ClustalW program (Larkin et al. 2007), and the more conserved regions were selected as amplification targets. Primers were designed using the Primer3Plus on-line application at the 3′-end region of these consensus sequences to ensure equal RT efficiencies (Supplementary Figs. S1–S3).
Two step real-time RT-PCR
cDNA was obtained by reverse transcription from 2 μg of total RNA using a M-MLV reverse transcriptase (Invitrogen) in combination with oligo (dT)12–18 and random nonamers in 20 μL of total reaction volume according to the manufacturer’s instructions. Amplification using either gDNA or cDNA as template and primers PSY1Fw2/Rev2 spanning an intron (Atienza et al. 2007a) were used to discard DNA contamination. These primers allowed discarding DNA contamination in all samples but rye. Subsequent amplifications with primer pair Tef1-α (Table 2), also spanning an intron, allowed to fully discard DNA contamination in all samples.
For each accession, cDNA of three plants was adjusted to the same concentration and pooled to minimize the individual plant variation in gene expression. Five serial dilutions [1:4] were established for each sample for the qPCR experiment. No template controls (NTC) were also included. In addition, a serially diluted sample of a bulk comprising the 22 samples (11 accessions × 2 biological repetitions) considered in the experiment was included for each gene as inter-run calibrator to detect and correct inter-run variation.
The qPCR was carried out using the SYBR® GREEN on an ABI PRISM 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The PCR conditions were 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. For every experiment, 5 μL of diluted cDNA solution was used for PCR amplification in a 25 μL volume reaction containing 12.5 μL of 2× FastStart Universal SYBR Green Master (Roche Applied Science, Mannheim, Germany) and 225 nM of each primer. All experiments were performed in 96-well optical reaction plates (P/N 4306737) with MicroAmp optical adhesive film (P/N 4311971) from Applied Biosystems. The specificity of the amplifications was confirmed by the presence of a single band of expected size for each primer pair in agarose gel (2% w/v, data not shown) and by the single-peak melting curves of the PCR products (Supplementary Fig. S4).
Data analysis
PCR efficiency of each primer pair was determined for all genotypes by the LinRegPCR quantitative PCR data analysis program (version 11.0) (Ruijter et al. 2009) using raw normalized fluorescence as input data. Expression of the reference genes for each sample (N 0) was determined using the equation N 0 = 0.2/E Cq, where E = PCR efficiency for each primer, Cq is the number of cycles needed to reach 0.2 arbitrary units of fluorescence. Since LinReg package allows inspecting the PCR efficiency in each PCR well, the mean PCR efficiency for each gene and species was determined on the basis of up to 10 data (five serial dilutions × 2 biological repetitions).
Bioinformatic analysis
An in silico approach was applied to identify the Unigenes of wheat and barley amplifiable with our set of primers. In a first step, the NCBI BLASTn software (http://blast.ncbi.nlm.nih.gov/) was used to match each primer pair against sequences of the organisms Triticum (taxid: 4564) and Hordeum (taxid:4512) included in the GenBank non-redundant nucleotide database (nr) and in the ESTs database (non-mouse and non-human ESTs collections). The Unigenes containing the matching sequences and their homologous Unigenes in both species were selected and their sequences downloaded. A consensus sequence for each UniGene was obtained using the EGassembler platform (http://egassembler.hgc.jp/). The second step consisted in matching the set of primers against the sequences of each Unigene to test for potential mispriming using the NCBI BLASTn software.
Stability analysis
Data were analyzed using geNorm (Vandesompele et al. 2002) as described in its manual. The geNorm software provides a measure of gene expression stability (M), using the principle that the expression ratio of two ideal internal control genes is identical in all tested samples. Genes with the lowest M values have the most stable expression. The geNorm program also computes a normalization factor (NF) based on the geometric mean of the expression levels of the best-performing reference genes.
Results
Selection of potential reference genes and primer design for comparative qPCR profiling in the Triticeae
The set of samples used in this work was selected in order to include different ploidy levels (diploid, tetraploid and hexaploid) in wild and cultivated species belonging to the Triticeae tribe (Table 1). Each of the five different genomes considered (A, B, D, H ch and R) are represented by at least three species. In addition, cultivated barley (genome H v) was included for bioinformatics analysis. The set of candidate genes evaluated in this work was selected on the basis of their good performance as reference genes in common wheat since most species used in this work share at least one genome with it. The Unigenes Ta54227 (CDC, cell division control protein, AAA-superfamily of ATPases), Ta2291 (ADP-RF, ADP-ribosylation factor) and Ta2776 (RLI, 68 kDa protein HP68 similar to Arabidopsis thaliana RNase L inhibitor protein) were selected since these genes outperformed other traditional reference genes in wheat (Paolacci et al. 2009). The orthologous of these genes in barley and rye were identified (Supplementary Tables S1–S3) and a consensus sequence was obtained and used to design the PCR primers CDC(a), RLI(a) and ADP-RF(a) to maximize cross-species amplification within the Triticeae (Supplementary Figs. S1–S3). The primer pairs CDC(b), RLI(b) and ADP-RF(m), designed by (Paolacci et al. 2009) were also used to evaluate the relative performance of wheat-specific versus consensus-specific primer pairs on the qPCR quantification of selected Triticeae species. According to their authors, the primer pair ADP-RF(m) was designed to amplify simultaneously the transcripts of two homologous genes in wheat. On the contrary, the ADP-RF(a) primer pair is Ta2291-specific (see below). Primer pairs amplifying both Ta1868 (Actin) and Ta659 (Tef-1α, translation elongation factor 1 alpha-subunit) were also included as a reference since they are routinely used in our lab for qPCR in common wheat. All primers used in this work are listed in Table 2.
Expression profiling of reference genes
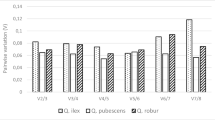
Gene-specific amplification of each of the eight candidate genes was confirmed by the appearance of a single, dominant peak in the qPCR dissociation curve analyses (see Supplementary Fig. S4). The PCR efficiency of each primer pair for all genotypes and the bulk samples was determined using LinReg package as explained in “Materials and methods”. For each primer pair, the mean, range and standard deviation of qPCR efficiency across species are shown in Fig. 1a. Significant differences in PCR efficiency were found between primer pairs amplifying the same locus as follows: CDC(a) (1.903) versus CDC(b) (1.846) and RLI(a) (1.918) versus RLI(b) (1.846) (Fig. 1a) and, therefore, both CDC(b) and RLI(b) primer pairs were excluded for the expression stability analysis using geNorm as discussed below.

Expression level of candidate reference genes and qPCR efficiencies. a Mean (square dot), range (whiskers) and standard deviation (box) of qPCR efficiency across species for each pair of primers used in this work. b The expression levels (Cq) values of each candidate reference gene are shown as means (square dot), standard deviation (boxes) and expression range (whiskers) for the samples analyzed
The quantification cycle (Cq) value denotes in a simplified way the level of the gene expression in the samples when similar qPCR efficiency is found. The Cq values were determined for each amplicon at the common threshold fluorescence value of 0.2. There was a difference of five cycles from the lowest mean value (20.47) in ADP-RF(a), to the highest (25.52) in Actin (Fig. 1b). The low mean Cq value of CDC and ADP-RF suggests that these genes are highly expressed. On the contrary, Actin had the lowest RNA levels in all the samples while RLI showed an intermediate expression (Fig. 1b).
Expression data analysis
The geNorm software was used to determine the expression stability of the candidate genes across selected species. It calculates a measure of gene expression stability (M), under the hypothesis that the expression ratio of two ideal reference genes is identical in all tested samples. Since a comparative transcriptomic analysis involving all the species considered in this work is not likely, four subsets of samples were defined as follows: all samples included; samples carrying H v genome excluded; samples with either H v or R genome excluded; samples carrying H v or H ch genomes excluded. Using geNorm, all the six genes analyzed showed high expression stability and had M values lower than the default limit of M ≤ 1.5 proposed by the geNorm program for any of the groups of samples considered. The ranking of the genes in terms of stability varied depending on the subgroup of samples considered. Both CDC(a) and RLI(a) were always among the three most stable genes with ADP-RF(m), ADP-RF(a) or Tef-1α completing this trio depending on the analysis (Fig. 2).

geNorm output charts of M values for the six selected reference genes. The x-axis indicates the ranking of the primer pair tested according to their expression stability (M). Four different analyses were performed with different subsets of samples. a All samples included. b Samples with H v genome excluded. c Samples with either H v or R genomes excluded. d Samples with either H v or H ch genome excluded
Two different approaches were considered to determine the optimal number of genes. First, the pairwise variation (V n/n+1) between two sequential normalization factors NF n and NFn+1 were calculated using geNorm for each of the group of samples considered (Fig. 3a). Second, the normalization factor considering the six stably expressed genes (NF6) was calculated. A stepwise reduction in the number of reference genes was successively performed to calculate the resulting NFs (NF5, NF4, NF3 and NF2) by excluding the least stable gene. Scatter plots between NF6 and each NF i were obtained for each of the subsets of samples (Fig. 3b). The coefficients of determination were higher than 95% in all cases but NF6 versus NF2 (R 2 = 0.9292) when considering all samples simultaneously (Fig. 3b).

Determination of the optimal number of control reference for normalization. a geNorm pairwise variation (V n/n+1) analysis between the normalization NF n and NFn+1. For each subset of samples, a V n/n+1 value below 0.15 indicates that “n” is the optimal number of reference genes. b Scatterplots of normalization factors with the maximum number of stable genes (NF6, x-axis) and after a stepwise exclusion of the least stable gene (NF5, NF4, NF3 and NF2, y-axis), for any subset of samples considered. R 2 = coefficient of determination)
In silico approach to identify wheat/barley unigenes amplifiable with the primers
The candidate reference genes selected in this work belong to different gene families. Differences in performance of the primer pairs could be explained in terms of number of priming targets and sequence mismatches. To check how many family members are amplified by each primer pair, a bioinformatics analysis was performed in wheat, barley and rye but the lower number of rye sequences did not allow obtaining significant conclusions for this species (data not shown). The degree of identity (match %) between each primer pair and the Unigene sequences of its respective gene family was determined. In wheat, both set of primers amplifying either CDC or RLI showed a perfect match over their respective target sequences (Table 3; Supplementary Table S4). For RLI the Unigenes Ta57374, Ta56294 and the sequence AK331207.1 would also be amplified although their relative abundance is low compared to the initial target sequence Ta2776 (based on its EST counts). Regarding barley, primers CDC(a) and RLI(a) also showed, as expected, a perfect match over their respective target sequences since barley sequences were considered in their design. However, CDC(b) primers and RLI(b) did not yielded a perfect match in neither case. Both primer pairs were not considered in geNorm analyses since they showed a poorer performance in terms of mean PCR efficiency (Fig. 1a), which may result from a poorer match to non-wheat genomes (Table 3). On the contrary, the bioinformatics analysis demonstrates that both ADP-RF(m) and ADP-RF(a) primers amplify different loci. In wheat, the primer pair ADP-RF(a) has two Unigenes targets (Ta2291 and Ta30873) while ADP-RF(m) has four Unigene targets. Regarding barley, different Unigenes are amplified by each primer set (Table 3), providing a further justification for its inclusion in geNorm analyses.
Discussion
For valid qPCR analysis, accurate normalization of gene expression against an adequate set of reference genes is needed. Accordingly, the number of studies identifying reference genes in plant species has increased over the last years (Hong et al. 2008; Cruz et al. 2009; Hu et al. 2009; Paolacci et al. 2009; Tong et al. 2009; Lee et al. 2010). For gene expression comparison among different Triticeae species, single reference genes, such as actin (T. aestivum beta-actin, accession number AB181991), have also been used (Campoli et al. 2009; Gil-Humanes et al. 2009) but a validation of their stability across species is lacking. It has been shown that qPCR results may significantly differ depending on the reference gene used for data normalization (Dheda et al. 2005). When amplifying orthologous genes across different species using wheat-specific primers the occurrence of mismatches is likely. Although the identity of the orthologous genes is usually demonstrated through sequencing of the PCR products, mismatches between the primers and the templates cannot be discarded by this mean since, as a result of amplification, the sequence of the amplicons will be complementary to the primer.
In this work we have investigated the existence of mismatches between primer sequences and wheat and barley Unigenes. Previous studies have demonstrated that single mismatches between the primer and the target sequence lead to an underestimation of the target gene expression (Bru et al. 2008; Ghedira et al. 2009). The bioinformatics analysis shows that the wheat-specific primers RLI(b) and CDC(b) do not perfectly match barley sequences, which would affect the qPCR quantification. Besides, although the existence of mismatches cannot be discarded from the bioinformatics analysis for any of the genomes with no sequence information available, such as H ch, it is more likely that mismatches would occur with wheat-specific primers than with consensus-specific ones as resulted for barley (Table 3). Indeed, the mean PCR efficiency was lower for wheat-specific primers for CDC and RLI transcripts. This may reflect the existence of mismatches between primer pairs and target sequences although other factors such as the amplicon length may affect the qPCR results (Gimenez et al. 2010). A different situation is found for ADP-RF set of primers since they amplify different loci. The use of primers targeting multiple members of some gene families has been exploited as an adequate strategy for normalization (Paolacci et al. 2009). Since several Triticeae species with no previous information were considered in this work, it was possible that this strategy performs better than using single reference genes. Therefore, we used the ADP-RF(m) primers proposed by Paolacci et al. (2009) which were designed to amplify the wheat Unigenes Ta45379 and Ta2291. Surprisingly, the bioinformatics analysis revealed the lack of a perfect match to Ta2291 and the amplification of two supplementary ADP-RF loci, Ta55524 and Ta55119 (Table 3). Since it was not possible to design primers simultaneously amplifying all these sequences plus the barley ones, we focused on the most stable gene of the ADP-RF family in wheat (Ta2291) (Paolacci et al. 2009) in order to find orthologous sequences in barley and rye and to design primers.
Although ADP-RF(a) primers are Ta2291-specific, a second Unigene (Ta30873) consisting of only two sequences would be amplified as revealed by the bioinformatics analysis (Table 3). However, this Unigene is annotated as similar to ADP-RF from Magnaphorte grisea. Both sequences included in this Unigene are derived from a cDNA library dbEST 9984 obtained from spikes inoculated with Fusarium graminearum. Therefore, it may be possible that these sequences belong to this pathogen. In any case, it may be concluded that both ADP-RF(a) and ADP-RF(m) amplify different Unigenes and therefore they can be considered for geNorm analyses. This consideration is important since co-regulated genes must be excluded from geNorm analyses since it may affect the ranking of stability of the candidate genes (Maccoux et al. 2007; Hibbeler et al. 2008).
The optimal number of reference genes must be experimentally determined. The original geNorm publication recommends a minimal use of the three most stable internal control genes for calculation of the normalization factor (NF3) (Vandesompele et al. 2002), and stepwise inclusion of more reference genes until the (n + 1)th gene does not significantly contribute to the newly calculated normalization factor (NFn+1) (Vandesompele et al. 2002). In this work we have followed two slightly different approaches to estimate the optimal number of genes. Under the hypothesis that the best NF would be obtained when all the stably expressed reference genes are included, we determined the effect of a stepwise reduction by discarding the least stable gene. Indeed, the geNorm procedure to determine the optimal number of genes is based on the assumption that “it is waste of resources to quantify more genes than necessary if all genes are relatively stably expressed and if the normalization factor does not significantly change whether or not more genes are included”. Therefore, we propose to follow a method of stepwise exclusion from the maximum number of stably expressed genes instead of a stepwise inclusion of genes. Thus, starting from the NF n calculated with the maximum number of stably expressed genes (n), a stepwise exclusion of the least stable reference gene would be performed and the subsequent NFn−1, NFn−2, …, NF2 would be determined. After this, the coefficient of determination between NF n and each NF i will be calculated. We propose to select the minimum number of reference genes (x) with R 2 between NF n and NF x over 0.95 since this would imply that the results obtained using both NF would not be significantly different.
In this work, the reduction from 6 to 3 reference genes (CDCa, RLIa and ADP-RFm) does not significantly affect to the NF when all samples are considered since the R 2 between NF6 and NF3 is 0.969. We could also consider three genes according to geNorm since the V3/4 value is 0.169 and the recommended threshold of 0.15 must not be taken as a too strict cut-off according the geNorm manual.
In any of the subsets of samples considered, according to the R 2 criteria, it would suffice with two reference genes. Thus, we suggest to choose CDC(a)/RLI(a), ADP-RF(a)/RLI(a) and CDC(a)/Tef-1α for data normalization of the subsets excluding H v, H v/R and H v/H ch samples, respectively. Similarly, according to geNorm, a third reference gene, ADP-RF(a) or CDC(a), should be added when H v or H v/R is excluded, respectively. In any case, a maximum number of three reference genes would provide adequate normalization independent of the subset of samples considered.
In conclusion, the six genes studied in this work are useful for normalization of qPCR results when comparing different Triticeae species but CDC(a) and RLI(a) were consistent among the most stable ones and thus they should be preferably used. The optimal number of reference genes does not exceed of 3 with independence of the species being compared or the procedure used (geNorm or coefficient of determination between NFs). However, the best choice of the reference genes would differ upon the species being compared since the ranking of the reference genes in terms of stability is not identical in all cases. Finally, the use of consensus primers should always be considered for comparative transcriptomics since they generally outperformed single-species-derived primers.
Abbreviations
- ADP-RF:
-
ADP-ribosylation factor
- CDC :
-
Cell division control protein, AAA-superfamily of ATPases
- NF:
-
Normalization factor
- RLI :
-
RNase L inhibitor-like protein
- qPCR:
-
Quantitative real-time PCR
- Tef-1α:
-
Translation elongation factor 1α subunit
References
Atienza SG, Avila CM, Martin A (2007a) The development of a PCR-based marker for PSY1 from Hordeum chilense, a candidate gene for carotenoid content accumulation in tritordeum seeds. Aust J Agric Res 58:767–773
Atienza SG, Martin AC, Ramirez MC, Martin A, Ballesteros J (2007b) Effects of Hordeum chilense cytoplasm on agronomic traits in common wheat. Plant Breed 126:5–8
Ballesteros J, Ramirez MC, Martinez C, Atienza SG, Martin A (2005) Registration of HT621, a high carotenoid content tritordeum germplasm line. Crop Sci 45:2662–2663
Ballesteros J, Cabrera A, Aardse A, Ramirez MC, Atienza SG, Martin A (2007) Registration of TS1, TS10 and TS41, three high biomass production tetraploid triticale germplasm lines. J Plant Reg 1:71–72
Barkworth ME, Bothmer Rv (2009) Scientific names in the Triticeae. In: Feuillet C, Muehlbauer GJ (eds) Genetics and genomics of the Triticeae. Springer, Heidelberg, pp 3–30
Becher M, Talke IN, Krall L, Krämer U (2004) Cross-species microarray transcript profiling reveals high constitutive expression of metal homeostasis genes in shoots of the zinc hyperaccumulator Arabidopsis halleri. Plant J 37:251–268
Bolot S, Abrouk M, Masood-Quraishi U, Stein N, Messing J, Feuillet C, Salse J (2009) The inner circle’ of the cereal genomes. Curr Opin Plant Biol 12:119–125
Bru D, Martin-Laurent F, Philippot L (2008) Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl Environ Microbiol 74:1660–1663
Bustin S (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23–39
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Campoli C, Matus-Cádiz MA, Pozniak CJ, Cattivelli L, Fowler DB (2009) Comparative expression of Cbf genes in the Triticeae under different acclimation induction temperatures. Mol Genet Genomics 282:141–152
Chain FJJ, Ilieva D, Evans BJ (2008) Single-species microarrays and comparative transcriptomics. PLoS One 3:e3279
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LMG, Romano E, Grossi-de-Sa MF, Vaslin M, Alves-Ferreira M (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Mol Breed 23:607–616
Dheda K, Huggett JF, Chang JS, Kim LU, Bustin SA, Johnson MA, Rook GAW, Zumla A (2005) The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal Biochem 344:141–143
Feuillet C, Salse J (2009) Comparative genomics in the Triticeae. In: Feuillet C, Muehlbauer GJ (eds) Genetics and genomics of the Triticeae. Springer, Heidelberg, pp 3–30
Ghedira R, Papazova N, Vuylsteke M, Ruttink T, Taverniers I, De Loose M (2009) Assessment of primer/template mismatch effects on real-time PCR amplification of target taxa for GMO quantification. J Agric Food Chem 57:9370–9377
Gil-Humanes J, Piston F, Martin A, Barro F (2009) Comparative genomic analysis and expression of the APETALA2-like genes from barley, wheat, and barley-wheat amphiploids. BMC Plant Biol 9:66
Gimenez MJ, Piston F, Martin A, Atienza SG (2010) Application of real-time PCR on the development of molecular markers and to evaluate critical aspects for olive oil authentication. Food Chem 118:482–487
Guenin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot 60:487–493
Gutierrez L, Mauriat M, Pelloux J, Bellini C, Van Wuytswinkel O (2008) Towards a systematic validation of references in real-time RT-PCR. Plant Cell 20:1734–1735
Gutierrez N, Giménez MJ, Palomino C, Avila CM (2010) Assessment of candidate reference genes for expression studies in Vicia faba L. by real-time quantitative PCR. Mol Breed. doi:10.1007/s11032-010-9456-7
He P, Friebe BR, Gill BS, Zhou J-M (2003) Allopolyploidy alters gene expression in the highly stable hexaploid wheat. Plant Mol Biol 52:401–414
Hibbeler S, Scharsack J, Becker S (2008) Housekeeping genes for quantitative expression studies in the three-spined stickleback Gasterosteus aculeatus. BMC Mol Biol 9:18
Hong SY, Seo PJ, Yang MS, Xiang F, Park CM (2008) Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol 8:112
Hu R, Fan C, Li H, Zhang Q, Fu Y-F (2009) Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol Biol 10:93
Huang XQ, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9:868–877
Inostroza L, del Pozo A, Matus I, Castillo D, Hayes P, Machado S, Corey A (2009) Association mapping of plant height, yield, and yield stability in recombinant chromosome substitution lines (RCSLs) using Hordeum vulgare subsp. spontaneum as a source of donor alleles in a Hordeum vulgare subsp. vulgare background. Mol Breed 23:365–376
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lee J, Roche J, Donaghy D, Thrush A, Sathish P (2010) Validation of reference genes for quantitative RT-PCR studies of gene expression in perennial ryegrass (Lolium perenne L.). BMC Mol Biol 11:8
Maccoux L, Clements D, Salway F, Day P (2007) Identification of new reference genes for the normalisation of canine osteoarthritic joint tissue transcripts from microarray data. BMC Mol Biol 8:62
Martin AC, Atienza SG, Ramirez MC, Barro F, Martin A (2008) Male fertility restoration of wheat in Hordeum chilense cytoplasm is associated with 6H(ch)S chromosome addition. Aust J Agric Res 59:206–213
Martin AC, Atienza SG, Ramirez MC, Barro F, Martin A (2009) Chromosome engineering in wheat to restore male fertility in the msH1 CMS system. Mol Breed 24:397–408
Martin AC, Atienza SG, Ramirez MC, Barro F, Martin A (2010) Molecular and cytological characterization of an extra acrocentric chromosome that restores male fertility of wheat in the msH1 CMS system. Theor Appl Genet. doi:10.1007/s00122-010-1374-x
Matus I, Corey A, Filichkin T, Hayes PM, Vales MI, Kling J, Riera-Lizarazu O, Sato K, Powell W, Waugh R (2003) Development and characterization of recombinant chromosome substitution lines (RCSLs) using Hordeum vulgare subsp. spontaneum as a source of donor alleles in a Hordeum vulgare subsp. vulgare background. Genome 46:1010–1023
Murray YHG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4326
Paolacci A, Tanzarella O, Porceddu E, Ciaffi M (2009) Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol 10:11
Poole R, Barker G, Wilson I, Coghill J, Edwards K (2007) Measuring global gene expression in polyploidy; a cautionary note from allohexaploid wheat. Funct Integr Genomic 7:207–219
Quackenbush J (2002) Microarray data normalization and transformation. Nat Genet 32:496–501
Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, Moorman AFM (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45
Salentijn E, Goryunova S, Bas N, van der Meer I, van den Broeck H, Bastien T, Gilissen L, Smulders M (2009) Tetraploid and hexaploid wheat varieties reveal large differences in expression of alpha-gliadins from homoeologous Gli-2 loci. BMC Genomics 10:48
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M (2010) A practical approach to RT-qPCR–Publishing data that conform to the MIQE guidelines. Methods 50:S1–S5
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol 10:71
Uauy C, Distelfeld A, Fahima T, Blechl A, Dubcovsky J (2006) A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314:1298–1301
van Ginkel M, Ogbonnaya F (2007) Novel genetic diversity from synthetic wheats in breeding cultivars for changing production conditions. Field Crops Res 104:86–94
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034–research0034.11
Villegas D, Casadesús J, Atienza SG, Martos V, Maalouf F, Karam F, Aranjuelo I, Nogués S (2010) Tritordeum, wheat and triticale yield components under multi-local mediterranean drought conditions. Field Crops Res 116:68–74
Warburton M, Crossa J, Franco J, Kazi M, Trethowan R, Rajaram S, Pfeiffer W, Zhang P, Dreisigacker S, Ginkel M (2006) Bringing wild relatives back into the family: recovering genetic diversity in CIMMYT improved wheat germplasm. Euphytica 149:289–301
Weber M, Harada E, Vess C, Roepenack-Lahaye Ev, Clemens S (2004) Comparative microarray analysis of Arabidopsis thaliana and Arabidopsis halleri roots identifies nicotianamine synthase, a ZIP transporter and other genes as potential metal hyperaccumulation factors. Plant J 37:269–281
Acknowledgments
This work was funded by projects (to S.G.A.) AGL2008-03720, P09-AGR-93 from Spanish Ministry of Science (MSI) and Innovation, Junta de Andalucía and FEDER. F. Pistón was the recipient of a Juan de la Cierva Research Grant from MSI.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Giménez, M.J., Pistón, F. & Atienza, S.G. Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta 233, 163–173 (2011). https://doi.org/10.1007/s00425-010-1290-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-010-1290-y