
Abstract
Two novel N-phosphinyl ureas containing different substituents were synthesized and characterized by 1H, 13C, and 31P NMR, IR, UV, mass spectroscopy, and elemental analysis. The crystal structures of these compounds were determined by X-ray crystallography. The structure of one compound exhibits the presence of two independent forms of the molecule with equal occupancy in the lattice and theoretical data reveal the same stabilization energies for these conformers. The title molecules have anti conformation with respect to the C=O and P=O bonds, whereas the other compound shows syn configuration. Quantum chemical calculations were applied to clarify this conformational behavior. Furthermore, the molecular geometry and vibrational frequencies of the new derivatives in the ground state were calculated by using the Hartree–Fock (HF) and density functional method (B3LYP) with 6-31+G** and 6-311+G** basis sets and compared with experimental values. The new derivatives were additionally tested in view of their antibacterial properties.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Substituted ureas are one of the most important classes of chemical compounds due to their various biological activities as anticancer [1], antibacterial [2], antiviral, antifungal, and anti-HIV agents [3], agricultural pesticides, herbicides, and plant growth regulators [4]. The stereochemical and pharmacological properties of six-membered phosphorus-containing heterocycles and their active role as cyclophosphamide analogs have also attracted attention, e.g., regarding their structural stability and conformational behavior [5–8]. However, the combination of urea and phosphoryl fragments can be interesting from different points of view such as conformational and biological properties and complexation.
Although syntheses of molecules with the general formula RNHC(O)NHP(O)R2 have been reported [9–12], little attention has been given to their biological properties [13] and structural studies from either experimental or theoretical points of view [14] so far. Besides, in our previous studies, the crystal structure of most of the compounds containing a –C(O)NHP(O)– backbone showed that the P=O and C=O bonds are in anti position with respect to each other [15–19] and the syn configuration is rare in the relevant studies [20, 21]. In this study, two new N-phosphinyl ureas were synthesized and characterized by IR, 1H, 13C, and 31P NMR, UV, mass spectroscopy, and elemental analysis. The molecular structure and conformational properties in the solid state were determined by X-ray diffraction analysis and vibrational spectroscopy. To further investigate the conformation, quantum chemical calculations were applied. Furthermore, we calculated geometric parameters of the title compounds in the ground state and compared the observed IR spectra of these molecules with calculated harmonic vibrations by Hartree–Fock (HF) and density functional theory (DFT) (B3LYP) methods. Finally, the antibacterial activities of the compounds against Staphylococcus aureus, Bacillus subtilis, Escherichia coli, and Salmonella typhi were measured.
Results and discussion
The reaction of diamines with N-arylureidophoshoryl dichlorides is a method for the formation of phosphorus-containing heterocycles. As indicated in Scheme 1, we synthesized two new heterocyclic compounds from the reaction of N-phenylureidophosphoryl dichloride (2) and N-(4-nitrophenyl)ureidophosphoryl dichloride (3) [22, 23] with 2,2-dimethyl-1,3-diaminopropane in the presence of an HCl scavenger (an excess amount of the corresponding diamine).

NMR study
Phosphorus–hydrogen and phosphorus–carbon coupling constants and δ (31P) data of compounds 4 and 5 are summarized in Table 1. The 31P NMR spectra demonstrate that substitution of a proton in 5 by a NO2 group results in a shielded phosphorus atom. The CH3 groups on the diazaphosphorinane ring in these compounds are diastereotopic, thus the 1H and 13C NMR spectra each show two separate methyl signals. The 1H NMR reveals large 3 J PNCH coupling constants of about 24.5 Hz, which is related to an equatorial proton with P–N–C–H torsion angle near 180°, as obtained from X-ray crystallography. These values are much larger than 3 J PNCH for acyclic phosphoramidates [17, 18, 24]. Also, the 13C NMR indicates that 3 J PC coupling constants are larger than 2 J PC coupling constants.
Crystal structure analysis
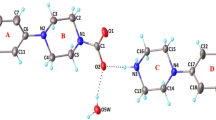
Single crystals of 5,5-dimethyl-2-(N-phenylureido)-1,3,2-diazaphosphorinane-2-oxide (4) and 5,5-dimethyl-2-[N-(4-nitrophenyl)ureido]-1,3,2-diazaphosphorinane-2-oxide (5) were grown from diffusion of diethyl ether into methanol solution and ethanol/acetonitrile at room temperature. Details of the crystallographic data collection and refinement parameters are presented in Table 2. Molecular structures are shown as Oak Ridge thermal ellipsoid plots (ORTEP) in Figs. 1 and 2.

Molecular structure of 4 showing the atom-labeling scheme and 50% probability level displacement ellipsoids

Molecular structure of 5 showing the atom-labeling scheme and 50% probability level displacement ellipsoids
Compound 4 exists as two crystallographically independent molecules in the crystalline lattice (A and B) at a 1:1 ratio due to the different spatial orientations of the two conformers relative to each other that cause different torsion angles (Table 3). Each conformer is connected to four molecules of the other conformer via P(O)···H–N and C(O)···H–N hydrogen bonds. Linking of these hydrogen bonds leads to form a two-dimensional polymeric chain in the crystalline lattice. Furthermore, there are intramolecular P=O···H–NPh hydrogen bonds in both conformers. The crystal packing of 5 is dominated by the occurrence of P=O···H–N and N–O···H–N hydrogen bonds, which lead to a two-dimensional polymeric chain.
The P–(endocyclic N) bond lengths in both compounds 4 and 5 are lower than the P–N bond of –C(O)N(H)P(O)– moieties (Tables 3, 4). All of these bonds are in the range 1.622(2)–1.694(2) Å and thus are significantly shorter than a typical P–N single bond (1.77 Å) [25]. The P=O bond lengths found in the molecules A and B (1.483(18) and 1.480(18) Å) and for compound 5 (1.47(2) Å) are slightly longer than the normal P=O bond length (1.45 Å) [25]. It is interesting to note that in conformers A and B the form with the stronger P=O bond, i.e., B, has weaker P–(endocyclic N) bonds.
The phosphorus atoms have slightly distorted tetrahedral configuration with the angles in the range of 102.16(10)–115.95(11)° (in A), 102.22(10)–115.95(11)° (in B), and 104.27(12)–114.34(12)° (in 5). Moreover, the P=O bond is placed in an equatorial position. The equatorial preference for the P=O bond was previously observed by Bentrude [26] and was attributed to the overlap of the endocyclic nitrogen p orbital with the P–(exocyclic N) antibonding orbital. The endocyclic nitrogen atoms in compounds 4 and 5 are distorted from planarity. The sum of angles around these atoms in 4 are 352.78° and 351.86° for N(3) and N(4) atoms, 352.35° and 350.41° for N(7) and N(8) atoms, and in 5 are 344.9° and 346.38° for N(4) and N(5). The sum of the surrounding angles for all the exocyclic nitrogen atoms are almost 360°, therefore the environment of the N atoms is practically planar.
Mass spectroscopy
Mass spectra of two synthetic derivatives 4 and 5 with formula  (R = H (4), 4-NO2 (5)) reveal the presence of the molecular ions at m/z = 282 and 327. In our previous studies, it was assumed that the fragmentation pathway of compounds with the general formula R–C6H4C(O)NHP(O)Ŕ2 involves P–N cleavage and P–O formation. Subsequently, the rearranged molecule is cleaved by using a pseudo-McLafferty mechanism to generate R–PhCN+ and the related amidophosphoric acid cations [17, 19, 27].
(R = H (4), 4-NO2 (5)) reveal the presence of the molecular ions at m/z = 282 and 327. In our previous studies, it was assumed that the fragmentation pathway of compounds with the general formula R–C6H4C(O)NHP(O)Ŕ2 involves P–N cleavage and P–O formation. Subsequently, the rearranged molecule is cleaved by using a pseudo-McLafferty mechanism to generate R–PhCN+ and the related amidophosphoric acid cations [17, 19, 27].
In the present study, mass spectra of the title compounds show that with insertion of NH between C=O and phenyl ring the pseudo-McLafferty pathway is observed with very weak intensity and the main pathway is C–N(1) bond cleavage of the –N(1)HC(O)N(2)H– moiety. Then, the cleavage pathway is the same as the constitution pathway of N-arylureidophoshoryl dichlorides 2 and 3 (Scheme 1). The fragmentation of synthesized products shows a peak at m/z = 189, corresponding to the loss of aniline or nitroaniline groups and formation of  . Fragment ions at m/z = 93 and 138 are base peaks and assigned to PhNH2
+ and 4-NO2PhNH2
+.
. Fragment ions at m/z = 93 and 138 are base peaks and assigned to PhNH2
+ and 4-NO2PhNH2
+.
UV–Vis study
The electronic spectra of compounds 4 and 5 were studied in DMSO solution. The results revealed that a strong absorbance at 275 nm corresponds to the π–π* transition for 4. This band shifts to 308 nm with higher intensity for 5. The red-shifted band evidently arises from a resonance effect of the 4-NO2 group. Besides, compound 5 has a weak absorbance at 370 nm which is consistent with the n–π* transition.
Computational study
The experimental and optimized geometric parameters (bond lengths, bond angles, and dihedral angles) by HF and DFT (B3LYP) with 6-31+G** and 6-311+G** basis sets are listed in Tables 3 and 4. Similar geometric parameters are obtained by the two applied methods. Although the biggest differences between calculated and experimental values of bond lengths and angles are about 0.024 Å and 0.048 Å and 1.99° and 1.54° for HF and DFT methods, respectively, the HF method at 6-311+G** for the bond length and the DFT method at 6-31+G** for the bond angles correlate slightly better than other levels for compound 4.
For compound 5, the HF method with 6-311+G** basis set correlates slightly better than the DFT method for bond lengths and bond angles. The considerable differences between the calculated and experimental data of the bond lengths are about 0.057 and 0.086 Å for the HF and DFT method and those of angle values are 2.02 and 1.84° for the DFT and HF method, respectively. The calculated energy for two conformers of compound 4 is summarized in Table 5. The calculated data in Tables 3 and 5 indicate that the bond lengths and angles are identical and the structural stability of conformer A is equal to those of conformer B. Also, the data show that the two conformers only have differences in their corresponding torsion angles.
On the basis of the previously reported structures [15–18, 28, 29] and the data obtained by theoretical calculations [30, 31], orientation of the P=O and C=O groups in –C(O)NHP(O)– skeleton is anti and the crystal structure of 4 is in agreement with the data given in the literature. In contrast, 5 is one of the few compounds that is close to syn configuration with O(4)–P(1)–C(7)–O(3) torsion angle of −52.73°, whereas calculations predict a structure with anti conformation as the most stable form for compound 5 (Table 6). This arrangement is attributed to the packing effect that allows a hydrogen bond between the phosphoryl oxygen atom and the two hydrogens of the –NHC(O)NH– moiety (Fig. 3). Table 6 shows that the anti conformer has a larger total dipole moment than the syn form, which is consistent with the increased relative stability of the anti conformer. In fact, some of the earlier studies on conformational analysis have shown that a conformation with the larger dipole moment becomes stabilized due to dipole–dipole interaction [32, 33].

Molecular structure of 5 showing hydrogen bond interactions
Vibrational analysis
We calculated the theoretical vibrational spectra of compounds 4 and 5 and compared these calculations with their experimental results. The most important absorptions together with the computed vibrational data are listed in Tables 7 and 8. Since two conformers of compound 4 have equal energy in the gas phase, only one molecular structure was selected to obtain harmonic vibrational frequencies.
Calculated vibrational frequencies of 4 and 5 were compared with the experimental data. Subsequently, a tentative assignment of the observed bands in the infrared spectra was carried out by comparison with theoretical wavenumbers, as well as with the relevant data reported in the literature for urea derivatives [34, 35] and related phosphoramidates [15, 30, 31, 36]. To discuss the vibrational assignments, we took the functional density (DFT) and HF calculated wavenumbers corrected by a scale factor of 0.960 and 0.902.
As can be seen from Table 7, the intense bands centered at 3,205 and 2,935 cm−1 in the IR spectrum of 4 are assigned to the N–H stretching vibration of amide and amine groups. For 5 these modes were assigned to the observed bands at 3,335, 3,090, and 2,920 cm−1 (Table 8). Well-defined absorptions at 1,677 and 1,699 cm−1 correspond to C=O stretching mode for 4 and 5. Intense absorptions observed at 1,184 and 1,200 cm−1 can be assigned with confidence to the characteristic P=O stretching mode for 4 and 5. It is interesting to note that ν(C=O) and ν(P=O) modes for compound 5 appear at higher frequencies than the corresponding modes of compound 4 due to substitution of the electron-withdrawing group on the phenyl ring in 5 that are in good agreement with X-ray data. In molecule 5, the ν as(NO2) mode is located at 1,547 cm−1.
The observed bands at 1,256 and 1,380 cm−1 are assigned to the N–C–N stretching vibration of compounds 4 and 5. The ring breathing normal mode was assigned to the bands at 1,019 cm−1 for 4 and 1,075 cm−1 for 5. From three medium absorptions centered at 1,090, 1,045, and 951 cm−1 for 4, the first can be assigned with confidence to the C–N stretching mode, whereas the other two bands can be assigned to ν(P–N)amine and ν(P–N)amide. For derivative 5, the bands located at 1,042, 951, and 922 cm−1 were attributed to the same modes. Moreover, the intense bands observed at 1,593, 1,448, and 1,019 cm−1 in the IR spectrum of 4 are assigned to modes associated with N–H, ring, and N–C–N deformations (Table 7). Similar assignments were provided for 5 to the observed very intense infrared bands at 1,596, 1,494, and 922 cm−1 (Table 8).
Antimicrobial activity
The compounds 4 and 5 were screened to evaluate their antimicrobial activity against S. aureus (ATCC 6538P), B. subtilis (ATCC 6633), E. coli (ATCC 35218), and S. typhi (ATCC 19430) using the disk diffusion method and MIC (minimum inhibitory concentration) experiments. The results of the assays are presented in Table 9. The screening data reveal that compound 5 exhibited higher activity towards the tested microorganisms (MIC 1.17–18.8 μg cm−3) than compound 4 (MIC 385 to >400 μg cm−3). The MIC values of 4 and 5 against certain bacterial strains indicate that S. aureus were more sensitive to the toxicity of the synthesized compounds than other microorganisms. The data show that compounds were inactive against S. typhi. Furthermore, derivative 5 was the more potent compound against B. subtilis; its activity was higher than gentamycin. It may be concluded that the structure of the tested compounds is the principal factor influencing the antimicrobial activity and changing the substituents in the phenyl ring leads to compounds with different antibacterial activity.
Conclusion
Two new N-phosphinyl ureas were synthesized and characterized by multinuclear (1H, 13C, and 31P) NMR, UV, IR spectroscopy, elemental analysis, and mass spectrometry techniques. X-ray crystallography confirmed the occurrence of two independent conformers for compound 4 with anti configuration around the dihedral angle of the –C(O)NHP(O)– skeleton. Quantum chemical calculations predicted that the structural stability of these molecules is equal. The crystal structure of compound 5 showed that the P=O and C=O double bonds are in syn position with respect to each other, whereas theoretical data showed that anti conformation is stable. Furthermore, the harmonic vibrations of the synthesized derivatives computed by the RHF and DFT methods were in good agreement with the experimental IR spectra values. The results of antimicrobial assays indicated that derivative 5 with an electron-withdrawing group has higher activity against the tested microorganisms.
Experimental
Materials and methods
All reactions were carried out under argon atmosphere. All chemicals and solvents were purchased from Merck and used without further purification. 1H, 13C, and 31P NMR spectra were recorded on a Bruker Avance DRS 500 MHz spectrometer. 1H and 13C chemical shifts were determined relative to TMS, 31P chemical shifts relative to 85% H3PO4 as external standards. Infrared spectra were obtained by using KBr pellets on a Shimadzu IR-60 spectrometer. Elemental analysis was performed by using a Heraeus CHN-O-RAPID apparatus. The experimental data were in good agreement with the calculated values. Melting points were determined on an electrothermal apparatus. Mass spectra were obtained with MS model 5973 Network apparatus using 70 eV as ionization energy. Electronic spectra were recorded on a Shimadzu UV-2100 spectrometer. Dichlorophosphinylureas 2 and 3 were prepared by using a method reported by Kirsanov et al. [22, 23] (Scheme 1). First, dichloroisocyanatophosphine oxide (1) was obtained from the reaction of phosphorus pentachloride and ethyl carbamate in ethylene chloride, then the treatment of aniline derivatives with 1 led to N-phenylureidophosphoryl dichloride (2) and N-(4-nitrophenyl)ureidophosphoryl dichloride (3).
General procedure for the synthesis of N-phosphinylureas 4 and 5
A solution of 1.23 g 2,2-dimethyl-1,3-diaminopropane (12 mmol) was added dropwise to a suspension of 2 or 3 (6 mmol) in 30 cm3 dry diethyl ether and stirred at 0 °C. After 5 h, the products were filtered off and washed with H2O.
5,5-Dimethyl-2-(N-phenylureido)-1,3,2-diazaphosphorinane-2-oxide (4, C12H19N4O2P)
Yield 85%; m.p.: 195–196 °C;1H NMR (DMSO-d 6): δ = 0.79 (s, 3H, CH3), 1.03 (s, 3H, CH3), 2.57 (ddd, 2 J HH = 11.92 Hz, 3 J HH = 5.26 Hz, 3 J PNCH = 24.21 Hz, 2H), 2.98 (d, 2 J HH = 11.93 Hz, 2H), 4.66 (d, 2 J PNH = 2.70 Hz, 2H, endocyclic NH), 6.95 (t, 3 J HH = 7.22 Hz, 1H), 7.25 (t, 3 J HH = 7.57 Hz, 2H), 7.36 (d, 3 J HH = 7.99 Hz, 2H), 7.63 (d, 2 J PNH = 6.76 Hz, 1H, NHP), 9.32 (s, 1H, PhNH) ppm;13C NMR (DMSO-d 6): δ = 23.29 (s, CH3), 24.84 (s, CH3), 30.46 (d, 3 J PC = 4.28 Hz), 52.35 (d, 2 J PC = 1.76 Hz), 118.11 (s), 121.94 (s), 128.70 (s), 139.29 (s), 153.33 (d, 2 J PC = 2.33 Hz) ppm;31P NMR (DMSO-d 6): δ = 3.78 (m) ppm; IR (KBr): \( \bar{\nu } \) = 3,205 (s, N–H), 2,935 (m, N–H), 1,677 (vs, C=O), 1,593 (s), 1,547 (s), 1,487 (m), 1,448 (s), 1,381 (w), 1,336 (m), 1,256 (w), 1,184 (s, P=O), 1,090 (m), 1,045 (m, P–N), 1,019 (w), 951 (m), 858 (m), 746 (s) cm−1; UV–Vis (DMSO): λ max = 275 nm; MS (70 eV): m/z = 282 (M+), 189 (M − C6H5NH2 +), 93 (C6H5NH2 +).
5,5-Dimethyl-2-[N-(4-nitrophenyl)ureido]-1,3,2-diazaphosphorinane-2-oxide (5, C12H18N5O4P)
Yield 70%; m.p.: 204–205 °C; 1H NMR (DMSO-d 6): δ = 0.79 (s, 3H, CH3), 1.04 (s, 3H, CH3), 2.58 (ddd, 2 J HH = 11.86 Hz, 3 J HH = 5.07 Hz, 3 J PNCH = 24.47 Hz, 2H), 3.01 (d, 2 J HH = 11.80 Hz, 2H), 4.79 (s, 2H, endocyclic NH), 7.62 (d, 3 J HH = 9.05 Hz, 2H), 8.17 (d, 3 J HH = 9.05 Hz, 2H), 7.92 (brd, 2 J PNH = 4.15 Hz, 1H, NHP), 10.06 (s, 1H, 4-NO2-PhNH) ppm; 13C NMR (DMSO-d 6): δ = 23.25 (s, CH3), 24.83 (s, CH3), 30.42 (d, 3 J PC = 4.53 Hz), 52.34 (br), 117.51 (s), 125.10 (s), 141.22 (s), 145.82 (s), 153.13 (s) ppm; 31P NMR (DMSO-d 6): δ = 3.23 (m) ppm; IR(KBr):\( \bar{\nu } \)=3,335 (m, N–H), 3,090 (m, N–H), 2,920 (m, N–H), 1,699 (s, C=O), 1,596 (m), 1,547 (m), 1,494 (s), 1,380 (w), 1,330 (s), 1,299 (m), 1,200 (s, P=O), 1,105 (m), 1,075 (m), 1,042 (m), 951 (w), 922 (w, P–N), 850 (s), 647 (w) cm−1; UV–Vis (DMSO): λ max = 370, 308 nm; MS (70 eV): m/z = 327 (M+), 189 (M −(4-NO2C6H4NH2)+), 138 (4-NO2C6H4NH2 +).
X-ray measurements
X-ray data of compounds 4 and 5 were collected on a Bruker SMART 1000 CCD single crystal diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were refined with SHELXL-97 [37] by full-matrix least-squares on F 2. The positions of hydrogen atoms were obtained from the difference Fourier map. Routine Lorentz and polarization corrections were applied and an absorption correction was performed by using the SADABS program for these compounds [38]. CCDC 742025 and 742031 contain the supplementary crystallographic data for 4 (C12H19N4O2P1) and 5 (C12H18N5O4P1). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
Biological evaluation
The synthesized compounds were tested for their antibacterial activities by the standardized disk diffusion method [39]. The assayed collection included the following microorganisms: S. aureus (ATCC 6538P), B. subtilis (ATCC 6633), E. coli (ATCC 35218), and S. typhi (ATCC 19430).
In the disk diffusion method, sterile paper discs (6.4 mm diameter) impregnated with compounds tested (solutions in DMSO) to a load 400 μg of a compound per disc were placed on the surface of the media inoculated with the microorganisms. Discs containing DMSO were used as negative control. Gentamycin was used as standard drug (positive control). The diameter of the growth inhibition zone was read after 24 h of incubation at 35 °C. These compounds were further examined by the broth dilution method to determine their MIC (minimal inhibitory concentration) [40]. Concentrations of the agents tested in solid medium ranged from 0.5 to 400 μg cm−3. Minimal inhibitory concentrations were read after 24 h of incubation at 35 °C.
Computational methods
All quantum chemical calculations were performed with the GAUSSIAN 98 [41] system of programs, implemented in a Pentium 4 computer. The calculations were performed for molecules in the gaseous phase. The molecular geometries were optimized by using B3LYP and HF methods with 6-31+G** and 6-311+G** basis sets. Harmonic vibrational frequencies were obtained by using B3LYP and HF methods with 6-311+G** basis sets and compared with the experimental data.
References
Li HQ, Lv PC, Yan T, Zhu HL (2009) Anticancer Agents Med Chem 9:471
Seth PP, Ranken R, Robinson DE, Osgood SA, Risen LM, Rodgers EL, Migawa MT, Jefferson EA, Swayze EE (2004) Bioorg Med Chem Lett 14:5569
Struga M, Kossakowski J, Kedzierska E, Fidecka S, Stefańska J (2007) Chem Pharm Bull 55:796
Vishnyakova TP, Golubeva IA, Glebova EV (1985) Russ Chem Rev 54:249
Zalán Z, Martinek TA, Lázár L, Fülöp F (2003) Tetrahedron 59:9117
Billman JH, Meisenheimer JL, Awl RA (1964) J Med Chem 7:366
Gholivand K, Shariatinia Z, Afshar F, Faramarzpour H, Yaghmaian F (2007) Main Group Chem 6:231
Frank É, Kazi B, Mucsi Z, Ludányi K, Keglevich G (2007) Steroids 72:446
Safiulina AM, Goryunov EI, Letyushov AA, Goryunova IB, Smirnova SA, Ginzburg AG, Tananaev IG, Nifant’ev EE, Myasoedov BF (2009) Mendeleev Commun 19:263
Goryunov EI, Nifant’ev EE, Myasoedov BF (2007) RF Patent 2 296 768
Khailova NA, Krepysheva NE, Saakyan GM, Bagautdinova RKh, Shaimardanova AA, Zyablikova TA, Azancheev NM, Litvinov IA, Gubaidullin AT, Zverev VV, Pudovik MA, Pudovik AN (2002) Russ J Gen Chem 72:1071
Papanastassiou ZB, Bardos TJ (1962) J Med Chem 5:1000
Zhao GF, Yang HZ, Wang LX (1998) Chem J Chin Univ 19:555
Tananaev IG, Letyushov AA, Safiulina AM, Goryunova IB, Baulina TV, Morgalyuk VP, Goryunov EI, Gribov LA, Nifant’ev EE, Myasoedov BF (2008) Dokl Chem 422:260
Gholivand K, Mostaanzadeh H, Koval T, Dusek M, Erben MF, Della Ve′dova CO (2009) Acta Cryst B 65:502
Gholivand K, Pourayoubi M (2004) Z Anorg Allg Chem 630:1330
Gholivand K, Shariatinia Z, Pourayoubi M (2006) Z Anorg Allg Chem 632:160
Gholivand K, Pourayoubi M, Shariatinia Z, Mostaanzadeh H (2005) Polyhedron 24:655
Gholivand K, Madani Alizadehgan A, Mojahed F, Soleimani P (2008) Polyhedron 27:1639
Gholivand K, Shariatinia Z, Ansar S, Mashhadi SM, Daeepour F (2009) Struct Chem 20:481
Gholivand K, Mostaanzadeh H, Koval T, Dusek M, Erben MF, Stoeckli-Evans H, Della Ve′dova CO (2010) Acta Cryst B 66:441
Kirsanov AV (1954) J Gen Chem USSR 24:1031
Kirsanov AV, Zhmurova IV (1956) J Gen Chem USSR 26:2642
Eliel E, Hutchins RO (1969) J Am Chem Soc 91:2703
Cogridge DEC (1995) Phosphorus, an outline of its chemistry, biochemistry, and technology, 5th edn. Elsevier, Amsterdam
Bentrude WG, Setzer WN, Khan M, Sopchik AE, Ramli E (1991) J Org Chem 56:6127
Gholivand K, Pourayoubi M, Shariatinia Z (2007) Polyhedron 26:837
Gholivand K, Hosseini Z, Pourayoubi M, Shariatinia Z (2005) Z Anorg Allg Chem 631:3074
Gholivand K, Shariatinia Z (2007) Struct Chem 18:95
Iriarte AG, Erben MF, Gholivand K, Jios JL, Ulic SE, Della Ve′dova CO (2008) J Mol Struct 886:66
Iriarte AG, Cutin EH, Erben MF, Ulic SE, Jios JL, Della Ve′dova CO (2008) Vib Spectrosc 46:107
Mizushima S (1954) Structure of molecules and internal rotation. Academic, New York
Watanabe IM, Mizuchima M, Masiko Y (1943) Sci Pap Inst Phys Chem Res Jpn 40:425
Badawi HM (2009) Spectrochim Acta A 72:523
Mido Y, Kitagawa I, Hashimoto M, Matsuura H (1999) Spectrochim Acta A 55:2623
Gholivand K, Madani Alizadehgan A, Arshadi S, Anaraki Firooz A (2006) J Mol Struct 791:193
Sheldrick GM (1998) SHELXTL V.5.10, Structure Determination Software Suite, Bruker AXS. Madison, WI
Sheldrick GM (1998) SADABS V. 2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS. Madison, WI
Greenwood D (1989) Antimicrobial chemotherapy. Oxford University Press, New York
Vincent JG, Vincent HW (1994) Proc Soc Exp Biol Med 55:162
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewsky VG, Montgomery JA Jr, Stratman RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Menucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslovski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RI, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzales C, Head-Gordon M, Replogle ES, Pople JA (1998) Gaussian 98, Revision A.6. Gaussian, Pittsburgh
Acknowledgments
The financial support of this work by the Research Council of Tarbiat Modares University is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Gholivand, K., Dorosti, N. Synthesis, spectroscopic characterization, crystal structures, theoretical studies, and antibacterial evaluation of two novel N-phosphinyl ureas. Monatsh Chem 142, 183–192 (2011). https://doi.org/10.1007/s00706-010-0436-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-010-0436-8