
Abstract
Background
Oral extended release (ER) delivery systems have quickly gained increasing importance because of their ability to maintain drug levels in the blood more consistently, reducing side effects and improving patient compliance. The complexity of ER formulation leads to additional development challenges in the fulfilment of quality-related regulatory requirements. Despite their challenging properties, the potential of ER system formulation and process development can be better exploited by applying quality by design (QbD) approaches and advanced modeling techniques such as machine learning (ML).
Area covered
This review provides a comprehensive overview of QbD concepts applied to oral ER delivery systems, clarifying the impact of raw materials and process variables on critical quality attributes (CQAs). Moreover, data science coupled with ML algorithms is also elucidated in this article as a potential tool for predicting and optimizing ER formulation design and manufacturing processes.
Expert opinion
QbD, as a scientific and risk-based approach, provides a comprehensive understanding of oral ER drug delivery systems improving product quality and reducing postapproval changes. Enabling QbD with ML-driven pharmaceutical development can provide an opportunity to move toward risk mitigation for efficient ER tablet formulation and process development. However, there are some barriers to overcome in the way of adopting QbD concepts. The key issues are the lack of understanding and the gap between industries and regulatory authorities concerning the scientific principles and terms beyond QbD, which can create an obstacle during the approval process. Furthermore, it is generally believed that the resources and time invested in applying QbD tools are not cost-effective during constant and continuous improvement. Today, it is time to realize that a multidisciplinary understanding of the formulation and manufacturing process is as important as achieving the final result.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The oral route is the most common route for drug administration due to its clear advantages and convenience. One of the strategies used in drug development includes the consideration of using different delivery systems and technologies to ensure the most appropriate pharmacokinetic and pharmacodynamic profiles (Walker 2008).
According to the generally accepted definition, modified release (MR) is a dosage form release pattern where the time, rate and/or location of release of the drug substance are chosen to fulfil therapeutic or compliance goals not offered by conventional dosage forms administered by the same route (FDA 1997b; EMA 2014). MR is a slightly ambiguous term embracing several types of formulations with distinct release patterns. Compendial publications such as the United States Pharmacopeia (USP) and European Pharmacopoeia (Ph. Eur.) or regulatory agencies such as the food and drug administration (FDA) and European medicines agency (EMA) do not provide a harmonized definition for MR or controlled release (CR) dosage forms.
Several types of MR systems have been recognized, including extended release (ER), delayed release (usually gastroresistant), targeted release and orally disintegrating tablets (Ding 2016). In this review, special attention is given to oral ER drug delivery systems. Since expressions such as “prolonged” (an equivalent term of extended release used by the EMA), “controlled”, “sustained”, “long-acting” and “repeat action” have also been interchangeably used to describe ER drug delivery systems, in the context of this article, ER will be used when referring to “extended release” and/or “modified release” formulations.
Although the concept of ER was introduced a few decades ago, its unique advantages and innovative technologies continue to provide pharmaceutical interest. When developing and manufacturing effective ER systems, it is crucial to ensure controlled and timed drug release with predictable kinetics, as revealed by recent research (Khan et al. 2020; Mohamed et al. 2020; Than et al. 2021; Akhtar et al. 2022). Therefore, critical raw material and process properties must be carefully selected and assessed to achieve the desired release profile.
Although ER drug product development faces several constraints in data acquisition, understanding of the drug release mechanisms, robustness and reproducibility, the potential of ER systems can be better exploited if formulation and process development are performed using the quality by design (QbD) approach.
The systematic QbD approach, supported by the International Council of Harmonisation (ICH) Q8, Q9 and Q10 guidelines, has been widely used by pharmaceutical industries to design, develop and manufacture high-quality drug products. QbD elements include the quality target product profile (QTPP), which enables the identification of critical quality attributes (CQAs); identification of critical material attributes (CMAs) and critical process parameters (CPPs), linking to the CQAs; risk assessment (RA); definition of the design space; and control strategy and continuous improvement. The application of these concepts can ensure safety, efficacy, and quality across the ER drug product lifecycle and streamline regulatory processes. Understanding the drug product and respective manufacturing process results in quality improvement and risk reduction (ICH Q9 2005, ICH Q10 2008, ICH Q8(R2) 2009). Design of experiments (DoE) and process analytical technology (PAT) are two useful tools applied in QbD. PAT tools could be fundamental to support real-time release testing (RTRT) as part of a control strategy (ICH Q8(R2) 2009).
Extended release of drugs can be achieved using numerous manufacturing technologies. Usually, strong efforts are allocated to eliciting the type of rate controlling polymers with a focus on their unique properties and respective amount. However, the performance of ER drug products based on their dissolution profiles can be more time-consuming and complex than conventional formulations, as models predicting drug release from ER systems eventually consider a high number of factors, which may lead to a high volume of data, thus hampering their fast and effective pharmaceutical development.
On the other hand, the increase in data volume and complexity generated in drug discovery and development has resulted in the growing application of efficient statistical and modeling tools. From this perspective, modern data analytics technology based on the concepts of multivariate data analysis (MVDA), artificial intelligence (AI) and machine learning (ML) algorithms, frequently coupled to the QbD approach, has guided pharmaceutical R&D assuring the desired product quality (Banner et al. 2021; Paul et al. 2021a).
While several previous studies have reported the critical points related to oral ER drug delivery systems (Maderuelo et al. 2011) as well as the importance of the use of the QbD strategy (Yu et al. 2014) and ML algorithms (Lou et al. 2021) in the pharmaceutical development context, no reviews in the literature were found comprising a multidisciplinary approach to link the three different strands: (1) different oral ER drug delivery systems; (2) a comprehensive approach of a well-structured QbD framework; and (3) application of advanced statistical modeling tools such as ML, applied to oral ER drug delivery system development.
The present review intends to detail the current state of applying QbD concepts to better understand the design and manufacture of oral ER delivery systems in pharmaceutical development. An outline of emerging opportunities in QbD implementation coupled to MVDA methods and AI/ML tools applied to oral ER drug products will also be discussed.
Applying the QbD framework to oral ER formulations
The development of ER formulations dates back to the 1960s. Since then, an increasing number of researchers from both industry and academia have allocated significant resources to a wide range of scientific domains to expand the scientific knowledge in the field of ER delivery systems (Hoffman 2008; Lee et al. 2010; Florence 2011).
The main drivers for the ground-breaking advances in controlled release were the clinical need to prolong action and improve patient benefit (Lee et al. 2010). Moreover, the first mathematical models to study the dissolution of drugs, the understanding of the behavior of delivery systems in vivo and advances in polymer sciences have also greatly contributed to this development in the pharmaceutical industry (Hoffman 2008).
Types of oral ER drug delivery systems
Oral ER drug delivery systems exhibit drug release patterns that are intentionally distinct from conventional immediate release. In fact, these specialized dosage forms allow a reduction in the dosage frequency compared to conventional dosage forms. Sustained release (SR) and CR are both definitions for drug delivery systems that can be used to achieve an ER pattern (FDA 1997a; Ding 2016). The emergence of ER systems has paved the way for significant advancements in safety and efficacy of drug release, whether by decreasing the risk of “dose-dumping” or incidence of adverse side effects or by maximizing therapeutic benefits in the maintenance of therapeutic blood levels and enhancement of patient compliance (Wen et al. 2010; Bruschi 2015; Ding 2016).
In oral ER drug delivery systems, several physical, chemical, and biological mechanisms can be strategically employed to control drug release, e.g., dissolution, diffusion, partitioning, solvent activation (osmosis and swelling), erosion and targeting. They may act simultaneously or at different stages of a delivery process. In a broad sense, the different drug release systems can incorporate different mechanisms. When different mechanisms take place simultaneously or sequentially, the dominant and rate limiting step process is the slowest (Wen et al. 2010; Nokhodchi et al. 2012; Siepmann et al. 2012a, b; Bruschi 2015).
Diffusion is one of the most common strategies for controlling drug release. It is a physical mechanism for the transport of drug molecules through a polymer under a concentration gradient and can be described by Fick’s law of diffusion. The basic designs for diffusion-controlled delivery systems are the reservoir and matrix systems where drug molecules are released through a polymer membrane or a polymer matrix, respectively (Siepmann et al. 2012a; Qiu et al. 2017; Bermejo et al. 2020).
Conversely, dissolution is the rate controlling step in dissolution-limited systems. If the polymer is quickly dissolved, the solvated drug is immediately available to diffuse from the surface, and zero-order kinetics are not achievable. Therefore, the solubility of the polymer carrier and thickness of the membrane (reservoir systems) are the key factors in controlling drug release (Siegel et al. 2012; Bermejo et al. 2020).
In dissolution- and diffusion-limited release systems, both processes often coexist. Drug release occurs by dissolution followed by diffusion through the matrix. First, the medium goes into the core, and hence, quick drug dissolution occurs by allowing diffusion of the dissolved drug out of the system. In this case, it is difficult to elicit the rate-limiting step, but commonly, the dissolution rate is controlled by the dominant mechanism—diffusion (Siegel et al. 2012; Bermejo et al. 2020).
A significant number of mathematical models were developed to aid in understanding the drug release kinetics and associated mechanisms. A review by Costa et al. (2001) describes some of the most common mechanisms, such as zero-order, first-order, Weibull, Higuchi and Korsmeyer-Peppas. Mathematical modeling of drug release can help researchers better understand and develop highly effective ER drug delivery systems (Peppas et al. 2014).
The most common oral ER drug delivery systems are matrix, reservoir polymeric and osmotic systems (Siepmann et al. 2012a; Ding 2016; Qiu et al. 2017). A brief overview of each system is provided below.
Matrix systems
Matrices are also defined as monolithic since the drug is dissolved or dispersed homogeneously through a release rate controlling polymeric matrix (Tiwari et al. 2011; Siepmann et al. 2012a; Qiu et al. 2017). Depending on the initial drug loading/drug solubility ratio, monolithic devices can be distinguished into two groups: monolithic solutions and monolithic dispersions (Siepmann et al. 2012a; Bermejo et al. 2020). The former refers to a nonsaturated drug solution—the initial drug loading is below its solubility—in which the release rate decreases with time, while the latter consists of a saturated or oversaturated drug solution comprising a dissolved and nondissolved drug fraction. In this case, the dissolved drug is first released, decreasing the concentration inside the polymer, and thereafter, the undissolved drug solid aggregates will be slowly released by diffusion after they are dissolved (Siepmann et al. 2012a; Bermejo et al. 2020). Since the mean distance traveled by the drug to the matrix surface increases with time, the geometry of monolithic systems has a substantial impact on drug release kinetics (Siepmann et al. 2000; Siegel et al. 2012; Bermejo et al. 2020).
Concerning the rate-controlling polymer properties, matrix systems may be broadly classified into hydrophilic, inert and lipid matrices (Bruschi 2015), with hydrophilic systems being the most widely utilized in marketed ER products. In these systems, the drug is dispersed or dissolved in water-soluble and/or swellable hydrophilic polymers (Vanhoorne et al. 2016; Parmar et al. 2018; Ilyes et al. 2021). Upon contact with the aqueous solution (water or physiological fluid), the hydrophilic matrix becomes hydrated, resulting in relaxation of the polymer chains and lowering of the glass transition temperature. These phenomena are responsible for the development of a ‘gel’ layer on the system surface controlling drug release (Colombo et al. 2000).
This process results in the formation of a series of fronts: the swelling front between the glassy polymer and the rubbery state, the erosion front that separates the swollen matrix of the surrounding solvent; and the diffusion front located between the swelling and erosion front, i.e., between undissolved and dissolved drug particles, respectively (Ford 2014). The gel layer thickness depends on several factors, such as the type and viscosity of the polymer, the penetration rate of the medium into the matrix, and the dissolution of drugs and excipients (Maderuelo et al. 2011; Tiwari et al. 2011; Siegel et al. 2012; Caccavo et al. 2014; Ford 2014; Timmins et al. 2016).
In hydrophilic systems, while water-soluble drugs may be released essentially by diffusion (Thapa et al. 2018), for drugs with low water solubility, matrix erosion is the predominant mechanism (Kim 1998; Chakraborty et al. 2009; Barmpalexis et al. 2018). Some examples of polymers used in hydrophilic matrices are hydroxyl propyl methyl cellulose (Hypromellose/HPMC) (Gavan et al. 2017; Barmpalexis et al. 2018), hydroxyl propyl cellulose (HPC) (Iurian et al. 2017; Than et al. 2021) and polyethylene oxide (PEO) (Nagy et al. 2019; Jang et al. 2021).
On the other hand, in inert matrix systems, the drug is incorporated into a water-insoluble polymer (Rus et al. 2020). Drug release occurs by permeation of the liquid into the polymeric matrix, dissolving the drug and/or creating pores and channels that facilitate solvent front penetration leading to dissolution and diffusion of the drug through the matrix (Frenning 2011). The drug release rate from inert matrix tablets is mainly governed by Higuchi’s equation. Ethyl cellulose (Sanoufi et al. 2020), polymethacrylates (Won et al. 2021) and polyvinyl acetate (Rus et al. 2020) are examples of water-insoluble polymeric materials used in inert matrices.
In lipid matrices, the rate-controlling polymers are hydrophobic and include waxes, glycerides, and fatty acids. Drug release from these matrices occurs through both diffusion and erosion (Petrovic et al. 2012; Bruschi 2015). Finally, the matrix systems can also be classified according to their porosity as microporous and nonporous systems (Wen et al. 2010).
Reservoir systems
In reservoir-based systems, drug diffusion is mediated by a functional controlling membrane. A drug-containing core is surrounded by a polymeric membrane, and the drug release rate is controlled by its attributes, such as thickness, composition, and physicochemical properties (Siepmann et al. 2012a). Once dissolved, the drug molecules diffuse across the membrane. As with monolithic systems, two types of reservoir systems can be found based on the polymeric membrane: nonporous, where drug molecules must diffuse through the polymer membrane, and microporous, when drug molecules are released through micropores (Wen et al. 2010).
Additionally, diffusion-controlled reservoir systems are also classified according to the drug loading as constant activity sources and nonconstant activity sources (Siepmann et al. 2012a). In the first case, when the reservoir comprises a drug concentration in the core above its solubility, the drug concentration gradient at the membrane remains constant, the diffusion of drug through the membrane is constant, and zero-order release kinetics can be achieved. In contrast, a nonconstant activity is characterized by a first-order release profile since the drug in the dosage form is completely and rapidly dissolved and drug molecules diffuse out through the controlling membrane (Siepmann et al. 2012a; Bermejo et al. 2020).
Osmotic pump systems
Osmotic drug delivery systems (ODDSs) are based on the osmosis phenomenon, where the inner core, filled with a mixture of the drug and osmotic agent, is surrounded by a semipermeable polymer membrane that has an orifice for drug release. Driven by the concentration gradient, the solvent tends to flow through the semipermeable membrane from the lower-concentration to a higher-concentration solution. Then, water influx by diffusion across the membrane dissolves the drug in an effort to achieve osmotic equilibrium and force the drug solution out through the orifice at a constant rate. ODDSs are characterized by a constant drug release with zero-order kinetics dependent on the osmotic pressure across the membrane and independent of the drug properties and the gastrointestinal environment (pH and motility) (Verma et al. 2002; Wen et al. 2010; Siegel et al. 2012; Ding 2016; Qiu et al. 2017).
Felix Theeuwes and contributors from the Alza Corporation (USA) had an important role in the development of oral osmotic devices, known as OROS® (osmotic controlled release oral delivery system) (Theeuwes 1975; Verma et al. 2002).
The different ODDSs can be classified based on their technology design as elementary osmotic pumps (Farooqi et al. 2020), push–pull osmotic pumps (PPOPs) (Malaterre et al. 2009; Missaghi et al. 2014; Liu et al. 2021), sandwiched osmotic pumps, push-stick osmotic pumps, controlled porosity osmotic pumps (Akhtar et al. 2022), asymmetric osmotic capsules (Yang et al. 2016), and liquid osmotic capsules (Verma et al. 2002; Qiu et al. 2017).
The polymers used in membrane-controlled systems, such as reservoir systems and ODDSs, are generally water insoluble. As mentioned above, examples of these polymers include cellulose acetate (Akhtar et al. 2022) and ethylcellulose (Hu et al. 2020).
Due to the variety of available polymers in the market and the significant number of drug delivery systems developed over the years, the selection of the most suitable polymer and delivery system for new drug product development requires a deep knowledge of the different controlled-release mechanisms.
Key factors in oral ER drug delivery system development
Based on QbD principles, the design of oral ER drug delivery systems involves consideration of potential high-impact factors (CMAs, CPPs and CQAs) that may be critical to product quality. Ensuring product knowledge and understanding the effect of such variables are essential to support the desired quality throughout the drug product lifecycle.
Some reviews have summarized the major factors affecting drug release from hydrophilic matrix tablets (Maderuelo et al. 2011; Vanza et al. 2020). The properties of the drug substance and polymer, formulation design and manufacturing process have been considered the key factors common to all oral ER drug delivery systems.
Drug release is influenced in different ways by physicochemical factors that essentially impact and determine the mechanism and rate of drug release. Regarding drug substances, although drug solubility and dose (Kim 1998; Li et al. 2008) are the most critical drug factors for generating ER delivery systems, the influence of parameters such as particle size and molecular weight should also be carefully evaluated (Maderuelo et al. 2011).
Notwithstanding, to better control drug release, it is crucial to define the polymer characteristics and understand their variability. The substitution pattern of cellulose derivatives (e.g., HPMC), which can present batch-to-batch differences, assumes an important role in the performance of hydrophilic matrices, and there may be a threshold where heterogeneity becomes critical for drug release (Viriden et al. 2009; Zhou et al. 2014). Compared to homogeneously substituted batches, the heterogeneous substitution pattern facilitates the formation of soluble gel-like components that increase the viscosity and extend the release rate of the drug (Viriden et al. 2010, 2011). Variations in the drug/polymer ratio and viscosity grade of polymers (Hiremath et al. 2008; Hu et al. 2020), as well as the particle size, can also affect the drug release rate (Heng et al. 2001; Crowley et al. 2004; Lakio et al. 2016).
An investigation of the three functionality-related characteristics of the carvedilol release profile from hydrophilic matrix tablets demonstrated that particle size plays a role in the first part of the drug release profile, while viscosity and degree of substitution play a determinant role in the later part of the drug release profile. An increased drug release can be obtained with a higher HPMC particle size, higher degree of substitution and lower viscosity (Kosir et al. 2018).
By identifying and understanding the CMAs related to drug substances and polymers as well as their performance, it is possible to tailor the CQAs, namely, the drug release rate, to achieve a robust and desired ER formulation.
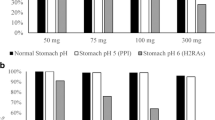
Regarding reservoir systems, as described above, polymeric membrane properties such as composition, thickness and permeability have a significant impact on drug release as well as on the occurrence of the burst effect (Siepmann et al. 2012a; Shah et al. 2022). The osmotic pressure gradient between the drug inner core and the external environment and the size of the delivery orifice coupled to the drug and semipermeable membrane properties are the major factors affecting the design of ODDSs (Malaterre et al. 2009). An increase in the tablet surface as well as an increase in the polymer molecular weight (both in the membrane and drug layer) showed an increase in the lag time. Otherwise, drug release was positively affected by the polymer proportion in the membrane and the proportion of the osmotic agent. An increase in the proportion of the osmotic agent in the tablet core increased the rate of water hydration and then decreased the lag time (Malaterre et al. 2009; Lin et al. 2022). On the other hand, the orifice size with a diameter ranging from 0.40 to 0.8 mm had no significant effect on drug release (Lin et al. 2022).
Considering the manufacturing process of oral ER delivery systems, depending on formulation properties, different methods can be elected. Direct compression (Sethi et al. 2018; Farooqi et al. 2020), dry granulation (Jang et al. 2021) and wet granulation (Kanwal et al. 2021) are the most common techniques used for manufacturing oral ER drug delivery systems. An increase in the applied compression force is generally translated into a higher degree of compactness and a greater density of the matrix, reducing the level of porosity and leading to a slower release rate of the drug (Crowley et al. 2004; Hiremath et al. 2008; Abu Fara et al. 2019). Additionally, a binary combination of two different polymers (Carbopol971P® NF and Eudragit®E100) improved the compaction properties (crushing strength) and SR properties of paracetamol matrix tablets (Obeidat et al. 2015).
Siepmann et al. (2000) and Reynolds et al. (2002) evaluated the influence of tablet size and geometry on drug release from HPMC matrices. These studies reported that lower values of the tablet surface area/volume (SA/Vo) ratio, achieved by increasing the initial radius of the tablets, have slower release profiles. SA/Vo, as a significant factor in controlling drug release, can be used as a tool to achieve target dissolution. Similar drug release profiles are typically reached with similar values of SA/Vo.
Overall, despite the study of various critical properties related to the drug substance by some authors, the functionality of polymers is key to the successful design of ER delivery systems.
Bridging QbD with solid oral ER formulations: the role in pharmaceutical development
The competitive quality environment, triggered by the emergence of quality concepts, is the major factor responsible for the high regulation of the pharmaceutical industry. The rationale behind the evolution of the concept of QbD, first outlined by Juran (1985) and based on three main pillars (planning, control, and improvement), gave rise to the pharmaceutical QbD (Davis et al. 2018).
In response to growing quality requirements and recurring quality issues in the pharmaceutical sector, regulatory agencies have implemented a new quality paradigm. The result was the publication of a set of ICH guidelines (ICHQ8-Q12) that make up the QbD ‘family’ and to provide a way to drive product and manufacturing processes to achieve the required quality (Davis et al. 2018).
According to the ICH Q8 guideline, QbD is defined as a systematic, scientific, and risk-based approach to pharmaceutical product development. It begins with predefined objectives and emphasizes product and process understanding and process control (ICH Q8(R2) 2009). QbD can be applied to all product types and normally starts from the earliest stage of development and progresses through the manufacturing and product lifecycle (Gibson et al. 2018).
Implementing QbD in the development of pharmaceutical products provides numerous advantages and opportunities to both industry and the regulatory authorities. Moreover, in-depth scientific knowledge based on the formulation and manufacturing process helps to minimize batch-to-batch variation and batch failures, enhances the production of a more robust and quality product and process, and streamlines postapproval regulatory submissions.
For instance, the goal of this new regulatory model is the creation of a process control strategy leading to continuous improvement over time and resulting in cost savings and efficiency for pharmaceutical industries, facilitating flexibility by the regulatory authorities (Gibson et al. 2018).
The application of quality risk management (QRM) principles is a valuable component of risk-based approach development. Keeping this in mind, ICH has issued the Q9 guideline, which describes a systematic process for the assessment, control, communication, and review of quality risks over the product lifecycle. The RA consists of risk identification followed by risk analysis and risk evaluation. The risk analysis objective is to rate the risk by linking the probability of occurrence, severity and sometimes detectability (ICH Q9 2005). The combination of QRM with prior scientific knowledge can help to identify and prioritize which material attributes and process parameters have a potential impact on product CQAs (Singh et al. 2010).
Different usefulness tools may be used for QRM. Cause and effect diagrams (also called Ishikawa or fishbone diagrams), failure mode effect analysis (FMEA), failure mode, effect and critical analysis, hazard analysis and critical control point and hazard operability analysis are some of the recommended risk analysis tools for use in the pharmaceutical industry (ICH Q9 2005).
In favor of ensuring innovation and continual improvement throughout the product lifecycle, the Q10 guideline, articulated with ICH Q8 and Q9, highlighted the importance of pharmaceutical quality systems (PQSs). Based on quality system-related documents such as International Standards Organization and Good Manufacturing Practices (GMP) guidelines, ICH Q10 lays out the major requirements of what a PQS should include. Knowledge management and QRM (ICH Q9 2005) are the enablers to establish the control strategy, i.e., a planned set of controls covering the process, its inputs and outputs, assuring that the product meets the required quality (ICH Q10 2008; Schmitt 2018).
On the whole, the comprehension of concepts depicted by ICH Q8, Q9 and Q10 are shifting the paradigm to better understanding, controlling and continually improving the manufacturing quality performance and efficiency of products throughout the product lifecycle (ICH Q9 2005, ICH Q10 2008, ICH Q8(R2) 2009). Figure 1 shows the relationship between ICH Q8, Q9 and Q10.

ICH Q8 (R2), Q9 and Q10 guidelines work together during the ER drug product lifecycle. ICH Q8 focuses on science and risk-based approaches for drug and process development, while ICH Q10 describes quality systems that facilitate the establishment of a control strategy and the continual improvement up to commercial scale manufacturing. QRM, described by ICH Q9 and applied over the product lifecycle, provides a structured way to assess and control risk (CMA critical material attribute, CPP critical process parameter, CQA critical quality attribute, ER extended release, ICH International Council for Harmonisation, QTPP quality target product profile)
ICH Q11 and Q12 are more recent and complementary guidelines to clarify other QbD-based concepts. ICH Q11 describes approaches to develop and understand the manufacturing process of drug substances linked to drug products (ICH Q11 2012), and ICH Q12 provides guidance on a framework to facilitate pharmaceutical life-cycle management concerning postapproval change management in CMCs (chemistry, manufacturing and controls) (ICH Q12 2019).
Hence, the development and manufacturing of oral ER delivery systems are associated with some complex features, and the alignment of the concepts of QbD becomes crucial to identify the critical factors impacting the performance of drug release (Singh et al. 2010). The ICH Q8 guideline establishes the elements and main steps of QbD to be considered during pharmaceutical development (ICH Q8(R2) 2009). To better understand the QbD-based development and manufacturing of ER delivery systems, the building blocks of the QbD flowchart will be addressed in the next sections.
Definition of a quality target product profile (QTPP)
The QTPP is a prospective summary of the main quality characteristics of pharmaceutical products that ensures the desired quality, safety, and efficacy targets. It describes the intended clinical use, route of administration, dosage form, delivery system, dosage strength and others (ICH Q8(R2) 2009; Gibson et al. 2018). The QTPP forms the basis of design for product development and should be regarded as a starting point for identifying CQAs.
In the development of oral ER delivery systems, depending on the formulation type, different QTPPs could be described as differentiating features of the drug to be developed. Drug product quality attributes such as the dosage form, floating lag time (Mirani et al. 2016; Chudiwal et al. 2018), mucoadhesion time (Chappidi et al. 2019) or drug release at the desired time (Vora et al. 2015; Desai et al. 2017; Chudiwal et al. 2018) should be set as the QTPP elements.
Critical quality attributes (CQAs)
After defining the QTPP, the second step is to identify the CQAs. The CQAs of ER drug products include physical, chemical, biological, or microbiological properties or characteristics of the drug substances, excipients and drug products that should be within an appropriate limit, range, or distribution to ensure the desired quality in the final product (ICH Q8(R2) 2009).
The drug product CQAs are selected to meet the QTPP and then assure product safety and efficacy. The CQAs for ER delivery systems are primarily associated with drug substances, polymers and other excipients and manufacturing. The percent cumulative drug release, erosion rate and swelling rate were identified as CQAs in the design and development of a hydrophilic matrix of metoprolol succinate (Shah et al. 2022).
The identification of the CQAs of rate controlling excipients (Parmar et al. 2018; Thapa et al. 2018) may also have special relevance because they can influence the mechanism and rate of drug release and allow accurate delivery of the necessary amount of drug over time.
The assessment of criticality can be difficult, and even when defined as critical in the initial development phase, not all CQAs will have the same effect on the QTPP. As recommended in ICH Q9, RA tools help to determine criticality and prioritize each quality attribute (ICH Q9 2005; Gibson et al. 2018). CQAs can be controlled through input factors such as CMAs and CPPs of the pharmaceutical formulation (materials) and manufacturing process, respectively. The initial array of potential CQAs can be large but is usually narrowed as formulation and manufacturing process activities progress, i.e., the CQA list can be dynamically modified (ICH Q8(R2) 2009).
The analysis of high-risk variables related to ER delivery systems could help to determine which material attributes and process parameters are critical and need further investigation to ensure drug product quality.
Linking of critical material attributes (CMAs) and critical process parameters (CPPs) to drug product CQAs
The development of ER delivery systems is associated with various challenges. Through the identification of potential CQAs linked to the properties of input materials (CMAs) and manufacturing process parameters (CPPs), it is possible to understand and identify formulation and process parameter ranges and controls (ICH Q8(R2) 2009). Yu and collaborators (Yu et al. 2014) reported a wide list of typical input material attributes, process parameters and quality attributes of tablet manufacturing unit operations.
QRM is one of the tools of the QbD approach to identify, evaluate, and control potential quality risks (ICH Q9 2005). An Ishikawa diagram is often used as a first step to identify the potential risk factors for CQAs. It is a systematic overview having a horizontal line with the underling CQAs and diagonal lines representing the major factors (Desai et al. 2017; Saydam et al. 2018; Zaborenko et al. 2019; Kovacs et al. 2021).
Based on experience and a thorough literature review, two examples of generalized Ishikawa diagrams for oral ER delivery systems manufactured by direct compression (a) and high shear wet granulation (b) were constructed (Fig. 2). In the presented diagrams, the formulation and process parameters, among others, are hierarchically organized to visualize and categorize the factors that may affect the CQAs.

Typical Ishikawa diagram for a direct compression of ER matrix tablets and b high shear wet granulation of ODDSs (CQA critical quality attribute, ER extended release)
The superior diagram (a) displays the cause-and-effect relationships for the ER matrix tablet formulation with the direct compression method. The major categories of factors included are environmental factors, raw material properties and process variables.
In ER matrix tablets, the excipient physical and chemical properties, mainly the polymer characteristics, have a significant impact on product manufacturing and performance. In fact, the variability of raw materials has been described by some authors (Dave et al. 2015; Zarmpi et al. 2017) and can lead to quality compliance issues. By studying the HPMC batch-to-batch and source-to-source variability through the determination of polymer characteristics (methoxy and hydroxypropyl substitution range, particle size and viscosity), it was possible to verify that the chemical heterogeneity of HPMC has an important effect on the drug release and erosion rate of a niacin CR formulation (Zhou et al. 2014).
The inferior diagram (b) is related to a complex dosage form, i.e., a bilayer osmotic pump tablet. As already mentioned above, in membrane-controlled systems, the coating composition and thickness are the factors that impact drug release, media uptake and push–pull patterns (Missaghi et al. 2014). Related to the orifice perforation, there are several types of processes that can be used to affect the osmotic pressure in the system and, consequently, the release kinetics. Laser drill (Kushner et al. 2020) modified punches (Liu et al. 2008) and pore formers (Yang et al. 2016; Yu et al. 2021; Akhtar et al. 2022) are some of the most well-known perforation techniques.
This tool is frequently associated with other RA techniques, such as the risk estimation matrix (REM) and FMEA. REM provides a simple color coding scheme and is commonly used on a summary chart to set priorities in risk management. The raw material, formulation and process properties can be categorized as critical or high risk (red), medium risk (yellow) and noncritical or low risk (green) to the product quality attributes (Mirani et al. 2016; Parmar et al. 2018). This classification can be justified based on prior knowledge, the literature, experience, preliminary screening results, and stability studies (Gibson et al. 2018). Alternatively, FMEA is a more formal risk management tool supported by the multiplication of three criteria: severity (S), probability (P) and detectability (D). The rank for risk quantification is defined through the risk priority number (RPN) score, which indicates the relative risk of each formulation and process variable (Vora et al. 2015; Gibson et al. 2018).
An FMEA could be performed to identify which formulation and process parameters have the highest impact on drug product attributes. To develop and optimize an ER enteric-coated tablet of isoniazid, the amount of PEO WSR 303, hardness and amount of ethyl cellulose exhibiting RPN ≥ 40 were considered high-risk factors that affect the core tablet formulation (Vora et al. 2015). In another study, the polymer and drug concentrations were considered high-risk factors in a formulation of a differential release fixed-dose matrix tablet of amlodipine besylate and simvastatin with an RPN above 15 (Kanwal et al. 2021). Thus, these main factors require further investigation and optimization by DoE to easily assess the interactions between factors. DoE should be performed to further establish the design space and define control strategies. Likewise, the application of tools such as the DoE and PAT can support the decision-making approach based on QRM (ICH Q9 2005).
Design of experiments (DoE)
DoE is a structured and useful tool in pharmaceutical development that applies statistical analysis to deploy the QbD framework in pharmaceutical industries. Experimental design establishes the relationship between formulation- and process-related input factors and output responses with a mathematical model. DoE enables the assessment of the statistical significance of input variables and the elucidation of mathematical interactions and helps to identify optimal conditions to improve product quality (Politis et al. 2017). Selection of the type of experimental design should consider some aspects, such as the main objective of the design, number of input factors, interactions to be studied and the available resources (Politis et al. 2017; Owen et al. 2018). Some examples of classical experimental designs are fractional or full factorial designs, screening designs and response surface designs (Owen et al. 2018).
Through DoE techniques, the controlled input factors (independent variables) are varied to determine their effects on the output responses (dependent variables), which allows the identification and determination of individual and interactive effects of factors on output results. As noted, the selection of the best experimental design should consider the defined objective. Based on the reported data in Table 1, DoE is applied both to screening (Ilyes et al. 2021) and optimization (Qazi et al. 2020; Gowthami et al. 2021; Won et al. 2021) purposes in oral ER drug delivery systems, although most of the literature studies described refer to optimization designs. Whereas screening designs, often used in the first part of drug development, allow the identification of CMAs and CPPs affecting the CQAs, the application of optimizing designs in QbD-based ER development allows the achievement of an optimized output response by changing the factors. Central composite designs (CCDs) (Saydam et al. 2018; Chappidi et al. 2019; Mohamed et al. 2020) and Box‒Behnken designs (BBDs) (Thapa et al. 2018; Jang et al. 2021), i.e., two response surface methodology models, and D-optimal designs (Lakio et al. 2016; Vanhoorne et al. 2016; Sanoufi et al. 2020) are the most commonly used optimization designs in ER drug product development. Factors such as the polymer amount and ratio, orifice size (for ODDSs) or compression pressure are optimized using DoE. The output responses include drug release over a period of time and tablet properties.
Although all the independent variables studied in the experimental designs are described in Table 1, only the relevant factors, i.e., the factors that showed an impact on the dependent variables, are depicted in the table results, which could be important in future studies.
The development of an ER formulation with paliperidone using a mixture of hydrophilic and hydrophobic polymers was studied by Iurian et al. (2017). After drug addition to an inert matrix made of Kollidon® SR, hydrophilic polymers were also included (NaCMC, sodium carboxymethyl cellulose; HPC; or HPMC). The mixture of these two types of polymers allowed a combined release mechanism through the formation of pores in the matrix and a gelled layer generated by the insoluble polymer and the hydrophilic polymer, respectively. In the study presented by Won et al. (2021), a bilayer tablet containing a high dose of metformin HCl in an SR layer and a low dose of evogliptin tartrate in an immediate release layer was developed. The appearance, friability, hardness, identification, assay, content uniformity, dissolution, degradation products, residual solvents and microbial limits were considered as potential CQAs. RA was used to determine which CMAs and CPPs were critical to the CQAs. Since the formulation and granulation process of each layer was based on the marketed tablet dosage form containing each single component, all formulation parameters were classified as low risk. Afterward, an REM was performed for the bilayer tableting process parameters, and those whose risk was identified as high were optimized through a face-centered CCD including five independent variables.
Therefore, the implementation of a DoE to optimize the formulation and/or process parameters has become an effective and successful QbD strategy to develop oral ER drug delivery systems. The evaluation of the DoE outcomes can justify predictions of the formulation and process behavior within the design space.
Design space and control strategy
The design space is a key concept in QbD defined by ICH Q8 as “the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters) that have been demonstrated to provide assurance of quality”. The design space describes the relationship between process inputs and CQAs (ICH Q8(R2) 2009) and begins with the definition of the QTPP. RA, as part of QRM (described above) and deep knowledge gained from process development experiments, can support the design space and guide the establishment of a control strategy.
The assessment of which variables are critical or not will help ensure consistent ER drug product quality. These critical factors are a very important tool to define the acceptable ranges of material attributes and process parameters and then limit and establish the design space. According to ICH Q8, operating within the design space is not considered a change from the regulatory point of view but a part of the control strategy, since it is expected that the final product has the same quality (ICH Q8(R2) 2009). The normal operating range or control space is a subset of the design space and is defined as the upper and/or lower limits for the CMAs and CPPs, i.e., a demarked region of the design space where parameters and materials are systematically controlled throughout production to assure reproducibility (Yu 2008). A design space can be established independently for one or more unit operations or constructed for the whole process through multidimensional interactions between CPPs (ICH Q8(R2) 2009).
Although the design space is optional and not required by regulatory authorities, it is an asset to the applicant, providing accurate and reliable product quality within specifications. Furthermore, the design space can be updated over the lifecycle of the product as additional knowledge is gained (ICH 2010).
Once the product and process understanding is achieved through the design space, the next step is the development of a control strategy to control the CMAs and CPPs. A control strategy is a planned set of controls derived from the current product and process understanding that ensures process performance and product quality. This can include parameters and attributes related to the drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control (ICH Q10 2008). A high level of process and product understanding and consequent identification of the sources of variability that can impact product quality will support the control of critical steps and can enable the shift of controls upstream.
A suitable control strategy justifies the use of PAT tools and RTRT, minimizing the need for end product testing (ICH Q8(R2) 2009). The control strategy model could include three levels of controls.
The design space, associated with the control strategy, is a driver for understanding the product and process, explaining and controlling the variability and ensuring that a given manufacturing process is robust enough to produce a product that meets the QTPP and CQAs (ICH 2010).
In the case of the design of inert and hydrophilic matrices, some critical factors must be considered, such as the polymer properties, drug particle size and compression pressure. The application of percolation theory along with critical points has been shown to be an important tool to establish the design space, based on ICH Q8 requirements, of this type of ER formulation (Aguilar-De-Leyva et al. 2017).
PAT and RTRT as part of a control strategy
The implementation of PAT tools helps to ensure a suitable level of risk control. PAT is a system for designing, analyzing, and controlling manufacturing through timely measurements of critical quality and performance attributes of raw and in-process materials and processes with the goal of ensuring final product quality. This definition is consistent with the FDA current drug quality system: “quality cannot be tested into products, it should be built-in or should be by design” (FDA 2004; ICH Q8(R2) 2009). A focus on raw materials, formulation and process control can reduce product and process variability and improve the robustness of product development and manufacturing with significant time and cost savings (FDA 2004; Lundsberg‐Nielsen et al. 2018).
The PAT framework includes four key elements to facilitate process understanding and control throughout the product lifecycle: multivariate tools for design, data acquisition and analysis, process analyzers, process control tools and continuous improvement and knowledge management tools (FDA 2004; Yu et al. 2014).
The introduction of PAT tools should be extended, beginning at development and continuing at commercial manufacturing. During the development phase, PAT can help in the identification of CPPs, CMAs and their interactions to control product CQAs and create opportunities to improve the scientific basis for setting regulatory specifications. The understanding and experience acquired at the laboratory scale can further aid in achieving reliable scale-up and technology transfer. In commercial manufacturing, the purpose of PAT is mainly process control and improvement and to provide the opportunity for RTRT application (FDA 2004; Lundsberg‐Nielsen et al. 2018).
Some authors have performed studies showing the applicability of different PAT tools in the design and development of ER tablets. Table 2 provides a summary of recent PAT applications in the development of oral ER drug delivery systems. Near infrared (NIR) and Raman spectroscopies have been used as in-line process analyzers in various applications, including the estimation of drug content (Sirbu et al. 2014; Muntean et al. 2017; Porfire et al. 2017; Rus et al. 2020; Gavan et al. 2022), tablet characterization (Porfire et al. 2017; Gavan et al. 2022), and coating operation endpoint and drug release determination (Gendre et al. 2011; Muller et al. 2012; Wirges et al. 2013; Wu et al. 2015). The obtained data were modulated using multivariate statistical tools such as principal component analysis (PCA) and/or partial least squares (PLS) (Van Snick et al. 2017; Nagy et al. 2019; Gavan et al. 2022).
For example, NIR-chemometric methods were used to chemically and pharmaceutically characterize indapamide SR tablets (Porfire et al. 2017). The combination of data provided by NIR spectrometry and off-line tablet press and PSD of the drug substance (drotaverine) and HPMC supported the development and implementation of an dissolution prediction model for matrix sustained-release tablets (Galata et al. 2021). The investigation of water penetration during the dissolution of nifedipine from CR PPOP by collection of NIR spectra allowed the researchers to understand the time dependency of water in different stages along the tablet dissolution process (Liu et al. 2021).
RTRT is the ability to evaluate and ensure the quality of in-process and/or final products based on process data (material attributes and process controls) (ICH Q8(R2) 2009). In other words, RTRT is a strategy implemented by some pharmaceutical companies where the process (manufacturing steps or unit operations) is continuously monitored—in real-time quality control—without the need for end-product quality tests. The basis for establishing an RTRT system involves the combination of ICH Q8, Q9 and Q10 principles and provides an opportunity for enhancing product and process understanding and increasing product quality assurance (EMA 2012).
Pawar et al. (2016) demonstrated for the first time the RTRT possibilities in the continuous manufacturing of an SR formulation. This study presented a method for dissolution prediction in direct compression continuous manufacturing with at-line transmission mode using NIR spectroscopy. The API concentration, compression force, blender speed and feed frame speed were the formulation and process variables included in the experimental design. PCA was performed between the NIR spectral data obtained for the DoE samples and the dissolution profile parameters (model dependent and model independent). The results obtained by the multilinear regression model showed the potential of NIR spectroscopy to predict tablet dissolution.
A recent review highlights the main challenges and opportunities of RTRT, focusing on the most prevalent CQAs for different manufacturing processes (direct compression and dry and wet granulation). The mixing homogeneity, tablet content and uniformity, moisture content, drug release, granule particle size, tablet porosity, tablet strength and coating thickness were published in the literature as drug product CQAs measured by PAT methods (Markl et al. 2020).
A control strategy for drug content and tablet uniformity on a commercial scale was recently developed, and three different options were considered in the development. The content uniformity methods included the use of individual tablet weight data (in process control); the use of estimated individual tablet content data (weight variation); and the application of at-line NIR spectroscopy to predict individual tablet content. At-line testing of the tablet content by NIR spectroscopy was selected as the most appropriate approach and could be applied as part of RTRT (Goodwin et al. 2018).
There is already a workflow available for developing and implementing a PAT strategy that supports real-time process control in continuous pharmaceutical manufacturing. From process analysis and the definition of monitoring tasks to technology selection, process integration and data acquisition, all these steps seem to be crucial to develop a robust continuous manufacturing process and to control the quality of drug products (Sacher et al. 2022).
Although it is clear that there is still a long way to go to achieve RTRT in solid oral ER tablets, the wide range of established in-line PAT applications to monitor CQAs and control, in real time, the CMAs and CPPs (Galata et al. 2021) confirms its potential to enable RTRT. The application of the QbD approach, based on deep scientific knowledge built through product development coupled with in-process monitoring of process parameters, should result in a robust control strategy to promote reproducible product quality and mitigate potential risks. The process understanding and control, method development and validation, and application of the method within the product control strategy are the basis for supporting RTRT development methods (Markl et al. 2020; Sacher et al. 2022).
Product lifecycle management and continual improvement
QbD strongly underlines the principle of continuous improvement in which the development must be updated as the understanding of the product and process increases during the product lifecycle. The combination of PAT methods, knowledge management and use of multivariate analysis can enhance the identification and understanding of CMAs and CPPs. Accordingly, product-related data acquired during routine commercial manufacturing should be considered and analyzed to refine knowledge and control strategies, improve statistical confidence, and consequently improve product quality in compliance with GMP regulations (ICH 2011). These strategies will help pharmaceutical industries minimize the risk of not meeting quality requirements by controlling the sources of raw material and process variability.
In summary, QbD tools are a crucial part of the modern approach to pharmaceutical quality. However, the pharmaceutical industry has not yet embraced QbD implementation, which has been facing some barriers. Currently, QbD is often implemented during late stages of development only to optimize the formulation and process, according to what was defined in the QTPP, or to generate data to support regulatory submission. Implementing QbD technologies as PAT involves a high level of investment in material and human resources. The lack of technology to execute and the need for an interdisciplinary strategy using different areas of expertise are key limitations. Furthermore, the lack of clarification regarding the scientific principles and terms beyond QbD causes a gap between industries and regulatory authorities that can be an obstacle during the approval process.
Although a trial-and-error approach can lead to the same results as a QbD-based approach, it does not generate product knowledge. Without an intrinsic knowledge of the process, problems can arise at the scale-up level and even throughout the product lifecycle. Therefore, using the QbD approach from the beginning of product development brings advantages when choosing the strategy to adopt next. In addition, since the results concerning oral ER drug delivery systems are more time-consuming, the implementation of these tools becomes even more relevant.
Insights on data science—MVDA, ML and ANN as tools to foster ER tablet development and lifecycle management
The QbD framework is based on the continuous improvement principle and provides a holistic understanding of the product and its manufacturing processes throughout the entire cycle, using risk management methodologies to ensure that the product fulfills the quality requirements (ICH Q8(R2) 2009). A deep understanding of the product and its manufacturing parameters mitigates the risk and enables a growing knowledge collection to offer a robust and reliable drug product. As considered by ICH Q10, knowledge management is one of the key enablers of a robust QRM and must be managed from development until the end of the product lifecycle (ICH Q10 2008). However, the emergence of a data-driven era and advancements in manufacturing sciences and technologies should be used to improve knowledge and risk management, providing a real opportunity to intensify process robustness and efficiency as well as increase time and cost savings. Accordingly, leveraging prior knowledge toward data-driven risk management is a key factor for a successful QbD application (Steinwandter et al. 2019).
As a consequence of scientific and technological advances and the resulting vast amount of data, the pharmaceutical industry continues to struggle to improve the processes used in drug development. Therefore, based on large complex datasets, tools offered by data science provide useful information to optimize processes, accelerate drug development and boost performance and results (Reinhardt et al. 2020). Data science is mainly referred to as the statistical field that applies advanced tools to derive useful information from complex data. Its process encompasses the identification of a problem, data collection, preparation and analysis, and model building through the combination of different fields, including statistics, data analytics and AI (Steinwandter et al. 2019). Therefore, data science and AI are strongly interconnected (Fig. 3).

The role of artificial intelligence in the data science lifecycle. The basic steps of data science from problem identification to model building using artificial intelligence
AI concepts have been increasingly used since the mid-twentieth century with a focus on mimicking human behavior (Haenlein et al. 2019) and have recently started to gear up its applications in different pharmaceutical areas, from drug discovery to clinical trials and postmarket product management (Vamathevan et al. 2019; Paul et al. 2021a). AI-related subfields can include ML, neural networks (one of the most important tools in ML), and expert systems (Fig. 3) (Haenlein et al. 2019). The focus of this topic is to discuss the potential of different ML models to predict solid oral ER tablet performance.
Conventional and multivariate statistical approaches in ER tablet QbD-based development
As mentioned earlier (Table 1), DoE is an efficient methodology used in QbD to understand the main effects and interactions and determine the relationship between multiple input variables and outputs. With a minimum number of experiments, it can be possible to gain formulation and process knowledge and define the design space.
In the context of oral ER drug delivery systems, their manufacturing processes are generally complex and can only be described by multifactorial relationships. Additionally, the introduction of PAT during the manufacturing process is associated with the generation of a large amount of data. Because huge amounts of data are generated, specialized data analysis tools, such as MVDA, are required to fully explore the multivariate outputs generated from DoE datasets or, for example, data acquired by PAT.
Integrated multivariate analysis methods have been widely implemented by pharmaceutical companies. These methods are data-driven statistical techniques to simultaneously analyze several variables in large/complex datasets and identify critical parameters that can then be controlled to improve process and product quality.
The majority of published research studies reporting the application of MVDA techniques in oral ER drug products is supported by PAT, dealing with the development of calibration models to predict and monitor CPPs and CQAs in real time (Wu et al. 2015; Rus et al. 2020; Gavan et al. 2022). Among the various MVDA techniques, MLR, PCA and PLS are the most common methods used in ER pharmaceutical development (Islam et al. 2014; Kosir et al. 2018; Diab et al. 2021).
PCA and PLS, as linear dimensionality reduction ML algorithms, are useful in pharmaceutical development to extract meaningful information from datasets. PCA orthogonally transforms the original dataset of observations of possible correlated variables into a lower dimensional set of axes named principal components. This conversion allows us to maximize the variance and find patterns in large datasets. On the other hand, PLS takes into account the covariance between the variables being applied based on correlation. PLS regression is also a projection technique where the original dataset is projected onto a low-dimensional set, followed by linear regression. Then, it is possible to identify and establish correlations between variables in the manufacturing of oral ER drug products based on QbD. The linear combination of variables is referred to as latent variables (Rajalahti et al. 2011; Lopes et al. 2018). A comparison between MLR and PLS multivariate regression models showed PLS as a more suitable model to determine which HPMC properties had the most significant role on the carvedilol release rate from hydrophilic matrix tablets (Kosir et al. 2018). However, each multivariate tool should be explored depending on the dataset and objective. While the PCA model might be a great choice for data exploration, PLS can be a better option for predictive purposes.
For pharmaceutical product and process development, although MVDA methods have started to be more applied, they are essentially used at the upstream phase of drug development either applied to analyze the historical data from the raw materials or combined with the DoE (Huang et al. 2009; Grangeia et al. 2020; Shi et al. 2021).
Various studies have selected PCA and PLS as complementary tools of DoE to evaluate the relationships between the input variables (CPPs and CMAs) and their impact on CQAs in oral ER drug delivery systems (Huang et al. 2009; Porfire et al. 2017; Rus et al. 2020). Other studies have used MVDA in PAT (Wu et al. 2015; Gavan et al. 2022) for interpreting the in-line measurements in ER drug product development and/or manufacturing (as depicted in Table 2). Overall, PCA has been used to visualize the relationships between the independent variables and classify the PAT spectral data files, whereas PLS is used to develop a calibration model to predict CQAs such as drug release (Banner et al. 2021). Diab et al. (2021) described the application of chemometric methods (PCA and PLS) to predict dissolution variation based on historical industrial batch data produced at the commercial scale. In this paper, the input data related to API and excipient attributes and each unit operation were correlated with ER tablet dissolution (output variable) via PLS. The PCA model was applied to evaluate the variability of the input dataset.
Despite the advantages of MDVA techniques in modeling complex relationships between CMAs/CPPs and CQAs in ER oral drug delivery systems, the dimensional reduction of data could lead to loss of some information from the original dataset. Moreover, since the formulation and process of oral ER drug delivery systems can be complex, these linear models could be insufficient when nonlinear relationships between CMAs/CPPs-CQAs are involved (Nagy et al. 2019). Therefore, more advanced and sophisticated approaches based on ML could be a good option to bridge these barriers.
Looking forward to advanced statistical models—ML
ML is a branch of AI technologies and a data science tool that focuses on using algorithms to automate the building of predictive models for data analysis. Coupled with AI, ML provides substantial advantages, enabling the recognition and identification of patterns within a large volume of datasets and the production of reliable results with continuous improvement (Ashenden et al. 2021; Paul et al. 2021a). The storage and processing of the massive amount of data gave rise to the term ‘big data’. Its definition is commonly based on the 5 V properties: Volume, Variety, Velocity, Veracity and Value (Demchenko et al. 2013).
Although drug discovery is the major field of application of ML in pharmaceuticals (Vamathevan et al. 2019; Reda et al. 2020; Paul et al. 2021a), ML strategies have emerged as a powerful solution for pharmaceutical scientists to improve the success rate and foster the development of high-quality products. A SWOT analysis of ML in pharmaceutical development is provided in Table 3, revealing the internal strengths and weaknesses coupled with the opportunities and threats faced by ML. Despite the ability to handle large datasets and model nonlinear relationships, the required amount and quality of data can be a limitation (Steinwandter et al. 2019). Insufficient and poor-quality data can limit the model’s accuracy, leading to a lack of interpretability and reproducibility.
The investment in new resources and technologies is generally a costly and time-consuming process that can limit the implementation of ML. However, the long-term application of these automated and efficient methods will accelerate product development and potentially save hefty costs. Moreover, ML is an opportunity to optimize manufacturing processes, where several PAT tools are used to enable real-time monitoring in continuous manufacturing. Therefore, the use of ML allows computing systems to identify patterns in data collected across the process and continuously improve the outcomes, following the QbD workplace.
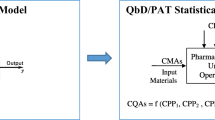
As summarized in a recent review, the growing trend toward solid oral dosage form development guided by QbD principles has been supported using ML methods to understand and relate CMAs and CPPs as input variables to achieve the desired outputs (CQAs) (Lou et al. 2021). Figure 4 provides an overview of how ML and other data analytics tools can transform raw big data providing useful models that can accurately ensure specification compliance to satisfy the CQAs with significant time and cost savings.

Overview of an integrated approach with QbD elements, PAT tools and different data analytics tools. Schematic visual representation of overlay and contour plots to understand the impact of CMAs and CPPs on drug product CQAs (CMA critical material attribute, CPP critical process parameter, CQA critical quality attribute, DoE design of experiments, ML machine learning, MVDA multivariate data analysis, PAT process analytical technology, PCA principal component analysis, PLS partial least squares)
In a typical ML approach, the datasets can be integrated for training, validation and testing. To address the question/problem, the process can be roughly divided into the following steps: (1) input the data collection (from various sources); (2) prepare, process and understand the dataset; (3) choose and build the ML model; (4) train the learning model on the training set; (5) tune and evaluate the ML model on the validation set; and (6) evaluate the final performance on the test set to confirm the results (Fig. 5) (Bannigan et al. 2021). The steps of gathering data and building a model by selecting the right ML algorithm are of special relevance to pharmaceutical development because the quality and quantity of the data as well as the selection of the most suitable algorithm will determine the accuracy and predictability of the ML model.

Basic steps involved in the ML process flow. Splitting data to training, validation and testing sets (ML machine learning)
Types of ML approaches
ML includes three types of approaches: supervised learning, unsupervised learning, and reinforcement learning, differing in the way the models are trained. Figure 6 shows an overview of the ML methods.

Brief explanation of supervised, unsupervised and reinforcement ML approaches (ML machine learning, SVM support vector machine, k-NN k-nearest neighbors, ANN artificial neural network, PCA principal component analysis)
Supervised learning comprises a set of labeled input data (training data). These already tagged data (historical learning and original input data) are used to train algorithms to find the specific structure for predicting the correct outcomes (Banner et al. 2021; Bannigan et al. 2021). Then, the test data are applied to validate the model and assess its predictive accuracy. Supervised learning can be performed in the context of classification or regression, depending on whether the output variable is discrete and qualitative or continuous and quantitative, respectively (Bannigan et al. 2021). Support vector machine (SVM) (Al-Zoubi et al. 2011), tree-based methods (decision tree, random forest and boosting) (Petrovic et al. 2012), K-nearest neighbors (k-NN) (Yang et al. 2019) and artificial neural networks (ANNs) (Galata et al. 2021) are some types of classification and regression algorithms used in oral ER drug delivery system development.
Two supervised regression ML models (MLR and SVM) were applied to the optimization of ER pentoxifylline matrix tablets based on a 32 full factorial experimental design where the drug weight ratio and percentage of the matrix former were selected as the independent variables and the drug release at four time points was selected as the dependent variables. Using SVM concepts, the training data comprising 11 experiments (32 + 2 replicated central points) were normalized by feature scaling, and these normalized factors were used as inputs for the SVM model construction. The suggested SVM model was externally validated with 6 checkpoints, and the experimental and predicted values were compared. The overall prediction ability was better for SVM than for MLR; thus, it is more suitable for optimizing drug release from ER matrix tablets (Al-Zoubi et al. 2011).
On the other hand, unsupervised learning algorithms can identify patterns, similarities and differences from a hidden structure in unlabeled data without any prior training or supervision. Clustering and dimensionality reduction are the key ML tasks used in unsupervised learning (Ashenden et al. 2021). As mentioned before, PCA has been used as an important unsupervised data analysis technique for reducing dimensionality in addressing the complex process data in oral ER drug delivery systems. Finally, the third type of ML is reinforcement learning, which works through the correlation between actions and delayed outcomes based on a reward system (Arden et al. 2021).
What does ML bring to the pharmaceutical industry?
In general, the application of ML algorithms can be a crucial tool to aid in deciding suitable starting materials, understanding formulations, product properties and processes (Akseli et al. 2017; Benedetti et al. 2019; Hayashi et al. 2019, 2021; Lou et al. 2019; Van Hauwermeiren et al. 2020; Djuris et al. 2021; Maki-Lohiluoma et al. 2021; Paul et al. 2021b; Thomas et al. 2021), and predicting dissolution (Galata et al. 2021) and drug stability (Ibric et al. 2007).
Hayashi et al. (2021) built a material library including 81 types of APIs, 20 types of API material properties, one type of process parameter and two types of tablet properties. Boosted tree (BT), random forest (RF) and PLS were applied to model relationships between the input variables (material properties and three levels of compression pressure) and output variables (tensile strength and disintegration time). BT and RF were demonstrated to be more suitable for modeling than multivariate models. The high R2 and low root mean square error (RMSE) values reported for the tree-based algorithms indicated accurate predictions. With regard to the input variables, the diameters at the tenth percentile of the cumulative percent undersize distribution (d10) and total surface energy (γs) were found to strongly impact on the tensile strength and disintegration time, respectively.
It has been confirmed that ML algorithms have the potential to help the pharmaceutical scientific community in the assessment and prediction of several factors involving large amounts of data and requiring more flexible analysis. Although there are few articles in which ML is applied to ER formulation development, Table 4 provides a summary of some of the available research.
In the future, additional scientific efforts are expected to understand and interpret ML models when handling large datasets. Likewise, the traditional models used in DoE are well established, and the high prediction accuracy of ML does not mean the end of using them. Traditional tools could remain a good approach for linear relationships between inputs and outputs.
Artificial neural networks (ANNs)
Artificial neural networks (ANNs) are the first and most widely used ML model in the study of ER formulations, frequently with the objective of characterizing and optimizing the formulations and modeling the dissolution (Lou et al. 2021).
ANNs are a useful tool for modeling input/output nonlinear functions inspired by the way neurons work in the human nervous system. In one of the simplest forms of ANN (called the feedforward neural network—FFNN), the data travel in one direction (Simoes et al. 2020; Wang et al. 2022). Multilayer perceptron (MLP) has been commonly used in pharmaceutical development, and the framework comprises three types of essential layers: input, hidden and output layers. Once the input layer receives the external data for the neural network, the hidden layer, located in the middle of the ANN, processes the information through several types of mathematical computation. Then, the output layer produces the final results for the given inputs (Wang et al. 2022).
FFNNs, including MLP and the generalized regression neural network (GRNN), with radial and regression layers are just some examples of common neural network architectures summarized elsewhere (Lou et al. 2021; Wang et al. 2022).
In the midst of other ML methods, ANNs have proven to be accurate in assessing tablet properties (Khan et al. 2020) and swelling and erosion mechanisms (Barmpalexis et al. 2018) as well as predicting the in vitro drug release (Al-Zoubi et al. 2015; Lefnaoui et al. 2018; Saracoglu et al. 2020; Galata et al. 2021) of oral solid ER tablets. For input variables, numerous CMAs and/or CPPs have been used to build multiple ML models to predict CQAs. Input materials such as drug (Yang et al. 2019) and polymer properties (Saracoglu et al. 2020) and process parameters (e.g., compression force (Galata et al. 2019), roller pressure (Pishnamazi et al. 2019) or crushing strength (Ivic et al. 2010)) were identified to predict the in vitro dissolution profile of ER tablets.
Nagy et al. (2019) built three-layer ANN models to predict the dissolution of ER anhydrous caffeine tablets and compared them to traditional PLS regression. In this work, the effects of the API and PEO content and compression force on drug dissolution were assessed. The scores obtained by the dimension reduction PCA from the FT-NIR and Raman spectra of each intact tablet were defined as the input variables, and the dissolution values at 35 sampling points were considered the output variables. The NIR and Raman spectroscopic tools demonstrated a complementary relationship. While NIR methods provide information on the effect of compression force, Raman provides better prediction of the effect of API and PEO on drug release. ANN-based models provided a lower RMSE for prediction than PLS. This is due to the ANN capability to determine the complex and nonlinear relationship between the input and output parameters.
Deep learning (DL)
Deep learning (DL), as a specific and more advanced subset of ML, engages ANNs in most cases (Paul et al. 2021a). When applied to DL, the architecture of ANN models can be very deep, with more than three layers—called deep neural networks (Ashenden et al. 2021). Due to the suitability of DL to deal with complex datasets, it could be a powerful tool in the future of ER delivery system development to improve the control strategy.
Yang et al. (2019) applied a deep neural network to predict the disintegration time and cumulative drug release of oral fast disintegrating films (OFDF) and oral SR matrix tablets (SRMT), respectively. For this purpose, the experimental dataset extracted from Web of Science was split into three datasets: training, validation, and testing. Six conventional ML methods (MLR, PLSR, SVM, ANNs, random forest and k-NN) were considered for comparison with DL. Deep neural networks showed higher accuracy (over 80%) on the OFDF and SRMT training, validation and test datasets compared to the ML algorithms. Two years later, based on these models, Yoo et al. (2022) proposed new DL approaches, using PCA and the Wasserstein generative adversarial network (WGAN), to maximize the prediction performance for OFDF and SRMT, respectively. The proposed models showed significantly higher performance than the existing models.
From QbD to pharma 4.0
In the last decade, the pharmaceutical industry has adopted several emerging technologies, techniques and processes, which offer high potential to change the landscape of drug development and production (Reinhardt et al. 2020; Arden et al. 2021; Wang et al. 2021).
To achieve high-quality and high-efficiency patterns, the pharmaceutical industry employs Pharma 4.0 (Arden et al. 2021), a concept that emerged to represent the era where different technologies and/or machines are converging to improve product quality through renewed digital solutions (Reinhardt et al. 2020). Additionally, “Smart Factories” are structures in which machines communicate with each other dealing autonomously with emerging problems and unexpected changes (Barenji et al. 2019). The essential technologies for data science, a cornerstone of Pharma 4.0, have already been established and are well known, aiming to control the process and product quality data in real time.
QbD, together with ICH guidelines, enables the requirements of Pharma 4.0 through a holistic development and manufacturing control strategy. The implementation of QbD, RTRT and PAT has provided several advances, offering systematic and quantitative approaches, reducing human interventions and therefore sharply reducing costs and time.
However, their application in industry is far from the potential presented by this new concept (Barenji et al. 2019; Steinwandter et al. 2019; Wang et al. 2021). The bigger challenges are related to technological gaps, namely, the lack of independence facing unexpected changes, the low level of interoperability and the low computational power (Barenji et al. 2019). To implement smart manufacturing systems, a cyber-physical-based PAT (CPbPAT) framework was proposed by Barenji et al. (2019). This framework, designed to obtain, record and monitor real-time data, combines several technologies, such as QbD and RTRT, to make autonomous decisions and determine improvement strategies. CPbPAT is developed in multiple levels, enabling the collection and continuous integration in the cloud of large amounts of data.
Due to the complex development and manufacturing of ER systems, the implementation of the Pharma 4.0 concept in pharmaceutical companies with continuous monitoring of product manufacturing will decrease product and process variability and consistently improve quality requirements.
Conclusion
Pharmaceutical companies have already started applying QbD concepts to the development of pharmaceutical products instead of traditional trial-and-error-based approaches, although their application remains far from what is expected. Because of the complexity of oral ER drug delivery system development and the relevance of polymer properties on drug product performance, the implementation of QbD tools is crucial to provide a better and complete understanding of the product and process parameters and optimize a control strategy. A design space can be established defining allowable operational ranges and providing flexible regulatory approaches. Furthermore, QbD is a cost-effective time-saving strategy that can be used throughout the product lifecycle ensuring compliance with regulatory quality requirements.
The increasing amount of generated data requires a greater ability to optimize formulations and processing parameters as well as accurately predict drug product performance. ML and DL algorithms will progressively be recognized, and we can be sure that they will be more widely used for effective pharmaceutical development through a Pharma 4.0 strategy to achieve greater robustness and help meet regulatory compliance. Certainly, these tools will allow us to move toward the possibility of RTRT application in the pharmaceutical industry.
References
Abu Fara D, Dadou SM, Rashid I, Al-Obeidi R, Antonijevic MD et al (2019) A direct compression matrix made from xanthan gum and low molecular weight chitosan designed to improve compressibility in controlled release tablets. Pharmaceutics 11:603
Aguilar-De-Leyva A, Campinez MD, Casas M, Caraballo I (2017) Design space and critical points in solid dosage forms. J Drug Deliv Sci Technol 42:134–143
Akhtar MF, Ashraf H, Uzair M, Ahmad S, Rasul A et al (2022) Development of leachable enalapril tablets by controlled porosity osmotic pump technique; a unique approach to enhance its sustained release effect. J Coat Technol Res 19:497–507
Akseli I, Xie JJ, Schultz L, Ladyzhynsky N, Bramante T et al (2017) A practical framework toward prediction of breaking force and disintegration of tablet formulations using machine learning tools. J Pharm Sci 106:234–247
Aktas E, Eroglu H, Kockan U, Oner L (2013) Systematic development of pH-independent controlled release tablets of carvedilol using central composite design and artificial neural networks. Drug Dev Ind Pharm 39:1207–1216
Al-Zoubi N, Kachrimanis K, Younis K, Malamataris S (2011) Optimization of extended-release hydrophilic matrix tablets by support vector regression. Drug Dev Ind Pharm 37:80–87
Al-Zoubi N, Alkhatib HS, Alobaidi G, Abdel-Rahim S, Obeidat W et al (2015) Optimization of pH-independent chronotherapeutic release of verapamil HCl from three-layer matrix tablets. Int J Pharm 494:296–303
Arden S, Fisher AC, Tyner K, Yu LCX, Lee SL et al (2021) Industry 4.0 for pharmaceutical manufacturing: preparing for the smart factories of the future. Int J Pharm 602:120554
Ashenden SK, Bartosik A, Agapow P-M, Semenova E (2021) Introduction to artificial intelligence and machine learning. In: Ashenden S (ed) The era of artificial intelligence, machine learning, and data science in the pharmaceutical industry, 1st edn. Academic Press, United Kingdom, pp 15–26
Banner M, Alosert H, Spencer C, Cheeks M, Farid SS et al (2021) A decade in review: use of data analytics within the biopharmaceutical sector. Curr Opin Chem Eng 34:100758
Bannigan P, Aldeghi M, Bao ZQ, Hase F, Aspuru-Guzik A et al (2021) Machine learning directed drug formulation development. Adv Drug Deliv Rev 175:113806
Barenji RV, Akdag Y, Yet B, Oner L (2019) Cyber-physical-based PAT (CPbPAT) framework for Pharma 4.0. Int J Pharm 567:118445
Barmpalexis P, Kanaze FI, Kachrimanis K, Georgarakis E (2010) Artificial neural networks in the optimization of a nimodipine controlled release tablet formulation. Eur J Pharm Biopharm 74:316–323
Barmpalexis P, Kachrimanis K, Malamataris S (2018) Statistical moments in modelling of swelling, erosion and drug release of hydrophilic matrix-tablets. Int J Pharm 540:1–10
Benedetti A, Khoo J, Sharma S, Facco P, Barolo M et al (2019) Data analytics on raw material properties to accelerate pharmaceutical drug development. Int J Pharm 563:122–134
Bermejo M, Sanchez-Dengra B, Gonzalez-Alvarez M, Gonzalez-Alvarez I (2020) Oral controlled release dosage forms: dissolution versus diffusion. Expert Opin Drug Deliv 17:791–803
Bruschi ML (2015) Strategies to modify the drug release from pharmaceutical systems. Woodhead Publishing, United Kingdom
Caccavo D, Cascone S, Lamberti G, Barba AA (2014) Modeling the drug release from hydrogel-based matrices. Mol Pharm 12:474–483
Chakraborty S, Khandai M, Sharma A, Patra CN, Patro VJ et al (2009) Effects of drug solubility on the release kinetics of water soluble and insoluble drugs from HPMC based matrix formulations. Acta Pharm 59:313–323
Chappidi SR, Bhargav E, Marikunte V, Chinthaginjala H, Vijaya Jyothi M et al (2019) A cost effective (QbD) approach in the development and optimization of rosiglitazone maleate mucoadhesive extended release tablets—in vitro and ex vivo. Adv Pharm Bull 9:281–288
Chudiwal VS, Shahi S, Chudiwal S (2018) Development of sustained release gastro-retentive tablet formulation of nicardipine hydrochloride using quality by design (QbD) approach. Drug Dev Ind Pharm 44:787–799
Colombo P, Bettini R, Santi P, Peppas NA (2000) Swellable matrices for controlled drug delivery: gel-layer behaviour, mechanisms and optimal performance. Pharm Sci Technol Today 3:198–204
Costa P, Manuel J, Lobo S (2001) Modeling and comparison of dissolution profiles. Eur J Pharm Sci 13:123–133
Crowley MM, Schroeder B, Fredersdorf A, Obara S, Talarico M et al (2004) Physicochemical properties and mechanism of drug release from ethyl cellulose matrix tablets prepared by direct compression and hot-melt extrusion. Int J Pharm 269:509–522
Dave VS, Saoji SD, Raut NA, Haware RV (2015) Excipient variability and its impact on dosage form functionality. J Pharm Sci 104:906–915
Davis B, Schlindwein WS (2018) Introduction to quality by design (QbD). In: Schlindwein WS, Gibson M (eds) Pharmaceutical quality by design: a practical approach, 1st edn. John Wiley & Sons, United States, pp 1–9
Demchenko Y, Grosso P, de Laat C, Membrey P (2013) Addressing big data issues in scientific data infrastructure. International Conference on Collaboration Technologies and Systems (CTS), San Diego
Desai N, Purohit R (2017) Design and development of clopidogrel bisulfate gastroretentive osmotic formulation using quality by design tools. AAPS PharmSciTech 18:2626–2638
Diab S, Bano G, Dhenge RM, Taylor J (2021) Impact of process parameters and formulation properties on dissolution performance of an extended release tablet: a multivariate analysis. J Pharm Innov 17:892–910
Ding H (2016) Modified-release drug products and drug devices. In: Shargel L, Yu AB (eds) Applied biopharmaceutics and pharmacokinetics, 7th edn. McGraw-Hill Education, New York
Djuris J, Cirin-Varadjan S, Aleksic I, Djuris M, Cvijic S et al (2021) Application of machine-learning algorithms for better understanding of tableting properties of lactose co-processed with lipid excipients. Pharmaceutics 13:663
European Medicines Agency (2012) Guideline on real time release testing (formerly guideline on parametric release). https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-real-time-release-testing-formerly-guideline-parametric-release-revision-1_en.pdf. Accessed 03 May 2022
European Medicines Agency (2014) Guideline on quality of oral modified release products. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-oral-modified-release-products_en.pdf. Accessed 05 May 2022
Farooqi S, Yousuf RI, Shoaib MH, Ahmed K, Ansar S et al (2020) Quality by design (QbD)-based numerical and graphical optimization technique for the development of osmotic pump controlled-release metoclopramide HCl tablets. Drug Des Dev Ther 14:5217–5234
Florence AT (2011) A short history of controlled drug release and an introduction. In: Wilson C, Crowley P (eds) Controlled release in oral drug delivery, 1st edn. Springer, pp 1–26
Food and Drug Administration (1997a) Extended release oral dosage forms: development, evaluation, and application of in vitro/in vivo correlations. https://www.fda.gov/media/70939/download. Accessed 03 May 2022
Food and Drug Administration (1997b) SUPAC-MR: modified release solid oral dosage forms - scale-up and postapproval changes: chemistry, manufacturing, and controls; in vitro dissolution testing and in vivo bioequivalence documentation. https://www.fda.gov/media/70956/download. Accessed 03 May 2022
Food and Drug Administration (2004) PAT—A framework for innovative pharmaceutical development, manufacturing, and quality assurance. https://www.fda.gov/media/71012/download. Accessed 03 May 2022
Ford JL (2014) Design and evaluation of hydroxypropyl methylcellulose matrix tablets for oral controlled release: A historical perspective. In: Timmins P, Pygall SR, Melia CD (eds) Hydrophilic matrix tablets for oral controlled release. Springer, pp 17–51
Frenning G (2011) Modelling drug release from inert matrix systems: from moving-boundary to continuous-field descriptions. Int J Pharm 418:88–99
Galata DL, Farkas A, Konyves Z, Meszaros LA, Szabo E et al (2019) Fast, spectroscopy-based prediction of in vitro dissolution profile of extended release tablets using artificial neural networks. Pharmaceutics 11:400
Galata DL, Konyves Z, Nagy B, Novak M, Meszaros LA et al (2021) Real-time release testing of dissolution based on surrogate models developed by machine learning algorithms using NIR spectra, compression force and particle size distribution as input data. Int J Pharm 597:120338
Gavan A, Porfire A, Marina C, Tomuta I (2017) Original research paper. Formulation and pharmaceutical development of quetiapine fumarate sustained release matrix tablets using a QbD approach. Acta Pharm 67:53–70
Gavan A, Sylvester B, Porfire A, Iurian S, Casian T et al (2022) NIR spectroscopy for monitoring of the critical manufacturing steps and quality attributes of paliperidone prolonged release tablets. J Mol Struct 1247:131326
Gendre C, Boiret M, Genty M, Chaminade P, Pean JM (2011) Real-time predictions of drug release and end point detection of a coating operation by in-line near infrared measurements. Int J Pharm 421:237–243
Gibson M, Carmody A, Weaver R (2018) Development and manufacture of drug product. In: Schlindwein WS, Gibson M (eds) Pharmaceutical quality by design: a practical approach, 1st edn. John Wiley & Sons, United States, p 117
Goodwin DJ, van den Ban S, Denham M, Barylski I (2018) Real time release testing of tablet content and content uniformity. Int J Pharm 537:183–192
Gowthami B, Krishna SVG, Rao DS (2021) Formulation of tablets in capsule system: statistical optimization for chronotherapeutic drug delivery of propranolol hydrochloride. J Drug Deliv Sci Technol 63:102398
Grangeia HB, Silva C, Simoes SP, Reis MS (2020) Quality by design in pharmaceutical manufacturing: a systematic review of current status, challenges and future perspectives. Eur J Pharm Biopharm 147:19–37
Guler GK, Eroglu H, Oner L (2017) Development and formulation of floating tablet formulation containing rosiglitazone maleate using Artificial neural network. J Drug Deliv Sci Technol 39:385–397
Haenlein M, Kaplan A (2019) A brief history of artificial intelligence: on the past, present, and future of artificial intelligence. Calif Manage Rev 61:5–14
Hayashi Y, Marumo Y, Takahashi T, Nakano Y, Kosugi A et al (2019) In silico predictions of tablet density using a quantitative structure-property relationship model. Int J Pharm 558:351–356
Hayashi Y, Nakano Y, Marumo Y, Kumada S, Okada K et al (2021) Application of machine learning to a material library for modeling of relationships between material properties and tablet properties. Int J Pharm 609:121158
Heiman J, Tajarobi F, Gururajan B, Juppo A, Abrahmsen-Alami S (2015) Roller compaction of hydrophilic extended release tablets-combined effects of processing variables and drug/matrix former particle size. AAPS PharmSciTech 16:267–277
Heng PWS, Chan LW, Easterbrook MG, Li XM (2001) Investigation of the influence of mean HPMC particle size and number of polymer particles on the release of aspirin from swellable hydrophilic matrix tablets. J Control Release 76:39–49
Hiremath PS, Saha RN (2008) Controlled release hydrophilic matrix tablet formulations of isoniazid: design and in vitro studies. AAPS PharmSciTech 9:1171–1178
Hoffman AS (2008) The origins and evolution of “controlled” drug delivery systems. J Control Release 132:153–163
Hu M, Zhu Z, Wu Y, Meng Q, Luo J et al (2020) Exploring the potential of hydrophilic matrix combined with insoluble film coating: preparation and evaluation of ambroxol hydrochloride extended release tablets. AAPS PharmSciTech 21:93
Huang J, Kaul G, Cai CS, Chatlapalli R, Hernandez-Abad P et al (2009) Quality by design case study: an integrated multivariate approach to drug product and process development. Int J Pharm 382:23–32
Ibric S, Jovanovic M, Djuric Z, Parojcic J, Solomun L et al (2007) Generalized regression neural networks in prediction of drug stability. J Pharm Pharmacol 59:745–750
International Conference on Harmonisation (2009) ICH guideline Q8 (R2) on pharmaceutical development. https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf. Accessed 03 May 2022
International Conference on Harmonisation (2010) Q8, Q9 and Q10: Questions and answers. https://database.ich.org/sites/default/files/Q8_Q9_Q10_Q%26As_R4_Q%26As_0.pdf. Accessed 06 May 2022
International Conference on Harmonisation (2011) Q8, Q9 and Q10: Points to consider. https://database.ich.org/sites/default/files/Q8_Q9_Q10_Q%26As_R4_Points_to_Consider_2.pdf. Accessed 06 May 2022
International Conference on Harmonisation (2012) ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological entities). https://database.ich.org/sites/default/files/Q11%20Guideline.pdf. Accessed 03 May 2022
Ilic M, Duris J, Kovacevic I, Ibric S, Parojcic J (2014) In vitro—in silico—in vivo drug absorption model development based on mechanistic gastrointestinal simulation and artificial neural networks: Nifedipine osmotic release tablets case study. Eur J Pharm Sci 62:212–218
Ilyes K, Casian T, Hales D, Borodi G, Rus L et al (2021) Applying the principles of quality by design (Qbd) coupled with multivariate data analysis (MVDA) in establishing the impact of raw material variability for extended release tablets. Farmacia 69:481–497
International Conference on Harmonisation (2005) ICH guideline Q9 on quality risk management. https://database.ich.org/sites/default/files/Q9%20Guideline.pdf. Accessed 03 May 2022
International Conference on Harmonisation (2008) ICH guideline Q10 on pharmaceutical quality system. https://database.ich.org/sites/default/files/Q10%20Guideline.pdf. Accessed 03 May 2022
International Conference on Harmonisation (2019) ICH guideline Q12 on technical and regulatory considerations for pharmaceutical product lifecycle management. https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf. Accessed 03 May 2022
Islam MT, Maniruzzaman M, Halsey SA, Chowdhry BZ, Douroumis D (2014) Development of sustained-release formulations processed by hot-melt extrusion by using a quality-by-design approach. Drug Deliv Transl Res 4:377–387
Iurian S, Turdean L, Tomuta I (2017) Risk assessment and experimental design in the development of a prolonged release drug delivery system with paliperidone. Drug Des Dev Ther 11:733–746
Ivic B, Ibric S, Betz G, Djuric Z (2010) Optimization of drug release from compressed multi unit particle system (MUPS) using generalized regression neural network (GRNN). Arch Pharm Res 33:103–113
Jang EH, Park YS, Choi D (2021) Investigation of the effects of materials and dry granulation process on the mirabegron tablet by integrated QbD approach with multivariate analysis. Powder Technol 382:23–39
Kanwal U, Mukhtar S, Waheed M, Mehreen A, Abbas N et al (2021) Fixed dose single tablet formulation with differential release of amlodipine besylate and simvastatin and its pharmacokinetic profile: QbD and risk assessment approach. Drug Des Dev Ther 15:2193–2210
Khan AM, Hanif M, Bukhari NI, Shamim R, Rasool F et al (2020) Artificial neural network (ANN) approach to predict an optimized pH-dependent mesalamine matrix tablet. Drug Des Dev Ther 14:2435–2448
Kim CJ (1998) Effects of drug solubility, drug loading, and polymer molecular weight on drug release from polyox (R) tablets. Drug Dev Ind Pharm 24:645–651
Kosir D, Ojstersek T, Baumgartner S, Vrecer F (2018) A study of critical functionality-related characteristics of HPMC for sustained-release tablets. Pharm Dev Technol 23:865–873
Kovacs B, Peterfi O, Kovacs-Deak B, Szekely-Szentmiklosi I, Fulop I et al (2021) Quality-by-design in pharmaceutical development: from current perspectives to practical applications. Acta Pharm 71:497–526
Kushner J, Lamba M, Stock T, Wang R, Nemeth MA et al (2020) Development and validation of a Level A in vitro in vivo correlation for tofacitinib modified -release tablets using extrudable core system osmotic delivery technology. Eur J Pharm Sci 147:105200
Lakio S, Tajarobi P, Wikstrom H, Fransson M, Arnehed J et al (2016) Achieving a robust drug release from extended release tablets using an integrated continuous mixing and direct compression line. Int J Pharm 511:659–668
Lee PI, Li JX (2010) Evolution of oral controlled release dosage forms. In: Wen H, Park K (eds) Oral controlled release formulation design and drug delivery: theory to practice, 1st edn. John Wiley & Sons, New Jersey, pp 21–31
Lefnaoui S, Rebouh S, Bouhedda M, Yahoum MM, Hanini S (2018) Artificial neural network modeling of sustained antihypertensive drug delivery using polyelectrolyte complex based on carboxymethyl-kappa-carrageenan and chitosan as prospective carriers. International conference on applied smart systems (ICASS), USA
Li H, Hardy RJ, Gu X (2008) Effect of drug solubility on polymer hydration and drug dissolution from polyethylene oxide (PEO) matrix tablets. AAPS PharmSciTech 9:437–443
Lin W, Li YK, Shi QZ, Liao XR, Zeng Y et al (2022) Preparation and evaluation of bilayer-core osmotic pump tablets contained topiramate. PLoS ONE 17:e0264457
Liu LX, Xu XN (2008) Preparation of bilayer-core osmotic pump tablet by coating the indented core tablet. Int J Pharm 352:225–230
Liu LL, Zhang KF, Sun ZY, Dong Q, Li L et al (2021) A new perspective in understanding the dissolution behavior of nifedipine controlled release tablets by NIR spectroscopy with aquaphotomics. J Mol Struct 1230:129872
Lopes JA, Sarraguca MC (2018) Data processing in multivariate analysis of pharmaceutical processes. In: Ferreira A, Menezes J, Tobyn M (eds) Multivariate analysis in the pharmaceutical industry, 1st edn. Elsevier, United Kingdom, pp 35–51
Lou H, Chung JI, Kiang YH, Xiao LY, Hageman MJ (2019) The application of machine learning algorithms in understanding the effect of core/shell technique on improving powder compactability. Int J Pharm 555:368–379
Lou H, Lian B, Hageman MJ (2021) Applications of machine learning in solid oral dosage form development. J Pharm Sci 110:3150–3165
Lundsberg-Nielsen L, Schlindwein WS, Berghaus A (2018) Process analytical technology (PAT). In: Schlindwein WS, Gibson M (eds) Pharmaceutical quality by design: a practical approach, 1st edn. John Wiley & Sons, United States, pp 227–255
Maderuelo C, Zarzuelo A, Lanao JM (2011) Critical factors in the release of drugs from sustained release hydrophilic matrices. J Control Release 154:2–19
Maki-Lohiluoma E, Sakkinen N, Palomaki M, Winberg O, Ta HX et al (2021) Use of machine learning in prediction of granule particle size distribution and tablet tensile strength in commercial pharmaceutical manufacturing. Int J Pharm 609:121146
Malaterre V, Ogorka J, Loggia N, Gurny R (2009) Approach to design push-pull osmotic pumps. Int J Pharm 376:56–62
Markl D, Warman M, Dumarey M, Bergman EL, Folestad S et al (2020) Review of real-time release testing of pharmaceutical tablets: state-of-the art, challenges and future perspective. Int J Pharm 582:119353
Mirani AG, Patankar SP, Kadam VJ (2016) Risk-based approach for systematic development of gastroretentive drug delivery system. Drug Deliv Transl Res 6:579–596
Missaghi S, Patel P, Farrell TP, Huatan H, Rajabi-Siahboomi AR (2014) Investigation of critical core formulation and process parameters for osmotic pump oral drug delivery. AAPS PharmSciTech 15:149–160
Mohamed MI, Al-Mahallawi AM, Awadalla SM (2020) Development and optimization of osmotically controlled drug delivery system for poorly aqueous soluble diacerein to improve its bioavailability. Drug Dev Ind Pharm 46:814–825
Momin MM, Kane S, Abhang P (2015) Formulation and evaluation of bilayer tablet for bimodal release of venlafaxine hydrochloride. Front Pharmacol 6:144
Muller J, Brock D, Knop K, Zeitler JA, Kleinebudde P (2012) Prediction of dissolution time and coating thickness of sustained release formulations using Raman spectroscopy and terahertz pulsed imaging. Eur J Pharm Biopharm 80:690–697
Muntean DM, Alecu C, Tomuta I (2017) Simultaneous quantification of paracetamol and caffeine in powder blends for tableting by NIR-chemometry. J Spectrosc 2017:7160675
Nagy B, Petra D, Galata DL, Demuth B, Borbas E et al (2019) Application of artificial neural networks for process analytical technology-based dissolution testing. Int J Pharm 567:118464
Nokhodchi A, Raja S, Patel P, Asare-Addo K (2012) The role of oral controlled release matrix tablets in drug delivery systems. Bioimpacts 2:175–187
Obeidat WM, Nokhodchi A, Alkhatib H (2015) Evaluation of matrix tablets based on Eudragit®E100/Carbopol®971P combinations for controlled release and improved compaction properties of water soluble model drug paracetamol. AAPS PharmSciTech 16:1169–1179
Owen M, Cox I (2018) Design of experiments. In: Schlindwein WS, Gibson M (eds) Pharmaceutical quality by design: a practical approach, 1st edn. John Wiley & Sons, United States, pp 157–199
Parmar C, Parikh K, Mundada P, Bhavsar D, Sawant K (2018) Formulation and optimization of enteric coated bilayer tablets of mesalamine by RSM: In vitro—In vivo investigations and roentogenographic study. J Drug Deliv Sci Technol 44:388–398
Paul D, Sanap G, Shenoy S, Kalyane D, Kalia K et al (2021a) Artificial intelligence in drug discovery and development. Drug Discov Today 26:80–93
Paul S, Baranwal Y, Tseng YC (2021b) An insight into predictive parameters of tablet capping by machine learning and multivariate tools. Int J Pharm 599:120439
Pawar P, Wang YF, Keyvan G, Callegari G, Cuitino A et al (2016) Enabling real time release testing by NIR prediction of dissolution of tablets made by continuous direct compression (CDC). Int J Pharm 512:96–107
Peppas NA, Narasimhan B (2014) Mathematical models in drug delivery: how modeling has shaped the way we design new drug delivery systems. J Control Release 190:75–81
Petrovic J, Ibric S, Betz G, Duric Z (2012) Optimization of matrix tablets controlled drug release using Elman dynamic neural networks and decision trees. Int J Pharm 428:57–67
Pishnamazi M, Ismail HY, Shirazian S, Iqbal J, Walker GM et al (2019) Application of lignin in controlled release: development of predictive model based on artificial neural network for API release. Cellulose 26:6165–6178
Politis SN, Colombo P, Colombo G (2017) Design of experiments (DoE) in pharmaceutical development. Drug Dev Ind Pharm 43:889–901
Porfire A, Filip C, Tomuta I (2017) High-throughput NIR-chemometric methods for chemical and pharmaceutical characterization of sustained release tablets. J Pharm Biomed Anal 138:1–13
Qazi F, Shoaib MH, Yousuf RI, Siddiqui F, Nasiri MI et al (2020) QbD based Eudragit coated meclizine HCl immediate and extended release multiparticulates: formulation, characterization and pharmacokinetic evaluation using HPLC-Fluorescence detection method. Sci Rep 10:14765
Qiu Y, Lee P (2017) Rational design of oral modified-release drug delivery systems. In: Qiu Y, Chen Y, Zhang G, Yu L, Mantri R (eds) Developing solid oral dosage forms, 2nd edn. Academic Press, London, pp 519–554
Rajalahti T, Kvalheim OM (2011) Multivariate data analysis in pharmaceutics: a tutorial review. Int J Pharm 417:280–290
Reda C, Kaufmann E, Delahaye-Duriez A (2020) Machine learning applications in drug development. Comput Struct Biotechnol J 18:241–252
Reinhardt IC, Oliveira JC, Ring DT (2020) Current perspectives on the development of industry 4.0 in the pharmaceutical sector. J Ind Inf Integr 18:100131
Reynolds TD, Mitchell SA, Balwinski KM (2002) Investigation of the effect of tablet surface area/volume on drug release from hydroxypropylmethylcellulose controlled-release matrix tablets. Drug Dev Ind Pharm 28:457–466
Rus LL, Casian T, Iovanov RI, Orzea RM, Onisor I et al (2020) Quantitative characterization of sustained release tablets with diclofenac sodium by means of near-infrares spectroscopy and chemometry. Farmacia 68:728–739
Sacher S, Poms J, Rehrl J, Khinast JG (2022) PAT implementation for advanced process control in solid dosage manufacturing-A practical guide. Int J Pharm 613:121408
Sanoufi MR, Aljaberi A, Hamdan I, Al-Zoubi N (2020) The use of design of experiments to develop hot melt extrudates for extended release of diclofenac sodium. Pharm Dev Technol 25:187–196
Saracoglu OK, Uludag MO, Ozdemir ED, Degim IT (2020) Development of controlled release dexketoprofen tablets and prediction of drug release using artificial neural network (ANN) modelling. Braz J Pharm Sci 56:e18540
Saydam M, Takka S (2018) Development and in vitro evaluation of pH-independent release matrix tablet of weakly acidic drug valsartan using quality by design tools. Drug Dev Ind Pharm 44:1905–1917
Schmitt S (2018) Quality systems and knowledge management. In: Schlindwein WS, Gibson M (eds) Pharmaceutical quality by design: a practical approach, 1st edn. John Wiley & Sons, United States, pp 47–60
Sethi S, Mangla B, Kamboj S, Rana V (2018) A QbD approach for the fabrication of immediate and prolong buoyant cinnarizine tablet using polyacrylamide-g-corn fibre gum. Int J Biol Macromol 117:350–361
Shah V, Khambhla E, Nivsarkar M, Trivedi R, Patel RK (2022) An Integrative QbD approach for the development and optimization of controlled release compressed coated formulation of water-soluble drugs. AAPS PharmSciTech 23:120
Shi GL, Lin LF, Liu YL, Chen GS, Luo YT et al (2021) Pharmaceutical application of multivariate modelling techniques: a review on the manufacturing of tablets. RSC Adv 11:8323–8345
Siegel RA, Rathbone MJ (2012) Overview of controlled release mechanisms. In: Siepmann J, Siegel RA, Rathbone MJ (eds) Fundamentals and applications of controlled release drug delivery, 1st edn. Springer, New York, pp 19–43
Siepmann J, Siepmann F (2012b) Swelling controlled drug delivery systems. In: Rathbone MJ, Siepmann J, Siegel RA (eds) Fundamentals and applications of controlled release drug delivery, 1st edn. Springer, New York, pp 153–170
Siepmann J, Kranz H, Peppas NA, Bodmeier R (2000) Calculation of the required size and shape of hydroxypropyl methylcellulose matrices to achieve desired drug release profiles. Int J Pharm 201:151–164
Siepmann J, Siegel RA, Siepmann F (2012a) Diffusion controlled drug delivery systems. In: Rathbone MJ, Siepmann J, Siegel RA (eds) Fundamentals and applications of controlled release drug delivery, 1st edn. Springer, New York, pp 127–152
Simoes MF, Silva G, Pinto AC, Fonseca M, Silva NE et al (2020) Artificial neural networks applied to quality-by-design: From formulation development to clinical outcome. Eur J Pharm Biopharm 152:282–295
Singh SK, Venkateshwarn T, Simmons SP (2010) Oral controlled drug delivery: quality by design (QbD) approach to drug development. In: Wen H, Park K (eds) Oral controlled release formulation design and drug delivery: theory to practice, 1st edn. John Wiley & Sons, New Jersey, pp 279–303
Sirbu C, Tomuta I, Achim M, Rus LL, Vonica L et al (2014) Quantitative characterization of powder blends for tablets with Indapamide by near-infrared spectrocopy and chemometry. Farmacia 62:48–57
Steinwandter V, Borchert D, Herwig C (2019) Data science tools and applications on the way to Pharma 4.0. Drug Discov Today 24:1795–1805
Than YM, Titapiwatanakun V (2021) Statistical design of experiment-based formulation development and optimization of 3D printed oral controlled release drug delivery with multi target product profile. J Pharm Investig 51:715–734
Thapa P, Jeong SH (2018) Effects of formulation and process variables on gastroretentive floating tablets with a high-dose soluble drug and experimental design approach. Pharmaceutics 10:161
Theeuwes F (1975) Elementary osmotic pumps. J Pharm Sci 64:1987–1991
Thomas S, Palahnuk H, Amini H, Akseli I (2021) Data-smart machine learning methods for predicting composition-dependent Young’s modulus of pharmaceutical compacts. Int J Pharm 592:120049
Timmins P, Desai D, Chen W, Wray P, Brown J et al (2016) Advances in mechanistic understanding of release rate control mechanisms of extended-release hydrophilic matrix tablets. Ther Deliv 7:553–572
Tiwari SB, DiNunzio J, Rajabi-Siahboomi A (2011) Drug–polymer matrices for extended release. In: Wilson CG, Crowley PJ (eds) Controlled release in oral drug delivery, 1st edn. Springer, New York, pp 131–159
Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E et al (2019) Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18:463–477
Van Hauwermeiren D, Stock M, De Beer T, Nopens I (2020) Predicting pharmaceutical particle size distributions using kernel mean embedding. Pharmaceutics 12:271
Van Snick B, Holman J, Cunningham C, Kumar A, Vercruysse J et al (2017) Continuous direct compression as manufacturing platform for sustained release tablets. Int J Pharm 519:390–407
Vanhoorne V, Vanbillemont B, Vercruysse J, De Leersnyder F, Gomes P et al (2016) Development of a controlled release formulation by continuous twin screw granulation: Influence of process and formulation parameters. Int J Pharm 505:61–68
Vanza JD, Patel RB, Dave RR, Patel MR (2020) Polyethylene oxide and its controlled release properties in hydrophilic matrix tablets for oral administration. Pharm Dev Technol 25:1–19
Verma RK, Krishna DM, Garg S (2002) Formulation aspects in the development of osmotically controlled oral drug delivery systems. J Control Release 79:7–27
Viriden A, Wittgren B, Andersson T, Larsson A (2009) The effect of chemical heterogeneity of HPMC on polymer release from matrix tablets. Eur J Pharm Sci 36:392–400
Viriden A, Larsson A, Wittgren B (2010) The effect of substitution pattern of HPMC on polymer release from matrix tablets. Int J Pharm 389:147–156
Viriden A, Abrahmsen-Alami S, Wittgren B, Larsson A (2011) Release of theophylline and carbamazepine from matrix tablets–consequences of HPMC chemical heterogeneity. Eur J Pharm Biopharm 78:470–479
Vora C, Patadia R, Mittal K, Mashru R (2015) Risk based approach for design and optimization of site specific delivery of isoniazid. J Pharm Investig 45:249–264
Walker RB (2008) Modified-release delivery systems for oral use. In: Rathbone M, Hadgraft J, Roberts MS, Lane ME (eds) Modified-release drug delivery technology, 2nd edn. CRC Press, New York, pp 131–141
Wang W, Ye ZYF, Gao HL, Ouyang DF (2021) Computational pharmaceutics-A new paradigm of drug delivery. J Control Release 338:119–136
Wang S, Di JW, Wang D, Dai XD, Hua YB et al (2022) State-of-the-art review of artificial neural networks to predict. Characterize and optimize pharmaceutical formulation. Pharmaceutics 14:183
Wen H, Park K (2010) Introduction and overview of oral controlled release formulation design. In: Wen H, Park K (eds) Oral Controlled release formulation design and drug delivery: theory to practice, 1st edn. John Wiley & Sons, New Jersey, pp 1–19
Wirges M, Funke A, Serno P, Knop K, Kleinebudde P (2013) Monitoring of an active coating process for two-layer tablets-model development strategies. J Pharm Sci 102:556–564
Won DH, Park H, Ha ES, Kim HH, Jang SW et al (2021) Optimization of bilayer tablet manufacturing process for fixed dose combination of sustained release high-dose drug and immediate release low-dose drug based on quality by design (QbD). Int J Pharm 605:120838
Wu HQ, Lyon RC, Khan MA, Voytilla RJ, Drennen JK (2015) Integration of near-infrared spectroscopy and mechanistic modeling for predicting film-coating and dissolution of modified release tablets. Ind Eng Chem Res 54:6012–6023
Yang Y, Zhao ZN, Wang YF, Yang L, Liu DD et al (2016) A novel asymmetric membrane osmotic pump capsule with in situ formed delivery orifices for controlled release of gliclazide solid dispersion system. Int J Pharm 506:340–350
Yang YL, Ye ZYF, Su Y, Zhao QQ, Li XS et al (2019) Deep learning for in vitro prediction of pharmaceutical formulations. Acta Pharm Sin B 9:177–185
Yoo S, Kim J, Choi GJ (2022) Drug properties prediction based on deep learning. Pharmaceutics 14:467
Yu LX (2008) Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res 25:781–791
Yu LX, Amidon G, Khan MA, Hoag SW, Polli J et al (2014) Understanding pharmaceutical quality by design. AAPS J 16:771–783
Yu JJ, Meng X, Dong X, Han MF, Li GT et al (2021) Synthesis and characterization of osmotic pump capsules containing polyoxyethylene and pH modifier to control the release of nifedipine. Eur J Pharm Biopharm 163:102–108
Zaborenko N, Shi Z, Corredor CC, Smith-Goettler BM, Zhang L et al (2019) First-principles and empirical approaches to predicting in vitro dissolution for pharmaceutical formulation and process development and for product release testing. AAPS J 21:32
Zarmpi P, Flanagan T, Meehan E, Mann J, Fotaki N (2017) Biopharmaceutical aspects and implications of excipient variability in drug product performance. Eur J Pharm Biopharm 111:1–15
Zhou DL, Law D, Reynolds J, Davis L, Smith C et al (2014) Understanding and managing the impact of HPMC variability on drug release from controlled release formulations. J Pharm Sci 103:1664–1672
Acknowledgements
This work was financially supported by the Drugs R&D Doctoral Program assigned by FCT (Fundação para a Ciência e Tecnologia), Portugal and Tecnimede Group [grant PD/BDE/150736/2020].
Author information
Authors and Affiliations
Contributions
SS: Conceptualization, Writing—original draft, Visualization. JS: Conceptualization, Supervision. CE: Writing—original draft. RC: Writing—review & editing. AR: Conceptualization, Writing—review & editing, Supervision.
Corresponding author
Ethics declarations
Conflict of interest
All authors (A.S. Sousa, J. Serra, C. Estevens, R. Costa, and A.J. Ribeiro) declare that they have no conflicts of interest.
Research involving human and animal particpants
This article does not contain any studies with human and animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sousa, A.S., Serra, J., Estevens, C. et al. A quality by design approach in oral extended release drug delivery systems: where we are and where we are going?. J. Pharm. Investig. 53, 269–306 (2023). https://doi.org/10.1007/s40005-022-00603-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40005-022-00603-w