
Abstract
Alcoholic liver disease (ALD) is associated with a spectrum of liver injury ranging from steatosis and steatohepatitis to fibrosis and cirrhosis. While multifactorial pathogenesis plays a role in the disease progression, enhanced inflammation in the liver during ethanol exposure is a major feature of ALD. Dysregulated cytokine metabolism and activity are crucial to the initiation of alcohol-induced liver injury. The pro-inflammatory cytokine tumor necrosis factor (TNF-α) has been demonstrated to be one of the key factors in the various aspects of pathophysiology of ALD. The immunomodulatory cytokines such as interleukin 10 and interleukin 6 play roles in exerting hepatic protective effects. Adiponectin is an adipose tissue–derived hormone, which displays protective actions on ethanol-induced liver injury. Treatment for mice with adiponectin decreases TNF-α expression, steatosis and prevents alcohol-induced liver injury. Adiponectin exerts its anti-inflammatory effects via suppression of TNF-α expression and induction of anti-inflammatory cytokines such as IL-10. Adiponectin attenuates alcoholic liver injury by the complex network of multiple signaling pathways in the liver, leading to enhanced fatty acid oxidation and reduced steatosis. Interactions between pro- and anti-inflammatory cytokines such as TNFα and adiponectin and other cytokines are likely to play important roles in the development and progression of alcoholic liver disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcoholic liver disease (ALD) is a syndrome of progressive inflammatory liver injury associated with chronic intake of ethanol. It progresses through different stages from steatosis to alcoholic hepatitis to fibrosis/cirrhosis (O’Shea et al. 2010). It is evident that multifactorial pathogenesis plays a role in the disease progression. Among these factors, impaired immune-mediated events lead to chronic inflammation. It is well established that ALD is associated with imbalanced immune responses and increased production of pro-inflammatory cytokines/chemokines (McClain et al. 2004; Tilg and Diehl 2000). Many of the processes related to alcohol-induced liver injury are mediated via cytokines.
Cytokines are low–molecular weight polypeptide mediators of cellular communication that are produced and released by different cell types in the liver (McClain et al. 2004). Cytokines display a wide range of effects such as modulation of inflammatory responses. In ALD, there is increased pro-inflammatory cytokine production by ethanol-induced LPS-activated Kupffer cells (Thurman 1998). TNF-α is believed to be one of the major pro-inflammatory cytokines in alcohol-induced liver injuries, which is involved in inflammatory response, steatosis and cell death (McClain et al. 2004; Tilg and Diehl 2000; Endo et al. 2007; Lawler et al. 1998). In addition to pro-inflammatory cytokines, there are hepatoprotective cytokines such as IL-6 and anti-inflammatory cytokines such as IL-10 (Gao 2005). These two cytokines are produced by ethanol-induced LPS-stimulated Kupffer cells and can attenuate alcohol-induced liver injury. The disease progression ultimately depends on in vitro interactions with these and other cytokines and a balance of the pro and anti-inflammatory factors in the system (Tilg and Diehl 2000). For example, when the gene for IL-10, the anti-inflammatory cytokine, is eliminated, TNF-α-mediated liver injury in mice is exacerbated (Tilg and Diehl 2000; Berg et al. 1995).
While there are many cytokines involved in the regulation of ALD, one cytokine, adiponectin, not only plays a role in regulating inflammation but also enhances lipid metabolism and fatty acid oxidation (Rogers et al. 2008).
Although the mechanisms contributing to the pathogenesis of ALD are not completely understood, significant research has shed light onto our understanding of some of the key cytokines involved in the pathophysiology of ALD.
Tumor necrosis factor-α
Tumor Necrosis Factor (TNF)-α is a pleiotropic cytokine produced by different types of cells in the body. However, in liver, TNF-α is mainly produced by activated Kupffer cells and is involved in the pathophysiology of various conditions including viral hepatitis, alcoholic liver disease, and nonalcoholic fatty liver disease (Tilg and Diehl 2000; Szabo et al. 2007). TNF-α is also an important mediator in various physiological processes such as cell proliferation, inflammation, and cell death (apoptosis; McClain et al. 2004).
LPS-induced Kupffer cell activation and TNF-α effect
Activated Kupffer cells are responsible for the release of pro-inflammatory cytokines including TNF-α. LPS/Toll-like receptor (TLR)-4–mediated activation of Kupffer cells is a central component in the initiation of the pro-inflammatory response in ALD (Szabo et al. 2007). TLRs are pattern recognition receptors that recognize and bind proteins and toxins released by pathogens (Szabo et al. 2006). The receptor most relevant to endotoxin-induced ALD is TLR-4, which is expressed on Kupffer cells as well as on other cell types in the liver (Hines and Wheeler 2004). The endotoxin molecule, also known as lipopolysaccharide (LPS), is a component of the cell walls of Gram-negative bacteria, which normally inhabits the lumen of the colon and terminal ileum. Endotoxin levels are normally extremely low due to the intestinal barrier and Kupffer cell-mediated detoxification function in the liver (Rao et al. 2004). Ethanol consumption promotes hepatic inflammation by increasing translocation of gut-derived endotoxins to the portal circulation and activating Kupffer cells through the LPS/TLR-4 pathway.
Intestinal sterilization with antibiotics prevents alcohol-induced liver injury (Adachi et al. 1995; Nanji et al. 1994a). Studies using macromolecular markers including LPS also show that there is a correlation between intestinal permeability and alcoholic liver damage in experimental animal models (Choudhry et al. 2002; Keshavarzian et al. 1994; Parlesak et al. 2000).
Two glycoproteins have been identified as receptors for LPS: LPS-binding protein (LBP) and CD14 (Uesugi et al. 2002; Wright et al. 1990). CD14 binds to the LPS-LBP complex, which subsequently attaches to the Kupffer cell. The LPS/CD14 complexes then interact with TLR-4. Ligand-induced engagement of the TLR-4 triggers different downstream signaling pathways, causing nuclear factor-κB (NF-κB) activation, which leads to TNF-α production and liver injury (Mandrekar and Szabo 2009).
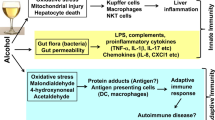
TLR4 acts through two distinct signaling pathways. In the MyD88-(myeloid differentiation primary response gene 88) dependent pathway, the common adaptor, MyD88, recruits interleukin 1 receptor-associated kinase (IRAK) and TNF-α receptor–associated kinase 6 (TRAF6) to the TLR 4 complex. This results in activation of NF-κB and subsequent stimulation of TNF-α production. The second pathway of TLR4 signaling, the MyD88-independent signaling pathway, is initiated by recruiting IL-1 receptor-associated kinase through the adaptor molecular toll/interleukin-1 receptor (TIR-) domain–containing adaptor inducing IFN-β (TRIF). TRIF activation results in production of type 1 interferons (IRFs) and delayed NF-κB activation (Mandrekar and Szabo 2009, Lu et al. 2008; Fig. 1).

Ethanol-induced LPS/TLR4-mediated TNF-α production in alcoholic liver disease. Kupffer cells (KCs) play a major role in development of ALD. KCs, as well as other cells in the liver, express the toll-like receptor 4 (TLR4). Ethanol consumption promotes hepatic inflammation by increasing intestinal tract leakiness, which causes translocation of gut-derived endotoxins (LPS) into the circulation, which then stimulates KCs to produce pro-inflammatory cytokines such as TNF-α through LPS/TLR-4 pathway. Signaling through the TLR-4 receptor complex results in activation of two distinct intracellular signaling cascades: MyD88-dependent pathway and MyD88-independent pathway (TRIF), both of which leads to TNF-α expression
A large number of studies have suggested that Kupffer cell-related cytokine production plays a key role in the pathogenesis of alcohol-induced liver injury. Kupffer cell depletion using gadolinium chloride significantly blunted early alcohol-induced liver injury (Thurman 1998). Activated Kupffer cells are responsible for the release of various mediators, including pro-inflammatory cytokines including TNF-α, IL-1, as well as ROS. ROS participates in inflammation and modulation of hepatocyte metabolism (Wheeler 2003). McClain and Cohen reported the first observation of TNF-α abnormality in cultured monocytes from patients with ALD (McClain and Cohen 1989). These monocytes produced large amounts of TNF-α in response to a LPS stimulus. Elevated serum TNF-α concentrations were also reported in alcoholic hepatitis patients (McClain et al. 1999; Bird et al. 1990). The increased serum TNF-α level has been shown to correlate with disease severity and mortality (Felver et al. 1990). These data were confirmed on animal and cellular studies. Chronically ethanol-fed rats had much higher LPS-stimulated plasma levels of TNF-α than control rats (Honchel et al. 1992). The development of liver injury in the intragastric alcohol-feeding model was also associated with increased TNF-α mRNA expression (Kamimura and Tsukamoto 1995; Nanji et al. 1994b). These results suggest that TNF-α is a principle mediator of early alcohol-induced liver injury leading to progression of the liver disease (Honchel et al. 1992; Hill et al. 1995). This hypothesis was confirmed by studies in animal models using TNF-α receptor knockout mouse. Mice lacking TNR-α receptor-1 showed diminished liver injury (Yin et al. 1999) compared with wild-type enteral ethanol-feeding mice. Anti-TNF-α antibodies prevented inflammation and necrosis in the alcohol-fed rats, further indicating that TNF-α plays an important role in inducing liver injury (Iimuro et al. 1997). Elevated serum levels of TNF-α-inducible cytokines/chemokines including IL-6, IL-8 and IL-18 were also reported in patients with alcoholic hepatitis (Hill et al. 1992; Sheron et al. 1993). In addition, there is increased production of ROS in ALD. ROS can be released from a variety of sources such as activated Kupffer cells, CYP2E1 and NADPH oxidase. NADPH oxidase can enhance activation of NF-κB and phosphorylate the ERK1/2 and p38 MAPK kinases that amplify Kupffer cell production of TNF-α (Albano 2008). CYP2E1 is the minor pathway of ethanol metabolism; after chronic ethanol consumption, there is 5- to 20-fold induction of CYP2E1 activity through both enzyme stabilization and increased gene expression (Lu and Cederbaum 2008). CYP2E1 also potentiates TNF-α-induced hepatotoxicity through increased oxidative stress and mitochondrial dysfunction (Lu and Cederbaum 2010).
There are fewer reports regarding TNF-α’s effects on steatosis (reviewed by Purohit et al. 2009). However, TNF-α has been shown to induce steatosis in the liver of mice by increasing mRNA expression of sterol regulatory element–binding protein-1c (SREBP-1c; Endo et al. 2007) and act as a mediator of SREBP-1 maturation in human hepatocytes (Lawler et al. 1998). TNF-α seems to positively mediate the development of alcoholic fatty liver (steatosis). This correlates with the histopathological observation of acute or chronic alcohol consumption in murine models that steatosis is the earliest stage of liver injury (Yin et al. 2007).
TNF-α-induced apoptosis
TNF-α can induce cell death by apoptosis. Kupffer cells can be stimulated by apoptotic hepatocytes to produce more TNF-α (Canbay et al. 2003). The apoptotic cells also release cytokines such as IL-8 and IL-18 and cause sustained inflammation (Joshi-Barve et al. 2003). TNF-α signals and acts through two distinct cell membrane receptors TNFα-R1 and TNFα-R2. Both TNF-α receptors are a subset of a wider family of receptors that include CD95 (Fas/APO 1), nerve growth receptor, and CD40 (Deaciuc et al. 1995). Hepatocytes have higher amounts of high-affinity, low-capacity TNF-α receptors in comparison with other cells such as Kupffer cells and endothelial cells (Deaciuc et al. 1995). TNFα-R1 is involved in the majority of TNF-α biological activities (McClain et al. 2004). The signaling starts with TNF-α trimer binding to the extracellular domain of TNFα-R1 (Schwabe and Brenner 2006). This binding causes a conformational change to occur in the receptor, leading to the dissociation of the inhibitory protein, silencer of death domains (SODD) from the intracellular death domain. This dissociation allows the death domain to interact with the adapter molecule TNF-α receptor–associated protein with death domain (TRADD). TNFα-R1-bound TRADD then serves as an assembly platform for subsequent protein binding of TNF-α receptor–associated factor (TRAF) 2, receptor-interacting kinase (RIP), and Fas-associated death domain (FADD; Wajant et al. 2003). FADD, which contains a death effector domain, then recruits procaspase-8. Activation of procaspase-8 through self-cleavage leads to a series of downstream events, leading to cell apoptosis.
TNF-α also activates JNK. JNK is a member of the MAPK super family implicated in the cell death pathway activated primarily by cytokines (Davis 2000). With TRAF2 and RIP binding to TRADD, JNK is activated. This is due to the phosphorylation of JNK kinases by upstream kinases such as MEK kinase 1 (MEKK1) and apoptosis signaling kinase (ASK1). JNK is phosphorylated efficiently by MKK 7 and then becomes activated (Tournier et al. 2001). Activated MAPK proteins move from the cytoplasm into the nucleus and activate transcription factors such as c-jun, ATF-2 and Jun D. The JNK pathway is involved in cell differentiation, proliferation and possibly plays proapoptotic roles in TNF-α-induced cell death. The death signaling induced by TNF-α plays a rather minor role compared with its functions in the inflammatory process.
Importantly, hepatocytes with ALD are more susceptible to TNF-α killing, whereas normal hepatocytes are more resistant to TNF-α cell apoptosis. Studies have shown that mice with chronic alcohol intake die quickly in exposure to excess TNF-α (Fernandez-Checa et al. 1997). TNF-α increases mitochondrial energy production, which leads to increased production of ROS and other factors that further induce cell death (Schulze-Osthoff et al. 1992; Higuchi et al. 1997). Alcohol significantly decreased GSH levels in mitochondria (Fernandez-Checa et al. 1997; Neuman et al. 1998). GSH depletion in chronic alcohol-fed rats sensitizes liver to TNF-α-induced hepatocyte killing (Chaisson et al. 2002; Fernandez-Checa et al. 1997).
Adiponectin
Adiponectin is a 30-kDa adipokine, a protein mostly synthesized and secreted by adipocytes. Adiponectin has an amino-terminal collagenous domain and C-terminal globular domain that is structurally similar to complement factor C1q (Kadowaki et al. 2006). Adiponectin circulates in serum as different oligomeric forms: a low–molecular weight (LMW) trimer, a medium–molecular weight (MMW) hexamer and a high–molecular weight (HMW) multimer (Pajvani et al. 2003; Wake et al. 2003; Neumeier et al. 2006). Full-length adiponectin can undergo proteolytic processing to liberate a fragment containing the C-terminal globular domain (gAd), which exists in serum in very small amounts and exhibits more potent biological activity (Fruebis et al. 2001). gAd, similar to its full-length counterpart, possesses anti-diabetic, anti-atherogenic and anti-inflammatory properties (Kadowaki et al. 2006).
Adiponectin is highly abundant in human serum, and its production by adipose tissue is inversely proportional to the body mass index (Arita et al. 1999). Both murine and human adiponectin exert their bioactivity by binding to the specific membrane-bound receptors (AdipoRs), designated as adiponectin receptor 1 (AdipoR1) and adiponectin receptor 2 (AdipoR2; Yamauchi et al. 2003). Initial studies using rodent tissues showed that AdipoR1, with high affinity for globular but low-affinity receptor for full-length adiponectin, is predominantly expressed in skeletal muscle. AdipR2, an intermediate affinity receptor for both forms, shows preference of expression in the liver. In human tissues, AdipoR1 and AdipoR2 mRNAs are most abundant in skeletal muscle and both are moderately expressed in the liver (Yamauchi et al. 2003). Further, AdipoR1 protein was easily detected in human hepatocytes, indicating that both AdipoR1 and AdipoR2 have liver-specific effects (Neumeier et al. 2005).
Role of adiponectin in lipid metabolism
Chronic ethanol feeding decreases circulating levels of adiponectin (Xu et al. 2003; You et al. 2005; Thakur et al. 2006). This is associated with accumulation of lipid. Treatment for mice with adiponectin during ethanol exposure increases fatty acid metabolism (Xu et al. 2003). This suggests that adiponectin plays a role in inducing lipid oxidation. While the exact mechanisms of the adiponectin effects are not quite clear, it appears that adiponectin has the ability to alleviate steatosis in alcoholic liver disease (Xu et al. 2003).
Dyslipidemia (high circulating triglycerides and low levels of high density lipoprotein (HDL) cholesterol) is frequently associated with steatosis, which is characteristic of an early stage of liver injury (Qureshi and Abrams. 2007). Adiponectin negatively correlates with serum triglycerides and apolipoprotein B (Matsubara et al. 2002). The lipid deposition in the liver can be decreased through additional expression of adiponectin (Awazasa et al. 2009). Studies in different animal models with liver injury due to various pharmacological compounds, such as LPS, have demonstrated the hepatoprotective effect of adiponectin (Thakur et al. 2006; Matsumoto et al. 2006; Masaki et al. 2004). Two mechanisms for how adiponectin regulates lipid metabolism are through an increase in insulin sensitivity and interaction with metabolism-regulating factors (reviewed by Rogers et al. 2008).
Insulin-sensitizing effect
Adiponectin is involved in modulation of insulin sensitivity, cardiovascular disease and inflammatory responses. Kadowaki and colleagues contributed to the earlier finding that adiponectin is an insulin-sensitizing adipokine in their animal model studies (Yamauchi et al. 2001). Adiponectin level is decreased in subjects with metabolic syndrome and type 2 diabetes in comparison with healthy individuals (Kadowaki et al. 2006). In obese animal models, lack of insulin sensitivity can be restored by treatment with adiponectin (Berg et al. 2001).
Adiponectin signaling and its protective effect against ALD: adiponectin in the S-adenosyl-l-methionine (SAM) cycle
Homocysteine plays a role in the regulation of adiponectin expression in the liver. In mice models with hyperhomocysteinemia, adiponectin levels were decreased (Song et al. 2008). Chronic ethanol lowers the synthesis of methinoine from homocysteine through inhibition of methionine synthase. The lack of methionine prevents conversion to S-adenosyl methionine in the SAM cycle. This is associated with lowered adiponectin levels and increased liver steatosis. In ethanol-fed animals, SAM supplement lowers the homocysteine levels and helps block ethanol-induced reduction in circulating adiponectin levels (Esfandiari et al. 2007).
AMPK and SREBPS signaling
AMP-activated protein kinase (AMPK) is a key regulator of lipid metabolism in liver. Its activation allows for the metabolism of fatty acids. Adiponectin stimulates AMPK to phosphorylate Acetyl-CoA carboxylase (ACC) and decreases ACC activity. This decrease in ACC blocks the production of malonyl-CoA, an allosteric inhibitor of carnitine palmitoyltransferase I (CPT1). Thus, activity of CPT1 increases, thereby promoting the transportation of fatty acids to the mitochondria for oxidation. In cultured hepatic cells or the livers of ethanol-fed mice, there is decreased AMPK activity and intensified steatosis in the liver, associated with increased ACC activity (You et al. 2004; Garcia-Villafranca et al. 2008). Treating ethanol-fed mice with recombinant adiponectin relieves the liver injury (Xu et al. 2003).
Sterol regulatory element-binding proteins (SREBPS) are transcriptional factors that regulate cholesterol and lipid synthesis. SREBP-1c in particular regulates fatty acid synthesis (Awazasa et al. 2009). Adiponectin activates AMPK, which leads to decreased mRNA and protein expression of SREBP-1c regulating genes, thus reducing lipid synthesis (Yamauchi et al. 2002). Ethanol increases nuclear SREBP-1c levels in cultured hepatic cell lines (You et al. 2002; Ji and Kaplowitz 2003) and different animal models (Esfandiari et al. 2007; Ji et al. 2006). This increased SREBP-1c activity by ethanol is associated with increased mRNA levels of lipogenic enzymes of lipid metabolism. In SREBP-1c knockout mice, there is no steatosis development in liver (Ji et al. 2006). AICAR, an activator of AMPK, has been shown to have antagonistic effects on SREBP-1 and can potentially be used to treat ethanol-activating effects on SREBP-1 (Tomita et al. 2005).
Sirtuin 1 (SIRT1) is a NAD+-dependent class III protein deacetylase that regulates gene expression of SREBP-1c. SIRT has been shown to be downregulated in chronically ethanol-fed animals. Ethanol inhibits SIRT1 activity, which allows for SREBP-1c activation. Resveratrol (agonist of SIRT1) increases adiponectin concentrations with associated increased levels of SIRT1 and/or FOXO1 in adipose tissue (You et al. 2008). The ethanol-mediated decrease in SIRT1 is associated with diminished adiponectin levels during alcoholic liver steatosis.
Treatment with S-adenosylmethionine (SAM) restores adiponectin levels and allows adiponectin to inhibit SREBP-1 activity through AMPK induction (Esfandiari et al. 2007). Globular adiponectin has been shown to increase SIRT1 expression levels. This suggests that adiponectin can normalizes SREBP-1 levels in ethanol exposure by upregulating SIRT1.
Adiponectin regulation of PPARα
Peroxisome proliferator-activated receptor alpha (PPARα) is a transcription factor that controls genes that encode fatty acid oxidation enzymes (Jay and Ren 2007). Adiponectin stimulates PPARα and increases the activity of many hepatic enzymes involved in fatty acid oxidation. Adiponectin activation of PPARα-responsive promoter activity requires the coexpression of PPARγ coactivator-1α (PGC-1α; Bouskila et al. 2005). This suggests that adiponectin induces PPARα activity through upregulation of PGC-1α. Ethanol inhibits PPARα transcription and increases steatosis in the liver. Treating mice with PPARα activators helps assuage the effect of alcohol by increasing fatty acid oxidation enzyme activity. Additional adiponectin enhances this treatment. Thiazolidinedione (TZD), a ligand of PPARγ, increases levels of gene expression and systemic levels of adiponectin (Maeda et al. 2001).
Regulation of adiponectin expression
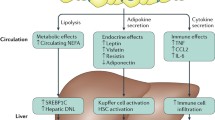
Gene expression and regulation of adiponectin in alcoholic and nonalcoholic liver injuries are highly complex. Ethanol decreases adiponectin secretion in adipocytes. This is associated with disruption in the intracellular trafficking of adiponectin (Thakur et al. 2006). Adiponectin and TNF suppress each other’s gene expression. Ethanol-induced gut-derived endotoxin (LPS) activates Kupffer cells to produce TNF-α. Circulating adiponectin and TNF-α levels have an inverse relationship in several ethanol-feeding animal models, suggesting that increased TNF-α might cause the depression of adiponectin level (Xu et al. 2003; You, et al. 2005; Thakur et al. 2006). Alcohol diminishes Adiponectin receptor 1 (adipoR1) expression. However, treatment with SAM supplements was able to restore this depression of adiponectin activity back to control values (Fig. 2).

The role of adiponectin on fatty acid synthesis and oxidation in the liver. Adiponectin can attenuate alcoholic fatty liver by stimulating AMP-activated protein kinase (AMPK), which inhibits acetyl-CoA carboxylase (ACC), increases CPT-1 activity resulting in increased fatty acid oxidation and decreased synthesis. Adiponectin modulates sterol regulatory element-binding proteins (SREBPS) via activating AMPK, which decreases SREBP-1 activity resulting in decreased lipid synthesis. AICAR, an activator of AMPK, possesses antagonistic effects on SREBP-1 and blunts the ethanol effects on increasing SREBP-1. SIRT-1, a NAD+-dependent class III protein deacetylase, is known to bind to SREBP-1, resulting in its inactivation. Adiponectin also upregulates peroxisome proliferator-activated receptor-α (PPAR-α), which regulates transcription of genes involving in fatty acid oxidation and attenuates alcoholic liver injury
Anti-inflammatory effects of adiponectin
Increasing evidence suggests that the imbalance between pro- and anti-inflammatory factors contributes to alcoholic liver injury. Ethanol intake promotes hepatic inflammation by increasing the translocation of gut-derived endotoxins to the portal circulation and activating the Kupffer cells through a LPS/TLR-4 pathway (Szabo et al. 2006; Hines and Wheeler 2004; Rao et al. 2004). Activated Kupffer cells are responsible for the release of pro-inflammatory cytokines and mediators, particularly TNF-α, interleukin 6 (IL6) and reactive oxygen species (ROS; McClain et al. 2004; Hines and Wheeler 2004), thus leading to progression of liver injury. Adiponectin regulates immune response through both AdipoR1 and AdipR 2 (Yamauchi et al. 2003; Neumeier et al. 2005). Earlier observations show that adiponectin inhibits the proliferation of myelomonocytic progenitor cells, reduces the phagocytic activity of human macrophages and suppresses cytokine production (Yokota et al. 2000). Adiponectin suppresses TNF-α-mediated inflammatory responses in human endothelial cells (Ouchi et al. 2000). In mice fed with alcohol chronically, treatment with adiponectin decreases hepatic steatosis and prevents liver injury (Xu et al. 2003). There is an improvement in fatty acid oxidation and also normalization of TNF-α by adiponectin treatment (Xu et al. 2003). Adiponectin treatment inhibits TLR-mediated NF-κB activation in murine macrophages (Yamaguchi et al. 2005). In a study using murine macrophage-like cells, globular adiponectin (gAd) showed inhibition of NF-κB activation and IκB phosphorylation by negatively regulating TLR signaling (Yamaguchi et al. 2005). LPS-induced NFκB activation and ERK1/2 activity are attenuated through anti-inflammatory effects of adiponectin (Thakur et al. 2006; Wulster-Radcliffe et al. 2004).
In several animal models of immune-mediated hepatitis, adiponectin reduces TNF-α but induces interleukin-10 (IL-10) release from Kupffer cells. Matsumoto and colleagues demonstrated the hepatoprotective effects of adiponectin in the galactosamine (GalN)/LPS mouse model (Matsumoto et al. 2006). After intraperitoneal injection of GalN/LPS in adiponectin deficient mice, plasma IL-10 levels were significantly diminished, while TNF-α levels were remarkably increased; this was associated with more serious liver injury and higher mortality. There was significantly less damage in the wild-type mice, indicating that deficiency of adiponectin could enhance LPS-induced liver injury through modulation of cytokine production by Kupffer cells (Matsumoto et al. 2006). Adiponectin increases IL-10 expression and decreases TNF-α expression through inhibition of NF-κB activation in porcine macrophages (Wulster-Radcliffe et al. 2004). Adiponectin, particularly the globular form, renders macrophages to develop resistance to further stimulation by adiponection (Tsatsanis et al. 2005). Nagy and associates further elaborated on the mechanism of adiponectin actions via pro- and anti-inflammatory cytokines. Their studies show that treatment of rat Kupffer cells with adiponectin can normalize LPS-stimulated TNF-α production after chronic ethanol feeding. This normalization of TNF-α is due to increased sensitivity of Kupffer cells to the inhibitory effects of gAd (Thakur et al. 2006), suggesting the hepatic protective effect of adiponectin. They further demonstrated that expression of IL-10 is required for the anti-inflammatory effects of adiponectin in the macrophages (Tsatsanis et al. 2005), as antibodies to IL-10 prevent gAd-mediated desensitization to LPS (Park et al. 2007). This suggests a possible link between adiponectin and IL-10, two critical anti-inflammatory mediators and their role in ethanol-induced liver injury.
Interleukin-6
Interleukin-6 (IL-6) is a cytokine with a variety of cellular functions in the human body. It belongs to a family of mediators involved in the regulation of acute-phase response to injury and infection (Heinrich et al. 2003). It has pro- as well as anti-inflammatory properties. In the liver, IL-6 is released along with IL-10, TNF-α and other cytokines by activated macrophages/Kupffer cells after alcohol consumption (reviewed in Gao 2005). IL-6 and IL-10 are two cytokines that play roles in reducing alcoholic liver injury and inflammation through activation of STAT 3 (Signal Transducer and Activator of Transcription; Gao 2005).
IL-6 type cytokines bind to plasma membrane receptor complexes containing the common signal-transducing subunit gp130 (glycoprotein 130), which is expressed on hepatocytes at high levels (Gao 2005). There are two signal transduction pathways involved in IL-6 type cytokine signaling: the activation of transcription factors of the STAT family via activating tyrosine kinase family member Janus kinase (JAK) and the mitogen-activated protein kinase (MAPK) cascade (Heinrich et al. 2003). IL-6 binds with IL-6 receptors, forming a complex that induces the gp130 to activate the receptor-associated JAK1, JAK2 and Tyk2 (Gao 2005), which then activates STAT. The activated STAT translocates into the nucleus to activate the transcription of many genes including acute-phase genes (Hibi et al. 1996). Disruption of the STAT3 gene impairs liver regeneration and causes insulin resistance (Li et al. 2002; Inoue et al. 2004).
Studies have demonstrated that IL-6 plays a role in alleviating alcoholic liver injury (reviewed by Miller et al. 2011). IL-6 has been shown to be involved in the acute-phase response, liver regeneration and protection against liver injury (Gao 2005). IL-6 level correlated with the severity of the alcoholic liver disease (Hill et al. 1992). Mice deficient of IL-6 became vulnerable to ethanol-induced steatosis (El-Assal et al. 2004). When mice that were affected by fatty liver disease associated with previously excessive alcohol intake were treated with IL-6, the disease was significantly relieved (Hong et al. 2004). Further, IL-6 pre-treatment for steatotic rat liver graft provided hepatoprotective effects on the transplantation. There is marked improved hepatic circulation and reduced mortality in the ethanol-induced fatty liver transplant in the rat model (Sun et al. 2003). This is due to the activation of the cell survival signal of STAT3. Chronic ethanol plus TNF-α-induced apoptosis is prominent in IL-6 deficient mice but not in wild-type mice (Hong et al. 2002). IL-6 treatment prevented the effects of ethanol-induced hepatic apoptosis.
In ethanol-fed mice, conditional deletion of STAT 3 in hepatocytes enhances hepatic steatosis (Horiguchi et al. 2008), whereas, conditional deletion of STAT 3 in endothelial cells markedly increases both endothelial and hepatocyte damage (Miller et al. 2010). There is clearly a cell type-specific STAT effect in alcoholic liver disease. From studies of cell type-dependent role of STAT 3, hepatocyte STAT3 may act as a pro-inflammatory cytokine. This may be caused by cytokines and other mediators produced by stimulated hepatocytes (Horiguchi et al. 2008). However, the anti-inflammatory effect of STAT3 from myeloid cell linage (macrophages and neutrophils) may dominate the pro-inflammatory effects of STATS 3 in hepatocytes (Miller et al. 2011).
Interleukin-10
Interleukin-10 (IL-10) is a major immunoregulatory cytokine with anti-inflammatory properties (Moore et al. 2001). IL-10 was originally identified as a molecule mainly produced from B cells, activated macrophages/monocytes and T-cell subsets including Th1 cells. IL-10 decreases production of pro-inflammatory cytokines from activated macrophages (Moore et al. 2001). IL-10 also possesses a hepatic protective effect on proliferation and fibrosis (McClain et al. 2002; Louis et al. 2003).
Protective effects of IL-10 on the liver have previously been demonstrated in several models. In these studies, IL-10 inhibits the release of pro-inflammatory cytokines such as TNF-α and Interleukin-6 (IL-6) by macrophages/monocytes. In a study of D-galactosamine (GalN)-sensitized mice, administration of LPS in GalN-sensitized mice caused lethal shock and massive hepatic necrosis in almost all of the mice. This effect was associated with a significant increase in plasma TNF-α level, upregulation of adhesion molecules, and neutrophil infiltration and lethality. Pre-treatment with IL-10 decreased the serum levels of TNF-α, LPS/GalN-induced liver injury and lethality (Santucci et al. 1996).
In the liver, Kupffer cells are the prominent sources of pro-inflammatory cytokines. Kupffer cells produce IL-10 in response to LPS challenge and downregulate the release of IL-6 and TNF-α (Knolle et al. 1995). This is evidenced in an animal model of partial hepatectomy, when TNF-α synthesis is counter-regulated by IL-10 production during the regeneration process (Rai et al. 1997).
Endotoxin administration is an extensively studied model to induce IL-10 from monocytes and macrophages (Fiorentino et al. 1991). Human monocytes activated by LPS are able to produce a high level of IL-10 in a dose-dependent manner (Waal Malefyt et al. 1991). The activated monocytes inhibit production of pro-inflammatory cytokines such as IL-6, IL-1 and TNF-α. Further, LPS enhanced alcohol-induced liver inflammation and injury in the IL-10 knockout mice fed with alcohol to a greater extent than that of the wild-type mice. Pro-inflammatory cytokine levels such as TNF-α and IL-1β were also increased by LPS treatment in the IL-10 knockout mice (Hill et al. 2002).
Ethanol-induced steatosis and lipid accumulation (also a common histological feature to nonalcoholic steatohepatitis-NASH) results from an increase in fatty acid synthesis via transcription factor SREBP-1, which encodes for lipogenic enzymes (Lawler et al. 1998); TNF-α and IL-1 have been implicated in promoting steatosis (Yin et al. 1999; Miura et al. 2010). It has been suggested that steatosis promotes inflammation, as lipid accumulation induces production of pro-inflammatory cytokines (Joshi-Barve et al. 2007; Feldstein et al. 2004), and lipotoxicity enhances hepatic damage and inflammation (Neushwander-Tetri 2010). However, the effects of inflammation on steatosis and liver damage are still unclear.
In a recent study (Miller et al. 2011), Miller and colleagues investigated the role of IL-10 on ALD and NASH by using IL-10 knockout (IL-10−/−) mice. IL-10−/− mice showed greater liver inflammatory response, but less steatosis and hepatocellular damage after alcohol or high fat diet (HFD). Interestingly, they found that IL-10−/− mice produce higher levels of IL-6/STAT3 activation in hepatocytes, which may be responsible for attenuation of steatosis and liver injury; an additional deletion of IL-6 or hepatic STAT 3 restored steatosis and exacerbated inflammation in IL-10−/− mice. In this model, inflammation response reduces steatosis via activation of hepatic IL-6/STAT3 that subsequently downregulates the lipogenic genes. These results further demonstrated the anti-inflammatory effects of IL-10 as well as its possible steatosis-reducing effects through producing IL-6/STATS.
IL-10 interacts with IL-10 receptors. The receptor complex activates the Janus kinases JAK1 and Tyk2. JAK1 initiates phosphorylation of the IL-10R chain. This results in the recruitment of STAT3 to the IL-10R, which activate the anti-inflammatory cascade (Weber-Nordt et al. 1996). IL-10 also stimulates the expression of SOCS3 (suppressor of cytokine signaling 3; Murray 2007), a regulator of gp130 (IL-6 receptor) and other cytokine receptors. SOCS3 is induced by IL-10 and exerts negative regulatory effects on other cytokines besides IL-10. IL-10R does not bind to SOCS and appears to be refractory to the effects of all members of the SOCS family (Williams et al. 2004). Deletion of SOCS3 increases STAT 3 signaling from gp130 (Croker et al. 2003). Therefore, SOCS3 controls the quality and quantity of STAT activation mediated via gp130 through IL-6, allowing interleukin-6 to exert anti-inflammatory effects (Yasukawa et al. 2003).
IL-10 treatment also causes diminished NF-κB activation in response to a variety of different stimuli (Williams et al. 2004). There is suppression of inhibitor of NF-κB (IκB) kinase (IKK) activity, inhibition of translocation of NF-κB from cytoplasm to nucleus and of binding to DNA (Schottelius et al. 1999). The inhibition of NF-κB by IL-10 decreases the effectiveness of immune responses.
Conclusion
The mechanisms underlying alcohol-induced liver injuries are complex, involving interactions of mediators and cytokines of inflammatory response, transcriptional regulators of hepatic lipid metabolism and various systems/pathways.
Ethanol exposure activates a gut-derived LPS-TLR4-Kupffer cell pathway and produces TNF-α, which plays a major role in inducing inflammation in NASH as well as ALD. Alcohol consumption induces steatosis by increasing SREBP-1 and decreasing PPAR-α. TNF-α seems to contribute to alcoholic fatty liver by upregulating SREBP-1 gene expression.
Adiponectin, a hormonal peptide, has been shown to suppress hepatic steatosis by positively affecting transcription regulating factors of lipid metabolism such as AMPK, PGC-1α and PPAR-α, thus enhancing fatty acid oxidation, decreasing fatty acid synthesis and decreasing SREBP-1 activity. Adiponectin has also demonstrated hepatic protective effects by suppressing TNF-α-mediated inflammatory response, liver injury and decreasing LPS-stimulated TNF-α production. IL-10 may be required for the anti-inflammatory effects of adiponectin.
Despite numerous studies, the exact molecular signaling mechanisms of ethanol-mediated inhibition of lipid regulators are still unknown. Further studies will help reveal these details and lead to a better understanding whether any functional modification is possible on these signaling proteins.
IL-10 is one of the most important anti-inflammatory cytokines in ameliorating liver inflammation in different models, whereas IL-6 may play a compensatory role in protecting against alcoholic liver injury. This was further demonstrated in a recent study (Miller et al. 2011) in that IL-10 knockout mice had greater inflammatory response but less steatosis after ethanol or HFD feeding, suggesting that inflammation induced during ethanol exposure may actually attenuate the fatty liver via inducing cytokines such as IL-6, which can activate STAT3 and reduce steatosis. It is probable that interactions between different cytokines and the balance between detrimental cytokines that promote steatosis and hepatoprotective ones that prevent steatosis may determine the disease progression. It will be of great interest to further study the effect of inflammation on steatosis and explore therapeutic options to treat ALD.
References
Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG (1995) Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108:218–224
Albano E (2008) Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Mol Aspects Med 29:9–19
Arita Y, Kihara S, Ouchi N, Takahasshi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraquchi M, Ohmoto Y, Funahashi T, Matsuzwa Y (1999) Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79–83
Awazasa M, Ueki K, Inabe K, Yamauchi T, Kaneko K, Okazaki Y, Bardeesy N, Ohnishi S, Nagai R, Kadowaki T (2009) Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem Biophys Res Commun 382:51–56
Berg DJ, Kuhn R, Rajewsky K, Muller W, Menon S, Davidson N, Grunig G, Rennick D (1995) Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest 96:2339–2347
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001) The adipocyte-secreted protein Acrp 30 enhances hepatic insulin action. Nat Med 7:947–953
Bird GLA, Sheron N, Goka AKJ, Alexander GL, Williams RS (1990) Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann Intern Med 112:917–920
Bouskila M, Pajvani UB, Scherer PE (2005) Adiponectin: a relevant player in PPAR-gamma-agonist-mediated improvements in hepatic insulin sensitivity? Int J Obes 29:S17–S23
Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ (2003) Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 38:1188–1198
Chaisson ML, Brooling JT, Ladiges W, Tsai S, Fausto N (2002) Hepatocyte-specific inhibition of NF-κB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J Clin Invest 110:193–202
Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM (2002) Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol 282:G937–G947
Croker BA, Krebs DL, Zhang JG, Wormald S, Wilson TA, Stanley EG, Robb L, Greengalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4:540–545
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103:239–252
Deaciuc IV, D’Souza NB, Spitzer JJ (1995) Tumor necrosis factor-α cell-surface receptors of liver parenchymal and nonparenchymal cells during acute and chronic alcohol administration to rats. Alcohol Clin Exp Res 19:332–338
El-Assal O, Hong F, Kim WH, Radaeva S, Gao B (2004) IL-6 deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol 1(3):205–211
Endo M, Masaki T, Seike M, Yoshimatsu H (2007) TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c. Exp Biol Med 232:614–621
Esfandiari F, You M, Villanueva JA, Wong DH, French SW, Halsted CH (2007) S-adenosylmethionine attenuates hepatic lipid synthesis in micropigs fed ethanol with a folate- deficient diet. Alcohol Clin Exp Res 31:1231–1239
Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Brugart LJ, Gores GJ (2004) Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40:185–194
Felver ME, Mezey E, McGuire M, Mitchell MC, Herlong HF, Veech VA, Veech RL (1990) Plasma tumor necrosis factor a predicts decreased long term survival in severe alcoholic hepatitis. Alcohol Clin Exp Res 14:255–259
Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, Ardite E, Morales A (1997) GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol 273:G7–G17
Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O’Garra A (1991) IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol 146:3444–3451
Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF (2001) Proteolytic cleavage product of 30-kDa adipocyte complement- related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Acad Sci USA 98:2005–2010
Gao B (2005) Cytokines, STATs and liver disease. Cell Mol Immunol 2:92–100
Garcia-Villafranca J, Guillen A, Castro J (2008) Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 90:460–466
Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Nuwen G, Schaper F (2003) Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J 374:1–20
Hibi M, Nakajima K, Hirano T (1996) IL-6 cytokine family and signal transduction: a model of the cytokine system. J Mol Med 74:1–12
Higuchi M, Aggarwal BB, Yeh ET (1997) Activation of CPP32-like protease in tumor necrosis factor-induced apoptosis is dependent on mitochondrial function. J Clin Invest 99:1751–1758
Hill DB, Marsano L, Cohen D, Allen J, Shedlofsky S, McClain CJ (1992) Increased plasma interleukin-6 concentrations in alcoholic hepatitis. J Lab Clin Med 119(5):547–552
Hill DB, Schmidt J, Sheldlofsky S, Cohen D, McClain CJ (1995) In vitro tumor necrosis factor cytotoxicity in HepG2 liver cells. Hepatology 21:1114–1119
Hill DB, D’Souza NB, Lee EY, Burikhanor R, Deaciuc IV, de Villiers WJ (2002) A role for interleukin-10 in alcohol-induced liver sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res 26(1):74–82
Hines IN, Wheeler MD (2004) Recent advances in alcoholic liver disease III. Role of the innate immune response in alcoholic hepatitis. Am J Physiol Gastrointest Liver Physiol 287:G310–G314
Honchel R, Ray MB, Marsano L (1992) Tumor necrosis factor in alcohol enhanced endotoxin liver injury. Alcohol Clin Exp Res 16:665–669
Hong F, Kim WH, Tian Z, Jaruga B, Ishac E, Shen X, Gao B (2002) Elevated interleukin- 6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: involvement of induction of Bcl-2 and Bcl-x(L) proteins. Oncogene 21(1):32–43
Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B (2004) Interlerlin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology 40(4):933–941
Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D, Moh A, Fu XY, Pacher P, Kunos G, Gao B (2008) Cell type-dependent pro-and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 134(4):1148–1158
Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG (1997) Antibodies to tumor necrosis factor-α attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology 26:1530–1537
Inoue H, Ogawa W, Ozaki M, Haga S, Matsumoto M, Furukawa K, Hashimoto N, Kido Y, Mori T, Sakaue H, Teshiquware K, Jin S, Iquch H, Hiramatsu R, LeRoith D, Takeda K, Akira S, Kasuga M (2004) Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med 10:168–174
Jay MA, Ren J (2007) Peroxisome proliferator-activated receptor (PPAR) in metabolic syndrome and type 2 diabetes mellitus. Curr Diabetes Rev 3:33–39
Ji C, Kaplowitz N (2003) Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 124:1488–1499
Ji C, Chan C, Kaplowitz N (2006) Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol 45:717–724
Joshi-Barve S, Barve SS, Butt W, Klein J, McClain CJ (2003) Inhibition of proteasome function leads to NF-κB-independent IL-8 expression in human hepatocytes. Hepatology 38:1178–1187
Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P, McClain CJ (2007) Palmitic acid induces production of proinflammatory cytokine Interleukin-8 from hepatocytes. Hepatology 46:823–830
Kadowaki T, Yamuchi T, Kubota N, Hara K, Ueki K, Tobe K (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116:1784–1792
Kamimura S, Tsukamoto H (1995) Cytokine gene expression by Kupffer cells in experimental alcoholic liver disease. Hepatology 21:1304–1309
Keshavarzian A, Fields JZ, Vaeth J, Holmes EW (1994) The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am J Gastroenterol 89:2205–2211
Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Buschenfelde KH, Gerken G (1995) Human Kupffer cells secrete IL-10 in response to lipopolysacharide (LPS) challenge. J Hepatol 22:226–229
Lawler JF Jr, Yin M, Diehl AM, Roberts E, Chatterjee S (1998) Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J Biol Chem 273:5053–5059
Li W, Liang X, Kellendonk C, Poli V, Taub R (2002) STAT3 contributes to the mitogenic response of heptocytes during liver regeneration. J Biol Chem 277:28411–28417
Louis H, Le Moine O, Goldman M, Deviere J (2003) Modulation of liver injury by interleukin-10. Acta Gastroenterol Belg 66(1):7–14
Lu Y, Cederbaum AI (2008) CYE2E1 and oxidative liver injury by alcohol. Free Radic Biol Med 44(5):723–738
Lu Y, Cederbaum AL (2010) CYP2E1 potentiation of LPS and TNF-α-induced hepatotoxicity by mechanisms involving enhanced oxidative and nitrosative stress, activation of MAP kinases and mitochondrial dysfunction. Genes Nutr 5:149–167
Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42:145–151
Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y (2001) PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 50(9):2094–2099
Mandrekar P, Szabo G (2009) Signalling pathway in alcohol-induced liver inflammation. J Hepatol 50:1258–1266
Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, Yoshimatsu H (2004) Adiponecin protects LPS-induced liver injury through modulation of TNF-α in KK-Ay obese mice. Hepatology 40:177–184
Matsubara M, Maruoka S, Katayose S (2002) Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur J Endocrinol 147:173–180
Matsumoto H, Tamura S, Kamada Y, Kiso S, Fukushima J, Wada A, Maeda N, Kihara S, Funahashi T, Matsuzawa Y, Shimomura I, Hayashi H (2006) Adiponectin deficiency exacerbates lipopolysaccharide/D-galactosamine-induced liver injury in mice. World J Gastroenterol 12:3352–3358
McClain CJ, Cohen DA (1989) Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 9:349–351
McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D (1999) Cytokines in alcoholic liver desease. Semin Liver Dis 19:205–219
McClain CJ, Hill DB, Song Z, Chawla R, Watson WH, Chen T, Barve S (2002) S-Adenosyl-methionine, cytokines, and alcoholic liver disease. Acohol. 27(3):185–192
McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I (2004) Recent advances in alcoholic liver diseses IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 287:G497–G502
Miller AM, Wang H, Park O, Horiguchi N, Lafdil F, Mukhopadhyay P, Moh A, Fu XY, Kunos G, Pacher P, Gao B (2010) Anti-inflammatory and anti-apoptotic roles of endothelial cell STAT3 in alcoholic liver injury. Alcohol Clin Exp Res 34(4):719–725
Miller AM, Wang H, Bertola A, Park O, Horiguchi M, Ki SH, Yin S, Lafdil F, Gao B (2011) Molecular mechanisms of alcoholic liver diseases: innate immunity and cytokines. Alcohol Clin Exp Res 35(5):787–793
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoynama T, Ohnish H, Olefsky JM, Brenner DA, Seki E (2010) Toll-like receptor 9 promotes steatohepatitis by induction of Interleukin-1 beta in mice. Gastroenterology 139(323–334):e7
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765
Murray PJ (2007) The JAK-STAT signaling pathway: input and output integration. J Immunol 178:2623–2629
Nanji AA, Zhao S, Sadrzadeh SM, Waxman DJ (1994a) Use of reverse transcription-polymerase chain reaction to evaluate in vivo cytokine gene expression in rats fed ethanol for long periods. Hepatology 19:1483–1487
Nanji AA, Khettry U, Sadrzadeh SM (1994b) Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver disease. Proc Soc Exp Biol Med 205:243–247
Neuman MG, Shear NH, Bellentani S, Tiribelli C (1998) Role of cytokines in ethanol-induced cytotoxicity in vitro in HepG2 cell. Gastroenterology 115:157–166
Neumeier M, Weigert J, Schaffler A, Weiss T, Kirchner S, Laberer S, Scholmerich J, Buechler C (2005) Regulation of adiponectin receptor 1 in human hepatocytes by agonists of nuclear receptors. Biochem Biophys Res Commun 334:924–929
Neumeier M, Weigert J, Schaffler A, Wehrwein G, Muller-Ladner U, Scholmerich J, Wrede C, Buechler C (2006) Different effects of adiponectin isoforms in human Monocytic cells. J Leukoc Biol 79:803–808
Neushwander-Tetri BA (2010) Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52:774–788
O’Shea RS, Dasarathy S, McCullough AJ (2010) Alcoholic liver disease. Hepatology 51(1):307–328
Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matwuzawa Y (2000) Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 102:1296–1301
Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE (2003) Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications for metabolic regulation and bioactivity. J Biol Chem 278:9073–9085
Park PH, McMullen MR, Huang H, Thakur V, Nagy LE (2007) Short-term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor-alpha (TNF-α) expression via ERK1/2 activation and Egr-1 expression: role of TNF-α in adiponectin-stimulated interleukin-10 production. J Biol Chem 282:21695–21703
Parlesak A, Schafer C, Schutz T, Bode JC, Bode C (2000) Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 32:742–747
Purohit V, Gao B, Song B-J (2009) Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res 33(2):191–205
Qureshi K, Abrams GA (2007) Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 13:3540–3553
Rai RM, Loffreda S, Karp CL, Yang SQ, Lin HZ, Diehl AM (1997) Kupffer cell depletion abolishes induction of interleukin-10 and permits sustained overexpression of tumor necrosis factor alpha messenger RNA in the regenerating rat liver. Hepatology 25:889–895
Rao RK, Seth A, Sheth P (2004) Recent advances in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 280:G881–G884
Rogers CQ, Ajmo J, You M (2008) Adiponectin and alcoholic fatty liver disease. IUBMB Life 60(12):790–797
Santucci L, Fiorucci S, Chiorean M, Brunori PM, Di Matteo FM, Sidoni A, Migliorati G, Morelli A (1996) Interleukin 10 reduces lethality and hepatic injury induced by lipopolysaccharide in galactosamine-sensitized mice. Gastroenterology 111:736–744
Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS Jr (1999) Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kapparB DNA binding. J Biol Chem 274:31868–31874
Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W (1992) Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions: evidence for the involvement of mitochondrial radical generation. J Biol Chem 267:5317–5323
Schwabe RF, Brenner DA (2006) Mechanisms of liver injury. I. TNF-α-induced liver injury: role of IKK, INK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 290:G583–G589
Sheron N, Bird G, Koskinas J, Portmann B, Ceska M, Lindley I, Williams R (1993) Circulating and tissue levels of the neutrophil chemotaxin interleukin-8 are elevated in severe acute alcoholic hepatitis, and tissue levels correlate with neutrophil infiltration. Hepatology 18:41–46
Song Z, Zhou Z, Deaciuc I, Chen T, McClain CJ (2008) Inhibition of adiponection production by homecysteine: a potential mechanism for alcoholic liver disease. Hepatology 47:867–879
Sun Z, Klein AS, Radaeva S, Hong F, El-Assal O, Pan HN, Jaruga B, Batkar S, Hoshino S, Tian Z, Kunos G, Diehl AM, Gao G (2003) In vitro interleukin- 6 treatment prevents mortality associated with fatty liver transplants in rats. Gastroenterology 125(1):202–215
Szabo G, Dolganiuc A, Mandrekar P (2006) Pattern recognition receptors: a contemporary view on liver disease. Hepatology 44:287–298
Szabo G, Mandrekar P, Dolganiuc A (2007) Innate immune response and hepatic inflammation. Semin Liver Dis 27:339–350
Thakur V, Pritchard MT, McMuller MR, Nagy LE (2006) Adiponectin normalizes LPS-stimulated TNF-α production by rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol 290:G998–G1007
Thurman RG (1998) II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol 275:G605–G611
Tilg H, Diehl AM (2000) Cytokines in alcoholic and nonalcoholic steatoheptohepatitis. N Eng J Med 343:1467–1476
Tomita K, Tamiya G, Ando S, Kitamura N, Koizumi H, Kato S, Horie Y, Kaneko T, Azuma T, Nagat H, Ishii H, Hibi T (2005) AICAR, an AMPK activator, has protective effects on alcohol induced fatty liver in rats. Alcohol Clin Exp Res 29:240S–245S
Tournier C, Dong C, Tourner TK, Jones SN, Flavell RA, Davis RJ (2001) MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev 15:1419–1426
Tsatsanis C, Zacharioudaki V, Androulidaki A, Dermitzaki E, Charalampopoulos I, Minas V, Gravanis A, Margioris A (2005) Adiponectin induces TNF-α and IL-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem Biophy Res Commun 335:1254–1263
Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E et al (2002) Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol 168:2963–2969
Waal Malefyt R, Abrams J, Bennett B, Figdor CG, Vries JE (1991) Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 174:1209–1220
Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ 10:45–65
Wake H, Yamuchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R (2003) Impaired multimerization of human adiponectin mutants associates with diabetes: molecular structure and multimer formation of adiponectin. J Biol Chem 278:40352–40363
Weber-Nordt RM, Riley JK, Greenlund AC, Moore KW, Darnell JE, Schreiber RD (1996) Stat3 recruitment by two distint ligand-induced, tyrosine-phosphorylated docking sites in the interleukin-10 receptor intracellular domain. J Biol Chem 271:27954–27961
Wheeler MD (2003) Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res Health 27:300–306
Williams LM, Ricchetti G, Sarma U, Smallie T, Foxwell BM (2004) Interleukin-10 suppression of myeloid cell activation—a continuing puzzle. Immunology 113:281–292
Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC (1990) CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431–1433
Wulster-Radcliffe MC, Ajuwon KM, Wang J, Chirstian JA, Spurlock ME (2004) Adiponectin differentially regulates cytokines in porcine macrophages. Biochem Biophys Res Commun 316:924–929
Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ (2003) The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver disease in mice. J Clin Invest 112:91–100
Yamaguchi N, Guillermo J, Argueta M, Masuhiro Y, Kagishita M, Nonaka K, Saito T, Hanazawa S, Yamashita Y (2005) Adiponectin inhibits toll-like receptor family-signaling. FEBS Lett 579:6821–6826
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yodo M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T (2001) The fat- derived hormone adiponectin reverses insulin resistence associated with both lipoatrophy and obesity. Nat Med 7:941–946
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8(11):1288–1295
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yakomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T (2003) Cloining of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769
Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M et al (2003) IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol 4:551–556
Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG (1999) Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 117:942–945
Yin HQ, Kim M, Kim JH, Kong G, Kang KS, Kim HI, Yoon BI, Lee MO, Lee BH (2007) Differential gene expression and lipid metabolism in fatty liver induced by acute ethanol treatment in mice. Toxicol Appl Phamacol 223(3):225–233
Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyam A, Oushi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y (2000) Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 96:1723–1732
You M, Fischer M, Deeg MA, Crabb DW (2002) Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem 277:29342–29347
You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW (2004) The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 127:1798–1808
You M, Considine RV, Leone TC, Kelly DP, Crabb DW (2005) Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 42:568–577
You M, Liang X, Ajmo JM, Ness GC (2008) Involvement of mammalian sirtuin I in the action of ethanol in the liver. Am J Physiol Gastrointest Liver Physiol 370:44–48
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
An, L., Wang, X. & Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch Toxicol 86, 1337–1348 (2012). https://doi.org/10.1007/s00204-012-0814-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-012-0814-6