
Abstract
A new series of 2-[N-(R-phenylureido)]-1,3,2-diazaphosphore-2-oxide derivatives (R = CH3, F, NO2, CN) were synthesized and characterized by 31P, 1H, 13C NMR and FT-IR spectral techniques. All the compounds were evaluated for their antibacterial activity against some Gram-positive, Gram-negative strains of bacteria, as well as for their cytotoxic effects on MCF-7, MDA-MB-231, PC-3, HeLa, and K562 human cell lines. In vitro activity results exhibited an important role for six-membered diaza ring in both assays as well as high effect of meta-methyl and ortho-fluoro substitutes on the aromatic ring against the studied human cell lines and B. subtilis bacteria, respectively. To understand the correlation between the anticancer activity and physicochemical properties of the synthesized compounds, the QSAR studies were carried out. Further, the crystal structure of compound 15 was investigated and revealed that the title derivative is composed of two symmetrically independent molecules in the solid state with anti configuration the C=O versus P=O. NBO and AIM analyses were performed to investigate electronic aspects of hydrogen bonding of the crystal cluster, which play an extremely important role in biochemical systems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The increasing issue of drug resistance has prompted an intensive search for new bioactive agents. Phosphor heterocyclic compounds are attractive in this regard because structural properties of these derivatives such as steric, electronic, and conformational interactions allow them to interact easily with the biopolymers of the living systems (Hua et al., 2009; Venkatachalam et al., 2006; Mohe et al., 2003). They inhibit acetylcholinesterase and butyrylcholinesterase enzymes (Elgorashi et al., 2006; Gholivand et al., 2014) and, as such, are useful for inhibiting of cell proliferation in the treatment of cancer (Voorde et al., 2011; Monteil et al., 2014; Gholivand et al., 2010). In addition, these compounds exhibit antimicrobial, antimalarial, and antiviral activities (Gholivand and Dorosti, 2013; Mara et al., 2011; Hocková et al., 2011). One representative phosphor group is oxazaphosphorine family, which is used as potent drugs in the treatment of human cancers (Fig. 1a) (Li et al., 2003; Borch and Canute, 1991; Ludeman et al., 1979). Modification of cyclophosphamide (CP), one of oxazaphosphorinane derivatives, has led to the synthesis of numerous phosphoramidate alkylating agents (Venkatachalam et al., 2006; Li et al., 2003; Moon et al., 1995) to find an antitumor agent that has fewer side effects than drugs now present. In all the research, analogs of CP have two important components including P ring and the substituent on phosphor (Jiang et al., 2006; Sun et al., 2006). In our previous studies, we have designed CP analogs based on the principle of conjugating the two pharmacophores of the phosphoryl and urea, with anticipation that the obtained hybrids could possess powerful biological activity (Gholivand et al., 2010, 2012). Our findings highlighted the importance of –NHC(O)NHP(O)– framework. In this paper, we describe the design and synthesis of 22 compounds with the general skeleton of RC6H5NHC(O)NHP(O)(NH)1(NH)2 (Fig. 1b). New desired derivatives 15, 17–19, 22, 24–34 were characterized by IR, 1H, 13C, and 31P NMR. Cytotoxic activities of 22 compounds, phenylurea, and standard drug (CP) were assayed against HeLa, PC-3, K562, MCF-7, and MDA-MB-231 human cancer cell lines to elucidate the importance of replacement of the 5- and 6-membered diazaphosphore rings (A-ring) instead of oxazaphosphorinane ring as well as the variation in the type of substituent and their position on the aromatic ring (B-ring). In the following, the selected compounds were screened for their antimicrobial activities. We also present quantitative structure–activity relationship (QSAR) studies for validation of the observed pharmacological properties of the investigated anticancer compounds and for determination of the most important parameters controlling these properties. In addition, the crystal structure of compound 15 was investigated and employed as reference for quantum mechanical (QM) calculations at the B3LYP level. It is well known that the hydrogen bond plays a key role in the chemistry of life, such as protein–ligand interactions and enzymatic activity (Jeffrey and Saenger, 1991; Carletti et al., 2010; Massiah et al., 2001). Moreover, the physicochemical properties of compounds depend on the presence of intermolecular hydrogen bonds. Hence, the electronic aspects of hydrogen bonds in the crystal structure of the compound 15 have been investigated by NBO and AIM analyses.

a Bioactive oxazaphosphorines, b designed diazaphosphore
Experimental
Chemistry
1H, 13C, and 31P spectra were recorded on a Bruker Avance DRX 500 spectrometer. 1H and 13C chemical shifts were determined relative to internal TMS, and 31P chemical shifts, relative to 85 % H3PO4 as an external standard. IR spectra were recorded on a Shimadzu model IR-60 spectrometer using KBr pellets. Melting points were obtained with an electrothermal instrument.
General procedure for the synthesis of diazaphosphorinanes 15, 17–19, and 22
To a suspension of related intermediates of 2–12 (6 mmol) in dry diethyl ether (20 mL), 2, 2-dimethyl-1, 3-diaminopropane (1.23 g, 12.0 mmol) at 0 °C was added with stirring. After 5 h, the products were filtered off and washed with H2O.
5,5-Dimethyl-2-(N-4-fluoro-phenylureido)-1,3,2-diazaphosphorinane-2-oxide ( 15 )
Yield: 60 %, m.p. 198–199 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 0.78 (s, 3H, CH3), 1.03 (s, 3H, CH3), 2.57 (ddd, 2 J(H, H) = 12.00 Hz, 3 J(H, H) = 5.30 Hz, 3 J(P, H) = 24.4 Hz, 2H, CH), 2.98 (dd, 2 J(H, H) = 8.4 Hz, 3 J(H, H) = 3.65 Hz, 2H, CH), 4.67 (d,2(P, H) = 3.8 Hz, 2H, NHendocyclic), 7.08 (t, 3 J(H, H) = 8.85 Hz, 2H, Ar–H), 7.38 (dd, 3 J(H, H) = 9.00 Hz, 3 J(H, F) = 4.90 Hz, 2H, Ar–H), 7.68 (s, 1H, NHP), 9.37 (s, 1H, 4-F-C6H4NH) ppm; 13C NMR (d6-DMSO): δ 23.31 (s, CH3), 24.88 (s, CH3), 30.49 (d, 3 J(P, C) = 4.2 Hz, CH2), 52.39 (d, 2 J(P, C) = 1.40 Hz, CH2), 115.25 (d, 1 J(F, C) = 22.2 Hz, CH2), 119.8 (d, 2 J(C, F) = 7.68 Hz), 135.7 (s), 153.49 (d, 2 J(P, C) = 2.38 Hz), 156.4 (s); 31P NMR (202.46 MHz, d6-DMSO) δ 3.72 (m). IR (KBr, cm−1): 3215 (νN–H), 3112, 1690 (νC=O), 1614, 1569, 1468, 1446, 1310, 1337, 1220, 1178 (νP=O), 1090, 1046 (νP–N), 954 (νP–N), 864, 829, 766.
5,5-Dimethyl-2-(N-4-cyano-phenylureido)-1,3,2-diazaphosphorinane-2-oxide ( 17 )
Yield: 65 %, m.p. 192–193 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 0.74 (s, 3H, CH3), 1.03 (s, 3H, CH3), 2.59 (ddd, 2 J(H, H) = 11.95 Hz, 3 J(H, H) = 5.15 Hz, 3 J(P, H) = 24.36 Hz, 2H, CH), 3.0 (d, 3 J(H, H) = 11.95 Hz, 2H, CH), 4.7 (d, 2 J(P, H) = 2.5 Hz, 2H, NHendocyclic), 7.2 (d, 3 J(H, H) = 8.7 Hz, 2H, Ar–H), 7.41 (d, 3 J(H, H) = 8.7 Hz, 2H, Ar–H), 7.75 (d, 2 J(P, H) = 3.65 Hz, 1H, NHP), 9.47 (s, 1H, 4-CN-C6H4NH) ppm; 13C NMR (d6-DMSO): δ 23.29 (s, CH3), 24.87 (s, CH3), 30.47 (d, 3 J(P, C) = 4.33 Hz), 52.38 (s), 119.64 (s), 125.48 (s), 128.61 (s), 138.35 (s), 153.39 (d, 2 J(P, C) = 1.94 Hz). 31P NMR (202.46 MHz, d6-DMSO) δ 3.60 (m). IR (KBr, cm−1): 3295 (νN–H), 1709 (νC=O), 1595, 1527, 1467, 1409, 1314, 1257, 1187(νP=O), 1089, 1044 (νP–N), 850, 761, 733, 545.
5,5-Dimethyl-2-(N-3-methyl-phenylureido)-1, 3, 2-diazaphosphorinane-2-oxide ( 18 )
Yield: 53 %, m.p. 163–164 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 0.79 (s, 3H, CH3), 1.03 (s, 3H, CH3), 2.18 (s, 3H, CH3), 2.56 (m, 2H, CH2), 2.98 (m, 2H, CH2), 4.74 (s, 2H, NH), 6.89 (m, 1H, Ar–H), 7.11 (m, 2H, Ar–H), 7.91 (d, 1H, NHP), 9.19 (s, 1H, 3-CH3-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 17.79 (s,CH3), 23.36 (s, CH3), 24.89 (s), 30.54 (d, 3 J(P, C) = 3.5 Hz), 52.21 (s), 119.93 (s), 122.26 (s), 126.08 (s), 126.49 (s), 130.02 (s), 137.49 (s), 153.44 (d, 2 J(P, C) = 2.54 Hz). 31P NMR (202.46 MHz, d6-DMSO) δ 4.62 (m) ppm. IR (KBr, cm−1): 3320 (νN–H), 1680 (νC=O), 1616, 1588, 1558, 1455, 1326, 1193 (νP=O), 1099, 1038, 953, 861, 764, 607.
5, 5-dimethyl-2-(N-3-fluoro-phenylureido)-1, 3, 2-diazaphosphorinane-2-oxide ( 19 )
Yield: 60 %, m.p. 198–199 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 0.78 (s, 3H, CH3), 1.03 (s, 3H, CH3), 2.58 (dd, 2 J(H, H) = 12.69 Hz, 3 J(H, H) = 5.30 Hz, 2H, CH), 2.96 (d, 2 J(H, H) = 11.4 Hz, 2H, CH), 4.69 (s, 2H, NHendocyclic), 7.08 (t, 3 J(H, H) = 8.6 Hz, 2H, Ar–H), 7.37 (b, 2H, Ar–H), 7.7 (d, 1H, 2 J(P, H) = 5.18 Hz, NHP), 9.35 (s, 1H, 4-F-C6H4NH) ppm; 13C NMR (d6-DMSO): δ 23.28 (s, CH3), 24.87 (s, CH3), 30.46 (d, 2 J(P, C) = 4.5 Hz, CH2), 52.38 (s), 115.12 (s), 115.41 (s), 119.82 (d, 2 J(C, F) = 7.7 Hz), 135.69 (d, 2 J(P, C) = 1.97 HZ), 153.49 (d, 2 J(P, C) = 2.49 Hz); 31P NMR (202.46 MHz, d6-DMSO) δ 3.8 (m). IR (KBr, cm−1): 3215 (νN–H), 1690 (νC=O), 1614, 1569, 1468, 1446, 1337, 1220, 1178(νP=O), 1090, 1046 (νP–N), 954 (νP–N), 864, 829, 766.
5,5-Dimethyl-2-(N-2-fluoro-phenylureido)-1,3,2-diazaphosphorinane-2-oxide ( 22 )
Yield: 50 %, m.p. 198-199 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 0.78 (s, 3H, CH3), 1.02 (s, 3H, CH3), 2.59 (ddd, 2 J(H, H) = 12.4 Hz, 3 J(H, H) = 5.27 Hz, 3 J(P, H) = 24.38 Hz, 2H, CH), 3.0 (d, 2 J(H, H) = 12.0 Hz, 2H, CH), 4.69 (d, 2 J(P, H) = 3.28 Hz, 2H, NHendocyclic), 7.09 (t, 3 J(H, H) = 8.76 Hz, 2H, Ar–H), 7.38 (dd, 3 J(H, H) = 8.46 Hz, 3 J(H, H) = 5.1 Hz, 2H, Ar–H), 7.67 (d, 2 J(P, H) = 7.1 Hz, 1H, NHP), 9.35 (s, 1H, 2-F-C6H4NH) ppm; 13C NMR (d6-DMSO): δ 23.34 (s, CH3), 24.94 (s, CH3), 30.5 (d, 3 J(P, C) = 4.5 Hz, CH2), 52.4 (s), 115.2 (s), 115.5 (s), 119.86 (d, 2 J(C, F) = 7.8 Hz), 135.72 (s), 153.53 (d, 2 J(P, C) = 2.57 Hz); 31P NMR (202.46 MHz, d6-DMSO) δ 3.79 (m). IR (KBr, cm−1): 3215 (νN–H), 1690 (νC=O), 1614, 1569, 1468, 1446, 1337, 1220, 1178(νP=O), 1090, 1046 (νP–N), 954 (νP–N), 864, 829, 766.
General procedure for the synthesis of diazaphospholanes 24–34
These compounds were synthesized from the reaction of (6.06 mmol) intermediates 2–12 suspended in 10 mL dichloromethane with (0.73 g, 12.12 mmol) ethylenediamine at 0 °C. The solution was stirred overnight at room temperature and then evaporated in vacuum. The residue was washed with cooled water, and the precipitate was filtered off and dried at room temperature.
2-(N-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 24 )
Yield: 50 %, m.p. 200–201 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.12 (d, 3 J(P, H) = 5.05 Hz, 2H, CH2), 3.27 (d, 3 J(P, H) = 7.1 Hz, 2H, CH2), 4.74 (d, 2 J(P, H) = 12.5 Hz, 2H, NH), 6.95 (t, 3 J(H, H) = 7.35 Hz, 1H, CH), 7.24 (t, 3 J(H, H) = 8.8 Hz, 2H, CH, Ar–H), 7.36 (d, 3 J(H, H) = 7.7 Hz, 2H, CH, Ar–H), 7.52 (s, 1H, NHP), 9.52 (s, 1H, C6H5NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.8 (d, 2 J(P, C) = 11.52 Hz), 118.1 (s), 121.9 (s), 128.7 (s), 139.3 (s), 153.0 (d, 2 J(P, C) = 2.3 Hz). 31P NMR (202.46 MHz, d6-DMSO) δ 23.54 (m). IR (KBr, cm−1): 3365 (νNH), 1695 (νC=O), 1600, 1540, 1482, 1443, 1409, 1309, 1285, 1213, 1155 (νP=O), 1103, 1083, 1057, 1027, 938, 847, 816, 776, 738, 689, 610, 557, 469.
2-(N-4-methyl-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 25 )
Yield: 80 %, m.p. 189–190 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 2.20 (s, 3H, CH3), 3.12 (d, 3 J(P, H) = 12.65 Hz, 2H, CH2), 3.26 (d, 3 J(P, H) = 7.4 Hz, 2H, CH2), 4.71 (d, 2 J(P, H) = 12.35 Hz, 2H, NHendocyclic), 7.04 (d, 3 J(H, H) = 8.20 Hz, 2H, CH, Ar–H), 7.25 (d, 3 J(H, H) = 8.20 Hz, 2H, CH, Ar–H), 7.50 (d, 2 J(P, H) = 6.00 Hz, 1H, NHP), 9.42 (s, 1H, 4-CH3-C6H4NH).13C NMR (125.76 MHz, d6-DMSO) δ 20.2 (s), 40.8 (d, 2 J(P, C) = 11.53 Hz), 118.2 (s), 129.1 (s), 130.8 (s), 136.7 (s), 153.0 (d, 2 J(P, C) = 2.454 Hz).31P NMR (202.46 MHz, d6-DMSO) δ 23.65 (m). IR (KBr, cm−1): 3385 (νN–H), 3255, 2900, 1694 (νC=O), 1599, 1536, 1490, 1404, 1281, 1211, 1159 (νP=O), 1059 (νP–N), 940 (νP–N), 809 (νP–N), 719, 464.
2-(N-4-fluoro-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 26 )
Yield: 70 %, m.p. 201–202 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.26 (d, 3 J(P, H) = 12.0 Hz, 2H, CH2), 3.27 (d, 3 J(P, H) = 7.8 Hz, 2H, CH2), 4.75 (d, 2 J(P, H) = 12.3 Hz, 2H, NHendocyclic), 7.08 (t, 3 J(H, H) = 8. 0 Hz, 2H, Ar–H), 7.38 (m, 2H, Ar–H), 7.58 (s, 1H, NHP), 9.55 (s, 1H, 4-F-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.88 (d, 2 J(P, C) = 11.55 Hz), 115.25 (d, 1 J(C, F) = 22.2 Hz), 119.87 (d, 2 J(P, C) = 7.6 Hz), 135.64 (s), 153.14 (d, 2 J(P, C) = 1.89 Hz), 156.43 (s).31P NMR (202.46 MHz, d6-DMSO) δ 23.58 (m). IR (KBr, cm−1): 3390(νN–H), 3250, 2955, 1701 (νC=O), 1612, 1546, 1509, 1484, 1405, 1292, 1217, 1153 (νP=O), 1101, 1055 (νP–N), 939, 825, 783, 747, 708, 460.
2-(N-4-nitro-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 27 )
Yield: 70 %, m.p. 195–196 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.16 (d, 3 J(P, H) = 12.5 Hz, 2H, CH2), 3.29 (d, 3 J(P, H) = 7.65 Hz, 2H, CH2), 4.83 (d, 2 J(P, H) = 12.8 Hz, 2H, NHendocyclic), 7.62 (d, 3 J(H, H) = 9.05 Hz, 2H, Ar–H), 7.83 (s, 1H, NHP), 8.14 (d, 3 J(H, H) = 9.00 Hz, 2H, Ar–H), 10.19 (s, 1H, 4-NO2-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.8 (d, 2 J(P, C) = 11.78 Hz), 117.6 (s), 125.1 (s), 141.3 (s), 145.7 (s), 152.8 (d, 2 J(P, C) = 2.64 Hz).31P NMR (202.46 MHz, d6-DMSO) δ 23.30 (m). IR (KBr, cm−1): 3375 (νNH), 3265, 2960, 1705 (νC=O), 1549, 1489, 1407, 1328 (νNO2), 1299, 1155 (νP=O), 1059 (νP–N), 940 (νP–N), 847 (νP–N), 809, 732.
2-(N-4-cyano-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 28 )
Yield: 73 %, m.p. 192–193 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.13 (d, 3 J(P, H) = 10.75 Hz, 2H, CH2), 3.27 (d, 3 J(P, H) = 7.55 Hz, 2H, CH2), 4.76 (d, 2 J(P, H) = 12.55 Hz, 2H, NHendocyclic), 7.28 (d, 3 J(H, H) = 8.8 Hz, 2H, Ar–H), 7.41 (d, 3 J(H, H) = 8.8 Hz, 2H, Ar–H), 7.62 (d, 2 J(P, H) = 5.6 Hz, 1H, NHP), 9.65 (s, 1H, 4-CN-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.9 (d, 2 J(P, C) = 11.56 Hz), 119.66 (s), 125.52(s), 128.61(s), 138.29(s), 153.03(d, 2 J(P, C) = 2.6 Hz).31P NMR (202.46 MHz, d6-DMSO) δ 23.53 (m). IR (KBr, cm−1): 3379(νN–H), 3140, 2918, 1706 (νC=O), 1592, 1525, 1485, 1413, 1365, 1297, 1217, 1157 (νP=O), 1106, 1054 (νP–N), 938, 772, 545, 488, 445.
2-(N-3-methyl-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 29 )
Yield: 73 %, m.p. 195–196 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 2.17 (s, 3H, CH3), 3.12 (d, 3 J(P, H) = 12.0 Hz, 2H, CH2), 3.23 (d, 3 J(P, H) = 7.95 Hz, 2H, CH2), 4.8 (d, 2 J(P, H) = 12.45 Hz, 2H, NHendocyclic), 6.89 (t, 3 J(H, H) = 7.25 Hz, 1H, Ar–H), 7.09 (d, 3 J(H, H) = 7.5 Hz, 1H, Ar–H), 7.11 (d, 3 J(H, H) = 7.65 Hz, 1H, Ar–H), 7.81 (s, 1H, PNH), 7.92 (d,3 J(H, H) = 8.1 Hz,1H, Ar–H), 9.38 (s, 1H, 3-CH3-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 17.73(s), 40.9 (d, 2 J(P, C) = 11.44 Hz), 119.94 (s), 122.3(s), 126.09(s), 126.49(s), 130.04(s), 137.48 (s), 153.24(d, 2 J(P, C) = 2.4 Hz).31P NMR (202.46 MHz, d6-DMSO) δ 24.1 (m). IR (KBr, cm−1): 3368(νN–H), 2957, 2894, 1694(νC = O), 1614, 1591, 1540, 1481, 1455, 1411, 1295, 1210, 1168 (νP=O), 1109, 1050(νP–N), 944, 854, 824, 796, 749, 622, 571, 480.
2-(N-3-fluoro-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 30 )
Yield: 75 %, m.p. 201–202 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.09 (d, 3 J(P, H) = 10.11 Hz, 2H, CH2), 3.18 (d, 3 J(P, H) = 5.4 Hz, 2H, CH2), 4.76 (d, 2 J(P, H) = 12.6 Hz, 2H, NHendocyclic), 6.76 (d, 1H, 3 J(H, H) = 4.28 Hz), 7.08 (t, 3 J(H, H) = 8. 9 Hz, 2H, Ar–H), 7.38 (dd, 3 J(H, H) = 8.9 Hz, 3 J(H, F) = 4.9 Hz, 1H, Ar–H), 7.56 (b, 1H, NHP), 9.54 (s, 1H, 3-F-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.88 (d, 2 J(P, C) = 11.43 Hz), 115.16 (s), 115.46 (s), 119.8 (s), 119.9 (s), 153.13 (s). 31P NMR (202.46 MHz, d6-DMSO) δ 23.58 (m). IR (KBr, cm−1): 3390(νN–H), 3250, 2955, 1701 (νC=O), 1612, 1546, 1509, 1484, 1405, 1292, 1217, 1153 (νP=O), 1101, 1055 (νP–N), 939, 825, 783, 747, 708, 460.
2-(N-3-nitro-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 31 )
Yield: 55 %, m.p. 187-188 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.17(d, 3 J(P, H) = 12.2 Hz, 2H, CH2), 3.27 (d, 3 J(P, H) = 7.91 Hz, 2H, CH2), 4.85 (d, 2 J(P, H) = 12.7 Hz, 2H, NHendocyclic), 7.01 (t, 3 J(H, H) = 8.2 Hz, 1H, Ar–H), 7.22 (d, 3 J(H, H) = 7.9 Hz, 1H, Ar–H), 7.31 (d, 3 J(H, H) = 7.5 Hz, 1H, Ar–H), 7.52 (s, 1H, PNH), 7.75 (d, 3 J(H, H) = 8.0 Hz, 1H, Ar–H), 9.5 (s, 1H, 3-NO2-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.9 (d, 2 J(P, C) = 11.4 Hz), 123.55 (s), 123.75(s), 125.17(s), 134.1(s), 134.5(s), 138.3(s), 153.1(d, 2 J(P, C) = 2.35 Hz).31P NMR (202.46 MHz, d6-DMSO) δ 23.53 (m). IR (KBr, cm−1): 3370(νN–H), 3135, 1700(νC=O), 1544, 1493, 1402, 1344, 1214 (νP=O), 1158, 1094, 1069, 905, 859, 836, 805, 732, 675, 610, 578, 547.
2-(N-2-methyl-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 32 )
Yield: 73 %, m.p. 195-196 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 2.15 (s, 3H, CH3), 3.12 (d, 3 J(P, H) = 12.13 Hz, 2H, CH2), 3.2 (d, 3 J(P, H) = 7.3 Hz, 2H, CH2), 4.75 (d, 2 J(P, H) = 12.35 Hz, 2H, NHendocyclic), 6.93 (t, 3 J(H, H) = 7.33 Hz, 1H, Ar–H), 7.1 (d, 3 J(H, H) = 7.5 Hz, 1H, Ar–H), 7.18 (d, 3 J(H, H) = 7.61 Hz, 1H, Ar–H), 7.31 (s, 1H, PNH), 7.52 (d,3 J(H, H) = 7.92 Hz,1H, Ar–H), 9.21 (s, 1H, 2-CH3-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 17.52(s), 40.92 (d, 2 J(P, C) = 11.31 Hz), 119.87 (s), 122.3(s), 126.1 (s), 126.53(s), 130.02(s), 137.26 (s), 153.24(d, 2 J(P, C) = 2.2 Hz).31P NMR (202.46 MHz, d6-DMSO) δ 24.12 (m). IR (KBr, cm−1): 3375(νN–H), 3150, 2895, 1701 (νC=O), 1693, 1612, 1588, 1537, 1490, 1452, 1408, 1290, 1209, 1165 (νP=O), 1106, 1048 (νP–N), 941, 850, 745, 618, 568, 476.
2-(N-2-fluoro-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 33 )
Yield: 70 %, m.p. 201–202 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.12 (d, 3 J(P, H) = 12.88 Hz, 2H, CH2), 3.26 (d, 3 J(P, H) = 9.89 Hz, 2H, CH2), 4.76 (d, 2 J(P, H) = 12.61 Hz, 2H, NHendocyclic), 6.81 (d, 2 J(H, H) = 4.43 Hz, 1H, Ar–H), 7.08 (t, 3 J(H, H) = 8.9 Hz, 2H, Ar–H), 7.39 (dd, 3 J(H, H) = 8.96 Hz, 3 J(H, F) = 4.9 Hz, 1H, Ar–H), 7.58 (s, 1H, NHP), 8.03 (b,1H, Ar–H), 9.54 (s, 1H, 2-F-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.89 (d, 2 J(P, C) = 11.59 Hz), 114.94 (s), 115.16 (s), 115.45 (s), 119.86 (d, 2 J(F, C) = 7.8 Hz), 135.66 (d, 2 J(F, C) = 2.3 Hz), 153.16 (d, 2 J(P, C) = 2.7 Hz). 31P NMR (202.46 MHz, d6-DMSO) δ 23.66 (m). IR (KBr, cm−1): 3390(νN–H), 3250, 2955, 1701 (νC=O), 1612, 1546, 1509, 1484, 1405, 1292, 1217, 1153 (νP=O), 1101, 1055 (νP–N), 939, 825, 783, 747, 708, 460.
2-(N-2-nitro-phenylureido)-1,3,2-diazaphospholane-2-oxide ( 34 )
Yield: 60 %, m.p. 187–188 °C. 1H NMR (500.13 MHz, d6-DMSO) δ 3.14(d, 3 J(P, H) = 12.6 Hz, 2H, CH2), 3.25 (d, 3 J(P, H) = 8.9 Hz, 2H, CH2), 4.7 (d, 2 J(P, H) = 12.7 Hz, 2H, NHendocyclic), 7.2 (t, 3 J(H, H) = 7.9 Hz, 1H, Ar–H), 7.65 (t, 3 J(H, H) = 8.0 Hz, 1H, Ar–H), 8.02 (d, 3 J(H, H) = 8.15 Hz, 1H, Ar–H), 8.2 (d, 3 J(H, H) = 8.4 Hz, 1H, Ar–H), 8.39 (s, 1H, PNH), 10.3 (s, 1H, 2-NO2-C6H4NH). 13C NMR (125.76 MHz, d6-DMSO) δ 40.9 (d, 2 J(P, C) = 11.52 Hz), 122.65 (s), 123.32(s), 125.12(s), 134.01(s), 134.5(s), 138.5(s), 153.06(d, 2 J(P, C) = 2.5 Hz, d).31P NMR (202.46 MHz, d6-DMSO) δ 22.94 (m). IR (KBr, cm−1): 3334(νN–H), 3135, 2935, 1715(νC=O), 1609, 1586, 1483, 1433, 1344, 1278, 1209, 1172 (νP=O), 937, 864, 742.
Crystal structure determination
Crystals suitable for X-ray crystallography were obtained from ethanol at room temperature. X-ray data of compound 15 were collected on a Bruker SMART 1000 CCD (Bruker, 1998) area detector with graphite monochromated Mo Kα radiation (k = 0.71073 Å). The structure was refined with SHELXL-97 (Sheldrick, 2008) by full-matrix least-squares on F 2. The positions of hydrogen atoms were obtained from the difference Fourier map. An absorption correction was performed using the SADABS program for the titled structure.
Cytotoxicity assay
The cytotoxic activity of the synthesized compounds was determined on five human cancer cell lines: cervical cancer (HeLa), breast cancer (MCF-7 and MDA-MB-231), prostate cancer (PC-3), and leukemia (K562) by MTT assay. The cells suspended in the corresponding culture medium were inoculated in 96-well microtiter plates at a density of 2000–8000 cells per well and incubated for 24 h at 37 °C in a humidified atmosphere with 95 % air and 5 % CO2. An equal volume of additional medium containing either the serial dilutions of the test compounds, positive control (cyclophosphamide), or negative control (2 %DMSO) was added to the desired final concentrations, and the microtiter plates were further incubated for 24, 48, and 72 h. Each assay was set up in triplicate wells and repeated one to three times. The IC50 value is defined as compound concentration that inhibits cell growth by 50 %. Also, to study the degree of selectivity in the cytotoxic activity of the compounds, separation of lymphocyte was down according to the following procedure (Bøyum, 1976): Defibrinated or anticoagulant-treated blood was diluted with an equal volume of PBS and layered carefully over Ficoll-Paque PLUS (without intermixing) in a centrifuge tube. After a short centrifugation at room temperature (typically at 400 gav for 30–40 min), lymphocytes, together with monocytes and platelets, are harvested from the interface between the Ficoll-Paque PLUS and sample layers. This material was then centrifuged twice in a balanced salt solution to wash the lymphocytes and to remove the platelets.
Antibacterial evaluation assay
The cup-plate agar method (Barry, 1977) was used to determine the antibacterial activity of compounds 13–34 against three Gram-positive bacteria, namely S. aureus (ATCC 25923), B. subtilis (ATCC 12711), and B. cereus (ATCC 11778), three Gram-negative bacteria, viz. E. coli (ATCC 25922), P. aeruginosa (ATCC 27853), and Proteus vulgaris (NCIMB 8066). The compounds dissolved in dimethyl sulfoxide (DMSO) at a concentration of 6000 µg/cm3 were used. Then, the solutions of the tested compounds were placed on the well of the media inoculated with the microorganisms. DMSO was used as a solvent control. Gentamicin and tetracycline were used as reference antibacterial drugs. The diameter of the growth inhibition zone was read after 24 h of incubation at 35 °C. These compounds were further examined by the broth dilution method to determine their MIC (minimal inhibitory concentration) (Vincent and Vincent, 1944). Minimal inhibitory concentrations were read after 24 h of incubation at 35 °C.
Computational details
The solid-state structures were used as the starting point for density functional theory (DFT) calculations in the gas phase and fully optimized at the B3LYP/6–31 + G* level. Whereas the X-ray crystallography cannot determine accurately the position of the H atoms, the optimization of H atom positions was performed to investigate the hydrogen bond characters in solid-state structures. To achieve this goal, the solid-state structure of a conformer was modeled as clusters in which the target molecule is surrounded by two neighboring conformers and a similar molecule (Fig. 5). The positions of H atoms were optimized for model clusters, while other atoms were kept frozen during the optimization. Such computational justifications have also been used to describe well the geometry and electronic aspects of X-ray structures (Mirzaei et al., 2006; Esrafili et al., 2007). The NBO and AIM analyses have been performed to compare the electronic features of the gas-phase structures of the compound with those of model clusters at the B3LYP/6–311 + G** level. The hydrogen-bonding energies have been calculated on the basis of the energy difference between the hydrogen-bonded tetramer and its fragments, as represented in the equation E HB(1) = [E tetramer − (E frag1 + E frag2)]/2, where fragment 1 is composed of three monomers (conformers A, B, and B′ on the left in Fig. 5) and fragment 2 is the right molecule (conformer A′). The [E tetramer − (E frag1 + E frag2)] term corresponds to the energies of the two symmetry-related contacts between the two pairs of molecules, and the value is therefore divided by 2. Also, other hydrogen-bonding energies have been calculated on the basis of the equation E HB(2), (3) = [E tetramer − (E frag1 + E frag2)], where fragment 1 is composed of three monomers (conformers A, A′, and B in E HB(2); conformers A, A′, and B′ in E HB(3)) and fragment 2 is the conformer B′ in E HB(2) and conformer B in E HB(3), respectively. The hydrogen-bonding energies were then corrected for basis set superposition error (BSSE) using the counterpoise method (Boys and Bernardi, 1970). All quantum chemical calculations have been carried out using the GAUSSIAN 03 package (Frisch et al., 2003) on a Pentium 4 (2.8 GHz CPU and 1024 MB RAM) work station.
Statistical analysis
In order to identify the effect of descriptors on the molecule’s anticancer activity, QSAR studies were performed using the model described (Hansch and Fujita, 1964). The stepwise multiple linear regression (MLR) procedure, which is a common method used in QSAR studies, was used for selection of the descriptors using SPSS 16.0. The electronic and structural descriptors were obtained by both the quantum chemical calculations. The electronic descriptors included the highest occupied and lowest unoccupied molecular orbital (E HOMO and E LUMO), after highest occupied and next lowest unoccupied molecular orbital (E H−1 and E L+1), the energy difference between the LUMO and HOMO (∆E L−H), electrophilicity (w) (Parr et al., 1999), polarizability (PL), and the net atomic charges (Q). Hydrophobic coefficient (log P), dipole moment (µ), molecular volume (Mv), and molecular total energy (TE) were the structural descriptors. E H, E L, E H−1, E L+1, w, PL, Q, µ, Mv, and TE (low optimized energy) values were obtained from the DFT results (Sharma et al., 2012). The logarithm of partition coefficient (log P) was calculated by the Viswanadhan’s fragmentation method (Viswanadhan, 1989). In this study, only the variables that contain the necessary information for modeling were used. The principal component analysis (PCA) was utilized to find the relationship between the dependent and independent variables, reducing the set of independent variables (Pinto et al., 2001). The toxicities of 22 compounds, CP, and phenylurea were expressed in terms of IC50, which is defined as necessary molar concentration of compound causing 50 % inhibition against human cell lines. All obtained IC50 values are usually converted into the negative logarithm of IC50 in QSAR study (log (1/IC50)).
Results and discussion
Spectroscopic study
Sixteen new derivatives of diazaphosphore with the general formula RC6H4NHC(O)NHP(O)R1R2 were synthesized (Scheme 1) (Kirsanov, 1954; Kirsanov and Zhmurova, 1956) and characterized by 31P, 13C, and 1H NMR and IR spectroscopy. Comparison of the δ(31P) data in Table 1 shows that chemical shift of compounds bearing nitro group is at the most upfield region rather than those of other groups (CH3, F, and CN). For instance, derivative 32 (2-CH3) shows 31P chemical shift value 24.12 ppm, whereas derivatives 33 (2-F) and 34 (2-NO2) exhibit 23.66 and 22.94 ppm, respectively. The 31P NMR spectra of compounds 13–34 also show a chemical shift in the range from 2.86 (in 23) to 24.12 (in 19) ppm. 31P nuclei in compounds 13–23 are shielded relative to those of other compounds. Inspection of 1H and 13C NMR spectra of the new derivatives 15, 17–19, and 22 reveals two separate signals for Haxial and Hequatorial and carbons of two methyl groups. Also, it is observed from Table 1 two different signals of Haxial and Hequatorial for compounds 24–33. The 3J(PNCH) coupling constant from 10.11 to 12.88 is related to the Hequatorial atom, and for Haxial atom, the coupling with phosphorus atom is from 5.4 to 9.89 ppm. This led us to conclude that the synthesized compounds are toward one chair conformation (Denmark et al., 1999) that adopt with X-ray crystallography for 15. The small values of 3J(H, P) indicate that the relevant dihedral angles are close to orthogonal. In the case of compound 15, the torsional angles of P–N–C–Haxial and P–N–C–Hequatorial are ±68 and ±173, respectively, obtained from X-ray crystallography. Considering the Karplus equation (Breitmaier and Voelter, 1990), the Hequatorial proton has the largest coupling with phosphorus atom. In these heterocycles, the highest value coupling constant is 24.76 and 12.88 Hz for diazaphosphorinane 20 and diazaphospholane 33. In addition to the δ(31P), 2J(PNH)endocyclic and 2J(P,C)endocyclic values in compounds 24–34 are larger than those of compounds 13–23. The 1H NMR spectrum of 24–34 exhibits a doublet signal for the two equivalent amino protons (of the five-membered ring) with a high value for phosphorus–hydrogen coupling constant about 12.8 Hz and a doublet signal with 2J(PNH) about 6.0 Hz for the amidic proton. 2J(PNH) coupling constants of endocyclic amino protons in compounds containing six-membered (13–34) are in the range 2.5–3.8 Hz. The major reduction in the 2J(PNH) coupling constant from 12.8 (in 27) to 3.8 (in 15) is due to the increase in ring size because the five-membered ring has high ring strain. Similar to the 2J(PNH) value, 2J(P, C)endocyclic was observed for diazaphospholanes 24–34 (about 11.51 Hz), which are larger than the values of diazaphosphorinanes 13–23. 3J(P, C) coupling constant was obtained for compounds 13–24 (3.5–4.62 Hz).

Preparation pathway for the synthesis of compounds 13–34
X-ray crystallography
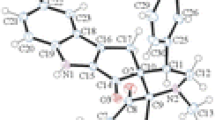
Single crystals of compound 15 were obtained by slow evaporation of the solvent at room temperature. Compound crystallizes in the monoclinic crystal system with space group P21/n, and the crystal system contains two independent molecules (15a and 15b). The crystallographic data, bond lengths, and angles are listed in Tables 2 and 3, respectively. Molecular structures are shown in Fig. 2. The molecules are significantly different as manifested by their torsion angles. For instance, the O1P1N1C1, O1AP1AN1AC1A, and O1P1N2C3, O1AP1AN2AC3A torsion angles are −162.26°, 160.29° and 165.49°, −164.53°, respectively. The P=O distances in molecules 15a and 15b are 1.4818 and 1.4823 Å and are larger than the normal P=O bond length (1.45 Å) (Corbridge 1995). These values are comparable to those reported for phosphoramidates (Gholivand and Dorosti 2011; Gholivand et al. 2009). The P atoms have slightly distorted tetrahedral configurations with the surrounding angles around the P atoms in the range of 101.92 (9)°–116.36 (10)° and 101.84 (9)°–116.14 (9)° for molecules 15a and 15b, respectively. The P–Nendocyclic distances are significantly shorter than the related P–NC(O)NHP(O) bond distance (Table 3). All of these bonds are in the range 1.6218(18)–1.6985(18) Å and thus are significantly shorter than a typical P–N single bond (1.77 Å) (Roy et al., 2006). The P=O bonds in molecules 15a and 15b are in an equatorial position of the related 1, 3-diazaphosphorinane rings. The endocyclic nitrogen atoms in compounds 15a and 15b are distorted from planarity. The sum of angles around these atoms in 15a is 353.15° and 353.69° for N(1) and N(2) atoms, and in 15b, is 351.39° and 352.72° for N(7) and N(8) atoms. The sum of the surrounding angles for all the exocyclic nitrogen atoms is almost 360°; therefore, the environment of the N atoms is practically planar. As shown in Fig. 3, the carboxyl and phosphoryl groups of ligands are involved in intermolecular N–H…O hydrogen bonding and form two types of robust hydrogen bond synthons, namely R 22 (8) I and R 66 (28) II. These intermolecular interactions connected the various components into a 2D network. There are also intramolecular P = O…H-NPh hydrogen bonds in both conformers. In addition to numerous strong hydrogen bonds, intermolecular interactions in 15 include weaker C9A-H9AA…F1Ai (i = 1/2 − x, −1/2 + y, −1/2 − z) interaction with C…F distance of 3.146(2). There are also C–H…π interactions [H… centroid distance = 2.82 (2), 2.72(2) Å] that occur between C1-H1B, C11AH11B, and Cg2 [Cg2i (i = −x, 2 − y, −z), Cg2ii (ii = x, y, z)] as well as [H… centroid distance = 2.96 (2), 2.96 (2) Å] that occur between C1A–H1AA, C11H11A, and Cg4 [Cg4i (i = 1 − x, 1 − y, −z), Cg4ii (ii = x, 1 + y, z)].

Molecular structure of 15 with its atom (50 % probability level) labeling scheme

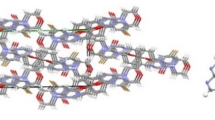
A view of the robust N–H…O hydrogen bond synthons, R 22 (8) I and R 66 (28) II, that link the molecules forming a two-dimensional network in 15 (the dashed lines show donor–acceptor distances of hydrogen bonds)
Cytotoxic activity
The goal of this study was to gain further insights into the structural requirements for the anticancer activity of diazaphosphore derivatives. Hence, the in vitro antitumor activities of the 22 synthesized compounds, namely diazaphosphorinane (13–23) and diazaphospholane (24–34) with –NHC(O)NHP(O)– functional group, were evaluated by MTT assay against five human cancer cell lines: cervical cancer (HeLa), breast cancer (MCF-7 and MDA-MB-231), prostate cancer (PC-3), and leukemia (K562), with cyclophosphamide (CP) as the positive control. Their IC50 values are listed in Table 4. The plots of IC50 values for all of the compounds are also shown in Fig. 4. As shown in Table 4, most of the compounds displayed higher activity than phenyl urea against the selected cell lines and even preferable cytotoxic activities than the commercial anticancer drug CP with slightly different capacity, which was consistent with our previous studies (Gholivand et al., 2010, 2012). On the basis of our previous work, –N1HC(O)N2HP(O)– scaffold was assayed as the optimum structure and the side chain of at P-position would be the key moiety to be explored. Thus, we attempted to replace the substituent on the phenyl ring attached to N1H group with electron-donating and electron-withdrawing groups which are usually considered to play a key role in cytotoxic via drug–target interactions. When H unit was replaced with methyl, fluoro, and nitro substituent, data showed that electron-donating substituents revealed better activities than electron-withdrawing substituents such as 18 (3-CH3, IC50, 1.33 µM), 19 (3-F, IC50, 4.75 µM), and 20 (3-NO2, IC50, 8.58 µM) against MCF-7 cells. Similarly, 18 (3-CH3, IC50, 2.25 µM), 21 (2-CH3, IC50, 2.66 µM), and 29 (3-CH3, IC50, 0.88 µM) exhibited higher activities than the other substituents such as 16 (4-NO2, IC50, 6.70 µM), 26 (4-F, IC50, 5.41 µM), and 31 (3-CN, IC50, 8.97 µM) against MDA-MB-231 cells. For this reason, synthesis of methyl analogs of this class compounds seems to be beneficial for anticancer drug design. Likewise, the above-mentioned structure–activity relationship was also found in the other titled cell lines. It is noteworthy that replacement of H with the selected groups at 4-position of aromatic ring did not follow a clear trend against the studied cell lines. In an attempt to explore the effects of substituent insertion in different positions of phenyl ring on the activity, the H in aromatic ring was replaced with 4-, 3-, and 2-methyl, fluorine, and nitro to afford the corresponding compounds (14–16, 25–27), (18–20, 29–31), and (21–23, 32–34), respectively. Table 4 demonstrates that the newly synthesized methyl analogs exhibited antitumor activity on the studied cell lines with the average activity order of 3-CH3 > 2-CH3 > 4-CH3 such as 29 (3-CH3, IC50, 1.74 µM), 32 (2-CH3, IC50, 6.08 µM), and 25 (4-CH3, IC50, 13.90 µM) against PC-3 cells, whereas cell lines from our panel respond to nitro and fluoro analogs significantly differently. Finally, replacement of the six-membered ring in 13–23 with a five-membered ring 24–34 resulted in an interesting structure–activity correlation in two carcinoma cells, HeLa and MCF-7. For instance, fluorine analogs (15; 4-F, IC50, 3.94 µM, 19; 3-F, IC50, 4.75 µM, and 22; 2-F, IC50, 3.14 µM) in six-membered ring displayed slightly higher average potency in comparison with these analogs in five-membered ring (26; 4-F, IC50, 16.64 µM, 30; 3-F, IC50, 16.06 µM, and 33; 2-F, IC50, 3.2 µM) against MCF-7. Also, among modified analogs, ortho substituent in diazaphosphorinane ring (21; 2-CH3, 2.13 µM, 22; 2-F, 3.14 µM, 23; 2-NO2, 5.64 µM) showed higher anticancer activity than similar counterpart in diazaphospholane ring (32; 2-CH3, 6.69 µM, 33; 2-F, 3.2 µM, 34; 2-NO2, 17.57 µM) against MCF-7, suggesting that the ring size plays a significant role for activity. It can be explained by reduction of the electron donation to the phosphorus atom in five-membered ring compared to six-membered ring. In contrast, as shown in Table 1, inhibitory activities of meta and para positions to the titled cancer cell lines were different. For example, phosphorinane derivatives 16 (4-NO2) and 18 (3-CH3) showed an IC50 value 8.92 and 1.33 µM against MCF-7 cell line, whereas phospholanes 27 (4-NO2) and 29 (3-CH3) exhibited IC50 values of 1.28 and 0.72 µM, respectively. These differences may result from a number of factors, in addition to the interaction with the target involved in the cytostatic/cytotoxic response. These factors include number of cell cycles during the incubation period, drug uptake, efflux, and metabolism. On the other hand, few differences among cell lines would suggest that some non-specific cytotoxic effects are responsible for the activity of the tested compounds. To study the degree of selectivity in the cytotoxic activity of the compounds, assays using lymphocyte isolation from whole human blood were carried out on some representative compounds such as 14, 20, and 33, which showed relatively high activity in tumor cells. The assay showed that survival values were 97 % for these compounds.


IC50 values of compounds 13–34 against HeLa, MCF-7, K562, PC-3, and MDA-MB-231
Antibacterial activity
The in vitro antibacterial activity of the synthesized derivatives 13–34 was tested against three Gram-positive bacteria, viz. S. aureus (ATCC 25923), B. subtilis (ATCC 12711), and B. cereus (ATCC 11778), and three Gram-negative bacteria, viz. E. coli (ATCC 25922), P. aeruginosa (ATCC 27853), and Proteus vulgaris (NCIMB 8066), using the cup test method and minimum inhibitory concentration (MIC) experiments. While carrying out antibacterial activity, gentamicin and tetracycline were used as reference compounds and MIC were determined in terms of µg/ml (Table 5). Twenty-two heterocyclic compounds with six- and five-membered rings of phosphor were prepared and evaluated. At first, the objective was to identify the effect of A-ring size on activity. A perusal of the results from Table 5 reveals that diazaphosphorinanes (13–23) with six rings showed higher activity toward the tested Gram-positive bacteria than diazaphospholane (24–34) with five rings. For example, 19 with six rings (3-F, MIC; 833 µg/ml) exhibited potent activity against B. subtilis than 30 with similar structure attached to five rings (3-F, MIC; > 1000 µg/ml). The second goal was to identify substitution effect on activity against the selected microorganisms. Among the synthesized analogs at para position, the most potent compound against S. aureus and B. subtilis was 16. It has NO2 group attached, whereas when CH3, F, and CN group substituents were attached to ring, the same as in case of 17 (4-CN), inhibition decreased (Table 5). Further, if NO2 group is attached to meta and ortho positions, activity decreased as observed in 20 and 23, respectively. Reduced activity was observed when CH3 group was attached at the titled positions as in compounds 18 and 21, whereas substitution of (3-F, 2-F) group to the phenyl group of 19 and 22 compounds increased inhibitory activity against B. subtilis. The most valuable finding is the inhibition of B. subtilis growth by compound 22 (MIC, 25 µg/ml) having electron-withdrawing group (F) at ortho position. The MIC values of compounds against certain bacterial strains indicate that B. subtilis were more sensitive to the toxicity of the synthesized compounds than other microorganisms. In addition, only derivatives 13, 14, and 16 showed negligible activities against E. coli and P. aeruginosa as compared to reference drugs.
NBO and AIM analyses
Fully optimized conformers in the gas phase
The geometries of conformers 15a and 15b were separately optimized as single molecules in vacuum in order to compare the structural and energy aspects of the two conformers in the gas phase. The optimized geometries were confirmed to be the minimum energy point with no imaginary vibrations. The P=O, P–N, C–N, and N–H bond lengths, charge densities, and vibrational data have been represented in Table S1 for two conformers. In both of the conformers, the weak intramolecular CH…OC hydrogen bond between the ortho-proton of the 4-FC6H4NH and the carbonyl O atom creates a six-membered ring via the O–C–N–C–C–H bond paths. This intramolecular contact stabilizes a gauche configuration between the C=O bond and the ortho-proton of 4-fluoroaniline. Furthermore, intramolecular 4-FC6H4N-H…O=P hydrogen bond creates a six-membered ring via the O–P–N–C–N–H bond paths. Based on the results of DFT calculations, we found that the change in basis set used for geometry optimizations has no significant effect on the measure of structural parameters and the two optimized conformers convert to a unique structure. Hence, only one molecular structure was selected to obtain electronic properties by AIM and NBO analyses. The NBO analysis reveals a weak electronic delocalization between the lone pair of the carbonyl oxygen, Lp(OC), and the vacant σ*(C-Hortho) orbital, whereas Lp(OP) and the vacant σ*(N–H) orbital strengthen NH…OP hydrogen bond between the proton of the 4-FC6H4NH and the phosphoryl O atom. Stabilization energies E2 of 4.7 and 8.2 kJ mol−1 were obtained for the Lp(OC) → σ *(C–Hortho) interaction in conformers 15a and 15b, respectively, whereas those of Lp(OP) → σ *(N–H) interaction were calculated as 21.1 and 40.0 kJ mol−1. At the same level, AIM analysis reveals charge density (ρ) values at the bcp of the CH…OC and NH…OP contacts in 0.0177 and 0.0321 e Å−3. The ρ value at the ring critical point (rcp) for the O–C–N–C–C–H and O–P–N–C–N–H six-membered rings (Fig. S1) is 0.0114 and 0.0138 e Å−3. This values confirm the formation of weak and strong interactions for CH…OC and NH…OP contacts.
Electronic parameters of the hydrogen-bonded clusters
For the compound 15, the conformers were modeled as hydrogen-bonded clusters in which the target molecule (A) is surrounded by the same conformer (A′) and two neighbors (B, B′) (Fig. 5). The electronic parameters of the hydrogen-bonded clusters of compound 15 were calculated by AIM and NBO methods. The results of AIM and NBO analyses for H-optimized clusters are presented in Table 6. The bond lengths in optimized clusters are equal to those obtained for X-ray structures (Table S1), except for C–H and N–H bonds, since the optimizations have only been performed for H atom positions. Also, the donor–acceptor distances for hydrogen bonds in model clusters are equal to the experimental values. NBO analysis reveals [LP(OC) → σ *(N–H)] and [Lp(OP) → σ *(N–H)] interactions among the subunits within the clusters. This electronic delocalization leads to the weakening of the N–H bond. This is in good agreement with the decrease in the ρ value at the bcp of the N–H bond from the monomers to their corresponding clusters (Table 6 and S1). Moreover, the D–H–A angles in the optimized clusters are more linear than those in the solid-state structures. These indicate that the hydrogen bonds in the optimized clusters are stronger than those in the X-ray structures. It has been previously explained that the charge density at the bcp of DH…A increased when the donor–acceptor distance shortens. The results of AIM analysis show that ρ value at bcp of intramolecular hydrogen bond A(NH)…(OP)A (0.0362 e Å−3) is larger in magnitude than that calculated for the intermolecular hydrogen bonds, i.e., A(NH)…(OP)B (0.0277 e Å−3), B′(NH)…(OP)A (0.0221 e Å−3), and A′ (NH)…(OC)A (0.0266 e Å−3). The smaller ρ values at the bcp of the N–H bond confirm the presence of the stronger interaction in intramolecular hydrogen bond in comparison with intermolecular hydrogen bonds. This is in good agreement with stabilizing energies E(2) for the title electronic delocalization in the model cluster. The stabilizing energies E(2) of 29.8 and 25.1 kJ mol−1 have been calculated for LP(OP) → σ *(N–H) electron density transfer in (OP) B …H A –N A and (OP) A …H B′ –N B′ hydrogen bonds, while it is 41.1 kJ mol−1 in the electronic delocalization LP(OC) → σ*(N–H) in the case of (OC) A …H Á –N Á . Furthermore, the stabilizing energy E 2 of 45.6 kJ mol−1 has been calculated for LP(OP) → σ *(N–H) in (OP) A …H A –N A hydrogen bond. This is in agreement with the values of distance for these hydrogen bonds in N A –H A …O A –P A (2.722(2)Å), (OP) B …H A –N A (2.933(2)Å), (OP) A …H B′ –N B′ (3.040(2)Å), (OC) A …H Á –N Á (2.969(2)Å) models. Besides, the hydrogen-bonding energy in the (OP) B …H A –N A and (OP) A …H B N B models (−9.7 and −7.1 kJ mol−1) is smaller than the value calculated for (OC) A …H Á –N Á (−39.9 kJ mol−1) model. It should be noted that the stabilizing energies E(2) differ from the hydrogen-bonding energy. The hydrogen-bonding energy is related to the sum of the total attractive and repulsive forces between two hydrogen-bonded fragments, and E2 refers to the stabilization energy of electronic delocalization between the donor–acceptor orbital.

Model hydrogen-bonded cluster for DFT calculations, in which the target molecule (conformer A) is in the center. A similar model was considered for the cluster in which conformer B in the center is the target molecule
QSAR analysis
In order to understand the observed pharmacological properties of the investigated analogs and determine the crucial factors governing these activities, QSAR studies were undertaken. The stepwise MLR procedure was used for model selection, which is a common method used in QSAR studies. Therefore, experimental IC50 values of 22 tested compounds with the general formula of RC6H5(NH)1C(O)(NH)2P(O)R1R2 (13–34), phenylurea, and CP were employed for constructing QSAR models against five data sets corresponding to the five tested cell lines, i.e., MDA-MB-231, PC-3, MCF-7, K562, and HeLa. For each data set, compounds with inactive cytotoxic activity were excluded from the analysis. For example, compound 30 was identified as an outlier for the MDA-MB-231 model and was subjected to removal, whereas none of the outliers were identified in MCF-7 model. The electronic and structural descriptors were obtained by quantum chemical calculations (Table 7). An optimal QSAR equation based on experimental data (δ, log P, and Mr) shown in Table 2 (eq. a), as well as eq. b is produced by replacement of experimental variables with the descriptors derived from the DFT calculations. Afterward, PCA method was used to reduce the independent variables (Pillai et al., 2005). The variable selection in PCA was performed by using the Fisher’s weights approach (Molfetta et al., 2005), and the results are summarized as the following Eqs. (1, 2).
The main variables were found from the principle scores of the normalized eigenvalue of the two principal components. The results showed that the first and second factor PC on the total variance were 46.7 and 18.3 %, respectively. Also, from the above equations, it was deduced that the electronic parameters E HOMO, E LuMO, W, Q P, Q C, Q N2, PLP=O, PLN–H, and T E predominated over those related to structural parameter (Mv).
MDA-MB-231 model
The MLR was performed using these thirteen descriptors and equations are shown in Table S2. This method led to an optimal QSAR Eq. 3:
In this equation, the inhibitory potency of MDA-MB-231 cell line is influenced mainly by the electronic parameters. Qc with the coefficient values of +77.247 has the highest contribution to log (1/IC50) rather than the other electronic parameters such as the net charge of P=O phosphorus (−1.078). The positive coefficient associated with the parameter Qc suggests that increases in the negative charge or electron density at the C=O carbon may be favorable to higher antitumor activity against MDA-MB-231 cell line. The correlation matrix was used to determine the interrelationship between the independent variables (Table 8). The high interrelationship observed between Q C descriptor and W (r = +0.721) showed that electrophilicity properties of the studied derivatives affect the inhibition of human MDA-MB-231. Compounds 16 (W = 0.205) and 36 (W = 0.062) are in order of the highest and the lowest inhibitory potentials.
MCF-7 model
Besides net charge of the N–H nitrogen Q N2, E H properties of compounds are highly involved in cytotoxic activity against the MCF-7 cell line as shown by the QSAR equation below:
Q N2 and E H with the coefficient values of −10.201 and 10.130 have the highest contribution to log (1/IC50) rather than the structural parameters. The negative and positive signs of Q N2 and E H in log (1/IC50) disclose that the compound with lower net charge Q N2 and higher molecular orbital E H is indicative of higher potency against MCF-7 cell line. The interrelationships between the variables and the correlation matrix results are presented in Table S3. The high and low interrelationships were observed between Q N2 and PLC=O (r = −0.840), and Q N2 and E H (r = −0.325), respectively. The high interrelationship between Q N2 and PLC=O (r = −0.840) shows that PLC=O controls the effect of net charge of the N–H nitrogen Q N2 on the inhibition of the titled cell line. Diazaphosphorinane with fluorine substituent at ortho position 22 (PLC=O = −1.476) and phenylurea 36 (PLC=O = −1.451) are in order of the highest and the lowest inhibitory potentials against MCF-7 cell line.
PC-3 and HeLa models
Similar results were found in both PC-3 and HeLa models in which the electronic descriptors were governing the cytotoxic activity. The QSAR equation of the PC-3 model is shown below:
In Eq. (5), Q N2 representing the net charge of the N–H nitrogen seems to be the most important descriptor as deduced from its highest regression coefficient value of 343.388. High colinearity (r > 0.5) was observed between different parameters (Table S4). The high interrelationship was observed between Q N2 and W (r = −0.841), and low interrelationship was observed between this descriptor and PLP=O (r = 0.087). From the correlation matrix, it was observed that the electrophilicity (W) was found to be most effective in describing the anticancer activity of the synthesized compounds. As shown in Table 7, compounds 16 and 36 with electrophilicity (W) values 0.205 and 0.062 have the highest and the lowest inhibitory potentials against the titled cell line.
The QSAR equation of the HeLa model is shown below:
Similarly the model described by Eq. 6 showed that the most effective variable in the inhibition of HeLa cell line by the tested derivatives was Q N2, with the coefficient value of 261.821. The interrelationships between the variables and the correlation matrix results are presented in Table S5. A high interrelationship was observed between Q N2 and E H (r = −0.758). Compounds 14 (E H = −0.2105) and 35 (E H = −0.268) are in order of the highest and the lowest inhibitory potentials. The positive coefficients of Q N2 at Eqs. 5 and 6 indicate that the increase in the negative charge or electron density at the N–H nitrogen may be favorable to higher antitumor activity.
K562 model
Distinct from other cell lines, the cytotoxic activity of compounds against the K562 cell line is dependent on the next lowest unoccupied molecular orbital shown in the equation below:
It can be seen that the E L+1 descriptor is responsible for prediction of the cytotoxic activity of K562 cell line as deduced from its high regression coefficient value of 22.06. The positive sign of E L+1 in log (1/IC50) discloses that the compound with higher molecular orbital (E L+1) may be conducive to the biological activity of these diazaphosphore compounds. The correlation matrix of different descriptors used in the QSAR models is shown in Table S6. The high interrelationship was observed between E L+1 and E H−1 (r = −0.553), and low interrelationship was observed for Q N2 (r = −0.01). Then, compound 21 having high E H−1 value (−0.240) displayed high anticancer activity. Similarly, compound 27 having low E H−1 value (−0.277) exhibited low anticancer potential. Obtained results from the above data can be summarized as follows:
-
1.
QSAR studies demonstrated that cytotoxic activities of compounds 13–34, CP, and phenylurea against the investigated cell lines were influenced by the electronic descriptors. This indicates that such distinct chemical properties are crucial for the cytotoxic activity against particular cell lines.
-
2.
QSAR studies also emphasize that higher probability of electrophilic attack at the nitrogen atom number 2 may be favorable to the biological activity compared to the nitrogen atom number 1. This is in agreement with the values of the electron density (ρ) for nitrogen atom in two amidic (8.383 eÅ−3) and aniline (8.174 eÅ−3) bonds obtained by AIM analysis. This may explain much interaction N1 with neighbor groups compared to the N2 atom. NBO analysis reveals an [Lp(N) σ *(C=O)] interaction with the stabilizing energies E 2 of 56.99 and 32.39 kcal mol−1 for lone pair N1 and N2, respectively. This electronic delocalization leads to reduction in the (ρ) value for nitrogen atom from N2–H bond to N1–H bond.
-
3.
Net charge of the N–H nitrogen (Q N2) seemed to be important descriptor for the cytotoxic activity against MCF-7, PC-3, and HeLa cell lines, whereas net charge of the C=O carbon (Qc) and E L+1 were shown to be crucial for MDA-MB-231 and K562, respectively. With respect to importance of the electronic descriptors against the studied cell lines, it could be hypothesized that the mechanism of cytotoxic activity of diazaphosphore compounds may be similar.
-
4.
Experimental and theoretical data demonstrate the importance of six-membered ring for cytotoxicity. This might be explained by the electronic nature of phosphor atom.
Validation of QSAR
In each cell lines, a set of compounds was used as a training set for QSAR modeling. The remaining compounds were adopted as a test set for validating the QSAR model. Considering the balance of the QSAR quality and the number of employed quantum chemical descriptors, an optimal equation was achieved for the selected compounds in the training set by MLR analysis for the studied cell lines (Table S7). The developed QSAR models were cross-validated with q 2 value (q 2 > 0.5) obtained by leave-one-out (LOO) method. The value of q 2 more than 0.5 indicates that the model developed is a valid one. According to the recommendations of Golbraikh and Tropsha, the only way to estimate the true predictive power of a model is to test their ability to predict accurately the biological activities of compounds. As the observed and predicted values are close to each other (Table 9), the QSAR models for anticancer activity are valid (Golbraikh and Tropsha, 2002).
Conclusion
In this study, new sixteen diazaphosphore derivatives, differing in the nature of the aliphatic and aromatic rings, were synthesized and investigated for their biological activity. Novel anticancer agents 18 and 29 with methyl group at meta position of aromatic ring exhibited higher cytotoxic activity against most of tested cell lines than other derivatives which may be due to size, steric, and electronic properties of the substituent. The models obtained through QSAR study gave a better prediction capability of anticancer activity against the studied human cell lines. Electronic parameters such as Q N2, QC, and E L+1 were major factors responsible for positively affecting the anticancer activity of the selected diazaphosphore derivatives. As evident from the experimental and QSAR data, the presence of six-membered ring of diazaphosphore is essential for higher antitumor activity. Besides, important of the nitrogen atom number 2 at anticancer activity compared with the nitrogen atom number 1 expressed by result of NBO and AIM analysis. Comparison of the results of antibacterial studies exhibited further emphasis on the importance of six-membered diaza ring in addition to the requirement of substitutions in the aromatic ring.
Supplementary data
Crystallographic data for structure 15 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication Nos. CCDC 1428625 (C12H18F1N4O2P). Copies of the data may be obtained free of charge upon request from CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk
References
Barry AL (1977) The Antimicrobial susceptibility test; principle and practice. Bio Abstr 64:25183
Borch RF, Canute GW (1991) Synthesis and antitumor properties of activated cyclophosphamide analogues. J Med Chem 34:3044–3052
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Bøyum A (1976) Isolation of lymphocytes, granulocytes and macrophages. Scand J Immunol 5:9–15
Breitmaier E, Voelter W (1990) Carbon-13 NMR spectroscopy. VCH, Weinheim, New York, pp 143, 153
Bruker S (1998) Bruker molecular analysis research tool, version 5.059, Bruker AXS: Madison, WI
Carletti E, Colletier JP, Dupeux F, Trovaslet M, Masson P, Nachon F (2010) Structural evidence that human acetylcholinesterase inhibited by tabun ages through o-dealkylation. J Med Chem 53:4002–4008
Corbridge DEC (1995) Phosphorus, an outline of its chemistry, biochemistry and technology, 5th edn. Elsevier, The Netherlands, pp 55–57
Denmark SE, Su X, Nishigaichi Y, Coe DM, Wong KT, Winter SBD, Choi JY (1999) Synthesis of phosphoramides for the lewis base-catalyzed allylation and aldol addition reactions. J Org Chem 64:1958–1967
Elgorashi EE, Malan SF, Stafford GI, Staden JV (2006) Quantitative structure-activity relationship studies on acetylcholinesterase enzyme inhibitory effects of Amaryllidaceae alkaloids. S Afr J Bot 72:224–231
Esrafili MD, Elmi F, Hadipour NL (2007) Density functional theory investigation of hydrogen bonding effects on the oxygen, nitrogen and hydrogen electric field gradient and chemical shielding tensors of anhydrous chitosan crystalline structure. J Phys Chem A 111:963–970
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JrE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003). GAUSSIAN03, Revision B. 03. Gaussian Inc., Pittsburgh, PA, USA
Gholivand K, Dorosti N (2011) Synthesis, spectroscopic characterization, crystal structures, theoretical studies, and antibacterial evaluation of two novel N-phosphinyl ureas. Monatsh Chem 142:183–192
Gholivand K, Dorosti N (2013) Some new compounds with P(E)NHC(O) (E = lone pair, O, S) linkage: synthesis, spectroscopic, crystal structures, theoretical studies, and antimicrobial evaluation. Monatsh Chem 144:1417–1425
Gholivand K, Shariatinia Z, Mahdieh S, Farzaneh Daeepour M, Farshidnasab N, Mahzouni HR, Taheri N, Amiri S, Ansar S (2009) Structural diversity in phosphoramidate’s chemistry: syntheses, spectroscopic and X-ray crystallography studies. Polyhedron 28:307–321
Gholivand K, Dorosti N, Shariatinia Z, Ghaziany F, Sarikhani S, Mirshahi M (2010) Cyclophosphamide analogues: synthesis, spectroscopic study, and antitumor activity of diazaphosphorinanes. Med Chem Res 21:2185–2195
Gholivand K, Dorosti N, Ghaziany F, Mirshahi M, Sarikhani S (2012) N-Phosphinyl ureas: synthesis, characterization, X-ray structure, and in vitro evaluation of antitumor activity. Heteroatom Chem. 23:74–83
Gholivand K, Ebrahimi Valmoozi AA, Bonsaii M (2014) Synthesis, biological evaluation, QSAR study and molecular docking of novel N-(4-amino carbonylpiperazinyl) (thio)phosphoramide derivatives as cholinesterase inhibitors. Pestic Biochem Physiol 112:40–50
Golbraikh A, Tropsha A (2002) Beware of q2! J Mol Graphics Model 20:269–276
Hansch C, Fujita T (1964) p-σ-π Analysis a method for the correlation of biological activity and chemical structure. J Am Chem Soc 86:1616–1626
Hocková D, Holý A, Andrei G, Snoeck R, Balzarini J (2011) Acyclic nucleoside phosphonates with a branched 2-(2-phosphonoethoxy) ethyl chain: efficient synthesis and antiviral activity. Bioorg Med Chem 19:4445–4453
Hua R, Doucet JP, Delamar M, Zhang R (2009) QSAR models for 2-amino-6-arylsulfonylbenzonitriles and congeners HIV-1 reverse transcriptase inhibitors based on linear and nonlinear regression methods. Eur J Med Chem 44:2158–2171
Jeffrey GA, Saenger W (1991) Hydrogen bonding in biological structures. Springer, Berlin
Jiang Y, Han J, Yu C, Vass SO, Searle PF, Browne P, Knox RJ, Hu L (2006) Design, synthesis, and biological evaluation of cyclic and acyclic nitrobenzylphosphoramide mustards for e. coli nitroreductase activation. J Med Chem 49:4333–4343
Kirsanov AV (1954) J Gen Chem USSR 24:1031
Kirsanov AV, Zhmurova IV (1956) J Gen Chem USSR 26:2642
Li Z, Han J, Jiang Y, Browne P, Knox RJ, Hu L (2003) Nitrobenzocyclophosphamides as potential prodrugs for bioreductive activation: synthesis, stability, enzymatic reduction, and antiproliferative activity in cell culture. Bioorg Med Chem 11:4171–4178
Ludeman SM, Zon G, Egan W (1979) Synthesis and antitumor activity of cyclophosphamide analogues. 2.1 Preparation, hydrolytic studies, and anticancer screening of 5-bromocyclophospha-mide, 3,5-dehydrocyclophosphamide, and related systems. J Med Chem 22:151–158
Mara C, Dempsey E, Bell A, Barlow JW (2011) Synthesis and evaluation of phosphoramidate and phosphorothioamidate analogues of amiprophos methyl as potential antimalarial agents. Bioorg Med Chem Lett 21:6180–6183
Massiah MA, Viragh C, Reddy PM, Kovach IM, Johnson J, Rosenberry TL, Mildvan AS (2001) Strong hydrogen bonds at the active site of human acetylcholinesterase: 1H-NMR studies. Biochemistry 40:5682–5690
Mirzaei M, Elmi F, Hadipour NL (2006) A systematic investigation of hydrogen-bonding effects on the 17O, 14N, and 2H nuclear quadrupole resonance parameters of anhydrous and monohydrated cytosine crystalline structures: a density functional theory study. J Phys Chem B. 110:10991–10996
Mohe NU, Padiya KJ, Salunkhe MM (2003) An efficient oxidizing reagent for the synthesis of mixed backbone oligonucleotides via the H-phosphonate approach. Bioorg Med Chem 11:1419–1431
Molfetta FA, Bruni AT, Honório KM, da Silva ABF (2005) A structure–activity relationship study of quinone compounds with trypanocidal activity. Eur J Med Chem 40:329–338
Monteil M, Migianu-Griffoni E, Sainte-Catherine O, Di Benedetto M, Lecouvey M (2014) Synthesis and biological evaluation in HuH7 hepatocarcinom a cells. Eur J Med Chem 77:56–64
Moon K-Y, Shirota FN, Baturay N, Kwon Ch-H (1995) Chemically stable N-methyl-4-(alkylthio)cyclophosphamide derivatives as prodrugs of 4-hydroxycyclophosphamide. J Med Chem 38:848–851
Parr RG, Szentpaly LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924
Pillai AD, Rani S, Rathod PD, Xavier FP, Vasu KK, Padh H, Sudarsanam V (2005) QSAR studies on some thiophene analogs as anti-inflammatory agents: enhancement of activity by electronic parameters and its utilization for chemical lead optimization. Bioorg Med Chem 13:1275–1283
Pinto MFS, Romero OAS, Pinheiro JC (2001) Pattern recognition study of structure-activity relationship of halophenols and halonitrophenols against fungus T. mentagrophytes. J Mol Struct THEO CHEM. 539:303–310
Roy DR, Sarkar U, Chattaraj PK, Mitra A, Padmanabhan J, Parthasarathi R, Subramanian V, Van Damme S, Bultinck P (2006) Analyzing toxicity through electrophilicity. Mol Divers 10:119–131
Sharma SK, Kumar P, Narasimhan B, Ramasamy K, Mani V, Mishra RK, Majeed AA (2012) Synthesis, antimicrobial, anticancer evaluation and QSAR studies of 6-methyl-4-[1-(2-substituted-phenylamino-acetyl)-1H-indol-3-yl]-2-oxo/thioxo-1,2,3,4-tetrahydropyrimidin e-5-carboxylicacid ethylesters. Eur J Med Chem 48:16–25
Sheldrick GM (2008) SHELXTL V.5.10, Structure determination software suite, Bruker AXS. Madison, WI
Sun Q, Li RT, Guo W, Cui JR, Cheng TM, Ge ZM (2006) Novel class of cyclophosphamide prodrug: cyclophosphamide spiropiperaziniums (CPSP). Bioorg Med Chem Lett 16:3727–3730
Venkatachalam TK, Qazi S, Uckun FM (2006) Synthesis and metabolism of naphthyl substituted phosphoramidate derivatives of stavudine. Bioorg Med Chem 14:5161–5177
Vincent JG, Vincent HW (1944) Filter paper disc modification of the Oxford cup penicillin determination. Proc Soc Exp Biol Med 55:162–164
Viswanadhan VN (1989) Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J Chem Inf Comput Sci 29:163–172
Voorde JV, Liekens S, McGuigan C, Murziani PGS, Slusarczyk M, Balzarini J (2011) The cytostatic activity of NUC-3073, a phosphoramidate prodrug of 5-fluoro- 20-deoxyuidine, is independent of activation by thymidinekinase and insensitive to degradation by phosphorolytic enzymes, the cytostatic activity of NUC-3073. Biochem Pharmacol 82:441–452
Acknowledgments
The financial support of this work provided by the Research Council of Tarbiat Modares University and the Razi Herbal Medicines Research Centre of Lorestan University of Medical Sciences is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dorosti, N., Delfan, B., Gholivand, K. et al. Synthesis, crystal structure, biological evaluation, electronic aspects of hydrogen bonds, and QSAR studies of some new N-(substituted phenylurea) diazaphosphore derivatives as anticancer agents. Med Chem Res 25, 769–789 (2016). https://doi.org/10.1007/s00044-016-1527-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1527-9