
Abstract
Key message
This study proposed the combination of PR26S and PP1 as a good choice for RT-qPCR normalization in pecan under abiotic stress, developing kernels, grafting, and various tissues.
Abstract
Reference gene selection is an essential pre-requisite to generate reliable results in real-time quantitative polymerase chain reaction (RT-qPCR) analysis. However, studies regarding systematic validation of suitable reference genes in pecan are still lacking. In this study, 17 candidate reference genes were selected and evaluated for their expression stabilities in pecan under various experimental conditions, including various tissues, developing kernels, grafting, and two plant tissues (leaves and roots) subjected to three abiotic stresses (salt, drought, and Zn deficiency). The stability of the candidate genes was assessed by geNorm, NormFinder, and BestKeeper, and their outputs were integrated to obtain a final comprehensive rank of stability based on the geometric mean. The results indicated that samples under different experimental conditions possessed their own best reference genes, and using two reference genes for RT-qPCR normalization was recommended for the tested experiments. Overall, the combination of 26S protease regulatory subunit 7A (PR26S) and serine/threonine-protein phosphatase-1 (PP1) was recognized as a good choice for RT-qPCR normalization in pecan across all the treatments. More importantly, the widely used alpha-tubulin (α-TUB), ubiquitin (UBQ), and actin (ACT) genes were not the best suitable reference genes in most of our experiments. Our results will be helpful for future gene-expression studies in pecan.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Real-time quantitative polymerase chain reaction (RT-qPCR) has been widely used in molecular biological studies as a sensitive and accurate technique for exploring gene-expression levels (Bustin 2002; Gachon et al. 2004; Valasek and Repa 2005). Gene expression detected by RT-qPCR could be influenced by several variations, such as initial sample amount, RNA integrity, cDNA quality, and amplification efficiency (Bustin 2002). It is necessary to select appropriate data normalization strategies to correct such biases. Several strategies have been introduced, such as normalization to sample size, RNA quantification, and internal reference genes (Huggett et al. 2005). Currently, the application of internal reference genes is the preferred method (Pfaffl 2001). Ideal reference genes should be expressed at constant levels regardless of the experimental contexts, and the expression levels of these genes could thus reflect the concentration of cDNA in each sample.
Over the past decades, a frequently used reference gene was a housekeeping gene, ubiquitously expressed in all cells, and its product was generally responsible for maintaining of cell structure or basic biochemical metabolism, such as actin (ACT), α-tubulin (α-TUB), β-tubulin (β-TUB), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ubiquitin (UBQ), ribosomal protein, and translation initiation factor (Kim et al. 2003; Nicot et al. 2005; Paolacci et al. 2009; Sun et al. 2009). Housekeeping genes were assumed to be stably expressed among different samples. However, extensive studies suggested that their expression could vary in certain conditions, no universally applicable reference genes existed (Bustin et al. 2005; Niu et al. 2017; Zhu et al. 2012). An inappropriate reference gene for RT-qPCR data normalization can lead to biased estimation of gene-expression patterns. Thus, the selection of suitable reference genes with relatively invariant expression is necessary for specific experimental condition. With the aim of suitable reference gene selection, a number of researches have been conducted in various species, such as Juglans regia (Zhou et al. 2018), Artemisia sphaerocephala (Hu et al. 2018), Taxodium ‘Zhongshanshan’ (Wang et al. 2017), Coffea arabica (Barsalobres-Cavallari et al. 2009), and Prunus persica (Tong et al. 2009).
Pecan (Carya illinoinensis), which belongs to the family Juglandaceae, is native to the United States and Mexico (Sparks 2002). Due to increasing interest in consumption of pecan nuts, this species has been cultivated in several countries, including China, South America, Australia, Argentina, Peru, and so on (Bilharva et al. 2018). Pecan has been introduced to China for more than 100 years. For a long time, a poor graft success was one of the factors that hampered the cultivation of pecan in China (Zhang et al. 2015). After years of efforts, researchers have made a technical breakthrough in improving the survival rate of grafting (Mo et al. 2017), which greatly facilitates the development of pecan planting in China. To date, however, the graft survival rates for some pecan cultivars are still seldom satisfied, and a relatively high rate of successful grafting is only observed with the ‘Pawnee’ cultivar (Mo et al. 2018a). Because pecan nuts have a large amount of oil (accounting for about 70% of the dry weight), it can be cultivated as a woody oil plant. This is also one of the reasons for the rapid development of pecan planting in China. Understanding the functional genes involved in the de novo biosynthesis of fatty acid in pecan will be helpful for future molecular breeding. Plants grown in non-native areas often encounter various adversities. Pecan was found to be sensitive to multiple stresses, such as drought, zinc deficiency, and salinity. These abiotic stresses could adversely affect the tree vigor and fruit yields of pecan (Miyamoto and Nesbitt 2011; Othman et al. 2014). For the healthy development of pecan cultivation, it is necessary to understand the molecular mechanisms underlying successful grafting, fatty acid biosynthesis, and abiotic stress response. Quantification assays by RT-qPCR are an important tool to study the molecular mechanisms of pecan in response to various experimental conditions. To the best of our knowledge, no systematic surveys have been conducted to identify suitable reference genes in pecan.
To identify suitable reference genes for normalization of RT-qPCR data in pecan, 17 candidate reference genes were selected and evaluated for their expression stabilities under a series of experimental conditions. To validate the reliability of identified reference genes, the expression trends of a cinnamyl alcohol dehydrogenase (CAD) gene and a stearoyl-ACP desaturase (SAD) gene were separately examined under salt stress and during kernel development.
Materials and methods
Plant materials and treatments
The experiment was performed at the Institute of Botany, Jiangsu province and Chinese Academy of Sciences. For abiotic treatments, seedlings of pecan were first grown under natural temperature and light conditions. At four-true-leaf stage, seedlings with uniform growth pattern were transferred to greenhouse and cultured in 1/4 Hoagland’s solution (air was continually supplied with an aquarium pump) for 3 days, followed by 1/2 Hoagland’s solution for 4 days. Then, the plants cultured in 1/2 Hoagland’s solution were subjected to multiple abiotic stresses. For drought or salt treatment, 10% polyethylene glycol (PEG) 6000 (drought) or 3‰ (w/v) NaCl (salt) were added to the solution. For zinc deficiency tolerance, no zinc was included in the nutrient solution. At 0, 6, 12, and 24 h of those treatments, roots and leaves were harvested. All samples with three biological replicates were immediately frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
Pecan cultivar ‘Mahan’ was chosen for the sampling of various tissues (leaves, stems, staminate flowers, pistillate flowers, and kernels) and kernels at different development stage. The developing kernels were collected 120, 140, and 170 days after full blossoming (DAF). For grafting, ‘Pawnee’ cultivar was used as scion and 2-year-old seedling was used as rootstock. Grafting was conducted in August using patch budding under field condition, and the scions and rootstocks were detached and collected 0, 3, 7, and 14 days after grafting.
RNA isolation and cDNA synthesis
Total RNA was extracted using the Universal Plant RNA Kit (BioTeke, Beijing, China), and the containing DNase I was used to remove genomic DNA following the manufacturer's instructions. RNA purity and concentration were measured with a Nanodrop 1000 spectrophotometer (NanoVue™ plus, Wilmington, USA). The integrity of RNA was further assessed by 1% agarose gel electrophoresis. Only RNA samples with A260/A280 ratios ranging from 1.8 to 2.2 and A260/A230 ratios larger than 2.0, as well as 28S:18S rRNA band intensity ratios of around 2:1 were retained. 0.9 μg of total RNA was reverse-transcribed using the PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, Kyoto, Japan). cDNA was diluted to the final concentration of 200 ng/μl for the subsequent analysis.
Selection of candidate reference genes
For reference selection, previously published transcriptome data sets (Jia et al. 2018; Mattison et al. 2017; Mo et al. 2018b) were downloaded, and then, the read counts of each gene were converted to fragments per kilobase of transcript per million mapped reads (FPKM) values. To evaluate the expression stability of each gene, maximum fold change (MFC, the ratio between the highest and lowest FPKM values) was calculated. Based on the previous literatures (Czechowski et al. 2005; Zhu et al. 2012), reference genes that have been validated were searched for their corresponding pecan orthologs. We selected those orthologs that have appropriate expression levels (FPKM > 20) and low MFC (MFC < 1.5) as candidate reference genes. Totally, 17 candidate reference genes in pecan were selected, comprising alpha-tubulin (α-TUB), beta-tubulin (β-TUB), Actin (ACT), ADP-ribosylation factor (ADP-RF), clathrin adaptor complex (CAC), cyclophilin (CYP), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), RNA helicase family (HEL), serine/threonine-protein phosphatase-1 (PP1), serine/threonine-protein phosphatase 2A (PP2A), 26S protease regulatory subunit 7A (PR26S), polypyrimidine tract-binding protein 1 (PTBP1), 26S proteasome non-ATPase (RPN6), translation factor (TLF), TIP41-like family protein (TIP41), ubiquitin (UBQ), and 60S ribosomal protein L22 (60S).
Primer design and validation
Since the release of pecan genome (Huang et al. 2019), intron-spanning primers could be designed to eliminate any possible amplification of contaminating genomic DNA. The criteria for primer design with Beacon Designer software (version 8) was the following: primer length of 18–24 bp, GC content of 40–60%, melting temperature (Tm) of 50–60 °C, and amplicon length of 75–200 bp. Generally, two or more primer pairs were designed for each gene. All primer pairs were checked by routine PCR with cDNA and DNA as templates. Only the primer pairs amplified a single band of expected size with cDNA but not with DNA templates were used for subsequent analysis.
RT-qPCR analysis
RT-qPCR was carried out in 96-well plates, and performed on an ABI 7500 Real-Time PCR System (Thermo Fisher Scientific, Inc., Waltham, USA). The volume of each reaction mix was 20 μl, containing 2 μl (400 ng) cDNA, 10 μl SYBR Premix Ex Taq™ (Takara), 0.6 μl each primer (10 μM), 0.4 μl ROX Reference Dye II, and 6.4 μl ddH2O. PCR amplification of each sample was performed in three experimental triplicates and three biological duplicates. A non-template control was also included in each plate for each primer pair. The reaction condition was as follows: 95 °C for 3 min, then 40 cycles of 95 °C for 10 s, and 60 °C for 30 s. A melting-curve analysis was also included to confirm the amplicon specificity of each gene. Standard curves with dilution series of mixed cDNA (10–1, 10–2, 10–3, 10–4, and 10–5) as templates were developed to calculate the amplification efficiency (E) of each primer pair by the LinRegPCR program (Ruijter et al. 2009).
Data analysis
Three computer programs were used to evaluate the expression stabilities of candidate reference genes under diverse experimental conditions: geNorm (Vandesompele et al. 2002), NormFinder (Andersen et al. 2004), and BestKeeper (Pfaffl et al. 2004). The final comprehensive ranking was determined by calculating the geometric means of the rank values obtained from each program, as suggested by RefFinder (Ma et al. 2016). For geNorm and NormFinder analysis, the raw cycle threshold (Ct) values were transformed into relative quantities (Q value) with the formula \(Q = 2^{{ - \Delta C_{{\text{t}}} }}\), in which ΔCt was each corresponding Ct value subtracting minimum Ct value. Then, the relative expression levels were imported into geNorm and NormFinder to calculate gene-expression stability. geNorm computes the expression stability of all the reference genes using M value. The M value has a negative relation with expression stability; the cut-off for estimating a gene as stable is proposed as M < 1.5 (Vandesompele et al. 2002). geNorm could also be applied to estimate the suitable number of reference genes for accurate normalization based on pairwise variation (Vn/n+1). This algorithm proposes 0.15 as the threshold of Vn/n+1. When Vn/n+1 < 0.15, it means that the number of n genes is sufficient for normalization without applying an additional gene (Vandesompele et al. 2002). The NormFinder program calculates a stability value (SV) for assessing expression variation. A lower SV indicates a less variable expression. For BestKeeper analysis, raw Ct values were imported to calculate the coefficient of variance (CV) and the standard deviation (SD) of the Ct values. The smaller the SD value is, the greater the stability of reference gene expression. Genes with SD > 1 are considered to be unacceptable (Zhu et al. 2013). The final comprehensive ranking was based on the geometric mean of each gene. A smaller geometric mean value means a higher ranking of the expression stability.
Validation of reference genes
CAD and SAD from pecan were cloned based on transcriptome data and were used as targets to examine the reliability of RT-qPCR data with \(2^{{ - \Delta \Delta C_{{\text{t}}} }}\) method. According to the cloned sequences, primer pairs of CAD (forward: 5′-GAGGATGAGGCAATCAACAG-3′, reverse: 5′-GGCTTATCAGGCAAACCGA-3′) and SAD (forward: 5′-AAGGATTAGGAAGTTACAG-3′, reverse: 5′-ATTTGACCTCCCTATTGA-3′) were designed. The relative expression levels of CAD under salt stress and SAD in different development-stage kernels were quantified and normalized to the most stable and the least stable reference genes.
Result
Primer specificity and amplification efficiency
Primers for the 17 reference genes were designed and tested by regular PCR (Online Resource 1, Supplementary Table 1 and Fig. 1). Agarose gel electrophoresis revealed that single products with the correct size were obtained with cDNA as templates, while either no bands or bands longer than the expected size were observed when tested on genomic DNA (Online Resource 1, Supplementary Fig. 1), indicating that the designed primers were intron spanning. Melting curve analysis by RT-qPCR further confirmed the specificity of amplifications (Online Resource 1, Supplementary Fig. 2). No signals were detected in no-template controls, suggesting the absences of primer dimmers and non-specific PCR products for each primer pairs. Amplification efficiencies (E) of the reference genes varied from 108.0 to 113.7%, which were within the acceptable range of 80–120% (Online Resource 1, Supplementary Table 1 and Fig. 3).

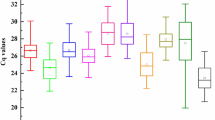
Expression levels of 17 candidate reference genes in all sample. Mean Ct values for each treatment are used for drawing the boxplot. The lower and upper ends of boxplot indicate the first and the third quartiles. The lower and upper fences indicate the minimum and maximum Ct values, respectively. The line across the box depicts the median. Points outside the fences are considered to be outliers
Expression profiles of reference genes
The raw cycle threshold (Ct) values extracted following RT-qPCR provide an overview of the variation in gene expression across all experimental samples. The lower Ct values reflect higher levels of mRNA abundance and vice versa. For the 17 reference genes, Ct values were generally ranged from 21.10 and 28.97 (Fig. 1), which were within the proposed range of 15–30 (Xu et al. 2015). GAPDH displayed the highest level of expression, with the Ct values in the range of 18.68–24.41, while HEL was the least expressed, with the Ct values larger than 23 across all samples. Ct values were most concentrated for 60S in the boxplot, suggesting the least variation in gene expression. Nevertheless, a relatively wide expression range (Ct values ranging from 20.12 to 25.33 cycles) of 60S indicated that no candidate reference genes had constant expression under varying conditions.
Evaluation of gene-expression stability
Generally, the stability rankings obtained by three different programs were different in pecan under three abiotic stresses. For example, GAPDH and ADP-RF were ranked as the most stable reference genes in salt-treated leaves, as assessed by geNorm analysis. However, in NormFinder analysis, β-TUB and PP1 were ranked at the top two positions. According to BestKeeper, CYP and TIP41 were identified as the two best references. To obtain a final comprehensive ranking, the geometric mean of each gene was calculated. Comprehensive ranking analysis indicated that β-TUB and ADP-RF were the two most stable reference genes in salt-treated leaves; 60S and PR26S in salt-treated roots; PR26S and GAPDH in PEG-treated leaves; PR26S and PP2A in PEG-treated roots; PR26S and PTBP1 in Zn deficiency-treated leaves; PP2A and PP1 in Zn deficiency-treated roots. Relatively, RPN6 was the least stable gene in salt-treated leaves, PEG-treated leaves, and Zn deficiency-treated leaves; ACT in salt-treated roots; GAPDH in PEG-treated roots and Zn deficiency-treated roots (Online Resource 1, Supplementary Table 2).
To find the overall applicable reference genes that might be used for multiple stresses, the expression data generated from salt, PEG, and Zn deficiency treatments were pooled together. Integrated analysis ranked 60S and PR26S as the top two reference genes, and RPN6 as the most unstable one under total abiotic stresses (Online Resource 1, Supplementary Table 3). For the kernels at different developmental stages, PTBP1 and 60S were identified as the two best reference genes, with RPN6 being the least stable one (Online Resource 1, Supplementary Table 4). For grafting, PR26S and PP1 were determined to be the most appropriate reference genes, while TIP41 was the least reliable reference gene (Online Resource 1, Supplementary Table 5). For the different tissues, RPN6 and PR26S were proposed as the two most appropriate reference genes, with α-TUB being the worst performance one (Online Resource 1, Supplementary Table 6).
As an intention to identify the generally applicable reference genes that would be used extensively for multiple experiments in pecan, all the expression data sets were pooled together. Comprehensive analysis showed that PR26S and PP1 were the two most stable reference genes in total samples, and α-TUB was the gene with the most variable expression (Table 1). The M values and SD values of PR26S and PP1 were separately below the proposed values of 1.5 and 1 under all the experimental conditions (Online Resource 1, Supplementary Tables 2, 3, 4, 5, and 6). Additionally, these two genes were always the top five ranked genes, expect for PP1, which ranked eleventh across different tissues (Online Resource 1, Supplementary Table 6).
To improve the accuracy of gene-expression analysis, we used geNorm to determine the optimal number of reference genes, with a proposed value of Vn/n+1 < 0.15. As shown in Fig. 2, four genes (V4/5 < 0.15) would be optimal for normalizing in total samples, three genes (V3/4 < 0.15) were essential for normalization under total abiotic stresses, and two genes (V2/3 < 0.15) were sufficient for individual treatments, including different tissues, grafting, developmental stages, leaves, and roots subjected to salt, drought, and Zn deficiency.

Pairwise variation calculated by geNorm to determine optimal number of reference genes. (a) Total samples; (b) different tissues; (c) grafting; (d) developing kernels; (e) total abiotic stresses; (f) salt-treated leaves; (g) salt-treated roots; (h) PEG-treated leaves; (i) PEG-treated roots; (j) Zn deficiency-treated leaves; (k) Zn deficiency-treated roots. Arrow in each treatment indicates the optimal number of genes for normalization
Reference gene validation
To illustrate the reliability of the selected genes, the expression patterns of CAD and SAD genes were used to normalize the results obtained under salt stress and during kernel development, respectively. According to the comprehensive ranking analysis, two of the most stable reference gene (β-TUB and ADP-RF for salt-treated leaves, 60S and PR26S for salt-treated roots, 60S and PTBP1 for developing kernels) and the least stable one (PRN6 for salt-treated leaves and developing kernels, ACT for salt-treated roots) were used for normalization. Results indicated that when using stable reference genes and their combination for normalization, similar expression profiles could be detected (Fig. 3). Contrastingly, when the most variable genes were used as reference genes, the relative expression values of CAD and SAD were both underestimated (Fig. 3).

Relative expression of CAD and SAD using validated reference genes for normalization. DAF days after full blossoming. Error bars show the standard error calculated from three biological replicates
Discussion
RT-qPCR has been considered to be the most commonly used technique for gene-expression analysis, due to its high specificity, accuracy, and sensitivity (Derveaux et al. 2010). To accurately normalize RT-qPCR data, optimal reference gene selection is critical. However, using previously published reference genes without verification may lead to inappropriate results (Ma et al. 2016). Thus, it is necessary to establish the expression stability of candidate reference genes in pecan under diverse experimental condition.
Primer design is of critical importance for RT-qPCR, and a perfect primer design should be intron spanning to effectively remove the contaminated DNA in cDNA samples (Dong et al. 2019). In the present study, at least one intron-spanning primer of a pair was designed based on the newly published pecan genome (Huang et al. 2019). Several primer pairs amplified longer fragments than the expected bands when DNA was used as a template, which might be due to the inclusion of intronic regions in the PCR products. However, for most of the primer pairs, no amplification products were obtained in DNA samples. The reason for this may be that intron-spanning primers extended from the upstream exon into the following downstream exon with long enough nucleotides (at least four), resulting in nucleotide mismatch when binding to DNA sequences. The developed primer pairs in this research could specifically amplify their corresponding reference genes on cDNA templates, and would be employed for future RT-qPCR analysis.
It is recommended to use various programs to minimize bias in the determination of gene-expression stability (Tong et al. 2009). Several programs have been developed to evaluate the expression stability of reference genes (Andersen et al. 2004; Garcıa-Vallejo et al. 2004; Haller et al. 2004; Pfaffl et al. 2004; Vandesompele et al. 2002). In our study, three widely used programs were adopted to estimate the expression stability of candidate reference genes, including geNorm, NormFinder, and BestKeeper. The previous reports have observed that different programs would generate different results (Li et al. 2015; Ma et al. 2016; Wang et al. 2013). In our research, differences in determining the stability rankings of reference genes were found, as well. This divergence may be attributed to the inconsistencies in mathematical approaches and analytical procedures for these programs.
Comprehensive ranking analysis revealed that samples under different experimental contexts in pecan possessed their own best reference genes. PR26S and PP1 were the top two most stable reference genes when all total samples were tested. Considering the low expression variations (M values < 1.5 and SD values < 1) and nearly all the top five ranked positions for these two reference genes under the tested experiments, PR26S and PP1 had the potential to be widely applicable in various RT-qPCR analysis of pecan. There is a strong consensus that increasing the number of reference genes could improve the accuracy of gene-expression analysis. It was reported that using two of the most stable reference genes was a valid normalization strategy for most case (Chen et al. 2011), and our study also indicated that using two reference genes would be sufficient for reliable normalization under all the examined treatments, except for the synthetic treatments (including total abiotic stresses and total samples). Therefore, a combination of PR26S and PP1 would be a good choice for accurate normalization in pecan.
PR26S encodes a 26S proteasome that plays a fundamental role in intracellular protein degradation (Jin et al. 2006). PP1 encodes a serine/threonine (Ser/Thr) protein phosphatase catalytic subunit, and the Ser/Thr protein phosphatase is implicated in cell growth and cellular stress responses (Farkas et al. 2007). The stable expressions of PR26S and PP1 in most of our analysis may be due to their essential roles in the normal and stressed cells. PR26S was also identified as one of the most stable genes in Populus bejingensis under biotic stress (Zhao et al. 2017) and in Solanum tuberosum under abiotic stress (Castro-Quezada et al. 2013).
Interestingly, the commonly used α-TUB, UBQ, and ACT genes were not the best choice in pecan under all the tested conditions. For those popular references, they performed differently in different plant species. α-TUB emerged as unstable reference gene in Poa pratensis under abiotic stress (Niu et al. 2017), which was different from the report made in Lentinula edodes (Luo et al. 2019). UBQ has been reported to perform unsatisfactorily in Litchi chinensis (Zhong et al. 2011); however, it was determined to be stably expressed in Paeonia ostia (Li et al. 2019). ACT exhibited poor expression stability in Arabidopsis thaliana (Czechowski et al. 2005), G. max (Hu et al. 2009), and Caragana intermedia (Zhu et al. 2013). In contrast, it has been evaluated as the most stable gene in Lilium davidii (Li et al. 2015) and Apium graveolens (Feng et al. 2019). Our results emphasized the necessity of systematic evaluation of reference genes based on the experimental conditions examined.
The reliability of reference genes was further validated by determining the expression levels of CAD and SAD. CAD protein is a key enzyme that catalyzes the last step of lignin precursor synthesis, and the produced lignin has important functions in response to abiotic stresses (Cheng et al. 2013; Liu et al. 2018). In Cucumis melo, CAD4 exhibited a fast induction and then declined in roots under 50 mM NaCl treatment, while it was decreased slightly and then induced significantly in leaves (Liu et al. 2018). The expressions of CAD in our study exhibited similar patterns to that in Cucumis melo when using the most stable reference genes for normalization. When the least stable reference gene was used, CAD was down-regulated throughout the entire treatment period in leaves, which might be unreliable. SAD protein is involved in the synthesis of polyunsaturated fatty acids (Huang et al. 2017). Pecan kernel is rich in fatty acid, of which above 90% is unsaturated fatty acids, suggesting that SAD might be involved in kernel development. In our study, the expression levels of SAD were similar to the results detected by transcriptome analysis (Huang et al. 2017; Jia et al. 2018) only when normalizations were made by the most stable reference genes. These results clearly indicated that inappropriate selection of reference gene would reduce precision.
Conclusion
In the present study, 17 candidate reference genes were evaluated for the normalization of RT-qPCR in pecan subjected to a series of experimental conditions. The stability of the genes was assessed by geNorm, NormFinder, and BestKeeper, and their results were further merged into a comprehensive ranking based on the geometric mean. The best reference genes for normalization should be chosen according to the experimental conditions. Using two reference genes would be a suitable normalization strategy for the tested experiments. Overall, the combination of PR26S and PP1 was a good choice for RT-qPCR normalization in pecan. More importantly, the commonly used α-TUB, UBQ, and ACT genes were not the best suitable reference genes in most of our analysis.
Abbreviations
- α-TUB:
-
Alpha-tubulin
- β-TUB:
-
Beta-tubulin
- ACT:
-
Actin
- ADP-RF:
-
ADP-ribosylation factor
- CAC:
-
Clathrin adaptor complex
- CAD:
-
Cinnamyl alcohol dehydrogenase
- CYP:
-
Cyclophilin
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- HEL:
-
RNA helicase family
- PEG:
-
Polyethylene glycol
- PP1:
-
Serine/threonine-protein phosphatase-1
- PP2A:
-
Serine/threonine-protein phosphatase 2A
- PR26S:
-
26S protease regulatory subunit 7A
- PTBP1:
-
Polypyrimidine tract-binding protein 1
- RPN6:
-
26S proteasome non-ATPase
- SAD:
-
Stearoyl-ACP desaturase
- TLF:
-
Translation factor
- TIP41:
-
TIP41-like family protein
- UBQ:
-
Ubiquitin
- 60S:
-
60S ribosomal protein L22
References
Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Barsalobres-Cavallari CF, Severino FE, Maluf MP, Maia IG (2009) Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC Mol Biol 10:1
Bilharva MG, Martins CR, Hamann JJ, Fronza D, De Marco R, Malgarim MB (2018) Pecan: from Research to the Brazilian Reality. J Exp Agric Int 23:1–16
Bustin S (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23–39
Bustin S, Benes V, Nolan T, Pfaffl M (2005) Quantitative real-time RT-PCR—a perspective. J Mol Endocrinol 34:597–601
Castro-Quezada P, Aarrouf J, Claverie M, Favery B, Mugniéry D, Lefebvre V, Caromel B (2013) Identification of reference genes for normalizing RNA expression in potato roots infected with cyst nematodes. Plant Mol Biol Rep 31:936–945
Chen L, Zhong HY, Kuang JF, Li JG, Lu WJ, Chen JY (2011) Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta 234:377
Cheng H, Li L, Xu F, Cheng S, Cao F, Wang Y, Yuan H, Jiang D, Wu C (2013) Expression patterns of a cinnamyl alcohol dehydrogenase gene involved in lignin biosynthesis and environmental stress in Ginkgo biloba. Mol Biol Rep 40:707–721
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
Derveaux S, Vandesompele J, Hellemans J (2010) How to do successful gene expression analysis using real-time PCR. Methods 50:227–230
Dong Z, Chen P, Zhang N, Huang S, Zhang H, Wang S, Li X, Guo Y, Wang Z (2019) Evaluation of reference genes for quantitative real-time PCR analysis of gene expression in Hainan medaka (Oryzias curvinotus). Gene Rep 14:94–99
Farkas I, Dombradi V, Miskei M, Szabados L, Koncz C (2007) Arabidopsis PPP family of serine/threonine phosphatases. Trends Plant Sci 12:169–176
Feng K, Liu JX, Xing GM, Sun S, Li S, Duan AQ, Wang F, Li MY, Xu ZS, Xiong AS (2019) Selection of appropriate reference genes for RT-qPCR analysis under abiotic stress and hormone treatment in celery. PeerJ 7:e7925
Gachon C, Mingam A, Charrier B (2004) Real-time PCR: what relevance to plant studies? J Exp Bot 55:1445–1454
Garcıa-Vallejo J, Van Het Hof B, Robben J, Van Wijk J, Van Die I, Joziasse D, Van Dijk W (2004) Approach for defining endogenous reference genes in gene expression experiments. Anal Biochem 329:293–299
Haller F, Kulle B, Schwager S, Gunawan B, von Heydebreck A, Sültmann H, Füzesi L (2004) Equivalence test in quantitative reverse transcription polymerase chain reaction: confirmation of reference genes suitable for normalization. Anal Biochem 335:1–9
Hu R, Fan C, Li H, Zhang Q, Fu YF (2009) Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol Biol 10:93
Hu X, Zhang L, Nan S, Miao X, Yang P, Duan G, Fu H (2018) Selection and validation of reference genes for quantitative real-time PCR in Artemisia sphaerocephala based on transcriptome sequence data. Gene 657:39–49
Huang R, Huang Y, Sun Z, Huang J, Wang Z (2017) Transcriptome analysis of genes involved in lipid biosynthesis in the developing embryo of pecan (Carya illinoinensis). J Agric Food Chem 65:4223–4236
Huang Y, Xiao L, Zhang Z, Zhang R, Wang Z, Huang C, Huang R, Luan Y, Fan T, Wang J (2019) The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition. GigaScience 8:giz036
Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6:279
Jia X, Li M, Luo H, Zhai M, Guo Z, Li Y, Qiao Y, Wang L (2018) Transcriptome survey reveals candidate genes involved in lipid metabolism of Carya illinoinensis. Int J Agric Biol 20:991–1004
Jin H, Li S, Villegas A (2006) Down-regulation of the 26S proteasome subunit RPN9 inhibits viral systemic transport and alters plant vascular development. Plant Physiol 142:651–661
Kim BR, Nam HY, Kim SU, Kim SI, Chang YJ (2003) Normalization of reverse transcription quantitative-PCR with housekeeping genes in rice. Biotechnol Lett 25:1869–1872
Li X, Cheng J, Zhang J, Ja TDS, Wang C, Sun H (2015) Validation of reference genes for accurate normalization of gene expression in Lilium davidii var. unicolor for real time quantitative PCR. PLoS One 10:e0141323
Li C, Hu L, Wang X, Liu H, Tian H, Wang J (2019) Selection of reliable reference genes for gene expression analysis in seeds at different developmental stages and across various tissues in Paeonia ostii. Mol Biol Rep 46:6003–6011
Liu W, Jin Y, Li M, Dong L, Guo D, Lu C, Qi H (2018) Analysis of CmCADs and three lignifying enzymes in oriental melon (‘CaiHong7’) seedlings in response to three abiotic stresses. Sci Hortic 237:257–268
Luo Y, Wang G, Wang C, Gong Y, Bian Y, Zhou Y (2019) Selection and validation of reference genes for qRT-PCR in Lentinula edodes under different experimental conditions. Genes 10:647
Ma R, Xu S, Zhao Y, Xia B, Wang R (2016) Selection and validation of appropriate reference genes for quantitative real-time PCR analysis of gene expression in Lycoris aurea. Front Plant Sci 7:536
Mattison CP, Rai R, Settlage RE, Hinchliffe DJ, Madison C, Bland JM, Brashear S, Graham CJ, Tarver MR, Florane C (2017) RNA-Seq analysis of developing pecan (Carya illinoinensis) embryos reveals parallel expression patterns among allergen and lipid metabolism genes. J Agric Food Chem 65:1443–1455
Miyamoto S, Nesbitt M (2011) Effectiveness of soil salinity management practices in basin-irrigated pecan orchards. HortTechnology 21:569–576
Mo Z, He H, Su W, Peng F (2017) Analysis of differentially accumulated proteins associated with graft union formation in pecan ( Carya illinoensis ). Sci Hortic 224:126–134
Mo Z, Feng G, Su W, Liu Z, Peng F (2018a) Identification of miRNAs Associated with Graft Union Development in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests 9:472
Mo Z, Feng G, Su W, Liu Z, Peng F (2018b) Transcriptomic analysis provides insights into grafting union development in pecan (Carya illinoinensis). Genes 9:71
Nicot N, Hausman JF, Hoffmann L, Evers D (2005) Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot 56:2907–2914
Niu K, Shi Y, Ma H (2017) Selection of candidate reference genes for gene expression analysis in Kentucky Bluegrass (Poa pratensis L.) under abiotic stress. Front Plant Sci 8:193
Othman Y, VanLeeuwen D, Heerema R, Hilaire RS (2014) Midday stem water potential values needed to maintain photosynthesis and leaf gas exchange established for pecan. J Am Soc Hort Sci 139:537–546
Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M (2009) Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol 10:11
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res 29:e45–e45
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Ruijter J, Ramakers C, Hoogaars W, Karlen Y, Bakker O, Van den Hoff M, Moorman A (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45–e45
Sparks D (2002) Rainfall governs pecan stand homogeneity in native, wild habitats. J Am Soc Hortic Sci 127:860–868
Sun HF, Meng YP, Cui GM, Cao QF, Li J, Liang AH (2009) Selection of housekeeping genes for gene expression studies on the development of fruit bearing shoots in Chinese jujube (Ziziphus jujube Mill.). Mol Biol Rep 36:2183
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol 10:71
Valasek MA, Repa JJ (2005) The power of real-time PCR. Adv Physiol Educ 29:151–159
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034-1
Wang L, Wang Y, Zhou P (2013) Validation of reference genes for quantitative real-time PCR during Chinese wolfberry fruit development. Plant Physiol Biochem 70:304–310
Wang Z, Gu C, Xuan L, Hua J, Shi Q, Fan W, Yin Y, Yu F (2017) Identification of suitable reference genes in Taxodium ‘Zhongshanshan’under abiotic stresses. Trees 31:1519–1530
Xu Y, Li H, Li X, Lin J, Wang Z, Yang Q, Chang Y (2015) Systematic selection and validation of appropriate reference genes for gene expression studies by quantitative real-time PCR in pear. Acta Physiol Plant 37:40
Zhang R, Peng F, Li Y (2015) Pecan production in China. Sci Hortic 197:719–727
Zhao J, Yang F, Feng J, Wang Y, Lachenbruch B, Wang J, Wan X (2017) Genome-wide constitutively expressed gene analysis and new reference gene selection based on transcriptome data: a case study from poplar/canker disease interaction. Front Plant Sci 8:1876
Zhong HY, Chen JW, Li CQ, Chen L, Wu JY, Chen JY, Lu WJ, Li JG (2011) Selection of reliable reference genes for expression studies by reverse transcription quantitative real-time PCR in litchi under different experimental conditions. Plant Cell Rep 30:641–653
Zhou L, Niu J, Quan S (2018) Identification of appropriate reference genes for RT-qPCR analysis in Juglans regia L. PLoS ONE 13:e0209424
Zhu X, Li X, Chen W, Chen J, Lu W, Chen L, Fu D (2012) Evaluation of new reference genes in papaya for accurate transcript normalization under different experimental conditions. PLoS ONE 7:e44405
Zhu J, Zhang L, Li W, Han S, Yang W, Qi L (2013) Reference gene selection for quantitative real-time PCR normalization in Caragana intermedia under different abiotic stress conditions. PLoS ONE 8:e53196
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 31901347), the Basic Talent Project of Institute of Botany, Jiangsu Province and Chinese Academy of Science (JSPKLB202002), and Scientific Research Staring Foundation for the Doctor of Institute of Botany, Jiangsu Province and Chinese Academy of Science.
Author information
Authors and Affiliations
Contributions
JX and ZG designed the research; ZM, YC, WL, and XJ performed the experiments and data analysis; ZM wrote the manuscript; MZ and YL prepared the materials. All authors both read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Francisco M. Cánovas .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mo, Z., Chen, Y., Lou, W. et al. Identification of suitable reference genes for normalization of real-time quantitative PCR data in pecan (Carya illinoinensis). Trees 34, 1233–1241 (2020). https://doi.org/10.1007/s00468-020-01993-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-020-01993-w