
Abstract
Liver fibrosis results from chronic damage to the liver in conjunction with various pathways and is mediated by a complex microenvironment. Based on clinical observations, it is now evident that fibrosis is a dynamic, bidirectional process with an inherent capacity for recovery and remodeling. The major mechanisms involved in liver fibrosis include the repetitive injury of hepatocytes, the activation of the inflammatory response after injury stimulation, and the activation and proliferation of hepatic stellate cells (HSCs), which represents the major extracellular matrix (ECM)-producing cells, stimulated by hepatocyte injury and inflammation. The microenvironment in the liver is synergistically regulated abnormal ECM deposition, scar formation, angiogenesis, and fibrogenesis. Moreover, recent studies have clarified novel mechanism in fibrosis such as epigenetic regulation of HSCs, the leptin and PPARγ pathways, the coagulation system, and even autophagy. Uncovering the mechanisms of liver fibrogenesis provides a basis to develop potential therapies to reverse and treat the fibrotic response, thereby improving the outcomes of patients with chronic liver disease. Although both scientific and clinical challenges remain, emerging studies attempt to reveal the ideal anti-fibrotic drug that could be easily delivered to the liver with high specificity and low toxicity. This review highlights the mechanisms, including novel pathways underlying fibrogenesis that may be translated into preventive and treatment strategies, reviews both current and novel agents that target specific pathways or multiple targets, and discusses novel drug delivery systems such as nanotechnology that can be applied in the treatment of liver fibrosis. In addition, we also discuss some current treatment strategies that are being applied in animal models and in clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A common pathological feature of chronic liver disease is fibrosis, which results from unregulated wound healing and is characterized by the progressive replacement of functional hepatic tissue with highly cross-linked collagen I/III-rich extracellular matrix (ECM) (Ellis and Mann 2012). Fibrosis results in a liver that is more resistant to subsequent injury by type I collagen, which is predominant in the fibrotic scar and protects hepatocytes against various toxic stimuli (Pellicoro et al. 2014). However, ECM protein accumulation distorts the hepatic architecture by forming a fibrous scar, and then nodules of regenerating hepatocytes develop; these features define cirrhosis. Cirrhosis produces hepatic dysfunction and increased intrahepatic resistance to blood flow, which results in hepatic insufficiency and portal hypertension. Fibrosis is also considered a precancerous state that provides the proper microenvironment for tumor development (Aravalli et al. 2013; Ellis and Mann 2012). The main causes of liver fibrosis include chronic viral infection, alcohol abuse, fatty liver, biliary track disease, autoimmune disease, metabolic etiologies, iron or copper overload, non-alcoholic steatohepatitis (NASH), and toxicant exposure (Gonsebatt et al. 2007; Loumbourdis 2005; Rockey 2013).
Studies of the mechanisms underlying fibrogenesis (excess ECM synthesis and deposition) using in vitro and in vivo models had noted several potential therapeutic approaches. Although therapies targeted to underlying disease processes such as viral infections have proven to be effective in reducing or reversing fibrosis, no drug has yet emerged as an effective anti-fibrotic agent in humans. Thus, it is necessary to provide an update on our understanding of fibrotic mechanisms as well as to acknowledge the evolving challenges faced in translating these discoveries into new treatment options (Rockey 2013). Here, we highlight the possible anti-fibrotic therapies for liver fibrosis used in the past 5 years in animal models and in human clinical trials based on the fundamental understanding of the mechanisms of liver fibrosis.
Liver fibrogenic process and mechanisms
Four prevailing mechanisms have been implicated in liver fibrogenesis. (1) The early and critical event in the fibrogenic response is that injury to hepatocytes and/or cholangiocytes stimulates the injury response (Rockey 2013). (2) Repetitive injury causes inflammation, which initiates the anti-fibrinolytic coagulation cascade. Leukocytes are then recruited to the injury site to phagocytose dead or apoptotic cells and to amplify the inflammatory response by generating pro-inflammatory cytokines. (3) Hepatic stellate cells (HSCs) are activated and transformed to ECM-producing myofibroblasts via the stimulation of mediators. In addition, the phagocytosis of hepatocytes or HSCs by lymphocytes directly triggers their fibrogenic activation. HSCs induce fibrosis by increasing their cell number and via ECM production, which is the central event in the hepatic fibrogenesis (Iredale 2007). (4) The complicated interaction in the microenvironment leads to the progression of fibrosis, including HSCs and myofibroblast gene expression regulation to control activation, proliferation, epigenetic regulation, signaling, microRNAs production, and angiogenesis. The detailed molecular mechanisms of liver fibrogenesis are discussed in some remarkable reviews written by Nova et al. (2014), Pellicore et al. (2014), and Luedde et al. (2014). The mechanisms of liver fibrogenesis are briefly recalled below.
Injury to hepatocytes and/or cholangiocytes
Chronic liver injury leads to hepatocyte necrosis, apoptosis, or necroptosis that is dependent on various stimuli and is mediated by various signaling pathways such as the JNK and p38 pathways, ER (endoplasmic reticulum) stress, and ROS (reactive oxidative species) production (Novo et al. 2014). Although cell death is a primarily protective response to stimuli, the persistence of cell death over decades dictates clinically adverse outcomes in the liver (Luedde et al. 2014). This stage is potentially reversible; therefore, blocking the pro-fibrogenic cell death response pathway by removing the cause of injury may allow for the tailored prevention or treatment of liver fibrosis (Novo et al. 2014).
Inflammatory response after liver injury
Repetitive injury responses in the liver induce oxidative stress and/or activated inflammatory responses (Luedde et al. 2014). ROS, the end product of lipid peroxidation, 4-hydroxy-2,3-nonenal (HNE), inflammatory chemokines such as interleukin-6 (IL-6), TNF-α (tumor necrosis factor α), IL-8, CCL4 (CC chemokine ligand), CCL5, and MCP-1, and pro-fibrogenic peptide mediators such as PDGF, ET-1, TGF-β (transforming growth factor-β), and CTGF are released by damaged hepatocytes, cholangiocytes, and activated inflammatory cells, leading to inflammatory response activation in a pro-fibrogenic environment (Novo et al. 2012). The most widely studied immune cell population in liver fibrosis is the macrophage population. The resident Kupffer cells (the largest population of resident macrophages in the body) likely play a role in the early response to injury; however, the number of Kupffer cells decreases during hepatic inflammation and fibrogenesis. In addition, the number of monocyte-derived hepatic macrophages markedly increases in response to tissue injury; thus, the pro-fibrogenic macrophages must be derived from this population (Holt et al. 2008). Macrophages produce chemokines to recruit myofibroblasts and facilitate leukocyte recruitment to sites of inflammation. Macrophages express TGF-β, galectin-3, TNF-α, and IL-1β to drive myofibroblast activation and ECM synthesis through activation of the nuclear factor kappa-light-chain enhancer of activated B cell (NFκB) pathway (Pradere et al. 2013).
Leukocytes are recruited to sites of injury to phagocytose dead or apoptotic cells and amplify the inflammatory response by releasing pro-inflammatory cytokines, which recruit T cells (Bataller and Brenner 2005). T cells such as TH2 cells are strongly pro-fibrogenic and produce IL-13 to stimulate TGF-β synthesis and matrix metalloproteinases 9 (MMP9) expression (Schuppan and Kim 2013); IL-4 and IL-13 stimulate the differentiation of fibrogenic myeloid cells and macrophages (Pellicoro et al. 2014). In contrast, TH1 cells have an anti-fibrogenic effect (Schuppan and Kim 2013). Other T cells such as TH17 cells secrete IL-17, which directly induces type I collagen production in HSCs through activation of the signal transducer and activator of transcription 3 (Stat3) signaling pathway (Meng et al. 2012). The role of other immune cells such as NK cells, Treg cells, neutrophils, other lymphoid cells, and mast cells in fibrosis has also been studied but remains unclear.
Among the inflammatory cytokines, TGF-β is the most potent fibrogenic cytokine; TGF-β binds to its receptor Smad (mothers against decapentaplegic homolog) to induce collagen production. TGF-β induces the phosphorylation of Smad3 and reduces formation of the Smad3/4 complex, thereby inducing Smad DNA-binding activity to stimulate the expression of collagen and suppress MMP expression (Inagaki et al. 2012). Therefore, nuclear accumulation of phosphorylated Smad3 is the most common feature observed in activated HSCs, and the inhibition of Smad3 accumulation or export to nuclear can suppress the transcriptional activity of the collagen gene (Inagaki et al. 2012).
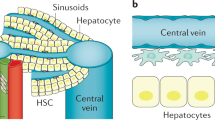
Activation of HSCs and myofibroblast in response to inflammation
HSC activation includes initiation and perpetuation phases. The initiation phase is also called pre-inflammation stage and is stimulated by oxidative stress signals, apoptotic bodies, and paracrine stimuli from neighboring cells (hepatic macrophages, sinusoidal endothelium cells), leading to changes in the gene expression and phenotypes of HSCs (Lee and Friedman 2011). Once the HSCs are primed for activation, the perpetuation phase results from the stimuli to maintain the activated phenotype, proliferation, and fibrogenesis by the regulation of inflammatory mediators, growth factors, and cytokines (Pellicoro et al. 2014). The perpetuation phase includes different responses such as scar production, contractility, altered matrix degradation, chemotaxis, and fibrosis generation to increase the accumulation of ECM (Pellicoro et al. 2014). Therefore, myofibroblasts and their upstream cellular elements represent major targets of fibrosis research in recent years (Pellicoro et al. 2014).
Microenvironmental interactions and signaling pathways in fibrosis progression
The complicated microenvironmental elements and mechanisms work together to accomplish fibrosis progression or regression (Friedman 2010). Several signaling pathways are involved in HSC activation and ECM production through the up-regulation of receptors (Friedman 2010). For instance, the activation of TGF-β receptors, PDGF-β receptors, and angiotensin II receptors is relevantly important. PDGF signaling is among the best-characterized pathways of HSC activation leading to PI3K (phosphatidylinositol 3-kinase)/Akt signaling activation, Ras/MAPK (mitogen-activated protein kinase) activation, and cellular proliferation (Lee and Friedman 2011). Angiogenic signaling is another key component in hepatic fibrosis, contributing to ECM production and portal hypertension through angiogenic mediators such as vascular endothelial growth factor (VEGF) (Friedman 2010). Oxidative stress has also been implicated in fibrogenic stimulation, and the NADPH enzyme is important for the generation of oxidative stress (Friedman 2010).
Recent studies also indicated that epigenetic events such as methyl-CpG-binding protein 2 (MeCP2)-induced chromatin structural repression were detected in myofibroblasts in diseased livers. MeCP2 is involved in silencing genes by methylating DNA and thereby preventing protein expression such as peroxisome proliferator-activated receptor-γ (PPARγ) gene expression contributed to myofibroblast differentiation and fibrosis (Mann et al. 2010). PPARγ is an important gene in the regulation of lipid and glucose metabolism, insulin sensitivity, and inflammation and is also important in hepatocyte lipid deposition (Moran-Salvador et al. 2013). Synthetic PPARγ ligands have been demonstrated to inhibit the pro-fibrogenic and pro-inflammatory effects of HSCs (Zhang et al. 2013). Another important example of epigenetic regulation in HSC activation involved the phosphorylation of NFκB subunit RelA at Ser536, which leads to its nuclear import, thereby increasing NFκB transcriptional activity and HSC survival (Friedman 2010). Inhibiting the phosphorylation of RelA in fibrotic rodents led to reduced survival of activated HSCs and fibrosis (Oakley et al. 2009). In another model, phosphorylation of the transcription factor C/EBPβ by the ribosomal S-6 kinase (RSK) induced HSC activation; this phosphorylation can be inhibited by cell-permeable peptides that block RSK (Buck and Chojkier 2007).
Taken together, understanding the complicated mechanisms of fibrosis is the exclusive route to developing effective therapeutic strategies for liver fibrosis. However, applying the novel mechanisms to the treatment of liver fibrosis still lags behind the demand for such treatments (Iredale 2007).
Strategies for the prevention and reversal of liver fibrosis in animals and humans
Recent clinical evidence has indicated that liver fibrosis can regress, and the regression of liver fibrosis was discussed further in the review article by Ellis et al. (Ellis and Mann 2012). Animal models, especially rodent models, remain important tools to confirm the relevance of targets and the efficacy of anti-fibrotic agents (Popov and Schuppan 2009). A promising agent should be studied in at least two mechanistically distinct models of fibrosis to avoid “model-specific” artifacts (Popov and Schuppan 2009). The model in which liver fibrosis is induced in animals using chemicals such as carbon tetrachloride (CCl4) is popular because it induces repetitive hepatocyte death, liver injury, and advanced fibrosis and is reversible (Paakko et al. 1996; Popov and Schuppan 2009). The disadvantages of this model included severe hepatocyte necrosis and its dependence on massive oxidative stress, which is not found to such a severe extent in human chronic liver disease (Popov and Schuppan 2009). Another chemical, TAA (thioacetamide), is useful to test the reversal effects of agents, and the pattern of fibrosis closely resembles parenchymal fibrosis in humans (Popov and Schuppan 2009). Genetic animal models such as those of PDGF or TGF-β overexpression can be used to confirm factors and mechanisms that drive fibrosis or fibrinolysis (the removal of excess ECM) and can also be used to test cell-specific or tissue-specific approaches (Popov and Schuppan 2009). Currently available mouse models for fibrogenesis or anti-fibrosis studies are discussed in a detailed review by Popov (Popov and Schuppan 2009). Based on mechanistic studies, we discuss the anti-fibrotic therapies divided into a few different categories, the novel preventive and reversal strategies for liver fibrosis, and the current clinical trials for the treatment of liver fibrosis in the following sections.
Eliminate primary disease using anti-viral drugs in chronic viral hepatitis
The most effective way to treat fibrosis is to eliminate the primary disease that induced injury such as by abstaining from alcoholic intake, removing excessive iron or copper, and clearing liver viral infection (Friedman 2008). Standard treatments for viral infections include interferon (IFN), PEGylated α-interferon (PEG-IFNα), and nucleoside analogues (NUCs) (Ellis and Mann 2012). It is evident that HCV patients treated with PEG-IFNα show successfully reduced fibrosis (Friedman 2010). Long-term therapy with NUCs such as entecavir, lamivudine, or adefovir (an acyclic nucleoside analogue) has been shown to improve liver fibrosis and disease progression, but resistance mutations are common with these agents (Ellis and Mann 2012). The recently identified novel agents and recently conducted clinical trials for the treatment of fibrosis that are targeted to eliminate viral infections are listed below.
-
1.
Patients coinfected with HIV and HCV progress more rapidly to liver fibrosis and cirrhosis compared with those infected with HCV alone (Ellis and Mann 2012). Rifaximin, a non-absorbable antibiotic with very few side effects that can reduce the toxic released by bacteria in the gut and therefore improve liver encephalopathy, has also been suggested to improve liver fibrosis in patients with HIV/HCV coinfection (Fuessl 2014).
-
2.
A recent clinical trial indicated that raltegravir (the first HIV integrase inhibitor) was safe and well tolerated in patients with HIV/HCV coinfection. Therefore, one clinical trial was conducted to evaluate whether a raltegravir-based regimen would reduce the progression rate of hepatic fibrosis in HIV/HCV coinfected patients (Hernandez-Novoa et al. 2014) (https://clinicaltrials.gov).
-
3.
Other strategies in animal models: Inactivated orf virus (iORFV), strain D1701, is a potent immune modulator that induces strong anti-viral activity in animal models of HCV and HBV infections. D1701 and the other strain NZ2 showed significant anti-fibrotic activity in rat models of liver fibrosis via unknown mechanisms (Paulsen et al. 2013).
-
4.
RNA interference (RNAi) for virus-specific genes offers the possibility for new drug development. A lentivirus-based RNAi system was used to deliver HBV-specific short hairpin RNA (shRNA) in a mouse model to suppress HBV replication. This effect was characterized by reduced HBsAg and HBeAg in the serum, suggesting this is a potential therapeutic strategy for treating viral infections (Deng et al. 2009; McCaffrey et al. 2003).
Anti-fibrotic strategies that reduce inflammation and the immune response
Inflammation always precedes or is accompanied by fibrosis, and drugs that target the inflammatory pathway may eliminate the stimuli to HSCs and therefore prevent or treat fibrosis (Friedman 2008). The agents mediating inflammation-related pathways via various mechanisms are listed and shown in Table 1.
-
1.
Modifying macrophage phenotypes in vivo: The equilibration between pro-fibrotic and pro-resolution macrophage populations determines whether the outcome of tissue injury is homeostatic or pathogenic scarring (Pellicoro et al. 2014). Conditionally depleting pro-fibrogenic macrophages results in decreased numbers of myofibroblasts and attenuated liver scaring in a CCl4-treated model (Duffield et al. 2005). In contrast, pro-resolution macrophages are rich sources of fibrinolytic MMPs (MMP12, MMP13, MMP9) and TNF-related apoptosis-inducing ligand (TRAIL) that can promote myofibroblast apoptosis and augment ECM degradation in rodent models (Pellicoro et al. 2012). The other cell therapy approach using bone marrow-derived macrophages could potentiate ECM degradation and promote regenerative effects (Sakaida et al. 2004). However, due to small numbers of heterogeneous patients tested, the potential effects of this therapy remain unclear (Pellicoro et al. 2014).
-
2.
Management of other immune cells: Previous reports indicated that mesenchymal stem cell (MSC) transplantation led to an improved liver microenvironment and reduced liver injury in mice treated with CCl4 (Nasir et al. 2013). Human umbilical cord-derived MSC (UC-MSC) transplanted to CCl4-treated rats indicated that liver fibrosis, TGF-β, and EMT were reduced and improved liver function (Jung et al. 2009; Li et al. 2013; Seo et al. 2014). A pilot study investigated the safety and efficacy of UC-MSC transfusion in primary biliary cirrhosis (PBC) patients and indicated that UC-MSC transfusion is feasible and well tolerated in patients (Wang et al. 2013). The other study indicated that transplantation of bone marrow-derived mesenchymal stem cell (BM-MSC) therapy for the treatment of liver fibrosis in CCl4-treated rats is effective (Motawi et al. 2014). Additionally, the clinical therapy in patients with alcoholic cirrhosis using BM-MSC induced a histological and quantitative improvement in hepatic fibrosis (Jang et al. 2014). However, the anti-fibrotic effect of MSCs must be confirmed by a larger clinical trial.
-
3.
Antagonizing the pro-fibrogenic cytokine pathways (TGF-β and CTGF): A small molecule inhibitor of the TGF-β type I receptor kinase (ALK5) EW-7197 was reported to block TGF-β stimulation of ROS, collagen, and α-SMA production in HSCs, in the livers of CCl4-treated mice, and in bile duct ligation (BDL) rats (Park et al. 2014). Treatment with recombinant human bone morphogenic protein-7 (rhBMP-7, an antagonist of TGF-β) alleviates renal fibrosis and liver fibrosis in a fibrotic rat model injected with porcine serum; this effect is dependent on the reduction of TGF-β overexpression and the inhibition of TGF-β-triggered intracellular hepatocyte signaling (Zhong et al. 2013). Another strategy using the traditional Chinese medicine Haobie Yangyin Ruanjian decoction (HYRD) inhibited liver fibrosis induced by CCl4 in rats, likely through down-regulation of the TGF-β/Smad fibrogenic signaling pathway (Yang et al. 2010). Another study indicated that Y-box protein-1 (YB-1) is a negative regulator of collagen expression by physically interacting with p300/Smad3, thereby abrogating the stimulatory effect of TGF-β and liver fibrosis induced by CCl4 in mice (Inagaki et al. 2012). The novel small compound HSc025 can bind to the C-terminal region of YB-1 and promote the nuclear import of YB-1, resulted in the suppression of collagen gene expression (Higashi et al. 2011). In addition, Wang et al. constructed Smad3 siRNA (small interfering RNA) encapsulated by liposomes targeted to the Smad3 gene in rats treated with TAA for 6 and 8 weeks. The results showed that Smad3 siRNA expression plasmids had anti-fibrotic effects, shown by the reduced liver fibrosis index (procollagen III, collagen IV, laminin, hyaluronic acid) (Wang et al. 2011). 4-Methyl-5(pyrazinyl-2)-1-2-dithiole-3-thione (oltipraz), a promising cancer-preventive agent and a nuclear factor erythroid 2-related factor 2 (Nrf2) activator, had ability to enhance biosynthesis of glutathione, the activities of phase II detoxification enzymes, and the functions of multidrug resistance-associated protein-mediated efflux transporter (Weerachayaphorn et al. 2014). Oltipraz can inhibit the phosphorylation of Smad3, thereby reducing Smad3/4 complex formation and TGF-β-induced Smad DNA-binding activity (Cho et al. 2006). Another Nrf2 activator NK-252 has a greater Nrf2-activating potential than oltipraz; NK-252 significantly reduced the progression of fibrosis in rats fed with choline-deficient l-amino acid-defined (CDDA) diet (Shimozono et al. 2013). The results of phase II clinical trial showed improved liver fibrosis in oltipraz treatment groups in a 24-week pilot study (Kim et al. 2011). However, a recent study indicated that although oltipraz protects against chemical-induced hepatotoxicity, it exacerbates the severity of liver injury following BDL. Therefore, oltipraz use should be avoided in bile duct obstruction, and its effect for the treatment of liver fibrosis remains to be studied (Weerachayaphorn et al. 2014). Because TGF-β is an important mediator in liver fibrosis, it is an attractive target for developing therapeutic strategies. However, targeting this pathway will be challenging because TGF-β also has anti-inflammatory and growth regulatory roles that are important for liver homeostasis (Iredale 2007).
In a CCl4-treated rat model, injecting siRNA targeted to another pro-fibrogenic cytokine, connective tissue growth factor (CTGF), could significantly attenuate type I/II collagen expression and inhibit ECM accumulation and liver fibrosis (Li et al. 2008). A monoclonal antibody to CTGF (FG-3019) was found to prevent and reverse fibrosis in CCl4-treated rats (Lipson et al. 2012). An ongoing phase II clinical trial is being conducted to evaluate the efficacy of FG-3019 for reversing liver fibrosis in subjects with HBV infection who are beginning anti-viral therapy with entecavir; the results are not yet published (http://search.centerwatch.com).
-
4.
Disrupting chemokine pathways: Increased IL-18 secretion, mainly by CD4(+) T cells and macrophages, was observed in HCV patients. IL-18 and IL-1β are secreted by inflammasomes to activate an acute inflammatory response via production of the inflammatory cytokines TNF-α, IFN-γ, the chemotaxis of immune cells, and the induction of tissue injury (Ouyang et al. 2013). The administration of concanavalin A (ConA) induced severe liver fibrosis in mice; this effect was aggravated by IL-18, but blocking IL-18 signaling in these mice reduced liver fibrosis (Zhang et al. 2007b).
S-adenosyl methionine (SAMe), which acts as a methyl donor for methylation reactions and participates in the synthesis of glutathione, has mostly been used in alcoholic liver disease to prevent TLR activation and thereby reduce inflammation (Oliva et al. 2011). The other mechanism of SAMe in the treatment of liver fibrosis involves increased ubiquitination and decreased type I collagen secretion in activated HSCs (Thompson et al. 2011). The clinical trial tested the efficacy of SAMe in treatment for alcoholic liver diseases showed overall improvement of AST, ALT, and bilirubin levels, but no differences or changes in liver histopathology scores for steatosis, inflammation, and fibrosis (Medici et al. 2011).
In contrast to pro-fibrogenic cytokines, recent evidence indicated that anti-fibrogenic cytokines such as IFNγ released by NK cells can kill early or senescent HSCs. The clinical studies revealed the ability of human NK cells isolated from HCV patients to kill HSCs; this killing effect is inversely correlated with the stage of liver fibrosis (Gao and Radaeva 2013). The animal study indicated that PEGylated IFNγ reduced the early fibrotic parameters more drastically than unmodified IFNγ in CCl4-treated mice. PEGylation significantly improved the pharmacokinetics, liver uptake, and anti-fibrotic effects of IFNγ (Bansal et al. 2011). The novel chimeric molecule contains the IFNγ signaling peptide (mimγ) that has the agonistic activity of IFNγ is coupled to PDGF-βR-binding peptide (BiPPB) by a PEG linker can inhibit early liver fibrosis in CCl4-treated mice (Bansal et al. 2014).
The “hepato-protectants” such as HGF (hepatocyte growth factor) and insulin growth factor (IGF) also have potent anti-apoptotic and mitogenic effects on hepatocytes during liver injury and essential roles in liver regeneration. The transplantation of HGF-overexpressing human umbilical cord blood-derived mesenchymal stem cells (hHGF-HUCB-MSCs) in CCl4-induced fibrotic rats improved liver function and induced liver regeneration (Seo et al. 2014). HGF and its receptor, c-mesenchymal–epithelial transition factor (c-Met), are essential for liver cell growth and proliferation. Liver-specific Met knockout BDL mice showed strong apoptosis, increased pro-inflammatory cytokine expression, and enhanced neutrophil recruitment. The results indicated that c-Met deletion in hepatocytes leads to liver cell damage and fibrosis because c-Met stimulates survival signals that are important for liver cell recovery (Geng et al. 2008). IGF was also reported to effectively block fibrosis. In contrast, in a chronic cholangiopathy mouse model, IGF1 overexpression stimulated fibrogenic processes by increasing the expression of TGF-β and collagen I, II, and IV (Sokolovic et al. 2013). Therefore, both HGF and IGF are hepatocyte mitogens that should be carefully monitored because of their potential role in hepatocarcinogenesis (Friedman 2008).
-
5.
Novel target of the SOCS (suppressor of cytokine signaling) pathway: Increasing evidence has indicated that the interaction of SOCS and molecular signaling pathways plays key roles in balancing cytokine activation and contributes to the termination of signals in innate immune cells during liver fibrosis (Cheng et al. 2014a). The expression of SOCS is regulated by epigenetic mechanisms including methylation, acetylation, or ubiquitination (Cheng et al. 2014a). Decreasing the expression of SOCS3 by hypermethylation results in increased expression of Stat3, granulocyte colony-stimulating factor (G-CSF), and secreted phosphoprotein 1 (SPP1) but reduced expression of PPAR, all of which might contribute to the development of liver fibrosis (Cheng et al. 2014a). In addition, altering microRNAs (miR) such as knocking down miR-155 can up-regulate SOCS1 protein expression and significantly decrease NO and inflammatory cytokine production (Cheng et al. 2014a). miR-9 effectively reduced SOCS5 expression leading to JAK/Stat pathway activation, thereby promoting endothelial cell migration and ECM generation in liver fibrosis (Cheng et al. 2014a). These studies suggested that methylation and some miRNA inhibitors could up-regulate SOCS expression to prevent the progression of liver fibrosis (Cheng et al. 2014a).
Down-regulating myofibroblast activation and ECM production
Following chronic liver injury, HSCs become activated and trans-differentiate into myofibroblast-like cells to secrete excessive ECM proteins and enhance contractility and pro-inflammatory and pro-fibrogenic factor release. Therefore, treatment strategies for fibrosis should dampen the fibrogenic activation of HSCs and induce the production of fibrinolytic enzymes (Schuppan and Kim 2013). Accordingly, several agents that block fibrogenic activation and ECM production by myofibroblasts work well in some rodent model but have unwanted side effects in humans due to their lack of specificity for myofibroblasts (Schuppan and Kim 2013). These agents and their mechanisms of inhibiting myofibroblast activation are listed and shown in Table 2.
-
1.
Targeting the mTOR pathway: HSC activation is mediated by PDGF and leads to activation of the MAPK/ERK (extracellular signal-regulated kinase) and PI3K/AKT/mTOR/p70S6 K (ribosomal S6 kinase) signaling pathways (Li et al. 2014a). Following low-dose treatment with the mTOR inhibitor rapamycin, TGF-β expression was down-regulated and EMT as well as HSC activation was attenuated in BDL rats (Bridle et al. 2009). Other mTOR inhibitors, sirolimus and everolimus, also reduced fibrosis progression and portal hypertension in BDL rats (Patsenker et al. 2011). Moreover, everolimus worked via multiple mechanisms, including inhibiting angiogenesis and preventing the tube formation and migration of liver sinusoidal endothelial cells (Piguet et al. 2014). The clinical study using everolimus monotherapy in liver transplantation recipients showed the less serum expression of TGF-β1, but no differences in inflammatory activity, in APRI test, or in liver elastography (Fernandez-Yunquera et al. 2014).
-
2.
Targeting to the heat shock protein 47 (Hsp47) pathway to inhibit ECM production: Several studies have indicated that HSCs are the sources of Hsp47, which is a collagen-specific chaperon protein that contributes to collagen maturation (Park et al. 2013). The administration of Hsp47-targeted shRNA remarkably reduced Hsp47 expression and collagen deposition in NIH3T3 cells and in liver tissue from Schistosoma japonicum-infected mice (Huang et al. 2014b). Because HSCs specifically take up vitamin A, the strategy using vitamin A-modified liposomes increased the delivery of Hsp47 siRNA to HSCs and caused anti-fibrogenic effects in the animals (Popov and Schuppan 2009). ND L02-s0201, an Hsp47 siRNA lipid nanoparticle conjugated to vitamin A, which preferentially targets HSCs, was applied in an ongoing phase I clinical trial started in 2013 (https://clinicaltrials.gov).
-
3.
Targeting the leptin and PPAR pathways:Leptin is an adipocyte-derived hormone that is produced by adipose tissues and plays a pivotal role in the pathogenesis of metabolic syndromes such as NASH, type 2 diabetes mellitus, and alcoholic cirrhosis (An et al. 2012; Ikejima et al. 2007). Leptin is a pro-fibrogenic factor that increases HSCs activation and collagen I production through TGF-β, JAK/Stat3, and PI3K/Akt signaling pathways (Elinav et al. 2009). In addition, leptin might inhibit PPARγ expression through the ERK1/2 pathway, therefore enhancing HSC activation and proliferation in a mouse model (Zhou et al. 2010). Elinav et al. (2009) demonstrated that the mouse leptin antagonist (MLA) markedly improved survival and attenuated liver fibrosis in chronic TAA-treated fibrosis model, suggesting that targeting leptin is a useful treatment strategy for fibrosis. However, leptin replacement in NASH patients showed significant improvement in metabolic profile, but fibrosis remained stable (Safar Zadeh et al. 2013).
PPARγ activation is also a potential strategy to block HSCs activation and differentiation. Synthetic PPARγ ligands such as 15d-PGJ2 and all-trans retinoic acid (ATRA) inhibited the pro-fibrogenic and pro-inflammatory effects of HSCs (Sharvit et al. 2013). In addition, kaerophyllin, a lignan, isolated from traditional Chinese herbs, up-regulated PPARγ expression, inhibited HSC activation in vitro, and protected rat livers from TAA-induced injury and fibrogenesis by inhibiting liver inflammation (Lee et al. 2012). A clinical trial indicated that PPARγ-agonists thiazolidinediones significantly improved lobular inflammation, steatosis, and inflammation in patients with NASH (Boettcher et al. 2012). Using nanoparticles carrying PPARγ agonist in BDL rats showed suppressing the activation of HSCs and decreasing inflammatory cytokines (Kumar et al. 2014). The novel active glitazone SKLB010 [(Z)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione] attenuated CCl4-induced liver fibrosis, collagen deposition, and α-SMA expression in rats (Chen et al. 2012). However, one study indicated that pioglitazone improved steatosis and transaminase levels but had no effect on other liver injury parameters (Ratziu et al. 2008). Therefore, larger, randomized, placebo-controlled clinical trials are still needed to examine the efficacy of PPARγ agonists in fibrosis.
-
4.
Reduced HSC activation by inhibiting oxidative stress: Oxidative stress can stimulate HSC activation; therefore, using antioxidant provides a rationale to suppress fibrosis. Several antioxidants extracted from plants and traditional Chinese medicines as well as those existing in nutrients were tested in vivo and in clinical trials. For instance, resveratrol is a phytoalexin, and oral administration of resveratrol prevented dimethylnitrosamine (DMN)-induced liver damage in rats (Lee et al. 2010). In BDL mice, resveratrol could reduce mortality, attenuate inflammation, decrease fibrosis, and promote hepatocyte regeneration (Chan et al. 2011). In cirrhotic rats, resveratrol-decreased hepatic fibrosis was associated with reduced collagen I, TGF-β and NFκB mRNA expression, and α-SMA protein expression (Di Pascoli et al. 2013). A clinical trial studying the effects of resveratrol supplementation on the lipid profile, liver enzymes, inflammatory factors, and hepatic fibrosis in patients with non-alcoholic fatty liver disease (NAFLD) indicated the beneficial effects of resveratrol on down-regulating inflammatory mediators and metabolic disorders. Mechanistically, resveratrol supplementation was associated with significantly reduced levels of the liver enzyme alanine aminotransferase, of inflammatory cytokines, and of NFκB, as well as a significant reduction in the hepatic steatosis grade, compared with placebo supplementation (Faghihzadeh et al. 2014). More studies are required to confirm the anti-fibrotic effects of resveratrol.
In a Schistosoma mansoni-injected model, the preventive and curative effects of liver fibrosis were evaluated by using antioxidants such as resveratrol, curcumin, and N-acetylcysteine (NAC); results indicated that curcumin had potent anti-fibrotic activity in both suppressing and reversing fibrosis, whereas resveratrol had a beneficial effect only in suppressing fibrosis (El-Agamy et al. 2011). In BDL- and CCl4- or TAA-treated rats, NAC prevented liver fibrogenesis associated with decreased oxidative stress in the liver (Nissar et al. 2013; Yang et al. 2008).
Another antioxidant, silybin (SB), has been used to treat liver disorders in DMN- or CCl4-treated rats (Ezhilarasan et al. 2012; Muriel et al. 2005). SB has been conjugated with vitamin E and phospholipids to improve its antioxidant activity. Treatment with silybin/vitamin E/phospholipids in patients with NAFLD decreased the indices of liver fibrosis (Loguercio et al. 2007). A phase III safety and efficacy clinical trial used a pharmaceutical complex of silybin/vitamin E/phospholipids in NAFLC patients with or without chronic hepatitis C infection and followed the fibrotic indices. The results indicated that the silybin/vitamin E/phospholipid complex was active, produced some therapeutic effects in patients with liver fibrosis, and improved insulin resistance (Trappoliere et al. 2005).
Promote the cell death mechanism or the quiescence of myofibroblasts
The resolution of fibrosis can occur following the induction of myofibroblast apoptosis, senescence, or quiescence (Luedde et al. 2014). Senescence is a phenotype associated with decreased matrix and cytokine synthesis, and senescent myofibroblasts can be removed by NK cells (Krizhanovsky et al. 2008). Several agents that induced HSC death through apoptosis or senescence were tested in animal models. For example, pentoxifylline (PTX) is a phosphodiesterase (PDE) inhibitor that prevented pig serum-induced rat liver fibrosis by inhibiting the proliferation of HSCs and the production of IL-6 (Toda et al. 2009). In a clinical trial, using PTX for the treatment of NAFLD significantly improved steatosis, lobular inflammation, and fibrosis but did not significantly affect the serum TNF-α levels; thus, larger well-designed studies are still required to confirm these results (Zeng et al. 2014). To induce the senescence of myofibroblasts, a statin with HMG-CoA reductase-inhibiting activity, atorvastatin, has been used in BDL rats (Klein et al. 2012). Other statins such as rosuvastatin administered in the early stage of cholestasis in BDL rats also provided beneficial effects by decreasing α-SMA level, suggesting the potential role of statins in preventing the progression of liver disease (Olteanu et al. 2012).
Activated HSCs mainly express specific receptors that are absent from the normal liver. Therefore, the anti-proliferative drug mycophenolate or the apoptosis inducer gliotoxin coupled to IGF type II receptor can target HSCs and sinusoidal endothelial cells (Popov and Schuppan 2009). Accordingly, the coupling of gliotoxin to the antibody against synaptophysin that is expressed on HSCs can induce apoptosis in HSCs and reduce fibrosis in CCl4-treated models (Popov and Schuppan 2009).
Stimulate the degradation or accumulation of extracellular matrix
Inhibiting matrix production, blocking matrix synthesis and processing, or increasing matrix lysis by altering the balance of tissue inhibitor of metalloproteinases (TIMP) and MMP in situ could resolve fibrosis (Friedman 2008). The MMP inhibitor CTS-1027, which has previously been studied in humans as an anti-arthritic agent, has well established anti-inflammatory and anti-fibrotic effects in BDL mice; CTS-1027 reduced hepatocyte apoptosis and liver injury, as characterized by decreasing the markers of HSC activation such as α-SMA and collagen I (Kahraman et al. 2009). A recent phase II clinical trial evaluating CTS-1027 in combination with PEGy-IFN-α2a and ribavirin in a treatment-experienced, HCV-null responder patient population was conducted (http://www.centerwatch.com).
Galectin-3 (Gal-3) is a multifunctional lectin that is mainly produced by macrophages and is involved in the integrin β1-induced EMT pathway, promotes fibroblast proliferation and transformation, and mediates collagen production (Li et al. 2014b). Gal-3 is activated and abnormally increased in patients with fibrosis, suggesting that Gal-3 may be a target for treating liver fibrosis (Li et al. 2014b). Using the antibody GR-MD-02 targeted to Gal-3 markedly reduced fibrosis in a NASH murine model (Traber and Zomer 2013). In TAA-induced fibrotic rats, treatment with Gal-3 inhibitors (GR-MD-02 and GM-CT-01) significantly reduced fibrosis, reversed cirrhosis, reduced macrophage numbers, and reduced portal pressure, suggesting a potential therapeutic role of Gal-3 inhibitors in liver fibrosis (Traber et al. 2013). Based on promising preclinical data with GR-MD-02, an investigational new drug (IND) application was submitted to the FDA, and researchers subsequently proceeded with a phase 1 clinical trial to evaluate the safety of GR-MD-02 in subjects with NASH and advanced hepatic fibrosis in 2013 (http://search.centerwatch.com).
ECM cross-linking is largely mediated by lysyl oxidase (LOX). LOX family members include LOXL-1, -2, -3, and -4, which catalyze the cross-linking of collagen and elastin in the ECM (Van Bergen et al. 2013). Targeting LOXL2 using the humanized antibody simtuzumab (GS-6624) has been shown to efficaciously block LOXL2 activity and fibrosis in a rodent model of liver fibrosis (Van Bergen et al. 2013). The pilot clinical study of simtuzumab in the treatment of liver fibrosis has been completed, but the results have not been published yet. A recent clinical study was conducted to evaluate whether simtuzumab is effective at preventing liver fibrosis from progressing to cirrhosis in NASH patients (https://clinicaltrials.gov).
Relaxin is a natural peptide hormone of the insulin superfamily that is involved in promoting ECM remodeling, decreasing collagen synthesis and increasing matrix degradation in cultured HSCs and rat livers (Williams et al. 2001). In CCl4-treated mice, relaxin reduced collagen deposition and HSC activation and increased MMP13 and collagen I expression (Bennett et al. 2013). Relaxin has not been used in a clinical trial to treat liver fibrosis, but was effective in the treatment of acute heart failure in a clinical trial (Teerlink et al. 2013).
The Chinese medicine Shaoqiduogan (SQDG) was also reported to significantly decrease collagen I levels in rats treated with CCl4 by controlling the levels of MMP-13 and TIMP-1 (Sun et al. 2010).
Angiotensin and related blocking agents
The renin-angiotensin system (RAS) regulates lipid and glucose homeostasis. Renin converts angiotensinogen into angiotensin I (Ang I), which is then converted to angiotensin II (Ang II) by angiotensin-converting enzyme (ACE). Ang II mediates biological responses through Ang II receptor type 1 (AT1) and Ang II receptor type 2 (AT2), but AT1 is the main receptor that mediates the biological effects of Ang II (Moreira de Macedo et al. 2014). Ang II is a pro-inflammatory, pro-oxidant, and pro-thrombotic protein that interferes with the insulin signaling. Previous studies have indicated that both the ACE and AT1 genes are up-regulated in areas of active hepatic fibrogenesis, and Ang II induces the activation and proliferation of HSCs through AT1. The anti-fibrotic effect of Ang II blocking agents has been shown in various animal models and hepatitis C patients, suggesting ACE/Ang II/AT1 could be a promising target for liver fibrosis therapy (Moreira de Macedo et al. 2014). A recent clinical study of the long-term oral administration of losartan in HCV patients with mild fibrosis showed that losartan treatment was associated with a significant decrease in the expression of procollagen I, IV, urokinase-type plasminogen activator, MMP-2, NOX activator 1 (NOXA-1) and organizer 1 (NOXO-1), and Rac-1. Losartan was well tolerated in all patients and was effective in attenuating systemic renin-angiotensin system activity. No significant toxicity was observed, suggesting that prolonged losartan administration is safe and is associated with the down-regulation of fibrogenic genes in patients with chronic hepatitis C (Colmenero et al. 2009; Friedman 2010). Candesartan (CAN) is another ATI antagonist. Early but not late initiation of therapy with CAN reduced mRNA levels of TGF-β, MMP2, and Smad2, which may be crucial for the prevention of cirrhosis. However, CAN treatment did not produce a significant effect on collagen I expression and fibrosis, possibly due to reduced A1 expression in the progression of fibrosis (Tox et al. 2007). One recent clinical trial using CAN combined with UDCA to treat patients with alcoholic liver fibrosis indicated that CAN reduced the area of fibrosis in the liver; additionally, the expression of TGF-β1, collagen-1, AT1, TIMP-1, MMP2, and Rac1 was decreased (Kim et al. 2012). Irbesartan is also an ATI antagonist that significantly reduced the mRNA expression of collagen I, TGF-β, and TNF-α and the levels of fatty acids in NASH mice (Kato et al. 2012). A phase III clinical trial was conducted to examine the efficacy of irbesartan on the progression of liver fibrosis in adult patients with chronic hepatitis C, but the results have not been published (https://clinicaltrials.gov).
Other relevant molecular targets and other possible anti-fibrogenic agents in animals and humans
With increasing understanding of the mechanisms of liver fibrosis, a number of novel targeted approaches for treating liver fibrosis are being explored, as discussed below (Table 3).
-
1.
Preventing hepatocyte cell death: Apoptotic or necrotic hepatocytes enhanced oxidative and ER stress, lysosomal activation, and mitochondrial damage, all of which are strong stimulators of fibrogenesis (Malhi and Gores 2008). The phagocytosis of apoptotic hepatocytes by myofibroblasts triggers their fibrogenic activation via the NADPH oxidase 2 (NOX2), JAK/Stat, and PI3K/Akt pathways (Jiang et al. 2009). The inhibition of hepatocyte apoptosis by a caspase inhibitor or cathepsin B antagonist ameliorated fibrosis in mice (Canbay et al. 2003, 2004). In C3H/HeN mice fed a high-fat diet (HFD), apoptosis was induced, whereas mice receiving the caspase inhibitor VX-166 showed inhibited apoptosis and reduced inflammation, suggesting that caspase inhibition may represent a valid therapeutic strategy (Anstee et al. 2010). Further studies to assess the long-term value of caspase inhibition are merited.
-
2.
Targeting the angiogenesis pathway: Angiogenesis is a fundamental part of liver fibrosis and is accompanied by massive inflammatory responses. The anti-angiogenic drug bevacizumab could block the effect of VEGF on HSCs, resulting in significantly reduced fibrosis in CCl4-treated rats (Huang et al. 2013). The endogenous angiogenesis inhibitor vasohibin-1 and VEGF are up-regulated in cirrhosis patients. The vasohibin-1/VEGF cascades are spatially coordinated through a negative feedback loop that drives pathological angiogenesis. The ectopic overexpression of vasohibin-1 disrupted the feedback loop, leading to lower VEGF synthesis, which was sufficient to maintain vascular homeostasis but not pathological angiogenesis. Therefore, increasing vasohibin-1 could represent a promising therapeutic target for fibrosis (Coch et al. 2014). The Notch pathway is also an important mediator in angiogenesis that enhances blood vessel stability and increases the aorta inner diameter by reducing the response of endothelial cells (ECs) to vascular growth factors. Recent reports indicated that VEGF can induce HSCs to express Notch ligands to promote angiogenic responses (Zhang et al. 2015). The transfer of Notch3 shRNA carried by recombinant adeno-associated virus type 1 (rAAV1) vector to livers improved liver fibrosis in rats treated with CCl4 by decreasing the expression of TGF-β and increasing the expression of E-cadherin, suggesting that Notch is a promising target for liver fibrosis therapy (Zheng et al. 2013).
-
3.
Targeted approaches using small molecule inhibitors and synthetic compounds: Small molecule inhibitors for the selected molecular targets in liver fibrosis have been used (Popov and Schuppan 2009). For example, the cannabinoid receptor CB2 is predominantly expressed in immune cells with anti-inflammatory and anti-fibrogenic effects. In CB2(−/−) mice, hepatic Th17 markers and IL-17 production increased after BDL compared with control mice. The CB2 agonist JWH-133 inhibits IL-17 production in a Stat5-dependent manner, suggesting the potential of CB2 in the treatment of liver fibrosis (Guillot et al. 2014). Some targeted approaches have been directed toward the pro-fibrogenic TGF-β signaling pathway using blocking antibodies, antisense oligonucleotides, or molecular interference (Popov and Schuppan 2009). The synthetic methylenedioxybenzene compound CW209292 prevented liver injury induced by DMN, therefore preventing liver fibrosis and inflammation in rats, possibly through the inhibition of TGF-β expression and subsequent inhibition of HSC proliferation (Oh et al. 2009).
Armepavine (Arm, C19H23O3N), an active compound from Nelumbo nucifera, exerts immunosuppressive activity on T lymphocytes and has been reported to attenuate the fibrotic index in BDL rats. The possible mechanism was possibly through preventing NFκB activation and through the expression of TGF-β, TIMP-1, ICAM-1, iNOS, and IL-6 (Weng et al. 2009). NFκB could mediate the inflammatory response through COX-2 expression; the Cox-2 inhibitor celecoxib exhibits antioxidant activity by restoring redox equilibrium, inhibiting TGF-β expression, inducing MMP2 activity, and preventing collagen accumulation. Celecoxib also exerts strong anti-fibrogenic and fibrinolytic effects in a CCl4-induced liver fibrosis model (Chavez et al. 2010).
The small molecule tyrosine kinase antagonist Gleevec and other inhibitors of Rho-mediated focal adhesions can reduce liver fibrosis in animal models (Yoshiji et al. 2005). Schistosomal cercaria-infected mice treated with Rho kinase inhibitor hydroxyfasudil showed decreased expression of CTGF, collagen IV, and laminin, and decrease phosphorylation of moesin (Zhang et al. 2007a). Another Rho kinase inhibitor, Y-27632, combined with the AT2 blocker TCV-116 has been used to treat choline-deficient L-amino acid-defined (CDAA)-administered rats, resulting in improved liver fibrosis and steatosis (Kitamura et al. 2007). Therefore, the Rho kinase pathway may represent a novel target for fibrosis therapy. These small molecule agents successfully underwent rapid proof of principle testing in vivo; however, due to unwanted side effects, most of these agents have not reached clinical phase studies (Popov and Schuppan 2009).
-
4.
Novel molecular targets: In vitro studies indicated that serum high mobility group box 1 (HMGB1) levels were positively correlated with TGF-β production and collagen deposition during fibrogenesis through receptor of advanced glycation end products (RAGE) pathway activation. CCl4-treated SD rats administered RAGE-specific siRNA twice weekly via tail vein injection for up to 6 weeks displayed significantly decreased levels of serum inflammatory cytokines, NFκB, procollagen III, and hepatic fibrosis, suggesting that HMGB1/RAGE may be a novel target to prevent liver fibrosis (Cai et al. 2014).
Tissue transglutaminase (tTG) was found to co-localize with collagen fibers to contribute to the development of liver fibrosis. One report indicated that the major component of propolis, pinocembrin (PIN), inhibited tTG activation and prevented TAA-induced liver cirrhosis (Chen et al. 2008). Previous reports indicated that cystamine has the ability to inhibit tTG activity. Garlic extracts contain many compounds related to cystamine that can reduce liver fibrosis and improve liver damage in CCl4-treated mice (D’Argenio et al. 2010).
Previous reports indicated that thrombin catalyzes the conversion of fibrinogen to fibrin and then mediates clot formation. Thrombin also activated platelet aggregation and induced HSCs activation via proteinase-activated receptor 1 (PAR1). The negative feedback loop of the thrombin pathway is mediated by the FV leiden protein, and mutation of FV leiden is associated with liver fibrosis. FV leiden-mutant mice treated with the anti-coagulative agent warfarin significantly reduced the progression of fibrosis induced by CCl4, suggesting that targeting the coagulation pathway (thrombin and PAR1) could be a potential novel therapeutic approach for liver fibrosis (Anstee et al. 2008; Sullivan et al. 2010).
Aldose reductase (AR) expression is known to mediate inflammation by affecting NFκB-dependent cytokine and chemokines expression. AR is also induced in hepatitis and hepatocellular carcinoma. Using the AR inhibitor MDA [(Z)2-(5-(4-methoxybenzylidene)-2, 4-dioxothiazolidin-3-yl) acetic acid] in CCl4-treated rats attenuated oxidative stress by increasing the glutathione content in association with suppressed NFκB activity, suggesting that AR could be a novel target for the treatment of liver fibrosis (Wang et al. 2012).
Secreted protein, acidic and rich in cysteine (SPARC) is a matricellular protein found to be overexpressed in cirrhotic livers. Fibrogenic models induced by TAA or BDL were developed on SPARC wild-type (SPARC(+/+)) and knockout (SPARC(−/−)) mice. SPARC(−/−) mice showed reduced inflammation, TGF-β expression, and myofibroblast activation and increased MMP2 expression, indicating that the inhibition of SPARC is a promising approach for liver fibrosis therapy (Atorrasagasti et al. 2013).
Recent research has uncovered that miRs (miR29-b) can inhibit or promote (miR-199, miR-200, and miR33a) fibrogenesis (Sekiya et al. 2011). miR33a is a novel modulator of lipid and cholesterol metabolism and is significantly increased in liver fibrosis, particularly in HSCs (Huang et al. 2014a). However, how to effectively deliver miRs to the liver remains a problem (Murakami et al. 2011).
Autophagy is a catabolic pathway regulated by autophagy-related genes and proceeds through numerous steps, including induction, cargo recognition/selection, autophagosome formation, fusion, and breakdown. Autophagy has a role in the induction of HSC activation, and the inhibition of autophagy by bafilomycin A1 resulted in significantly decreased activation and proliferation of HSCs (Thoen et al. 2011). Another study implicated that ER stress, particularly the IRE1/Xbp1 pathway, may be essential for HSC activation, suggesting that autophagy might be a therapeutic target for liver fibrosis (Hernandez-Gea et al. 2013). However, the specific autophagy mechanisms that are involved in liver fibrogenesis remain unclear.
-
5.
Nanotechnology applications for the treatment of liver fibrosis: The conventional anti-fibrotic treatments are still limited largely due to non-specific drug delivery. The major aim of using nanotechnology to treat liver fibrosis is to deliver anti-fibrotic drugs directly to the fibrotic region. Activated HSCs express specific receptors that could be used as targets for nanoparticles (NPs). Adrian et al. (2007) using MSP-HSA (human serum albumin) coupled to liposomes and hemagglutinating virus of Japan (HVJ) increased the delivery to HSCs, therefore offering a new possible method for treating liver fibrosis. MSP-HSA-conjugated liposomes have also been used to deliver the PPARγ ligand rosiglitazone to HSCs and were shown to decrease the grade of fibrosis in rats (Patel et al. 2012). The use of arginine-glycine-aspartic acid (RGD)-labeled sterically stabilized liposomes (SSLs) encapsulating IFNα-1b could also block fibrogenesis (Du et al. 2007). Sato et al. (2008) evaluated the effects of vitamin A-coupled liposomes containing siRNA targeted to gp46 in liver fibrosis; this treatment decreased collagen deposition and apoptosis induction in HSCs and improved liver functions in an animal model. Liposomes decorated with p-aminophenyl δ-d galactopyranoside, which specifically binds to galactosyl receptor, can be used as carriers to deliver the antioxidant flavonoid quercetin (QC) as a treatment of sodium arsenite (NaASO2)-induced liver fibrosis (Ghosh et al. 2010). Nanotechnology approaches have also been developed to deliver plant extracts to the specific targets in the liver. For example, the alkaloid extract oxymatrine (OM) can inhibit HBV and HCV replication and reduce collagen production and deposition in rats treated with CCl4 (Giannitrapani et al. 2014). Bisht et al. (2011) developed a polymeric nanoparticle formulation of curcumin (NanoCurc™), which accumulates in the environment containing pro-fibrotic HSCs and myofibroblasts; this formulation showed better bioavailability and fibrosis-reducing effects compared with control NPs in CCl4-treated animals. Therefore, due to the beneficial effects of NPs, including their bioavailability, target region specificity, and lower number of side effects, NPs may represent a novel delivery approach for anti-fibrotic drugs to treat liver fibrosis. However, more studies are required to assess the toxicity of NPs; the therapeutic benefits of NPs must be balanced against the potentially harmful risks (Giannitrapani et al. 2014).
-
6.
Nutrients: Previous study indicated that improvement of the nutritional state by a branched chain amino acid (BCAA)-enriched elemental diet combined with a liver diet (restricted energy and protein) could improve the clinical symptoms of liver failure patients and ameliorate DEN (diethylnitrosamine)-induced liver fibrosis by lowering the levels of α-SMA, VEGF, phospho-β-catenin, p-38, and PCNA, and increasing the activation of caspase-3 in animals (Cha et al. 2013; Matsuoka et al. 2014). α-lipoic acid (LA), an organosulfur compound derived from octanoic acid which is made from fatty acid synthesis and is an antioxidant present in almost all foods, acts as a cofactor in enzyme systems. In the study conducted by Foo and his colleague, the administration of LA to TAA-treated rats reduced the incidence of cirrhosis, attenuated hepatic fibrosis, and improved AST/ALT activities. Mechanistic studies indicated that LA/DHLA (dihydrolipoic acid, the reduced form of LA) may be involved in the interruption of ROS-related MAPK and PI3K/Akt signaling pathways to inhibit TGF-β and PDGF-induced HSC activation, suggesting that LA/DHLA also has therapeutic potential in patients with chronic liver injury (Foo et al. 2011). The potential effects of omega-3, olmesartan, and their combination on liver fibrosis induced by CCl4 in rats indicated the beneficial effects of omega-3 in attenuating liver fibrosis by reducing liver oxidative stress, augmenting antioxidants, preventing HSC activation, inhibiting HSC proliferation and chemotaxis, and inhibiting HSC fibrogenic response. The results suggested the beneficial effects of omega-3 in the treatment of liver fibrosis (Shaaban et al. 2014).
-
7.
Alternative, complementary, and traditional Chinese medicines: Traditional Chinese medicines (TCMs), especially those medicines for heat-clearing and detoxification, are often used to treat liver disease. The effective TCMs for the treatment of liver fibrosis are reviewed in detail by Zhao et al. (2014). Here we discussed the anti-fibrotic effects of some TCMs tested in the different animal models.
The famous herbal preparations of Fuzheng Huayu capsules/tablets (FZHYC) are typically used to treat liver disease, to improve liver function, and to decrease the expression of fibrosis biomarkers in cirrhotic or fibrotic patients with chronic HBV (CHB) infection (Zhao et al. 2014). Fibrotic patients significantly improved after treatment with FZHYC combined with anti-viral drugs for 24 and 48 weeks (Zhao et al. 2014). In addition, fibrotic patients with CHB who received nucleoside analogues for 2 years or longer showed significant improvements in their liver fibrosis stages, suggesting that Fuzheng Huayu capsules along with continued NAs therapy may represent a safe and effective treatment for fibrotic patients (Tian et al. 2013).
In a CCl4-induced fibrosis model, various TCMs were effective in treating fibrosis. For instance, Panax notoginseng (PNS), a species of the genus Panax, was reported to balance the pro- and anti-fibrotic cytokines by attenuating the fibrotic index degree, the collagen level, and the collagen area in the liver and reducing TGF-β, TNF-α, and IL-6 levels (Peng et al. 2009b). Piper betle leaves inducing active MMP2 expression though Ras/ERK pathway, and inhibiting TIMP2 level followed by attenuated the fibrosis (An et al. 2012). A gradient of Huisheng oral solution (HOS) attenuated collagen deposition and liver fibrosis by suppressing TGF-β signaling pathways (Li et al. 2014c). A similar effect was observed by saikosaponin-d (SSd), an extract from Bupleurum falcatum L., which significantly reduced collagen deposition, decreased TGF-β contents, down-regulated TNF-α, IL-6, and NFκB p65 subunit expression, and increased IκB activity in the liver (Dang et al. 2007; Fan et al. 2007). In the same model, hydroxysafflor yellow A (HSYA) significantly decreased fibrosis, HSC activation, and α-SMA expression, and decreased TGF-β, MEKK3, MEK5 expression, and ERK5 phosphorylation (Zhang et al. 2012). The anti-fibrotic effects of Paeonia lactiflora and Astragalus membranaceus (PAE) extract may be associated with its abilities to act as an antioxidant, to decrease TGF-β and collagen levels, and to inhibit HSC proliferation (Sun et al. 2007). Da Huang Zhe Chong pill (DHZCP) can reverse and alleviate hepatic fibrosis by decreasing the secretion of TNF-α and IL-13 through the down-regulation of p38 and ERK phosphorylation (Cai et al. 2010). Gomisin A is one of the major dibenzocyclooctadiene lignans isolated from Schisandra chinensis Baill; this treatment decreased liver oxidative stress by increasing superoxide dismutase activity and ameliorating inflammatory mediators (Teraoka et al. 2012).
In an alcohol-induced liver fibrosis model, Chunggan extract (CGX) inhibited VEGF, indicating that CGX exerts anti-fibrotic effects. Another study also indicated that CGX significantly decreased TGF-β, inactivated HSCs, and restored the GSH (glutathione) system in a TAA-induced chronic liver injury model in SD rats (Kwak et al. 2011).
In DMN-administered fibrotic rats, treatment with Tet (tetrandrine, an alkaloid isolated from Stephania tetrandra) reduced the expression of α-SMA, NFκB, ICAM-1, and TGF-β (Hsu et al. 2007). Therefore, the advantages of using TCMs to treat liver fibrosis may be associated with the multi-target regulation by these medicines. However, due to the lack of controlled studies and the liberal use of poorly standardized or complex mixtures of botanicals, which raises the risk of liver toxicity, only a few clinical trials using Chinese herbs have been conducted (Zhao et al. 2014) (https://clinicaltrials.gov). More studies are required to further understand the actions, mechanisms, and toxicities of TCMs.
-
8.
Natural products with multi-target approaches: Because of the complexity of pathways and microenvironments involved in the cross talk among different cell types during the fibrogenic process, approaches that target multiple mechanisms may reach optimal therapeutic effects compared with those targeting a single component of fibrogenesis. In this context, natural products with various pharmacological activities have increased therapeutic potential for liver fibrosis. Moreover, most of the natural products are contained in foods that have been used in the human diet for a long time; therefore, their toxicities should be low. The natural products used in the treatment of fibrosis are also discussed in the context of different animal models.
In CCl4-treated animals, the phytophenolic compound curcumin possessed anti-inflammatory activity and was reported to reduce TGF-β expression, induce apoptosis, and suppress proliferation in HSCs, as well as to inhibit ECM formation by enhancing MMP expression through PPARγ activation and to inhibit CTGF expression in HSCs by inhibiting ERK and NFκB activation (O’Connell and Rushworth 2008; Reyes-Gordillo et al. 2008). Bee venom (BV) has long been used to control pain and inflammation in chronic disease. Kim et al. (2010) indicated that BV regulated pro-inflammatory and fibrosis-related genes against liver fibrosis in this model through inhibiting the secretion of IL-1β, TNF-α, α-SMA, and fibronectin. The other peptide component of bee venom, apamin, was reported to attenuate pathological changes and decrease the expression of TGF-β and fibronectin in rats treated with CCl4 (Lee et al. 2014b). In addition, BV melittin, a major bioactive component in Apis mellifera venom, is well-known cytolytic, antimicrobial, and pro-inflammatory peptide that inhibits liver inflammation and fibrosis in TAA-induced liver fibrosis by interrupting NFκB signaling pathway and inflammation (Park et al. 2011). Baicalin is a flavonoid monomer derived from Huangqin that has been shown in China to have potential therapeutic effects on inflammatory diseases. In CCl4-induced fibrosis, administration of baicalin attenuated the liver fibrosis degree, collagen area, and collagen percentage in the liver; additionally, the levels of serum TGF-β, TNF-α, and IL-6 were significantly reduced (Peng et al. 2009a). The major active component of the medicinal Nigella sativa thymoquinone (TQ) obviously reversed liver damage in TAA- and CCl4-treated mice accompanied by α-SMA, collagen I, and TIMP-1 expression and NFκB pathway inactivation (Bai et al. 2014). The anti-fibrotic effects of methanolic black bean extract were also evaluated using a CCl4-induced liver injury model in rats; these antioxidants ameliorated collagen expression (Lopez-Reyes et al. 2008).
In HFD-fed and streptozotocin (STZ)-induced diabetic rats, the other polyphenolic antioxidant salvianolic acid could prevent the pathological progression of liver fibrosis by reducing ROS production, inhibiting α-SMA and TGF-β expression, and exerting mitochondria-protecting effects (Qiang et al. 2014). Oleuropein has antioxidant and anti-inflammatory properties and has also been reported to prevent the progression of fibrosis in HFD-fed mice; the expression of α-SMA and collagen I and the histopathological features of fibrosis were reduced (Kim et al. 2014).
In DMN-treated rats, morin, a member of the flavonoid family, inhibited liver fibrosis. A mechanistic study indicated that morin altered cell cycle distribution in LX-2 cells and down-regulated the expression of GSK-3β-catenin and cyclin D1 (MadanKumar et al. 2014). Choi et al. (2010) investigated the protective effects of the anthocyanin fraction (AF) obtained from the purple-fleshed sweet potato on DMN-induced liver fibrosis in rats and found that AF reduced the incidence of liver fibrotic lesions and the expression of α-SMA, collagen I, and III, demonstrating the preventive and therapeutic effect of AF against liver fibrosis. In a DMN-induced fibrosis model, the aqueous extract of the Platycodi Radix root exerted anti-fibrotic activities through the activation of Nrf2-mediated antioxidant enzymes, leading to increased antioxidant enzyme activities (Choi et al. 2013). In addition, the naturally occurring resveratrol analogue pterostilbene alleviated DMN-induced fibrosis in rats, likely by inhibiting the TGF-β1/Smad signaling pathway (Lee et al. 2013).
In TAA-treated mice, administration of AP (andrographolide, a diterpenoid lactone) inhibited liver neutrophil infiltration, decreased TNF-α and COX-2 expression, and down-regulated hypoxia-inducible genes such as VEGF, suggesting that AP can be considered as a treatment of liver fibrosis (Lee et al. 2014a).
Future prospects
Recent studies that clinically and serially assess biopsy samples from patients with chronic liver disease of diverse etiologies who have been successfully treated have indicated that liver fibrosis is a dynamic, bidirectional process that has an inherent capacity for recovery and remodeling (Ellis and Mann 2012). UDAC (dihydroxylated bile acid) is now the only standard treatment for primary biliary cirrhosis (PBC) patients who present with progressive destruction of small intrahepatic bile ducts, impaired biliary secretion, hepatocellular retention of toxic endogenous bile acids, and the development of fibrosis leading to cirrhosis, which commonly requires liver transplantation. UDAC is also one therapeutic option currently used for cystic-related fibrosis (Cheng et al. 2014b) and has anti-inflammatory effects that can reduce inflammation, fibrosis, and portal pressure in animal models (Fiorucci et al. 2003). However, with all of the tremendous progress that has been made in understanding the mechanisms of hepatic fibrosis, there remains a lack of effective preventive or therapeutic agents for other types of liver fibrosis in patients with chronic liver disease. Therefore, there is an urgent need to develop anti-fibrotic therapies that can prevent, arrest, and reverse liver fibrosis.
Several difficulties exist in developing therapeutic and preventive agents for liver fibrosis using animal models, particularly regarding the studies evaluating anti-fibrotic compounds, for which the efficacy in rodents has not yet been translated to humans (Abu Dayyeh et al. 2011; McHutchison et al. 2010). One possible issue is that the densely cross-linked collagen develops over decades in humans rather than over weeks in rodents (Pellicoro et al. 2014). Advanced liver fibrosis is much less reversible in humans than it is in rodents. In addition, animal models have variable relevance based on the model used, the dosing, and the therapeutic targets being used (Friedman 2010). Therefore, it is unknown whether an anti-fibrotic therapy that is successful in an animal model will result in positive clinical outcomes (Abu Dayyeh et al. 2011). Hence, the animal models used should more closely mimic the key features of the human disease, and the cellular or molecular target and its role in fibrosis should be similar between the animal model and the human disease (Friedman 2010). Moreover, the anti-fibrotic therapies applied in animal models should also be tested for efficacy and safety in humans in clinical settings (Hayashi and Sakai 2011). Therefore, the translation of anti-fibrotic therapies from animals to humans may provide public health benefits (Hayashi and Sakai 2011). For an elaborate review of the animal models applied in the liver fibrosis studies, please refer to the review written by Hayashi and Sakai (2011).
In previous studies, most focus on a single target without considering the complexity of the process of fibrosis and the interactions among cells, soluble mediators, the pro-fibrogenic microenvironment, and the fibrogenic signaling pathways. Therefore, this might represent another limitation for developing therapeutics in animal models; these therapies are far from ready for use as anti-fibrotic strategies in human (Schuppan and Pinzani 2012). Accordingly, due to the complexity of cross talk in the microenvironment, a combination therapy approach or agents against multiple targets may be more effective in the treatment of liver fibrosis (Schuppan and Kim 2013).
Although our understanding of the mechanisms in liver fibrosis is increasing, there remain many questions that must be studied in the future. For example, when and why the liver regenerates after injury, which is a unique quality from other organs, and which mechanism control the advances in fibrosis and decreases in regeneration remain unclear. Although challenges still exist, large-scale studies have investigated novel pathways, targets, and delivery methods to treat liver fibrosis. The goal is to attenuate the development of fibrosis in patients with CLD and to prevent organ failure induced by CLD. These efforts may reveal an ideal anti-fibrotic drug that could be easily delivered with high liver specificity and low liver toxicity in the future. Moreover, the identification of better markers of the fibrosis stage and activity that could be used to predict which patients with CLD will progress to liver cirrhosis or reverse their fibrosis and the performance of a proof of concept clinical trial that establishes a rationale for targeting fibrosis in patients with CLD must be conducted.
References
Abu Dayyeh BK, Yang M, Dienstag JL, Chung RT (2011) The effects of angiotensin blocking agents on the progression of liver fibrosis in the HALT-C Trial cohort. Dig Dis Sci 56:564–568. doi:10.1007/s10620-010-1507-8
Adrian JE, Kamps JA, Poelstra K, Scherphof GL, Meijer DK, Kaneda Y (2007) Delivery of viral vectors to hepatic stellate cells in fibrotic livers using HVJ envelopes fused with targeted liposomes. J Drug Target 15:75–82. doi:10.1080/10611860601141481
An L, Wang X, Cederbaum AI (2012) Cytokines in alcoholic liver disease. Arch Toxicol 86(9):1337–1348. doi:10.1007/s00204-012-0814-6
Anstee QM, Goldin RD, Wright M, Martinelli A, Cox R, Thursz MR (2008) Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J Thromb Haemos 6:1336–1343. doi:10.1111/j.1538-7836.2008.03015.x
Anstee QM, Concas D, Kudo H et al (2010) Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J Hepatol 53(3):542–550. doi:10.1016/j.jhep.2010.03.016
Aravalli RN, Cressman EN, Steer CJ (2013) Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol 87:227–247. doi:10.1007/s00204-012-0931-2
Atorrasagasti C, Peixoto E, Aquino JB et al (2013) Lack of the matricellular protein SPARC (secreted protein, acidic and rich in cysteine) attenuates liver fibrogenesis in mice. PLoS ONE 8:e54962. doi:10.1371/journal.pone.0054962
Bai T, Yang Y, Wu YL et al (2014) Thymoquinone alleviates thioacetamide-induced hepatic fibrosis and inflammation by activating LKB1-AMPK signaling pathway in mice. Int Immunopharmacol 19:351–357. doi:10.1016/j.intimp.2014.02.006
Bansal R, Post E, Proost JH, de Jager-Krikken A, Poelstra K, Prakash J (2011) PEGylation improves pharmacokinetic profile, liver uptake and efficacy of Interferon gamma in liver fibrosis. J Control Release 154:233–240. doi:10.1016/j.jconrel.2011.05.027
Bansal R, Prakash J, De Ruiter M, Poelstra K (2014) Interferon gamma peptidomimetic targeted to hepatic stellate cells ameliorates acute and chronic liver fibrosis in vivo. J Control Release 179:18–24. doi:10.1016/j.jconrel.2014.01.022
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218. doi:10.1172/jci24282
Bennett RG, Heimann DG, Singh S, Simpson RL, Tuma DJ (2013) Relaxin decreases the severity of established hepatic fibrosis in mice. Liver Int. doi:10.1111/liv.12247
Bisht S, Khan MA, Bekhit M et al (2011) A polymeric nanoparticle formulation of curcumin (NanoCurc) ameliorates CCl4-induced hepatic injury and fibrosis through reduction of pro-inflammatory cytokines and stellate cell activation. Lab Invest 91:1383–1395. doi:10.1038/labinvest.2011.86
Boettcher E, Csako G, Pucino F, Wesley R, Loomba R (2012) Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 35:66–75. doi:10.1111/j.1365-2036.2011.04912.x
Bridle KR, Popa C, Morgan ML et al (2009) Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transpl 15:1315–1324. doi:10.1002/lt.21804
Buck M, Chojkier M (2007) A ribosomal S-6 kinase-mediated signal to C/EBP-beta is critical for the development of liver fibrosis. PLoS ONE 2:e1372. doi:10.1371/journal.pone.0001372
Cai HB, Sun XG, Liu ZF et al (2010) Effects of dahuangzhechong pills on cytokines and mitogen activated protein kinase activation in rats with hepatic fibrosis. J Ethnopharmacol 132:157–164. doi:10.1016/j.jep.2010.08.019
Cai XG, Xia JR, Li WD et al (2014) Anti-fibrotic effects of specific-siRNA targeting of the receptor for advanced glycation end products in a rat model of experimental hepatic fibrosis. Mol Med Rep 10:306–314. doi:10.3892/mmr.2014.2207
Canbay A, Guicciardi ME, Higuchi H et al (2003) Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Cin Invest 112:152–159. doi:10.1172/jci17740
Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ (2004) The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. The J Pharmacol Exp Ther 308:1191–1196. doi:10.1124/jpet.103.060129
Cha JH, Bae SH, Kim HL et al (2013) Branched-chain amino acids ameliorate fibrosis and suppress tumor growth in a rat model of hepatocellular carcinoma with liver cirrhosis. PLoS ONE 8:e77899. doi:10.1371/journal.pone.0077899
Chan CC, Cheng LY, Lin CL, Huang YH, Lin HC, Lee FY (2011) The protective role of natural phytoalexin resveratrol on inflammation, fibrosis and regeneration in cholestatic liver injury. Mol Nutr Food Res 55:1841–1849. doi:10.1002/mnfr.201100374
Chavez E, Segovia J, Shibayama M et al (2010) Antifibrotic and fibrolytic properties of celecoxib in liver damage induced by carbon tetrachloride in the rat. Liver Int 30:969–978. doi:10.1111/j.1478-3231.2010.02256.x
Chen CS, Wu CH, Lai YC et al (2008) NF-kappaB-activated tissue transglutaminase is involved in ethanol-induced hepatic injury and the possible role of propolis in preventing fibrogenesis. Toxicology 246:148–157. doi:10.1016/j.tox.2008.01.009
Chen ZZ, Wang ZL, Deng CY et al (2012) (Z)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione protects rats from carbon tetrachloride-induced liver injury and fibrogenesis. World J Gastroenterol 18:654–661. doi:10.3748/wjg.v18.i7.654
Cheng C, Huang C, Ma TT et al (2014a) New surprises of suppressor of cytokine signalling in liver fibrosis. Expert Opin Ther Targets 18:415–426. doi:10.1517/14728222.2014.885953
Cheng K, Ashby D, Smyth RL (2014b) Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev 12: CD000222. doi:10.1002/14651858
Cho IJ, Kim SH, Kim SG (2006) Inhibition of TGFbeta1-mediated PAI-1 induction by oltipraz through selective interruption of Smad 3 activation. Cytokine 35:284–294. doi:10.1016/j.cyto.2006.10.001
Choi JH, Hwang YP, Choi CY, Chung YC, Jeong HG (2010) Anti-fibrotic effects of the anthocyanins isolated from the purple-fleshed sweet potato on hepatic fibrosis induced by dimethylnitrosamine administration in rats. Food Chem Toxicol 48:3137–3143. doi:10.1016/j.fct.2010.08.009
Choi JH, Jin SW, Kim HG et al (2013) Platycodi Radix attenuates dimethylnitrosamine-induced liver fibrosis in rats by inducing Nrf2-mediated antioxidant enzymes. Food Chem Toxicol 56:231–239. doi:10.1016/j.fct.2013.02.033
Coch L, Mejias M, Berzigotti A et al (2014) Disruption of negative feedback loop between vasohibin-1 and vascular endothelial growth factor decreases portal pressure, angiogenesis, and fibrosis in cirrhotic rats. Hepatology 60:633–647. doi:10.1002/hep.26995
Colmenero J, Bataller R, Sancho-Bru P et al (2009) Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am J Physiol Gastroint Liver Physiol 297:G726–G734. doi:10.1152/ajpgi.00162.2009
Dang SS, Wang BF, Cheng YA, Song P, Liu ZG, Li ZF (2007) Inhibitory effects of saikosaponin-d on CCl4-induced hepatic fibrogenesis in rats. World J Gastroenterol 13(4):557–563
D’Argenio G, Amoruso DC, Mazzone G et al (2010) Garlic extract prevents CCl(4)-induced liver fibrosis in rats: the role of tissue transglutaminase. Dig Liver Dis 42:571–577. doi:10.1016/j.dld.2009.11.002
Deng L, Li G, Xi L et al (2009) Hepatitis B virus inhibition in mice by lentiviral vector mediated short hairpin RNA. BMC Gastroenterol 9:73. doi:10.1186/1471-230x-9-73
Di Pascoli M, Divi M, Rodriguez-Vilarrupla A et al (2013) Resveratrol improves intrahepatic endothelial dysfunction and reduces hepatic fibrosis and portal pressure in cirrhotic rats. J Hepatol 58:904–910. doi:10.1016/j.jhep.2012.12.012
Du SL, Pan H, Lu WY, Wang J, Wu J, Wang JY (2007) Cyclic Arg-Gly-Asp peptide-labeled liposomes for targeting drug therapy of hepatic fibrosis in rats. J Pharmacol Exp Ther 322:560–568. doi:10.1124/jpet.107.122481
Duffield JS, Forbes SJ, Constandinou CM et al (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65. doi:10.1172/jci22675
El-Agamy DS, Shebl AM, Said SA (2011) Prevention and treatment of Schistosoma mansoni-induced liver fibrosis in mice. Inflammopharmacology 19:307–316. doi:10.1007/s10787-011-0092-6
Elinav E, Ali M, Bruck R et al (2009) Competitive inhibition of leptin signaling results in amelioration of liver fibrosis through modulation of stellate cell function. Hepatology 49:278–286. doi:10.1002/hep.22584
Ellis EL, Mann DA (2012) Clinical evidence for the regression of liver fibrosis. J Hepatol 56:1171–1180. doi:10.1016/j.jhep.2011.09.024
Ezhilarasan D, Karthikeyan S, Vivekanandan P (2012) Ameliorative effect of silibinin against N-nitrosodimethylamine-induced hepatic fibrosis in rats. Environ Toxicol Pharmacol 34:1004–1013. doi:10.1016/j.etap.2012.07.004
Faghihzadeh F, Adibi P, Rafiei R, Hekmatdoost A (2014) Resveratrol supplementation improves inflammatory biomarkers in patients with nonalcoholic fatty liver disease. Nutr Res 34:837–843. doi:10.1016/j.nutres.2014.09.005
Fan J, Li X, Li P et al (2007) Saikosaponin-d attenuates the development of liver fibrosis by preventing hepatocyte injury. Biochem Cell Biol 85:189–195. doi:10.1139/o07-010
Fernandez-Yunquera A, Ripoll C, Banares R et al (2014) Everolimus immunosuppression reduces the serum expression of fibrosis markers in liver transplant recipients. World J Transplant 4:133–140. doi:10.5500/wjt.v4.i2.133
Fiorucci S, Antonelli E, Morelli A (2003) Nitric oxide and portal hypertension: a nitric oxide-releasing derivative of ursodeoxycholic acid that selectively releases nitric oxide in the liver. Dig Liver Dis 35(Suppl 2):S61–S69
Foo NP, Lin SH, Lee YH, Wu MJ, Wang YJ (2011) alpha-Lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-beta. Toxicology 282:39–46. doi:10.1016/j.tox.2011.01.009
Friedman SL (2008) Hepatic fibrosis—overview. Toxicology 254:120–129. doi:10.1016/j.tox.2008.06.013
Friedman SL (2010) Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol 7:425–436. doi:10.1038/nrgastro.2010.97
Fuessl HS (2014) Rifaximin improves prognosis liver cirrhosis. MMW Fortschr Med 156:35
Gao B, Radaeva S (2013) Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta 1832:1061–1069. doi:10.1016/j.bbadis.2012.09.008
Geng WX, Ding HY, Yi YS (2008) Prevention and treatment of experimental liver fibrosis in rats by Semen Hoveniae extracts. Zhong Yao Cai 31:1550–1552
Ghosh D, Ghosh S, Sarkar S et al (2010) Quercetin in vesicular delivery systems: evaluation in combating arsenic-induced acute liver toxicity associated gene expression in rat model. Chem Biol Interact 186:61–71. doi:10.1016/j.cbi.2010.03.048
Giannitrapani L, Soresi M, Bondi ML, Montalto G, Cervello M (2014) Nanotechnology applications for the therapy of liver fibrosis. World J Gastroenterol 20:7242–7251. doi:10.3748/wjg.v20.i23.7242
Gonsebatt ME, Del Razo LM, Cerbon MA, Zuniga O, Sanchez-Pena LC, Ramirez P (2007) Arsenite induced oxidative damage in mouse liver is associated with increased cytokeratin 18 expression. Arch Toxicol 81:619–626. doi:10.1007/s00204-007-0192-7
Guillot A, Hamdaoui N, Bizy A et al (2014) Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology 59:296–306. doi:10.1002/hep.26598
Hayashi H, Sakai T (2011) Animal models for the study of liver fibrosis: new insights from knockout mouse models. Am J Physiol Gastroint Liver Physiol 300:G729–G738. doi:10.1152/ajpgi.00013.2011
Hernandez-Gea V, Hilscher M, Rozenfeld R et al (2013) Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol 59:98–104. doi:10.1016/j.jhep.2013.02.016
Hernandez-Novoa B, Moreno A, Perez-Elias MJ et al (2014) Raltegravir pharmacokinetics in HIV/HCV-coinfected patients with advanced liver cirrhosis (Child-Pugh C). J Antimicrob Chemother 69:471–475. doi:10.1093/jac/dkt386
Higashi K, Tomigahara Y, Shiraki H et al (2011) A novel small compound that promotes nuclear translocation of YB-1 ameliorates experimental hepatic fibrosis in mice. J Biol Chem 286:4485–4492. doi:10.1074/jbc.M110.151936
Holt MP, Cheng L, Ju C (2008) Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol 84:1410–1421. doi:10.1189/jlb.0308173
Hsu YC, Chiu YT, Cheng CC, Wu CF, Lin YL, Huang YT (2007) Antifibrotic effects of tetrandrine on hepatic stellate cells and rats with liver fibrosis. J Gastroenterol Hepatol 22:99–111. doi:10.1111/j.1440-1746.2006.04361.x
Huang Y, Feng H, Kan T et al (2013) Bevacizumab attenuates hepatic fibrosis in rats by inhibiting activation of hepatic stellate cells. PLoS ONE 8:e73492. doi:10.1371/journal.pone.0073492
Huang CF, Sun CC, Zhao F, Zhang YD, Li DJ (2014a) miR-33a levels in hepatic and serum after chronic HBV-induced fibrosis. J Gastroenterol. doi:10.1007/s00535-014-0986-3
Huang JQ, Tao R, Li L et al (2014b) Involvement of heat shock protein 47 in Schistosoma japonicum-induced hepatic fibrosis in mice. Int J Parasitol 44:23–35. doi:10.1016/j.ijpara.2013.08.009
Ikejima K, Okumura K, Kon K, Takei Y, Sato N (2007) Role of adipocytokines in hepatic fibrogenesis. J Gastroenterol Hepatol 22(Suppl 1):S87–S92. doi:10.1111/j.1440-1746.2007.04961.x
Inagaki Y, Higashiyama R, Higashi K (2012) Novel anti-fibrotic modalities for liver fibrosis: molecular targeting and regenerative medicine in fibrosis therapy. J Gastroenterol Hepatol 27(Suppl 2):85–88. doi:10.1111/j.1440-1746.2011.07006.x
Iredale JP (2007) Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 117:539–548. doi:10.1172/jci30542
Jang YO, Kim YJ, Baik SK et al (2014) Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: a pilot study. Liver Int 34:33–41. doi:10.1111/liv.12218
Jiang JX, Mikami K, Venugopal S, Li Y, Torok NJ (2009) Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol 51:139–148. doi:10.1016/j.jhep.2009.03.024
Jung KH, Shin HP, Lee S et al (2009) Effect of human umbilical cord blood-derived mesenchymal stem cells in a cirrhotic rat model. Liver Int 29:898–909. doi:10.1111/j.1478-3231.2009.02031.x
Kahraman A, Bronk SF, Cazanave S et al (2009) Matrix metalloproteinase inhibitor, CTS-1027, attenuates liver injury and fibrosis in the bile duct-ligated mouse. Hepatol Res 39:805–813. doi:10.1111/j.1872-034X.2009.00541.x
Kato J, Koda M, Kishina M et al (2012) Therapeutic effects of angiotensin II type 1 receptor blocker, irbesartan, on non-alcoholic steatohepatitis using FLS-ob/ob male mice. Int J Mol Med 30:107–113. doi:10.3892/ijmm.2012.958
Kim SJ, Park JH, Kim KH et al (2010) Bee venom inhibits hepatic fibrosis through suppression of pro-fibrogenic cytokine expression. Am J Chin Med 38:921–935. doi:10.1142/s0192415x10008354
Kim SG, Kim YM, Choi JY et al (2011) Oltipraz therapy in patients with liver fibrosis or cirrhosis: a randomized, double-blind, placebo-controlled phase II trial. J Pharm Pharmacol 63:627–635. doi:10.1111/j.2042-7158.2011.01259.x
Kim MY, Cho MY, Baik SK et al (2012) Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis—a randomized open-label controlled study. Liver Int 32:977–987. doi:10.1111/j.1478-3231.2012.02774.x
Kim SW, Hur W, Li TZ et al (2014) Oleuropein prevents the progression of steatohepatitis to hepatic fibrosis induced by a high-fat diet in mice. E Exp Mol Med 46:e92. doi:10.1038/emm.2014.10
Kitamura K, Tada S, Nakamoto N et al (2007) Rho/Rho kinase is a key enzyme system involved in the angiotensin II signaling pathway of liver fibrosis and steatosis. J Gastroenterol Hepatol 22:2022–2033. doi:10.1111/j.1440-1746.2006.04735.x
Klein S, Klosel J, Schierwagen R et al (2012) Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab Invest 92:1440–1450. doi:10.1038/labinvest.2012.106
Krizhanovsky V, Yon M, Dickins RA et al (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134:657–667. doi:10.1016/j.cell.2008.06.049
Kumar V, Mundra V, Mahato RI (2014) Nanomedicines of Hedgehog inhibitor and PPAR-gamma agonist for treating liver fibrosis. Pharm Res 31:1158–1169. doi:10.1007/s11095-013-1239-5
Kwak KG, Wang JH, Shin JW, Lee DS, Son CG (2011) A traditional formula, Chunggan extract, attenuates thioacetamide-induced hepatofibrosis via GSH system in rats. Human Exp Toxicol 30:1322–1332. doi:10.1177/0960327110389502
Lee UE, Friedman SL (2011) Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol 25:195–206. doi:10.1016/j.bpg.2011.02.005
Lee ES, Shin MO, Yoon S, Moon JO (2010) Resveratrol inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Arch Pharm Res 33:925–932. doi:10.1007/s12272-010-0616-4
Lee TF, Lin YL, Huang YT (2012) Protective effects of kaerophyllin against liver fibrogenesis in rats. Eur J Clin Invest 42:607–616. doi:10.1111/j.1365-2362.2011.02625.x
Lee MF, Liu ML, Cheng AC et al (2013) Pterostilbene inhibits dimethylnitrosamine-induced liver fibrosis in rats. Food Chem 138:802–807. doi:10.1016/j.foodchem.2012.11.094
Lee TY, Chang HH, Wen CK, Huang TH, Chang YS (2014a) Modulation of thioacetamide-induced hepatic inflammations, angiogenesis and fibrosis by andrographolide in mice. J Ethnopharmacol 158 Pt A:423–430. doi:10.1016/j.jep.2014.10.056
Lee WR, Kim KH, An HJ et al (2014b) Apamin inhibits hepatic fibrosis through suppression of transforming growth factor beta1-induced hepatocyte epithelial-mesenchymal transition. Biochem Biophy Res Commun 450:195–201. doi:10.1016/j.bbrc.2014.05.089
Li GM, Li DG, Xie Q et al (2008) Effect of silencing connective tissue growth factor on rat liver fibrosis and the accumulation of extracellular matrix. Chin J Hepatol 16:188–192
Li T, Yan Y, Wang B et al (2013) Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cell Dev 22:845–854. doi:10.1089/scd.2012.0395
Li J, Li X, Xu W et al (2014a) Antifibrotic effects of luteolin on hepatic stellate cells and liver fibrosis by targeting AKT/mTOR/p70S6K and TGFbeta/Smad signalling pathways. Liver Int 35:1222–1233
Li LC, Li J, Gao J (2014b) Functions of galectin-3 and its role in fibrotic diseases. J Pharmacol Exp Ther 351:336–343. doi:10.1124/jpet.114.218370
Li W, Wu Y, Zhu C, Wang Z, Gao R, Wu Q (2014c) Anti-fibrosis effects of Huisheng oral solution in CCl4-induced hepatic fibrosis in rat. Indian J Pharmacol 46:216–221. doi:10.4103/0253-7613.129323
Lipson KE, Wong C, Teng Y, Spong S (2012) CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 5:S24. doi:10.1186/1755-1536-5-s1-s24
Loguercio C, Federico A, Trappoliere M et al (2007) The effect of a silybin-vitamin e-phospholipid complex on nonalcoholic fatty liver disease: a pilot study. Dig Dis Sci 52:2387–2395. doi:10.1007/s10620-006-9703-2
Lopez-Reyes AG, Arroyo-Curras N, Cano BG et al (2008) Black bean extract ameliorates liver fibrosis in rats with CCl4-induced injury. Ann Hepatol 7:130–135
Loumbourdis NS (2005) Hepatotoxic and nephrotoxic effects of cadmium in the frog Rana ridibunda. Arch Toxicol 79:434–440. doi:10.1007/s00204-005-0652-x
Luedde T, Kaplowitz N, Schwabe RF (2014) Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterol 147(765–783):e4. doi:10.1053/j.gastro.2014.07.018
MadanKumar P, NaveenKumar P, Manikandan S, Devaraj H, NiranjaliDevaraj S (2014) Morin ameliorates chemically induced liver fibrosis in vivo and inhibits stellate cell proliferation in vitro by suppressing Wnt/beta-catenin signaling. Toxicol Appl Pharmacol 277:210–220. doi:10.1016/j.taap.2014.03.008
Malhi H, Gores GJ (2008) Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis 28:360–369. doi:10.1055/s-0028-1091980
Mann J, Chu DC, Maxwell A, et al (2010) MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterol 138:705–714, 714 e1–4, doi:10.1053/j.gastro.2009.10.002
Matsuoka S, Tamura A, Nakagawara H, Moriyama M (2014) Improvement in the nutritional status and clinical conditions of patients with liver failure using a liver diet combined with a branched chain amino acids-enriched elemental diet. Hepatogastroenterology 61:1308–1312
McCaffrey AP, Nakai H, Pandey K et al (2003) Inhibition of hepatitis B virus in mice by RNA interference. Nat Biotechnol 21:639–644. doi:10.1038/nbt824
McHutchison J, Goodman Z, Patel K, et al (2010) Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterol 138:1365–73, 1373 e1–2. doi:10.1053/j.gastro.2009.12.003
Medici V, Virata MC, Peerson JM et al (2011) S-adenosyl-l-methionine treatment for alcoholic liver disease: a double-blinded, randomized, placebo-controlled trial. Alcohol Clin Exp Res 35:1960–1965. doi:10.1111/j.1530-0277.2011.01547.x
Meng F, Wang K, Aoyama T et al (2012) Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterol 143(765–76):e1–e3. doi:10.1053/j.gastro.2012.05.049
Moran-Salvador E, Titos E, Rius B et al (2013) Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J Hepatol 59:1045–1053. doi:10.1016/j.jhep.2013.06.023
Moreira de Macedo S, Guimaraes TA, Feltenberger JD, Sousa Santos SH (2014) The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 62:189–196. doi:10.1016/j.peptides.2014.10.005
Motawi TM, Atta HM, Sadik NA, Azzam M (2014) The therapeutic effects of bone marrow-derived mesenchymal stem cells and simvastatin in a rat model of liver fibrosis. Cell Biochem Biophys 68:111–125. doi:10.1007/s12013-013-9698-1
Murakami Y, Toyoda H, Tanaka M et al (2011) The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS ONE 6:e16081. doi:10.1371/journal.pone.0016081
Muriel P, Moreno MG, Hernandez Mdel C, Chavez E, Alcantar LK (2005) Resolution of liver fibrosis in chronic CCl4 administration in the rat after discontinuation of treatment: effect of silymarin, silibinin, colchicine and trimethylcolchicinic acid. Basic Clin Pharmacol 96:375–380. doi:10.1111/j.1742-7843.2005.pto_06.x
Nasir GA, Mohsin S, Khan M et al (2013) Mesenchymal stem cells and Interleukin-6 attenuate liver fibrosis in mice. J Transl Med 11:78. doi:10.1186/1479-5876-11-78
Nissar AU, Farrukh MR, Kaiser PJ et al (2013) Effect of N-acetyl cysteine (NAC), an organosulfur compound from Allium plants, on experimentally induced hepatic prefibrogenic events in Wistar rat. Phytomedicine 20:828–833. doi:10.1016/j.phymed.2013.03.009
Novo E, Povero D, Busletta C et al (2012) The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J Pathol 226:588–597. doi:10.1002/path.3005
Novo E, Cannito S, Paternostro C, Bocca C, Miglietta A, Parola M (2014) Cellular and molecular mechanisms in liver fibrogenesis. Arch Biochem Biophys 548:20–37. doi:10.1016/j.abb.2014.02.015
Oakley F, Teoh V, Ching ASG et al (2009) Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterol 136(2334–2344):e1. doi:10.1053/j.gastro.2009.02.081
O’Connell MA, Rushworth SA (2008) Curcumin: potential for hepatic fibrosis therapy? Br J Pharmacol 153:403–405. doi:10.1038/sj.bjp.0707580
Oh SW, Kim DH, Ha JR, Kim DY (2009) Anti-fibrotic effects of a methylenedioxybenzene compound, CW209292 on dimethylnitrosamine-induced hepatic fibrosis in rats. Biol Pharm Bull 32:1364–1370
Oliva J, Bardag-Gorce F, Li J, French BA, French SW (2011) S-adenosylmethionine prevents the up regulation of Toll-like receptor (TLR) signaling caused by chronic ethanol feeding in rats. Exp Mol Path 90:239–243. doi:10.1016/j.yexmp.2011.01.005
Olteanu D, Nagy A, Dudea M et al (2012) Hepatic and systemic effects of rosuvastatin on an experimental model of bile duct ligation in rats. J Physiol Pharmacol 63:483–496
Ouyang X, Ghani A, Mehal WZ (2013) Inflammasome biology in fibrogenesis. Biochim Biophys Acta 1832:979–988. doi:10.1016/j.bbadis.2013.03.020
Paakko P, Anttila S, Sormunen R et al (1996) Biochemical and morphological characterization of carbon tetrachloride-induced lung fibrosis in rats. Arch Toxicol 70:540–552
Park JH, Kum YS, Lee TI et al (2011) Melittin attenuates liver injury in thioacetamide-treated mice through modulating inflammation and fibrogenesis. Exp Biol Med 236:1306–1313. doi:10.1258/ebm.2011.011127
Park SJ, Sohn HY, Park SI (2013) TRAIL regulates collagen production through HSF1-dependent Hsp47 expression in activated hepatic stellate cells. Cell Signal 25:1635–1643. doi:10.1016/j.cellsig.2013.04.001
Park SA, Kim MJ, Park SY et al (2014) EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-beta/Smad and ROS signaling. Cell Mol Life Sci. doi:10.1007/s00018-014-1798-6
Patel G, Kher G, Misra A (2012) Preparation and evaluation of hepatic stellate cell selective, surface conjugated, peroxisome proliferator-activated receptor-gamma ligand loaded liposomes. J Drug Target 20:155–165. doi:10.3109/1061186x.2011.610800
Patsenker E, Schneider V, Ledermann M et al (2011) Potent antifibrotic activity of mTOR inhibitors sirolimus and everolimus but not of cyclosporine A and tacrolimus in experimental liver fibrosis. J Hepatol 55:388–398. doi:10.1016/j.jhep.2010.10.044
Paulsen D, Urban A, Knorr A et al (2013) Inactivated ORF virus shows antifibrotic activity and inhibits human hepatitis B virus (HBV) and hepatitis C virus (HCV) replication in preclinical models. PLoS ONE 8:e74605. doi:10.1371/journal.pone.0074605
Pellicoro A, Aucott RL, Ramachandran P et al (2012) Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 55:1965–1975. doi:10.1002/hep.25567
Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA (2014) Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 14:181–194. doi:10.1038/nri3623
Peng XD, Dai LL, Huang CQ, He CM, Chen LJ (2009a) Correlation between anti-fibrotic effect of baicalin and serum cytokines in rat hepatic fibrosis. World J Gastroenterol 15:4720–4725
Peng XD, Dai LL, Huang CQ, He CM, Yang B, Chen LJ (2009b) Relationship between anti-fibrotic effect of Panax notoginseng saponins and serum cytokines in rat hepatic fibrosis. Biochem Biophys Res Commun 388:31–34. doi:10.1016/j.bbrc.2009.07.099
Piguet AC, Majumder S, Maheshwari U et al (2014) Everolimus is a potent inhibitor of activated hepatic stellate cell functions in vitro and in vivo, while demonstrating anti-angiogenic activities. Clin Sci 126:775–784. doi:10.1042/cs20130081
Popov Y, Schuppan D (2009) Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology 50:1294–1306. doi:10.1002/hep.23123
Pradere JP, Kluwe J, De Minicis S et al (2013) Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58:1461–1473. doi:10.1002/hep.26429
Qiang G, Yang X, Xuan Q et al (2014) Salvianolic Acid a prevents the pathological progression of hepatic fibrosis in high-fat diet-fed and streptozotocin-induced diabetic rats. Am J Chin Med 42:1183–1198. doi:10.1142/s0192415x14500748
Ratziu V, Giral P, Jacqueminet S et al (2008) Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 135:100–110. doi:10.1053/j.gastro.2008.03.078
Reyes-Gordillo K, Segovia J, Shibayama M et al (2008) Curcumin prevents and reverses cirrhosis induced by bile duct obstruction or CCl4 in rats: role of TGF-beta modulation and oxidative stress. Fund Clin Pharmacol 22:417–427. doi:10.1111/j.1472-8206.2008.00611.x
Rockey DC (2013) Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin Gastroenterol Hepatol 11(224–31):e1–e5. doi:10.1016/j.cgh.2013.01.005
Safar Zadeh E, Lungu AO, Cochran EK et al (2013) The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol 59:131–137. doi:10.1016/j.jhep.2013.02.007
Sakaida I, Terai S, Yamamoto N et al (2004) Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology 40:1304–1311. doi:10.1002/hep.20452
Sato Y, Murase K, Kato J et al (2008) Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol 26:431–442. doi:10.1038/nbt1396
Schuppan D, Kim YO (2013) Evolving therapies for liver fibrosis. J Clin Invest 123:1887–1901. doi:10.1172/jci66028
Schuppan D, Pinzani M (2012) Anti-fibrotic therapy: lost in translation? J Hepatol 56(Suppl 1):S66–S74. doi:10.1016/s0168-8278(12)60008-7
Sekiya Y, Ogawa T, Yoshizato K, Ikeda K, Kawada N (2011) Suppression of hepatic stellate cell activation by microRNA-29b. Biochem Biophys Res Commun 412:74–79. doi:10.1016/j.bbrc.2011.07.041
Seo KW, Sohn SY, Bhang DH, Nam MJ, Lee HW, Youn HY (2014) Therapeutic effects of hepatocyte growth factor-overexpressing human umbilical cord blood-derived mesenchymal stem cells on liver fibrosis in rats. Cell Biol Int 38:106–116. doi:10.1002/cbin.10186
Shaaban AA, Shaker ME, Zalata KR, El-kashef HA, Ibrahim TM (2014) Modulation of carbon tetrachloride-induced hepatic oxidative stress, injury and fibrosis by olmesartan and omega-3. Chem Biol Interact 207:81–91. doi:10.1016/j.cbi.2013.10.008
Sharvit E, Abramovitch S, Reif S, Bruck R (2013) Amplified inhibition of stellate cell activation pathways by PPAR-gamma, RAR and RXR agonists. PloS one 8:e76541. doi:10.1371/journal.pone.0076541
Shimozono R, Asaoka Y, Yoshizawa Y et al (2013) Nrf2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol Pharmacol 84:62–70. doi:10.1124/mol.112.084269
Sokolovic A, Rodriguez-Ortigosa CM, Bloemendaal LT, Oude Elferink RP, Prieto J, Bosma PJ (2013) Insulin-like growth factor 1 enhances bile-duct proliferation and fibrosis in Abcb4(−/−) mice. Biochim Biophys Acta 1832:697–704. doi:10.1016/j.bbadis.2013.02.005
Sullivan BP, Weinreb PH, Violette SM, Luyendyk JP (2010) The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. Am J Pathol 177:2837–2849. doi:10.2353/ajpath.2010.100425
Sun WY, Wei W, Wu L, Gui SY, Wang H (2007) Effects and mechanisms of extract from Paeonia lactiflora and Astragalus membranaceus on liver fibrosis induced by carbon tetrachloride in rats. J Ethnopharmacol 112:514–523. doi:10.1016/j.jep.2007.04.005
Sun W, Gui S, Wu L, Wang H, Wei W (2010) Effects of Shaoqiduogan on MMP-13, TIMP-1 expression in liver and hepatic stellate cells of hepatic fibrosis rats. Chin J Chin Mater Med 35(11):1447–1451
Teerlink JR, Cotter G, Davison BA et al (2013) Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 381:29–39. doi:10.1016/s0140-6736(12)61855-8
Teraoka R, Shimada T, Aburada M (2012) The molecular mechanisms of the hepatoprotective effect of gomisin A against oxidative stress and inflammatory response in rats with carbon tetrachloride-induced acute liver injury. Biol Pharm Bull 35:171–177
Thoen LF, Guimaraes EL, Dolle L et al (2011) A role for autophagy during hepatic stellate cell activation. J Hepatol 55:1353–1360. doi:10.1016/j.jhep.2011.07.010
Thompson KJ, Lakner AM, Cross BW et al (2011) S-adenosyl-l-methionine inhibits collagen secretion in hepatic stellate cells via increased ubiquitination. Liver Int 31:891–901. doi:10.1111/j.1478-3231.2011.02512.x
Tian YL, Zhu XY, Yin WW, Zang ZD, Wang L, Fu XL (2013) Supplemental Fuzhenghuayu capsule therapy for improving liver fibrosis markers in patients with chronic hepatitis B following unsatisfactory outcome of nucleos(t)ide analogue monotherapy. Chin J Hepatol 21(7):514–518. doi:10.3760/cma.j.issn.1007-3418.2013.07.010
Toda K, Kumagai N, Kaneko F et al (2009) Pentoxifylline prevents pig serum-induced rat liver fibrosis by inhibiting interleukin-6 production. J Gastroenterol Hepatol 24:860–865. doi:10.1111/j.1440-1746.2008.05749.x
Tox U, Scheller I, Kociok N et al (2007) Expression of angiotensin II receptor type 1 is reduced in advanced rat liver fibrosis. Dig Dis Sci 52:1995–2005. doi:10.1007/s10620-006-9133-1
Traber PG, Zomer E (2013) Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 8:e83481. doi:10.1371/journal.pone.0083481
Traber PG, Chou H, Zomer E et al (2013) Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 8:e75361. doi:10.1371/journal.pone.0075361
Trappoliere M, Federico A, Tuccillo C et al (2005) Effects of a new pharmacological complex (silybin + vitamin-E + phospholipids) on some markers of the metabolic syndrome and of liver fibrosis in patients with hepatic steatosis. Preliminary study. Minerva Gastroenterol Dietol 51:193–199
Van Bergen T, Marshall D, Van de Veire S et al (2013) The role of LOX and LOXL2 in scar formation after glaucoma surgery. Invest Ophth Vis Sci 54:5788–5796. doi:10.1167/iovs.13-11696
Wang ZR, Wang JH, Hu CL et al (2011) The effect of down-regulation of Smad3 by RNAi on hepatic stellate cells and a carbon tetrachloride-induced rat model of hepatic fibrosis. Braz J Med Biol Res 44:91–99
Wang ZL, Deng CY, Zheng H et al (2012) (Z)2-(5-(4-methoxybenzylidene)-2, 4-dioxothiazolidin-3-yl) acetic acid protects rats from CCl(4)-induced liver injury. J Gastroenterol Hepatol 27:966–973. doi:10.1111/j.1440-1746.2011.06913.x
Wang L, Li J, Liu H et al (2013) Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J Gastroenterol Hepatol 28(Suppl 1):85–92. doi:10.1111/jgh.12029
Weerachayaphorn J, Luo Y, Mennone A, Soroka CJ, Harry K, Boyer JL (2014) Deleterious effect of oltipraz on extrahepatic cholestasis in bile duct-ligated mice. J Hepatol 60:160–166. doi:10.1016/j.jhep.2013.08.015
Weng TC, Shen CC, Chiu YT, Lin YL, Kuo CD, Huang YT (2009) Inhibitory effects of armepavine against hepatic fibrosis in rats. J Biomed Sci 16:78. doi:10.1186/1423-0127-16-78
Williams EJ, Benyon RC, Trim N et al (2001) Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut 49:577–583
Yang YY, Lee KC, Huang YT et al (2008) Effects of N-acetylcysteine administration in hepatic microcirculation of rats with biliary cirrhosis. J Hepatol 49:25–33. doi:10.1016/j.jhep.2008.02.012
Yang FR, Fang BW, Lou JS (2010) Effects of Haobie Yangyin Ruanjian decoction on hepatic fibrosis induced by carbon tetrachloride in rats. World J Gastroenterol 16:1458–1464
Yoshiji H, Noguchi R, Kuriyama S et al (2005) Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am J Physiol Gastrointest Liver Physiol 288:G907–G913. doi:10.1152/ajpgi.00420.2004
Zeng T, Zhang CL, Zhao XL, Xie KQ (2014) Pentoxifylline for the treatment of nonalcoholic fatty liver disease: a meta-analysis of randomized double-blind, placebo-controlled studies. Eur J Gastroenterol Hepatol 26:646–653. doi:10.1097/meg.0000000000000068
Zhang AL, Yang Z, Xiao L, Li DJ, Niu LW (2007a) Regulatory effects of RhoGTPase on transition of liver sinusoidal capillarization: experiment with mice of schistosomal hepatic fibrosis. Zhonghua yi xue za zhi 87:1564–1569
Zhang Y, Li P, Li G et al (2007b) The mechanism of how anti-IL-18 prevents concanavalin-A-induced hepatic fibrosis on a mouse model. J Surg Res 142:175–183. doi:10.1016/j.jss.2007.01.024
Zhang YB, Dong HY, Zhao XM et al (2012) Hydroxysafflor yellow A attenuates carbon tetrachloride-induced hepatic fibrosis in rats by inhibiting ERK5 signaling. Am J Chin Med 40:481–494. doi:10.1142/s0192415x12500371
Zhang F, Kong D, Lu Y, Zheng S (2013) Peroxisome proliferator-activated receptor-gamma as a therapeutic target for hepatic fibrosis: from bench to bedside. Cell Mol Life Sci 70:259–276. doi:10.1007/s00018-012-1046-x
Zhang Z, Zhang F, Lu Y, Zheng S (2015) Update on implications and mechanisms of angiogenesis in liver fibrosis. Hepatol Res 45:162–178. doi:10.1111/hepr.12415
Zhao CQ, Zhou Y, Ping J, Xu LM (2014) Traditional Chinese medicine for treatment of liver diseases: progress, challenges and opportunities. J Integr Med 12:401–408. doi:10.1016/s2095-4964(14)60039-x
Zheng SP, Chen YX, Guo JL et al (2013) Recombinant adeno-associated virus-mediated transfer of shRNA against Notch3 ameliorates hepatic fibrosis in rats. Exp Biol Med 238:600–609. doi:10.1177/1535370213480698
Zhong L, Wang X, Wang S, Yang L, Gao H, Yang C (2013) The anti-fibrotic effect of bone morphogenic protein-7(BMP-7) on liver fibrosis. Int J Med Sci 10:441–450. doi:10.7150/ijms.5765
Zhou Y, Jia X, Qin J et al (2010) Leptin inhibits PPARgamma gene expression in hepatic stellate cells in the mouse model of liver damage. Mol Cell Endocrinol 323:193–200. doi:10.1016/j.mce.2010.03.005
Acknowledgment
This study was supported by the Ministry of Science and Technology, Taiwan (MOST 103-2314-B-006-060-MY2 and MOST 103-2321-B-006-019-MY3).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, RJ., Wu, HH. & Wang, YJ. Strategies to prevent and reverse liver fibrosis in humans and laboratory animals. Arch Toxicol 89, 1727–1750 (2015). https://doi.org/10.1007/s00204-015-1525-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-015-1525-6