
Abstract
The title compound C19H17F3N6 was synthesized and structurally characterized by infrared and mass spectroscopy, 1H NMR, elemental analyses and single crystal X-ray diffraction. The compound crystallizes in monoclinic system, space group P21/c with a = 17.097(4) Å, b = 7.1668(16) Å, c = 18.389(3) Å, β = 118.251(15)°, V = 1984.8(8) Å3, Z = 4, Dc = 1.293 g cm–3, F(000) = 800, μ(MoKα) = 0.10 mm–1, R1 = 0.0667, and wR2 = 0.2084 for reflections with I > 2σ(I). Pyrazolo[1,5-a]pyrimidine and phenyl ring are almost coplanar, and the piperazine ring is in a chair conformation. The crystal structure is stabilized by C–H⋅⋅⋅N hydrogen interactions and a number of weak π⋅⋅⋅π interactions. In addition, the results of the determination of biological activity showed that the compound exhibited significant inhibitory activity against K562 and MKN45 cancer cell lines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
In the past few decades, there is an urgent need to pay much attention to the design, synthesis and production of more potent and effective therapeutic agents to treat human cancer diseases, which is for the cause of major deaths worldwide [1, 2]. Various derivatives of pyrazolopyrimidines attract the attention of medicinal chemists because of their biological and chemotherapeutic importance [3]. Among them, analogues of pyrazolo[1,5-a]pyrimidine were found to have a number of biological activities, such as antitumor [4, 5], antifungal [6], antibacterial [7], analgesics [8], anti-inflammatory [9]. Based on the previous findings of biological effectiveness of pyrazolo[1,5-a]pyrimidine derivatives and the continuation of our research program for the synthesis of new compounds exhibiting biological activity [10–12], we synthesized a new pyrazolo[1,5-a]pyrimidine derivative, 7-(4-methylpiperazin-1-yl)-5-[4-(trifluoromethyl)phenyl]pyrazolo[1,5-a]pyrimidine-3-carbonitrile by three-step synthesis and evaluated its anticancer properties against two human cancer cell lines (K562 and MKN45) using MTT assay.
EXPERIMENTAL
Materials and Methods
Unless otherwise specified, all starting materials and reagents were obtained from commercial supplies without further purification. All melting points were obtained with a Beijing Taike X-4 microscopy melting point apparatus and were uncorrected. 1H NMR spectra were recorded on a Bruker Biospin 600 MHz or Bruker Biospin 300 MHz instrument using TMS as the internal standard. All chemical shifts were reported in ppm. Infrared (IR) spectra were recorded using KBr pellets on a PerkinElmer Spectrum One FT-IR spectrometer. Mass spectra were obtained on the analytical system of a 6460 QQQ mass spectrometer (Agilent, USA). Elemental analysis of newly synthesized compounds was carried out on a Carlo Erba 1108 analyzer and concentrations were within the theoretical values.
Synthesis of 7-Hydroxy-5-[4-(Trifluoromethyl)phenyl]pyrazolo[1,5-a]pyrimidine- 3-Carbonitrile (3)
A solution of 5-amino-1H-pyrazole-4-carbonitrile (2.0 g, 18.5 mmol) and methyl 3-oxo-3-[4-(trifluoromethyl)phenyl]propanoate (5.5 g, 22.3 mmol) in acetic acid (50 mL) was refluxed for 24 h. After cooling to room temperature, the formed precipitate was filtered off, washed with water, and dried to give 2.4 g compounds 3 in 42.6% yield; IR (KBr): 3430, 2964, 2221, 1631, 1553, 1487, 1384, 1326, 1215, 1165, 1112, 1065, 1016 cm–1; 1H NMR (DMSO-d6, 600 MHz): δ 12.78 (s, 1H), 8.06 (d, 2H, J = 8.4 Hz), 7.81 (s, 1H), 7.62 (d, 2H, J = 8.4 Hz), 6.32(s, 1H); (ESI) m/z: 305.1% [M + H]+.
Synthesis of 7-Chloro-5-[4-(Trifluoromethyl)phenyl]pyrazolo[1,5-a]pyrimidine- 3-Carbonitrile (4)
A mixture of 7-hydroxy-5-[4-(trifluoromethyl)phenyl]pyrazolo[1,5-a] pyrimidine-3-carbonitrile (2.0 g, 6.6 mmol), POCl3 (20 mL) was heated at reflux for 6 h. After cooling to room temperature, the reaction mixture was slowly added to ice/water with vigorous stirring. The mixture was then filtered, washed with cold water and dried to yield compounds 4 as yellow solid (1.3 g, 61.3%). The solid was used for the next step without further purification. IR (KBr): 2231, 1736, 1609, 1544, 1496, 1457, 1372, 1328, 1202, 1166, 1120, 1071, 1015 cm–1; (ESI) m/z: 323.2% [M + H]+.
Synthesis of 7-(4-Methylpiperazin-1-yl)-5-[4-(Trifluoromethyl)phenyl]pyrazolo[1,5-a]pyrimidine- 3-Carbonitrile (5)
A mixture of intermediate compounds 4 (0.80 g, 2.48 mmol), 1-methylpiperazine (0.50 g, 5.0 mmol), and potassium carbonate (0.79 g, 7.44 mmol) in acetonitrile (20 mL) was stirred at 50°C overnight. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure and then partitioned between water (50 mL) and CH2Cl2 (50 mL). The organic layer was separated and the aqueous layer was then extracted with CH2Cl2 (40 mL × 2). The combined organic extracts were sequentially washed with water (50 mL × 3), brine (80 mL × 3) and dried over anhydrous Na2SO4. After evaporation of the organic solvent, the residue was purified by column chromatography to afford 0.41 g title compound in 42.8% yield. Melting point 243–245°C; IR (KBr) ν: 2939, 2850, 2800, 2219, 1610, 1560, 1521, 1446, 1405, 1326, 1293, 1234, 1213, 1198, 1161, 1145, 1115, 1069, 1016, 1002 cm–1; 1H NMR (DMSO-d6, 600 MHz): δ 8.72 (s, 1H), 8.46 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.20 (s, 1H), 3.95 (br, 4H), 2.55 (br, 4H), 2.26 (s, 3H); (ESI) m/z: 387.2% [M + H]+; Calculated elemental concentrations for C19H17F3N6: C 59.06, H 4.43, N 21.75 at %; found C 59.18, H 4.48, N 21.83 at %.
Single Crystal X-Ray Crystallography
The powder of the title compound was dissolved in ethylanol/ethyl acetate/tetrahydrofuran mixed solvents in a ratio of 5 : 2 : 3 (vol/vol/vol). After slowly evaporation of the solvents for several days, some colourless single crystals suitable for X-ray analysis were obtained. A single crystal was mounted on glass fiber for data collection at 293 K on a Bruker APEX-II CCD automatic diffractometer equipped with a graphite monochromator (MoKα radiation, λ = 0.71073 Å). The data collected were integrated using the SAINT program [13]. Multi-scan absorption corrections were applied using the SADABS program [14]. The structure was solved by direct methods and refined on F2 by full matrix least squares methods using the SHELXTL package [15]. Hydrogen atoms were located from geometric calculations, and their positions and thermal parameters were fixed during the structure refinement. CCDC 1901456 contains the supplementary crystallographic data that can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
In Vitro Anticancer Activity Test on A549 and H460 Cell Lines
The compound 5 was evaluated for its in vitro cytotoxic activity against two cancer cell lines (K562 and MKN45) by the MTT-based assay method under standard conditions [16] using sorafenib tosylate as a positive control (Table 2). Cells were seeded in 96-well plate. After seeding for 24 h, the medium was removed. The test compounds were dissolved in DMSO and diluted with culture medium to different concentrations (the final concentration of DMSO was 0.1%). The test compound solution in amount of 20 μL was added in duplicates, and incubation continued for 72 h in a humidified atmosphere of 5% CO2 at 37°C. By removing the medium, 20 μL MTT solution (5 mg/mL, pH 7.4, PBS was the solvent) was added to each well and incubated for additional 3–4 h. The medium was replaced by 150 mL DMSO to solubilize the purple formazan crystals produced and the absorbance was measured on a microplate reader at 490 nm. The cellular proliferation inhibition rate was calculated as follows: inhibition rate = [1 – OD490 (treated)/OD490(control)] × 100%.
RESULTS AND DISCUSSION
The synthesis route of the title compound was depicted in Scheme 1. This compound was isolated by silica gel column chromatography and its structure was elucidated by IR, 1H NMR and elemental analyses. IR shows the peak at about 2219 cm–1 resulting from the cyano group stretching vibration at pyrazole ring. The 1H NMR spectrum exhibits a sharp singlet at 2.26 ppm, corresponding to the methyl proton of piperazinyl ring. In the mass spectrum, the peak appeared at m/z 387.2 ([M + H]+, 100%), which is in accordance with its molecular formula. IR and mass spectrometry, 1H NMR, elemental and single crystal X-ray analyses of the target compounds confirmed their structural integrity.

Synthetic route of the title compound.
The single crystal of the title compound was cultured by using ethylanol/ethyl acetate/ tetrahydrofuran mixed solvents in the ratio of 5 : 2 : 3 (vol/vol/vol) at room temperature and studied by X-ray diffraction method to confirm its structure. Crystallographic data and experimental details of structural analysis are summarized in Table 1. Selected geometric parameters (bond lengths, angles and torsion angles) and hydrogen bond parameters are listed in Tables 3 and 4, respectively.
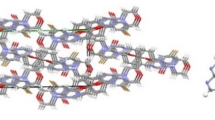
The title compound crystallizes in monoclinic symmetry, space group P21/c. In the crystal (Fig. 1), the average bond lengths and bond angles of pyrazolo[1,5-a]pyrimidine, piperazinyl and phenyl ring are in normal ranges. The distances N(1)–C(1), N(1)–C(4), N(2)–C(4), N(2)–C(3), and N(5)–C(3) are 1.341(3), 1.335(3), 1.388(3), 1.380(3), and 1.354(3) Å, respectively. They are remarkably shorter than the typical C(sp2)–N bond (1.426 Å), but closer to the C=N double bond (1.33 Å) [17]. As shown in Fig. 1, in the structure of the title compound, the piperazine ring is in a chair conformation with the Ph–N and N–methyl groups in the equatorial positions. The dihedral angles between pyrazolo[1,5-a]pyrimidine and phenyl ring is are 2.21°, which indicates that phenyl planes are nearly coplanar with pyrazolo[1,5-a]pyrimidine. Furthermore, the intermolecular hydrogen bonds C(2)–H(2)⋅⋅⋅N(4), C(18)–H(18)⋅⋅⋅N(4) play a major role in the stabilizing of the molecule. It should be noted that the crystal packing is further stabilized by weak π–π interactions (3.451(4) Å). These interactions together with intermolecular hydrogen bonds result in the formation of a two-dimensional layer framework along bc plane in the crystal (Fig. 2).

The structure of the C19H17F3N6 compound with all atoms exept hydrogen atoms. Ellipsoids are drawn at the 30% probability level.

A packing diagram of the C19H17F3N6 compound.
In addition, the bioassay results showed the title compound exhibited remarkable inhibitory activity against K562 and MKN45 cell lines, which was similar to that of the positive control sorafenib. As shown in Table 2, the title compound exhibited significant inhibitory activity against K562 cell lines and slightly more potent than Sorafenib Tosylate, but slightly less potent than sorafenib tosylate against MKN45 cell lines. Further structure optimization may result in formation of more active anticancer compounds.
REFERENCES
L. D. Via, O. Gia, V. Gasparotto, et al., Eur. J. Med. Chem. 43, 429 (2008).
H. G. Ding, Z. Q. Cai, L. Hou, et al., J. Chem. Soc. Pak. 41, 186 (2019).
J. Xiaohui, Z. Juan, L. Jiufu, J. Lingxia, and G. Hongguang, Chin. J. Struct. Chem. 38, 1889 (2019).
U. Asghar, A. K. Witkiewicz, N. C. Turner, et al., Nat. Rev. Drug Discov. 14, 130 (2015).
Y. Li, W. Gao, F. Li, et al., Mol. Biosyst. 9, 2266 (2013).
T. Novinson, R. K. Robins, and T. R. Matthews, J. Med. Chem. 20, 296 (1977).
M. A. Gouda, M. A. Berghot, A. I. Shoeib, et al., Eur. J. Med. Chem. 45, 1843 (2010).
M. R. Shaaban, T. S. Saleh, A. S. Mayhoub, et al., Biorg. Med. Chem. 16, 6344 (2008).
G. Auzzi, F. Bruni, L. Cecchi, et al., J. Med. Chem. 26, 1706 (1983).
L. Ju, G. Hongguang, and L. Jiufu, J. Chem. Res. 39, 4 (2015).
L. Ju, Z. Xinwei, S. Duanzheng, et al., J. Chem. Res. 40, 92 (2016).
L. Ju, S. Jiantao, H. Xuechen, et al., J. Chem. Res. 42, 486 (2018).
Bruker AXS, SMART & SAINT, Software Reference Manuals, Version 6.22 (Bruker AXS Analytic X-ray Systems Inc., Madison, WI, USA, 2000).
G. M. Sheldrick, SADABS, Software for Empirical Absorption Correction (Univ. of Göttingen, Göttingen, 2000).
G. M. Sheldrick, Acta Crystallogr. C 71, 3 (2015).
L. Ju, L. Yutong, H. Xuechen, et al., Arch. Pharm. 5, e201800338 (2019).
L. Jiufu, J. Lingxia, S. Juan, et al., Chin. J. Struct. Chem. 37, 1814 (2018).
ACKNOWLEDGMENTS
This work was performed as part of the Shenyang Science and Technology project (project no. 18-013-0-03).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yi-Lin Gong, Dai, P., Chen, Y. et al. Synthesis, Crystal Structure and Biological Activity of 7-(4-Methylpiperazin-1-Yl)-5-[4-(Trifluoromethyl)Phenyl]pyrazolo[1,5-a]Pyrimidine-3-Carbonitrile. Crystallogr. Rep. 65, 1111–1116 (2020). https://doi.org/10.1134/S1063774520070068
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774520070068