
Abstract
MicroRNAs (miRNAs) are a class of small non-coding, single-stranded RNA sequences that regulate gene expression at the post-transcriptional level and also reported to function in stress responses, but their role has not been studied in Camelina (Camelina sativa L.), an emerging oil crop. In this study, we predicted conserved as well as putative novel miRNAs from a Camelina drought stress cDNA library using comprehensive genomic approaches. Based on the sequence homology, we predicted 145 miRNAs, of which 61 were conserved, and 84 putative novel miRNAs were found to belong to 26 and 72 different miRNA families, respectively. In silico expression analysis indicated that 20 miRNAs were really expressed in Camelina genome, and several of them have tissue-specific expression character. We found that the 60 putative novel miRNA families target 117 genes. Most of the miRNA targets were predicted to genes including that regulate stress response, transcription factors, and fatty acid and lipid metabolism-related genes. Expression patterns of 6 randomly selected miRNAs under drought stress were validated by real-time quantitative polymerase chain reaction analysis. Coordinated expression changes between 6 randomly selected miRNAs and their target genes, suggested that the predicted miRNAs could be drought-responsive and that they would likely be directly involved in stress regulatory networks of Camelina. These results indicate that, in C. sativa, under drought stress, a large number of new miRNAs could be discovered, and the predicted stress-responsive miRNAs and their target transcripts will serve as valuable resources for future studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Camelina (Camelina sativa L.), also known as false flax or gold of pleasure, is an oil seed crop belonging to the family Brassicaceae. Camelina is widely cultivated for its seed oil, which contains high levels of essential fatty acids (omega-3 and omega-6 fatty acids) (Hixson et al. 2014). Due to its high fatty acid composition, seed oil of Camelina has emerged as a new biofuel with broad applications in industry, including jet fuel, biodiesel, as well as multiple livestock feeds (Collins-Silva et al. 2011). Camelina has many agronomic attributes, including short lifecycle, low nutritional requirements, resistance to various pathogens (Séguin-Swartza et al. 2009; Gehringer et al. 2006), as well as strong potential to grow under high salt and drought stress conditions (Moser 2012; Zubr 1997). Camelina is genetically similar to Arabidopsis (Beilstein et al. 2008), and its recently published complete genome sequence (Kagale et al. 2014) can be studied to identify stress-responsive genes and their regulators to improve cultivation of Camelina crops. Recent high-throughput microarray technologies such as new-generation sequencing platforms offer a wide range of programs to utilize genomic resources for Camelina.
De novo transcriptome expression profiles of Camelina plant have been recently studied with the goal of discovering useful genes (Wang et al. 2015; Mudalkar et al. 2014; Liang et al. 2013). However, the transcriptome profile of Camelina under stress has never been reported, although studies have been performed to understand the stress responsiveness of C. sativa and characterize the genes involved in stress responses (Kwak et al. 2013; Kim et al. 2013). When salt, cold, and dehydration stresses were previously applied to Camelina, upregulated glycine-rich RNA-binding proteins were observed to act as RNA chaperones during the stress adaptation process (Kwak et al. 2013). Another study reported an association between the cold tolerance of Camelina and the high influx rate of plasma membrane H+ATPase, which can be controlled by specific translational or post-translational modifications (Kim et al. 2013). However, the regulatory networks governing these genes and the overall response of Camelina under stress still need to be understood.
MicroRNAs (miRNAs) are a class of small (18–26 nucleotides) non-coding, endogenous, and single-stranded RNA sequences. These molecules bind to target mRNAs to induce RNA cleavage, inhibition of mRNA translation, or negative regulation of corresponding gene expression at the post-transcriptional level (Jones-Rhoades et al. 2006; Bartel 2004). In plants, miRNA gene processing (Dicer-like), transportation, and target mRNA degradation by the RNA-induced silencing complex (RISC) have been demonstrated in earlier reports (Baumberger and Baulcombe 2005; Kurihara and Watanabe 2004; Bartel 2004). A growing number of reports has shown that miRNAs play an important role in plant growth and development, including morphogenesis, transition from vegetative to reproductive phase, and root, stem, leaf, and flower initiations (Rubio-Somoza and Weigel 2011; Chen 2005). These miRNAs have also been reported to function in stress responses (Sunkar et al. 2012). To date, there are no Camelina-specific miRNAs in miRBASE and PMRD (Kozomara and Griffiths-Jones 2014; Zhang et al. 2010). Recently, Poudel et al. (2015) identified 63 miRNA families, including 207 conserved and five novel miRNAs. Also, all of these miRNAs were found to be differentially expressed in various tissues and developmental stages of Camelina sativa. There are 385 Arabidopsis miRNAs registered in the database, even though Camelina was shown to be similar to the Arabidopsis genome (Kagale et al. 2014; Liang et al. 2013). This result suggested that both conserved and novel miRNAs still need to be identified in Camelina sativa. Previous reports have identified conserved and novel miRNAs under stress conditions in several plants, including Oryza sativa (Jian et al. 2010), Brassica rapa (Yu et al. 2012), Populus euphratica (Li et al. 2013), Solanum tuberosum (Zhang et al. 2014), and Saccharina japonica (Liu et al. 2015). Computational approaches for identification of novel miRNAs are considered as reliable methods for both plants and animals based on expressed sequence tags (ESTs), genome survey sequences (GSSs), and high-throughput genomics sequences (HTGSs) (Panda et al. 2015; Dhandapani et al. 2011; Yin et al. 2008). The stress responsiveness as well as novel stress response-related miRNA genes of Camelina, an emerging biofuel crop, have never been reported. A computational approach for identification of miRNAs and their targets under drought stress will facilitate understanding of the post-transcriptional regulation networks involved in C. sativa drought stress tolerance. Recently, we developed an EST library from Camelina leaves exposed to four different levels of relative soil moisture content (RSMC) (Kanth et al. 2015). The present study utilized this EST library to identify the drought-responsive miRNAs in Camelina.
Using a computational approach, we identified a total of 145 miRNAs, including 61 conserved and 84 putative novel miRNAs belonging to 28 and 72 different miRNA families, respectively. In silico prediction indicated that the 50 putative novel miRNAs targeted 62 genes involved in resistance to stress, lipid metabolism, and transcription factors. Our study results have significant implications on the gene regulatory network in Camelina under drought stress and also help to understand the functions of stress-responsive miRNAs.
Materials and methods
Reference set of miRNAs and C. sativa sequences
A total of 10,898 and 8746 mature miRNA sequences were downloaded from PMRD: plant microRNA database (http://bioinformatics.cau.edu.cn/PMRD/) (Zhang et al. 2010) and miRBase 21 (http://www.mirbase.org/) (Kozomara and Griffiths-Jones 2014), respectively. After removing the redundant sequences using perl script (http://www.perl.org/), a total of 9863 sequences were used as the final reference set. For homology-based miRNA identification, 60,171 unique expressed sequence tag (EST) sequences of Camelina were used. The ESTs were obtained from a drought-induced leaf transcriptome of Camelina [Control, 10 kilopascal (kPa), 100 kPa, and Rehydration (Rehyd)], which is available in the laboratory (Kanth et al. 2015).
Computational identification of miRNAs
Non-redundant miRNA reference set was subjected to BLAST against Camelina EST sequences using Bowtie1 (Langmead et al. 2009). The miRNAs showing zero and 0–3 mismatches with Camelina sequences were considered as conserved and putative novel miRNAs, respectively, according to previous reports (Poudel et al. 2015). The entire process for prediction of miRNAs is shown in Supplementary Fig. S1. The precursor miRNA sequences (pre-miRNA) were excised from Camelina sequences using an in house perl script, in which 150 nucleotides (nt) up and downstream of the matched sequences were extracted. For removal of protein coding sequences, pre-miRNA sequences were subjected to BLASTX, and only non-coding sequences were chosen for further analysis. The hairpin secondary structures of the identified pre-miRNAs were predicted using Mfold web server with default parameters (Zuker 2003) by BLASTing pre-miRNA sequences one at a time. The selection of best hairpin folds of candidate miRNAs was done by manual analysis of predicted secondary structures with the following criteria: pre-miRNAs fold into stem-loop hairpin secondary structures and possess mature miRNA within one arm of the hairpin, <6 mismatches between the predicted mature miRNA sequence and other arm of the structure, hairpin secondary structures of predicted pre-miRNAs have high negative minimal folding free energies (MFEs) and minimal folding free energy indexes (MFEIs), and the percentage of A + U in the predicted mature miRNAs is between 30 and 80 %.
Prediction of identified miRNA targets
The miRNAs bind to targets with perfect or near-perfect complementarity and regulate corresponding gene expression at the post-transcriptional level, suggesting that prediction of potential targets of the candidate miRNAs could be possible through in silico BLAST against C. sativa coding sequence (CDS) databases at http://www.camelinadb.ca (Kagale et al. 2014). 0–4 mismatches between miRNAs and Camelina CDSs without any gaps are allowed in the BLAST alignment. Selected target genes were BLASTed in TAIR (http://www.arabidopsis.org/Blast/index.jsp) to determine their corresponding Arabidopsis homologs, and Gene Ontology (GO) terms for cellular components, biological processes, and molecular functions of miRNA targets were identified.
Real-time quantitative PCR validation of miRNA and changes in target expression
Small RNAs were extracted from control and various drought-treated (10 kPa, 100 kPa and Re-hyd) leaf tissues of Camelina using a mirVana™ miRNA isolation kit (Ambion, United States) according to the manufacturer’s instructions. Isolated small RNAs were subjected to polyadenylation with ATP by poly (A) polymerase and reverse-transcribed with poly(T) adaptor primer using a Mir-X™ miRNA First-Strand synthesis kit (Clontech, USA) according to the manufacturer’s instructions. For RT–qPCR, a 20 μl reaction was carried out containing 2 μl of reverse-transcribed cDNA, 2X quantispeed SYBR® Green mix (PhileKorea), 0.5 μM miRNA-specific forward and universal reverse primers provided with the kit; all primers used in this study are listed in Supplementary Table S4. Three technical and three biological replicates were performed for each gene, and the PCR run was carried out using an Eco™ Real-Time PCR System (Illumina). The PCR reaction included one cycle at 50 and 95 °C for 5 and 3 min, respectively, followed by 39 cycles at 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 15 s. The relative miRNA expression levels and their target genes were normalized to expression levels of internal standard genes such as 5.8 s ribosomal RNA (rRNA) using the comparative threshold 2−ΔΔCT method (Livak and Schmittgen 2001). Transcriptional levels of the target genes were obtained by calculating expression fold change values (log2base) from the number of EST reads in the drought stress-treated Camelina leaf transcriptome data (Kanth et al. 2015). Hierarchical clustering and visualization of target gene as well the differential expression analysis of miRNA were carried out by Cluster 3.0 and TreeView (Eisen et al. 1998).
Results
Identification of C. sativa miRNAs
The miRNAs have been reported to be highly conserved within the plant kingdom and share similar regulatory functions (Dhandapani et al. 2011). This fact means that miRNAs can be identified from a target plant species based on a homology search using computational approaches. The strategies used to identify the miRNAs in the current study are shown in Supplementary Fig. S1. A total of 9863 non-redundant reference miRNAs sequences were searched against C. sativa ESTs using Bowtie1, of which 2309 sequences were found to contain miRNAs. After removal of protein coding sequences by BLASTX, a total of 1084 sequences were noted as candidate miRNAs. The number of candidate miRNAs decreased to 145 after the secondary structure prediction and observation of considerable stem-loop formation using Mfold web server (Supplementary Fig. S2). Of the 145 identified C. sativa miRNAs, 61 conserved (zero mismatches) and 84 putative novel (0–4 mismatches) miRNAs were shown to belong to 26 and 72 different miRNA families, respectively.
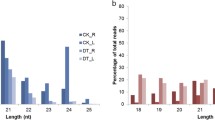
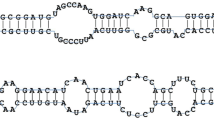
Eight miRNA families (mir156, 157, 160, 168, 171, 172, 390, and 860) were identified in both conserved and putative novel miRNAs. Therefore, in total, 92 different miRNA families including both conserved and novel miRNAs were identified. Variations in the number of miRNAs were noted. For example, in conserved miRNAs, maximum and minimum numbers of single miRNA families were 11 and 1, respectively (Fig. 1a and Supplementary Table S1). In the case of putative novel miRNAs, maximum and minimum numbers of single miRNA families were 4 and 1, respectively (Fig. 1b and Supplementary Table S2). The observed nucleotide (nt) length of mature miRNA sequences ranged between 19 and 24 nt (Fig. 2a). Most of the miRNAs were 21 nt long (54.4 %), corroborating that the obvious length of miRNAs in Camelina was similar to that of previously reported miRNAs by computational approaches (Fig. 2a). Furthermore, 50 % of the putative novel miRNAs possessed uracil at the 5′ terminus (Supplementary Table S2), which is important for interaction as well as reorganization between miRNAs and their target genes (Felice et al. 2009). Some of the predicted stem-loop hairpin structures of novel miRNA families from Camelina were shown in Fig. 3. It was found that mature miRNA regions were located either in 5′ or 3′ direction (Fig. 3). Among the 84 newly identified miRNAs, 57 and 27 miRNAs were located near the 5′ and 3′ stem loops of the precursor sequences, respectively (Supplementary Table S2).

Number of miRNAs identified in Camelina sativa. a 28 different conserved miRNA families. b 72 different putative novel miRNA families

Sequence characteristics of putative novel miRNAs and their precursor sequences. a Number of Camelina sativa miRNAs versus different lengths of mature miRNAs. b Number of matured miRNAs versus different lengths of precursor miRNAs

Stem-loop structures of putative Camelina miRNA precursors. The miRNA sequence regions are highlighted in green. The actual sizes of the precursor sequences may be slightly shorter or longer than those presented here
Sequence characteristics of novel miRNA precursors
The lengths of the excised precursor miRNAs are shown in Fig. 2b. The precursor sequences of the 84 novel miRNAs derived from EST sequences ranged from 50 to 240 nt in length with an average of 106.5 nt (Supplementary Table S2). This suggests that the predicted Camelina miRNA precursors have various sequence lengths, as in previous reports (Dhandapani et al. 2011; Yin et al. 2008). The estimated MFE values of the 84 putative novel miRNAs ranged from −8.3 to 84.6 kcal/mol with an average of 33.83 kcal/mol (Supplementary Table S2). According to previous studies, an MFEI value greater than 0.8 helps to distinguish miRNAs from other coding RNAs (Zhang et al. 2006a, b). The present study also calculated MFEI values for all of the identified novel miRNAs. MFEI values ranged from 0.33 to 1.446 kcal/mol; 31 (36.9 %) of 84 novel miRNAs showed MFEI values ≥0.8, and 39.3 % of miRNAs showed MFEI values ≥0.6.
Identification of novel, sense, and antisense miRNAs in Camelina
The present study identified 98 different miRNA families, including both conserved (26) and novel miRNA families (72). Eight miRNA families (mir156, 157, 160, 168, 171, 172, 390, and 860) were detected in both conserved and novel miRNAs. Twenty different miRNA families, including the aforementioned eight miRNA families (mir156, 157, 160, 168, 171, 172, 390, and 860), four conserved miRNA families (mir159, 167, 396, and 3435), and eight novel miRNA families (mir158, 161, 164, 169, 319, 395, 403, and 10,010) were reported already (Poudel et al. 2015). Thus, the remaining 72 miRNA families (16 conserved and 56 putative novel) are considered to be newly identified in our study. The newly predicted putative novel miRNAs were not identical with previously reported plant miRNAs and contained 1–3 nt variations when compared with other plant species-specific miRNAs. Although miRNAs are highly conserved in all plant species, some of the typical single nucleotide variations in the mature miRNAs were reported to be species-specific (Dhandapani et al. 2011). To identify the Camelina-specific nucleotide variations in mature miRNAs, Camelina miRNA families were compared with corresponding miRNA families in other plant species. One such comparison is the mir169e, which was shown to contain a cytosine at the 17th nt position of the mature miRNA in Camelina instead of the conserved guanine observed in other plant species, such as Arabidopsis, soybean, rice, and sorghum (Fig. 4). This suggests that single nucleotide mismatch variation in mir169e seems to be camelina-specific. The miRNAs were reported to be transcribed and processed from both sense and antisense transcripts derived from the same precursor (Xie et al. 2010; Zhang et al. 2008a). The present study did not detect the same miRNA family from the same precursor. However, we identified two different miRNA families (mir11736 and mir2401) from different precursor sequences separated by 64 nt on the same transcript sequence (Supplementary Fig. S3).

Camelina putative novel miRNA169e showing single nucleotide difference at the 17th nucleotide position of the mature miRNA in comparison to miRNA 169e of other plants: ath, Arabidopsis thaliana; gma, Glycine max; sbi, Sorghum bicolor; osa, Oryza sativa; mes, Manihot esculenta; tcc, Theobroma cacao; nta, Nicotiana tabacum; lus, Linum usitatissimum; mtr, Medicago truncatula
Expression characteristics of predicted novel miRNAs in camelina
The current study measured the expression of novel Camelina miRNA genes in different tissues and seed developmental stages from the Camelina transcriptome data (Poudel et al. 2015) in the NCBI database. In silico BLAST expression analysis revealed that out of 84 novel miRNAs, 24 miRNAs were expressed in different tissues of Camelina, including leaves, flower buds, and seed developmental stages (13 and 19 days after flowering) (Supplementary Table S3). Based on the number of read hits during BLAST searching, six different miRNA families (mir158b, 160a-3p, 168a, 168a-3p, 390, and 10639) accumulated at high level in all tissues (Supplementary Table S3). To find out the tissue-specific expression pattern of miRNAs, a heat map was constructed using the number of read hits as expression folds (Supplementary Table S3). Based on the heat map differential expression pattern, some of the miRNAs showed tissue-specific expression, including leaves (156b, 157c, and 171c), buds (mir390-3p, 2876 and 10010), and seeds (mir156h, 164b, 10104, 10698, and 11143), and buds as well as seeds of 19DAF (mir156h, mir10104, and mir11143). Also, it was also noted that some of the miRNAs had ubiquitous expression pattern, particularly mir168a-3P, 168a, and 390 (Supplementary Table S3). Our in silico BLAST expression analyses suggest that the predicted Camelina miRNAs were expressed in the genome.
Using computational approaches, putative novel miRNAs were identified in this study using EST sequences of cDNA libraries from drought stress-treated (Control, 10 kPa, 100 kPa and Re-hyd) Camelina leaves. To validate the predicted miRNAs as well as to confirm their drought responsiveness in Camelina, we carried out real-time quantitative PCR (RT-qPCR) for six miRNAs (Supplementary Table S4), as the remaining two (mir161-3P and mir6026) showed low primer efficiency or poor derivative melt peaks (data not shown). RT-qPCR demonstrated that all of the miRNAs were abundantly expressed under drought conditions (Fig. 5a). Relatively low miRNA expression was noted in control and re-hydrated camelina plants compared with treated plants. In particular, all miRNA genes, except mir151, showed higher expression at 100 kPa than 10 kPa, suggesting their significant roles in plants under extreme drought conditions. Among the six miRNA genes, mir319 showed significant changes in expression (about 140-fold) at 100 kPa, which was the most remarkable among all other miRNAs at 100 kPa (Fig. 5a). In contrast, mir151 showed higher expression at 10 kPa than 100 kPa, indicating that some mirRNA families were either downregulated or upregulated during prolonged stress conditions (Fig. 5a). Our results from the RT-qPCR suggest that predicted miRNAs could be drought-responsive as well as further validate the accuracy of computational identification of miRNAs from Camelina.

Expression of six randomly selected putative novel miRNAs from Camelina by real-time qPCR and expression levels of target genes from the transcriptome data. a Expression levels of miRNAs were normalized to that of the internal standard 5.8s rRNA. b Transcript expression levels of target genes in control and drought-treated Camelina leaf cDNA library. Color scale indicating up (red) or down (green) regulation of expression. Combined table of expression hot spot figure showing the Camelina CDS and EST accession numbers of target genes and their homologs in Arabidopsis and the miRNA family targeting their transcripts
Prediction of novel miRNA targets and stress-responsive miRNAs
miRNAs are reported to regulate gene expression at the post-transcriptional level by perfect or nearly perfect complementarity to coding regions of target genes involved in a wide range of biological and metabolic processes. Accordingly, previous reports have predicted target genes of miRNAs by allowing zero or 0–4 mismatches between miRNAs and target genes (Poudel et al. 2015; Dhandapani et al. 2011; Xie et al. 2010; Zhang et al. 2008a). The present study also identified targets of predicted miRNAs by a simple BLAST against Camelina CDS databases (http://www.camelinadb.ca/). The target genes were collected according to the criteria mentioned in the Materials and Methods section. Out of 72, 60 miRNA families were paired with 117 different target genes, as we could not identify the target genes for the remaining 12 miRNA families (Supplementary Table S5).
A total of 117 potential target genes were identified for 60 miRNA families involved in various biological processes. Identified target genes showed high homology with Arabidopsis homologs (Fig. 6 and Supplementary Table S5). Gene ontology (GO) analysis indicated that approximately 85, 80, and 84 % of target genes were mapped with cellular component, molecular function, and biological process, respectively. It was noted that most of the target genes are coding for proteins with functions in nucleotide binding, DNA-binding, catalytic activity, and transporter and transferase activity (Fig. 6). The profound location of these genes was observed in the nucleus and cell membranes. Furthermore, these target genes are noted as involving in various developmental, metabolic and cellular processes, signal transduction, and as regulators and transcription factors (Fig. 6 and Supplementary Table S5).

Potential targets of all putative novel miRNAs identified in C. sativa
The present study have identified number of miRNAs which were observed to be targeting genes that are involved in stress tolerance, transcription factors as well as fatty acid and lipid metabolism. The target genes of mir151, mir160, mir161, mir390, mir909, and mir11093 were observed to be stress-related genes namely RING/U-box superfamily protein, auxin response factor 17 (ARF17), NOD26-like intrinsic protein 7, Concanavalin A-like lectin protein (LPK), K+ uptake permease 7 (KUP7), and Leucine-rich repeat (LRR) family protein, respectively. The genes of SNF2 domain-containing (chromatin DNA remodeling process) and Ubiquitin-conjugating enzyme E2C-binding (ubiquitination process) were targets of mir528, while F-box family proteins, a gene plays multiple roles in plant growth and development, were targeted by mir319, 390, 1048, and 10698. mir104 and mir156 both targeting spermidine synthase 1, one of the key enzymes involved in polyamine biosynthesis (Supplementary Table S5). The miRNA families that target transcription factors (TF) of GRAS TF family, squamosa promoter-binding protein-like (SPL), and Transcription elongation factor (TFIIS) were identified as follows: mir171, 529, and 6026. Apart from this, it was also noted the presence of several fatty acid metabolism- and lipid metabolism-related genes such as alpha/beta-Hydrolases superfamily protein, phosphatidylinositol 3- and 4-kinase family protein, jasmonic acid carboxyl methyltransferase (JMT), fatty alcohol oxidase 3 (FAO3), and ABC-2 family transporter protein which were targeted by mir161, 319, 3437, 9472, and 11317, respectively (Supplementary Table S5).
miRNAs have been reported to be involved in various biotic and abiotic stress conditions, and some of them could be responsive to specific stresses (Liu et al. 2008). The present study have identified novel miRNAs from drought-treated transcriptome data (Kanth et al. 2015) which speculates that some of them might be drought- or other abiotic stress-responsive miRNAs. Among the identified novel Camelina miRNAs, the miRNA families mir156, mir158, mir168, mir169, mir171, and mir319 were already confirmed as drought-specific miRNAs according to the previous studies (Liu et al. 2008). Apart from this, we have also noted the presence of several other miRNA families (mir160, 390, 482 and 528) which also have been observed as drought-responsive miRNAs in various studies (Zhao et al. 2007; Kulcheski et al. 2011; Kantar et al. 2011). The current study has identified several drought-responsive miRNA members along with potential miRNA target genes which are also involved in the process of stress tolerance in plants. These result findings indicate that some of the identified Camelina miRNAs might be abiotic stress responsive. However, further studies are merited to elucidate the role of remaining miRNAs and their gene targets in Camelina under drought stress.
Verification of miRNA targets by reverse expression analysis
To verify correlations between miRNA expression profiles and their target genes, we performed reverse expression analysis of six corresponding target genes (Putative Protein, GRAS family transcription factor, RING/U-box superfamily protein, F-box/RNI-like/FBD-like domains-containing protein, Concanavalin A-like lectin protein kinase family protein, and Ubiquitin-conjugating enzyme E2C-binding protein) of the miRNAs analyzed in the above section (Fig. 5b). This was based on the fact that the upregulated/induced miRNAs could possibly downregulate/repress their corresponding target genes. The present study analyzed expression fold (log2base) change values of target gene transcripts from the drought stress-treated Camelina leaf transcriptome data (Kanth et al. 2015) and carried out comparison with miRNA expression levels of the reverse expression profile (Fig. 5b). Almost all miRNA-target pairs showed reverse expression patterns. All target genes were highly upregulated in untreated control leaves (Fig. 5b), whereas miRNAs showed relatively low expression (Fig. 5a). In contrast, target genes were downregulated upon drought treatment at 10 and 100 kPa (Fig. 5b), whereas miRNAs showed upregulated expression (Fig. 5a). However, upon rehydration, only two target genes, Putative protein and Ubiquitin-conjugating enzyme E2C-binding protein, showed upregulation and similar expression profiles as the untreated control (Fig. 5b), while the rest of the target gene expressions were considered to be repressed at rehydration where miRNAs could be induced at low level (Fig. 5a).
Discussion
Known and newly predicted miRNAs
Growing evidence suggests that miRNAs play a crucial role in plant growth and development, metabolism, and responses to biotic and abiotic stresses (Zhang et al. 2014; Rubio-Somoza and Weigels 2011; Chen 2004). miRNAs are evolutionarily conserved within plants and animals (Taylor et al. 2015; Quah et al. 2015). Therefore, identification of miRNAs has become feasible by a simple homology search. Several studies have already carried out identification of novel miRNAs from plants using comprehensive comparative genomics-based expressed sequence tag (EST) analysis (Panda et al. 2015; Dhandapani et al. 2011). Based on this (Supplementary Fig. S1), the present study also identified several conserved (Supplementary Table S1) and potential putative novel miRNAs from a drought-treated Camelina leaf EST library (Supplementary Table S2, and Supplementary Fig. S2). A previous study reported the presence of 207 miRNAs belonging to 63 different miRNA families in Camelina (Poudel et al. 2015). Currently, 133 miRNA families were reported to be present in Arabidopsis. In comparing the Camelina genome with Arabidopsis (Kagale et al. 2014; Liang et al. 2013), the number of miRNA families in Camelina (Poudel et al. 2015) was shown to be relatively less. Apart from these 63 different miRNA families (Poudel et al. 2015), the present study predicted 98 different miRNA families, including conserved (26) and putative novel (72) ones (Fig. 1), which increases the number of miRNAs and their families in Camelina. Among these 98 families, there are 20 different miRNA families (mir156, 157, 158, 159, 160, 161, 164, 167, 168, 169, 171, 172, 319, 390, 395, 396, 403, 860, 3435, and 10010) reported previously (Poudel et al. 2015). Thus, the remaining miRNA families are reported for the first time (Supplementary Table S1, S2).
The number of miRNA family members varied greatly. A maximum number of miRNA members were noted in mir172 and mir156. The mir172 family had a total of 12 members (11 miRNAs from conserved and 1 miRNA from novel group). While mir156 had 11 members including 7 miRNAs from conserved group, rest of them were from novel group (Fig. 1a, b). Both in conserved and novel miRNAs, most of the miRNA families consisted of only a single member. A possible explanation is that the present study used stress-induced EST sequences. Therefore, these miRNAs are stress-specific, which means that the number of miRNAs is relatively low compared with an earlier report (Poudel et al. 2015) that identified more miRNAs involved in tissue development of Camelina. In the present study, the predicted Camelina mature miRNAs (19–24 nt) and their precursor length ranges (50–200 nt) (Fig. 2a, b) as well as the predicted secondary hairpin fold structures (Fig. 3 and Supplementary Fig. S2) were in accordance with the sequence characteristics of miRNAs in previous reports (Zhang et al. 2008a). Further analysis of the mature miRNA sequences revealed that Camelina mir169e family had a specific mismatch variation of cytosine at 17th nt position whereas other plant species had guanine (Fig. 4). Similar kind of mismatch variations have been observed between mir162 families of B. rapa and B. napsus along with other plant species (Dhandapani et al. 2011). According to the previous report, mir169 was considered as moderately conserved miRNA which was found in 5–9 different plant families and might have conserved functions in leaf and flower development (Zhang et al. 2006b). This suggests that mismatch variations in mature miRNA binding region of Camelina mir169e may not have conserved functions with other plant mir169e families. The occurrence of same families of miRNA clusters has been reported previously (Dhandapani et al. 2011; Xie et al. 2010; Zhang et al. 2008a). Current study found no such miRNA clusters, but it was noted that two different miRNA families occur in the same transcript sequences: mir11736 and mir3698 were produced from different precursor sequences in the same direction and were separated by 64 nt on the same transcript sequences (Supplementary Fig. S3). It has been reported that several Manihot esculenta miRNAs namely mes-6, mes-22, and mes-29 were produced by different hairpins, and all of these hairpins were located in the same transcript sequences (Rogans and Rey 2016). Similarly, Marco et al. (2012) reported that the same transcript sequence often represents different miRNA members. This suggests that these miRNAs could not be the miRNA clusters since they were two different miRNA families derived from same transcript sequences (CaSativa1SL019889t001).
Expression patterns of Camelina novel miRNAs
The present study determined the expression levels of computationally identified miRNAs using an in silico BLAST search of novel miRNAs against a Camelina sequence read archive (SRA) database derived from different tissues such as leaves, flower buds, and seeds (Supplementary Table S3). Results indicate that some miRNAs were ubiquitously (mir168a-3P, 168a, and 390) expressed in all tissues, and some miRNAs showed tissue-specific expression pattern in leaves (mir156b, 157c, and 171c), buds (mir390-3p, 2876, and 10010), and seeds (mir156h, 164b, 10104, 10698, and 11143). These findings suggest that certain miRNAs are expressed either in a tissue-specific manner or in the whole plant, as in previous reports on other plant species as well as Camelina (Poudel et al. 2015; Dhandapani et al. 2011). In our BLAST analysis, out of 84 novel miRNAs, we found expression of 24 miRNAs. Remaining miRNAs are thought to be induced only during stress conditions, since these miRNAs were derived from EST sequences of drought stress-treated Camelina transcriptome. These findings confirm the expression of computationally predicted miRNAs in the Camelina genome.
The current study also performed RT-qPCR for experimental validation of the predicted novel miRNAs as well as to test their drought responsiveness. Eight miRNAs showing no expression during in silico BLAST analyses (mir151h, 156b, 161-3P, 171i, 319a, 390-3P, 528, and 6026) were chosen randomly (Supplementary Table S4). RT-qPCR analyses revealed that all of the miRNA genes were strongly upregulated by drought stress (10 and 100 kPa) compared with the control as well as upon rehydration (Fig. 5a). This result demonstrates that the predicted novel miRNAs in this study could be drought responsive, which is consistent with previous reports that miRNAs are upregulated by stress conditions (Zhang et al. 2014; Jian et al. 2010). mir528 was found to be upregulated about 12-fold in Camelina under drought stress (100 kPa), which is consistent with previous findings that mir528 is highly upregulated under drought stress (Bertolini et al. 2013). mir319a had an expression fold about 135 under drought stress (100 kPa) (Fig. 5a) and is regarded as a multi-stress responsive miRNA in Arabidopsis (Barciszewska-Pacak et al. 2015). Similarly, other miRNA families in RT-qPCR analysis such as mir156, mir171, and mir390 were upregulated under short-term water logging conditions in tolerant maize lines (Liu et al. 2012). Upregulated expression patterns of novel miRNAs in this study suggest functional roles in the stress response pathways of Camelina (Fig. 5a). The present study only characterized limited miRNA genes. Thus, comprehensive characterization of more miRNA genes in different tissues may elucidate the regulatory roles of miRNAs with respect to different tissues and stress conditions.
Target prediction of candidate miRNAs and functional analysis
The functional role of miRNAs can be elucidated by identification of their counterpart target genes, which helps to direct further study of miRNA candidates. miRNAs usually regulate their target genes by perfect or near-perfect complementarity binding (Poudel et al. 2015; Dhandapani et al. 2011; Zhang et al. 2008a). To further understand our novel miRNAs, the present study used BLAST search by aligning miRNAs with Camelina CDS databases (http://www.camelinadb.ca/). We identified a total of 117 potential target genes in C. sativa for the 60 identified novel miRNA families (Fig. 6 and Supplementary Table S5). Since Arabidopsis and Camelina are close relatives (Kagale et al. 2014), target genes align with Arabidopsis orthologs and are listed in Supplementary Table S5.
In the present study, we have identified various kinds of genes including stress responsive and transcription factors which were found as the most pronounced targets of miRNAs in the previous studies (Poudel et al. 2015; Dhandapani et al. 2011; Zhang et al. 2008a). mir161 was reported to be expressed under drought stress in Arabidopsis (Liu et al. 2008), and it targets the aquaporin gene called NOD26-like intrinsic protein 7, which facilitates the transportation of molecules through membrane, and is shown to confer tolerance to Arabidopsis under drought conditions (Yu et al. 2015). Similarly, the drought-responsive mir160 and mir390 (Zhao et al. 2007) targeted the ARF17 and Concanavalin A-like lectin protein kinase (LPK), respectively (Supplementary Table S5). ARF17 is shown to be involved in auxin-mediated cell signaling pathways, and under drought stress, it was post-transcriptionally controlled by mir160 in Arabidopsis (Ding et al. 2013). Concanavalin A-like LPK is type of a plant lectin, which is implicated in wide range of biological processes. In Arabidopsis, overexpression of a L-type lectin-like protein kinase 1 (AtLPK) is resulted in increased rate of seed germination and cotyledon growth under salt stress (Huang et al. 2013). On the other hand, mir909 reported as biotic stress-responsive miRNA in canola (Verma et al. 2014). In the present study, mir909 binds to the genes of Camelina KUP7, which is a high-affinity potassium transporter. An enhanced rate of drought tolerance was achieved in transgenic rice by the overexpression of KUP3 in rice (Song et al. 2014).
The other drought-responsive miRNAs, such as mir156 and mir319, (Liu et al. 2008) were found to be paired with Camelina CDS of spermidine Synthase 1 and F-box family proteins, respectively. Spermidine synthase 1, a polyamine biosynthesis enzyme, was already reported to associate with plant defense against various kinds of abiotic stresses (Kasukabe et al. 2004). Similarly, F-box proteins have also been noted to play essential roles in plant growth and development through hormone-mediated signaling pathways under drought stress (Zhang et al. 2008b). SNF2 domain-containing protein gene was targeted by a drought-responsive mir528 (Kantar et al. 2011). SNF2 proteins are implicated in DNA repair, recombination, and chromatin remodeling system, and they have been reported to function in drought response (Li et al. 2011; Han et al. 2012). In the present study, we have also noted several miRNAs targeting transcription factors. For example, Ubiquitin-conjugating enzyme E2C-binding protein was reported to be responsible for drought tolerance in monocots, and this enzyme was targeted by mir528 in a previous study (Bertolini et al. 2013) as similar to the present study (Supplementary Table S5). Similarly, the target genes of mir171 and mir529b were shown to be GRAS family transcription factor and squamosa promoter-binding protein (Supplementary Table S5) which was similar to the case of earlier reports (Bari et al. 2013; Zhang et al. 2015). These findings suggest that some of the identified novel miRNAs in Camelina might be evolutionarily and functionally conserved in plant species.
Since miRNAs have been reported to play a crucial role in lipid metabolism, identification of novel miRNAs in oil seed crops is of particular interest (Galli et al. 2014; Körbes et al. 2012; Chi et al. 2011). Apart from previous findings on miRNAs involved in lipid metabolism in Camelina (Poudel et al. 2015), the present study also observed several other miRNAs (mir161, 319, 3437, 9472, and 11317) involved in fatty acid and lipid metabolism in Camelina based on the target gene prediction (Supplementary Table S5). The alpha/beta-Hydrolases superfamily protein was the predicted target of mir161, which is a lipoprotein isoform of Arabidopsis monoacylglycerol lipase (MAGL). This enzyme facilitates the breakdown of triacylglycerol (TAG) into free fatty acids at the target tissues in fatty acid metabolism. Phosphatidylinositol 3- and 4-kinase family protein belongs to the target of mir319, and it has been reported to function in plant growth and development (Fujimoto et al. 2015) as well as phospholipid signaling pathways (Xue et al. 2009). Jasmonic acid carboxyl methyltransferase (JMT) is target gene for mir3437, this enzyme converts the jasmonate into methyl jasmonate in the oxylipin metabolism, which has been implicated in the drought response of plants (Xue et al. 2007). Similarly, another enzyme called fatty alcohol oxidase 3 (FAO3) is a target of mir9472. FAO3 has been found to be involved in β-oxidation of long chain fatty alcohols into fatty acids in seeds during wax-ester mobilization (Rajangam et al. 2013).
ABC-2 family transporter protein was noted to be the target of mir11317, which is also known as ATP-binding cassette, implicated in transmembrane movement of fatty acid and their related derivatives into peroxisomes for β-oxidation of free fatty acids (Theodoulou et al. 2006). In the present study, we also noted that some of the miRNA families (mir473, mir833, mir2876, mir2938, mir3698, mir5252, mir7728, mir9775, mir10169, mir10618, mir10639, and mir12315) were found to have no gene targets. Similar kind of observations have also been noted in a previous study (Dhandapani et al. 2011). In soybean, some of the conserved and non-conserved miRNAs have been reported to not have any of their target genes, although miRNAs without targets have been suggested to regulate genes by repression of their expression at translational level (Song et al. 2011). This suggests that further study is required to find out what could be the possible functional role of these miRNAs in Camelina.
miRNAs are reported to have a functional role in biotic and abiotic stress conditions in a wide range of plant species (Sunkar et al. 2012). Since we used Camelina EST resources derived from drought treatment in this study, several predicted miRNAs could be abiotic stress responsive (Supplementary Table S5). In Arabidopsis, the mir156, mir168, mir169, mir171, and mir319 were found to be more responsive to drought stress conditions (Sunkar and Zhu 2004; Liu et al. 2008), while in rice, mir160 and mir390 were reported to be drought-responsive miRNAs (Zhao et al. 2007). Furthermore, in wheat and soybean, mir482 and mir528 were found to be highly upregulated under drought stress, respectively (Kantar et al. 2011; Kulcheski et al. 2011). In addition to this, the present study also noted the high responsiveness of these mirRNAs (mir156, mir171, mir319, mir390, and mir528) in Camelina under drought stress which confirms their potential role in abiotic stress (Fig. 5a).
To validate coordinated expression changes between miRNA and their targets, the current study performed reverse expression analysis (Fig. 5a, b). Of six miRNA-target pairs, all of them showed changes in expression in this study (Fig. 5b), and all were newly verified in C. sativa. As shown in Fig. 5b, target gene transcription was upregulated under normal conditions (Control) but downregulated under drought conditions (10 and 100 kPa). On the other hand, miRNAs were upregulated under drought conditions and downregulated/suppressed under normal conditions. This result suggests that the identified miRNAs in this study could be drought responsive and participate in regulation of the mRNA expression levels of their counterpart target genes. This is consistent with previous findings on other plant species showing opposite expression between miRNAs and their targets under abiotic stress conditions (Liu et al. 2012; Li et al. 2013). The findings of the present study will provide a basis toward understanding the role of miRNAs in plant development and abiotic stress tolerance in Camelina.
Conclusions
In the present study, we identified 61 conserved and 84 putative novel miRNAs for the first time using a computational approach from drought EST data on Camelina. In silico expression analysis indicated that 20 different miRNA families were expressed in various tissues of Camelina. RT-qPCR revealed that the six novel miRNAs were strongly upregulated under drought conditions compared to normal conditions, further validating the accuracy of miRNA prediction. Sixty novel miRNA families targeted 117 potential genes involved in various biological and physiological processes in Camelina, including response to abiotic stress, transcription factors, and fatty acid and lipid metabolism. Coordinated expression changes between validated miRNAs and their counterpart targets varied under drought conditions, suggesting that the predicted miRNAs in this study could be stress responsive and contribute to gene regulatory frameworks in response to drought stress in Camelina. The present study will provide insights into the roles of miRNAs and their target genes in regulating C. sativa tolerance to drought stress.
References
Barciszewska-Pacak M, Milanowska K, Knop K, Bielewicz D, Nuc P, Plewka P, Am Pacak, Vazquez F, Karlowski W, Jarmolowski A, Szweykowska-Kulinska Z (2015) Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front Plant Sci 6:410
Bari A, Orazova S, Ivashchenko A (2013) miR156- and miR171-binding sites in the protein-coding sequences of several plant genes. BioMed Res Int 2013:1–7
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Baumberger N, Baulcombe DC (2005) Arabidopsis ARGONAUTE1 is an RNA slicer that selectively recruits microRNAs and short interfering RNAs. P Natl Acad Sci-Biol USA 102:11928–11933
Beilstein MA, Al-Shehbaz IA, Mathews S, Kellogg EA (2008) Brassicaceae phylogeny inferred from phytochrome A and ndhF sequence data: tribes and trichomes revisited. Am J Bot 95:1307–1327
Bertolini E, Verelst W, Horner DS, Gianfranceschi L, Piccolo V, Inzé D, Pè ME, Mica E (2013) Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol Plant 6:423–443
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Chen X (2005) MicroRNA biogenesis and function in plants. FEBS Lett 579:5923–5931
Chi X, Yang Q, Chen X, Wang J, Pan L, Chen M, Yang Z, He Y, Liang X, Yu S (2011) Identification and characterization of microRNAs from peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS ONE 6:e27530
Collins-Silva JE, Lu C, Cahoon EB (2011) Camelina: a designer biotech oilseed crop. Inform 22:610–613
Dhandapani V, Ramchiary N, Paul P, Kim J, Choi SH, Lee J, Hur Y, Lim YP (2011) Identification of potential microRNAs and their targets in Brassica rapa L. Mol Cells 32:1–37
Ding Y, Tao Y, Zhu C (2013) Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot 64:3077–3086
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95:14863–14868
Felice KM, Salzman DW, Shubert-Coleman J, Jensen KP, Furneaux HM (2009) The 5′ terminal uracil of let-7a is critical for the recruitment of mRNA to Argonaute2. Biochem J 422:329–341
Fujimoto M, Suda Y, Vernhettes S, Nakano A, Ueda T (2015) Phosphatidylinositol 3-kinase and 4-kinase have distinct roles in intracellular trafficking of cellulose synthase complexes in Arabidopsis thaliana. Plant Cell Physiol 56:287–298
Galli V, Guzman F, de Oliveira LF, Loss-Morais G, Körbes AP, Silva SD, Margis-Pinheiro MM, Margis R (2014) Identifying microRNAs and transcript targets in Jatropha seeds. PLoS One 9:e83727
Gehringer A, Friedt W, Luhs W, Snowdon RJ (2006) Genetic mapping of agronomic traits in false flax (Camelina sativa subsp. sativa). Genome 49:1555–1563
Han SK, Sang Y, Rodrigues A, Biol F, Wu MF, Rodriguez PL, Wagner D (2012) The SWI2/SNF2 chromatin remodeling ATPase BRAHMA represses abscisic acid responses in the absence of the stress stimulus in Arabidopsis. Plant Cell 24:4892–4906
Hixson SM, Parrish CC, Anderson DM (2014) Changes in tissue lipid and fatty acid composition of farmed rainbow trout in response to dietary camelina oil as a replacement of fish oil. Lipids 49:97–111
Huang P, Ju HW, Min JH, Zhang X, Kim SH et al (2013) Overexpression of L-type lectin-like protein kinase 1 confers pathogen resistance and regulates salinity response in Arabidopsis thaliana. Plant Sci 203–204:98–106
Jian X, Zhang L, Li G, Zhang L, Wang X, Cao X, Fang X, Chen F (2010) Identification of novel stress-regulated microRNAs from Oryza sativa L. Genomics 95:47–55
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kagale S, Koh C, Nixon J, Bollina V, Clarke WE, Tuteja R, Spillane C, Robinson SJ, Links MG, Clarke C, Higgins Erin E, Huebert T, Sharpe AG, Parkin IAP (2014) The emerging biofuel crop Camelina sativa retains a highly undifferentiated hexaploid genome structure. Nat Commun 5:3706
Kantar M, Lucas SJ, Budak H (2011) miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 233:471–484
Kanth BK, Kumari S, Choi SH, Ha HJ, Lee GJ (2015) Generation and analysis of expressed sequence tags (ESTs) of Camelina sativa to mine drought stress-responsive genes. Biochem Biophys Res Commun 467:83–93. doi:10.1016/j.bbrc.2015.09.116
Kasukabe Y, He LX, Nada K (2004) Overexpression of spermidine synthase enhances tolerance to multiple environmental stresses and up-regulates the expression of various stress-regulated genes transgenic Arabidopsis thaliana. Plant Cell Physiol 45:712–722
Kim HS, Oh JM, Luan S, Carlson JE, Ahn SJ (2013) Cold stress causes rapid but differential changes in properties of plasma membrane H+-ATPase of camelina and rapeseed. J Plant Physiol 170:828–837
Körbes AP, Machado RD, Guzman F, Almerão MP, de Oliveira LF, Loss-Morais G, Turchetto-Zolet AC, Cagliari A, dos Santos Maraschin F, Margis-Pinheiro M, Margis R (2012) Identifying conserved and novel microRNAs in developing seeds of Brassica napus using deep sequencing. PLoS ONE 7:e50663
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Kulcheski FR, de Oliveira LF, Molina LG, Almerão MP, Rodrigues FA, Marcolino J, Barbosa JF, Stolf-Moreira R, Nepomuceno AL, Marcelino-Guimarães FC, Abdelnoor RV, Nascimento LC, Carazzolle MF, Pereira GA, Margis R (2011) Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom 12:307
Kurihara Y, Watanabe Y (2004) Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA 101:12753–12758
Kwak KJ, Kang H, Han KH, Ahn SJ (2013) Molecular cloning, characterization, and stress-responsive expression of genes encoding glycine-rich RNA-binding proteins in Camelina sativa L. Plant Physiol Biochem 68:44–51
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Li XY, Wang C, Nie PP, Lu XW, Wang M, Liu W, Yao J, Liu YG, Zhang QY (2011) Characterization and expression analysis of the SNF2 family genes in response to phytohormones and abiotic stresses in rice. Biol Plant 55:625–633
Li B, Duan H, Li J, Deng XW, Yin W, Xia X (2013) Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol Biol 81:525–539
Liang C, Liu X, Yiu SM, Lim BL (2013) De novo assembly and characterization of Camelina sativa transcriptome by paired-end sequencing. BMC Genom 14:146
Liu H-H, Tian X, Li YJ, Wu CA, Zheng CC (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14:836–843
Liu Z, Kumari S, Zhang L, Zheng Y, Ware D (2012) Characterization of miRNAs in Response to Short-Term Waterlogging in Three Inbred Lines of Zea mays. PLoS One 7:e39786
Liu F, Wang W, Sun X, Liang Z, Wang F (2015) Conserved and novel heat stress-responsive microRNAs were identified by deep sequencing in Saccharina japonica (Laminariales, Phaeophyta). Plant Cell Environ 38:1357–1367
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Marco A, Macpherson JI, Ronshaugen M, Griffiths-Jones S (2012) MicroRNAs from the same precursor have different targeting properties. Silence 3:8
Moser BR (2012) Biodiesel from alternative oilseed feed stocks: camelina and field pennycress. Biofuels 3:193–209
Mudalkar S, Golla R, Ghatty S, Reddy AR (2014) De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol Biol 84:159–171
Panda D, Dehury B, Sahu J, Barooah ZM, Sen P, Modi MK (2015) Computational identification and characterization of conserved miRNAs and their target genes in garlic (Allium sativum L.) expressed sequence tags. Gene 537:333–342
Poudel S, Aryal N, Lu C (2015) Identification of MicroRNAs and transcript targets in Camelina sativa by deep sequencing and computational methods. PLoS One 10:e0121542
Quah S, Hui JHL, Holland PWH (2015) A burst of miRNA innovation in the early evolution of butterflies and moths. Mol Biol Evol 32:1161–1174
Rajangam AS, Gidda SK, Craddock C, Mullen RT, Dyer JM, Eastmond PJ (2013) Molecular characterization of the fatty alcohol oxidation pathway for wax-ester mobilization in germinated jojoba seeds. Plant Physiol 161:72–80
Rogans SJ, Rey C (2016) Unveiling the micronome of Cassava (Manihot esculenta Crantz). PLoS ONE 11:e0147251
Rubio-Somoza I, Weigel D (2011) MicroRNA networks and developmental plasticity in plants. Trends Plant Sci 16:258–264
Séguin-Swartza G, Eyncka C, Gugel RK, Strelkovb SE, Oliviera CY, Lic JL, Klein-Gebbinckd H, Borhana H, Caldwellc CD, Falka KC (2009) Diseases of Camelina sativa (false flax). Can J Plant Pathol 31:375–386
Song QX, Liu YF, Hu XY, Zhang WK, Ma B, Chen SY (2011) Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol 11:5
Song ZZ, Yang SY, Zuo J, Su YH (2014) Over-expression of ApKUP3 enhances potassium nutrition and drought tolerance in transgenic rice. Biol Plant 58:649–658
Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17:196–203
Taylor RS, Tarver JE, Hiscock SJ, Donoghue PC (2015) Evolutionary history of plant microRNAs. Trends Plant Sci 19:175–182
Theodoulou FL, Holdsworth M, Baker A (2006) Peroxisomal ABC transporters. FEBS Lett 580:1139–1155
Verma SS, Rahman MH, Deyholos MK, Basu U, Kav NN (2014) Differential expression of miRNAs in Brassica napus root following infection with Plasmodiophora brassicae. PLoS One 9:e86648
Wang F, Chen H, Li X, Wang N, Wang T, Yang J, Guan L, Yao N, Du L, Wang Y, Liu X, Chen X, Wang Z, Dong Y, Li H (2015) Mining and identification of polyunsaturated fatty acid synthesis genes active during camelina seed development using 454 pyrosequencing. BMC Plant Biol 15:147
Xie F, Frazier TP, Zhang B (2010) Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicum virgatum). Planta 232:417–434
Xue RG, Zhang B, Xie HF (2007) Overexpression of a NTR1 in transgenic soybean confers tolerance to water stress. Plant Cell Tiss Organ Cult 89:177–183
Xue HW, Chen X, Mei Y (2009) Function and regulation of phospholipid signalling in plants. Biochem J 421:145–156
Yin Z, Li C, Han X, Shen F (2008) Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum). Gene 414:60–66
Yu X, Wang H, Lu Y, De Ruiter M, Cariaso M, Prins M, van Tunen A, He Y (2012) Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J Exp Bot 63:1025–1038
Yu G, Li J, Sun X, Zhang X, Liu J, Pan H (2015) Overexpression of AcNIP5;1, a novel nodulin-like intrinsic protein from halophyte Atriplex canescens, enhances sensitivity to salinity and improves drought tolerance in Arabidopsis. Plant Mol Biol Rep 33:1864–1875
Zhang BH, Pan XP, Anderson TA (2006a) Identification of 188 conserved maize microRNAs and their targets. FEBS Lett 580:3753–3762
Zhang BH, Pan XP, Cobb GP, Anderson TA (2006b) Plant microRNA: a small regulatory molecule with big impact. Dev Biol 289:3–16
Zhang BH, Pan XP, Stellwag EJ (2008a) Identification of soybean microRNAs and their targets. Planta 229:161–182
Zhang Y, Xu W, Li Z, Deng XW, Wu W, Xue Y (2008b) F-box protein DOR functions as a novel inhibitory factor for abscisic acid-induced stomatal closure under drought stress in Arabidopsis. Plant Physiol 148:2121–2133
Zhang Z, Yu J, Li D, Zhang Z, Liu F, Zhou X, Wang T, Ling Y, Su Z (2010) PMRD: plant microRNA database. Nucleic Acids Res 38:D806–D813
Zhang N, Yang J, Wang Z, Wen Y, Wang J, He W, Liu B, Si H, Wang D (2014) Identification of novel and conserved MicroRNAs related to drought stress in potato by deep sequencing. PLoS One 9:e95489
Zhang SD, Ling LZ, Zhang QF, Xu JD, Cheng L (2015) Evolutionary comparison of two combinatorial regulators of SBP-box genes, MiR156 and MiR529, in plants. PLoS One 10:e0124621
Zhao B, Liang R, Ge L, Li W, Xiao H, Lin H, Ruan K, Jin Y (2007) Identification of drought-induced microRNAs in rice. Biochem Biophys Res Commun 354:585–590
Zubr J (1997) Oil-seed crop Camelina sativa. Ind Crops Prod 6:113–119
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
This research was supported by Bio-industry Technology Development Program (No. 312033-5), iPET (Korea Insitute of Planning and Evaluation for Technology in Agriculture, Food and Rural Affairs) and Radiation Technology R&D program through the National Research Foundation of Korea funded by the ministry of Science, ICT & Future Planing (NRF-2013M2A2A6043621).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
This manuscript has no financial or non-financial competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11816_2016_395_MOESM1_ESM.docx
Procedure of potential Camelina sativa miRNA gene search by identifying homologs of previously known plant miRNAs (DOCX 13 kb)
11816_2016_395_MOESM2_ESM.pdf
Predicted hairpin secondary structures of the 84 putative novel miRNAs identified in this study. Mature miRNA sequences are highlighted in green. The lengths of the accurate miRNA precursors may be slightly longer than that presented here (PDF 1079 kb)
11816_2016_395_MOESM3_ESM.tif
miRNA mir11736–mir3698 cluster in Camelina EST CaSativa1SL019889t001. a Schematic diagram of organization of the cluster. b EST sequence containing miRNAs encoded within the cluster. Underlined sequences indicate the pre-miRNAs; bold red- and green-colored nucleotides represent the mature miRNAs. c The predicted hairpin structures of mir11736 and mir3698 (TIFF 46083 kb)
11816_2016_395_MOESM4_ESM.xlsx
Conserved miRNAs identified by homolog search and secondary structure in the present study. mir: mature miRNA (XLSX 19 kb)
11816_2016_395_MOESM5_ESM.xlsx
Putative novel miRNAs identified by homolog search and secondary structure in the present study. ML: mature sequence length; MS: mature miRNA sequence arm side; LP: length of pre-miRNAs; NM: number of nucleotide mismatches (XLSX 30 kb)
11816_2016_395_MOESM6_ESM.xlsx
In silico blast expression analysis of novel miRNA genes in different tissues and seed developmental stages from the Camelina transcriptome data (Poudel et al. 2015). Number in the columns indicates number of miRNA hits during BLAST as well as expression in that particular tissue (leaves, buds, and seeds). Absence of number describes miRNA with no BLAST hit or absence of expression. On the right side panel, heatmap depicting the tissue-specific transcript accumulation of miRNAs in various tissues which is produced using the number of miRNA hits during BLAST. A gradient color bar scale represents the transcript accumulation level of high (red) or low (green) (XLSX 23 kb)
11816_2016_395_MOESM7_ESM.docx
List of primer sequences used for qRT-PCR analyses in the present study. miRNA-specific forward primers in combination with universal reverse primer from Mir-X™ miRNA First-Strand synthesis kit (Clontech, USA) were used (DOCX 12 kb)
11816_2016_395_MOESM8_ESM.xlsx
Potential targets of putative novel identified Camelina sativa miRNAs and similar Arabidopsis thaliana genes (XLSX 32 kb)
Rights and permissions
About this article

Cite this article
Subburaj, S., Kim, A.Y., Lee, S. et al. Identification of novel stress-induced microRNAs and their targets in Camelina sativa using computational approach. Plant Biotechnol Rep 10, 155–169 (2016). https://doi.org/10.1007/s11816-016-0395-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-016-0395-6