
Abstract
Camelina sativa L. is an emerging biofuel crop with potential applications in industry, medicine, cosmetics and human nutrition. The crop is unexploited owing to very limited availability of transcriptome and genomic data. In order to analyse the various metabolic pathways, we performed de novo assembly of the transcriptome on Illumina GAIIX platform with paired end sequencing for obtaining short reads. The sequencing output generated a FastQ file size of 2.97 GB with 10.83 million reads having a maximum read length of 101 nucleotides. The number of contigs generated was 53,854 with maximum and minimum lengths of 10,086 and 200 nucleotides respectively. These trancripts were annotated using BLAST search against the Aracyc, Swiss-Prot, TrEMBL, gene ontology and clusters of orthologous groups (KOG) databases. The genes involved in lipid metabolism were studied and the transcription factors were identified. Sequence similarity studies of Camelina with the other related organisms indicated the close relatedness of Camelina with Arabidopsis. In addition, bioinformatics analysis revealed the presence of a total of 19,379 simple sequence repeats. This is the first report on Camelina sativa L., where the transcriptome of the entire plant, including seedlings, seed, root, leaves and stem was done. Our data established an excellent resource for gene discovery and provide useful information for functional and comparative genomic studies in this promising biofuel crop.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With increased fuel concerns due to abating fossil fuels, rise in population and industrialization, a demand for the alternative renewable energy source has been increasing in the recent years. Various plants such as Jatropha, Pongamia, Brassica, Glycine, Helianthus, Moringa and Ricinus species have been exploited as sources for biofuel production (Atabani et al. 2012); Camelina is one such crop which is not only cost-effective but also a promising bioenergy and bio product feedstock (Moser 2010).
Camelina sativa L. Crantz, also known as gold of pleasure or false flax, is a potential biofuel crop belonging to the family Brassicaceae (Soriano and Narani 2012). It has been used in Europe for thousands of years as a vegetable oil and as animal feed. It is an annual low input crop, reaching a height of up to 3 feet, adaptable to different climatic conditions, with a short growing season of 85–100 days. The seeds are an important source of oil; with an oil content of 35–45 % dry seed weight. The oil is known for excellent food and medicinal properties since it is rich in omega-3, omega-6 poly unsaturated fatty acids and Vitamin E (Zubr and Matthaus 2002). Camelina, in addition to being an excellent food source, is also being studied as an emerging biodiesel crop. Camelina oil-based jet fuel has been used by the commercial airlines and private jets. Studies have shown Camelina-based jet fuel to reduce carbon emissions from jets by about 80 percent. Despite its importance as food, feed and biofuel, the plant is underexploited at molecular level due to the lack of information on its genomic and transcriptomic data.
The high-throughput next generation sequencing acts as a promising and powerful tool to acquire the genomic and transcriptomic data for non-model organisms and non-sequenced genomes. Recently, deep-sequencing technologies that deliver millions of short reads from platforms such as Roche/454, AB SOLiD and Illumina were used for de novo assemblies and transcriptome analysis of the organisms where reference sequence is absent (Collins et al. 2008; Vera et al. 2008; Morozova et al. 2009; Wang et al. 2009; Grabherr et al. 2011; Xia et al. 2011; Natarajan and Parani 2011).In our study, we have performed paired end RNA sequencing (RNA-seq) through Illumina GAIIX platform with short reads from pooled samples of 3 days and 7-day-old seedlings, young and mature leaves, inflorescence (florets, immature and mature pods), stem, root and seeds (Fig. 1). To our knowledge, this is the first report of de novo sequencing and transcriptome analysis of Camelina which will serve for the discovery of different genes involved in various metabolic pathways, especially lipid metabolism and development of markers for enhancing the oil related traits in this biofuel crop.

Camelina sativa tissues chosen for transcriptome analysis: a. Seeds, b. Seedlings, c. Root, d. Leaves and Stem, e. Inflorescence
Materials and methods
Plant material
The seeds of Camelina sativa L. were obtained from Tree oils India Limited (TOIL), Zaheerabad, Andhra Pradesh, India. The seeds were germinated in petri plates containing sterile wet blotting paper and later were transferred to pots containing red soil and sand (1:1). Plants were maintained in the green house under controlled conditions at 28 ± 2 °C and ~70 % humidity. To maximize the number of genes included in the transcriptome, plant tissues at various stages of development such as 3 days and 7-day-old seedlings, young and mature leaves, inflorescence (florets, immature and mature pods), stem, root and seeds were sampled and flash frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
RNA isolation
Total RNA from all the tissues was extracted using Agilent plant RNA isolation kit (Agilent Technologies, USA), according to manufacturer’s instructions. The purity and concentration of the isolated RNA were determined by using Agilent 2100 bioanalyzer (Agilent Technologies, USA). Samples with 260/280 ranging from 1.9 to 2.1 and RNA integrity number (RIN) of more than 8 were used for the analysis. RNA samples from different tissues were pooled to prepare an equimolar concentration of total RNA and were used for cDNA library construction.
cDNA library construction and Illumina sequencing
Transcriptome library for sequencing was constructed according to the Illumina TruSeq RNA library protocol outlined in “TruSeq RNA Sample Preparation Guide” (Illumina 2009). Briefly, mRNA was purified from 1 microgram of intact total RNA using oligodT beads (TruSeq RNA Sample Preparation Kit, Illumina, USA). The purified mRNA was fragmented for 4 min at 94 °C in presence of divalent cations and reverse transcribed with Superscript II Reverse transcriptase by priming with random hexamers. Second strand cDNA was synthesized by using DNA polymerase I and RnaseH. The cDNA was cleaned up using Agencourt Ampure XP SPRI beads (Beckman Coulter, USA). Illumina Adapters were ligated to the cDNA molecules after end repair and addition of single A base. SPRI cleanup was performed after ligation. The library was amplified using 11 cycles of PCR for enrichment of adapter ligated fragments. The prepared library was quantified using Nanodrop and validated for quality by running an aliquot on high sensitivity Bioanalyzer Chip (Agilent Technologies, USA). The library was sequenced on the Illumina Genome Analyzer II platform as paired end 100 bp reads following the manufacturer’s recommendations (Genotypic technology, Bangalore, India).
Sequence assembly and data analysis
The raw data from the images acquired after sequencing was transformed by base calling into raw reads and stored in FASTQ format. FASTQ reads were subjected to quality check using SeqQC V2.1 (Genotypic Proprietary Tool). The adaptor sequences, homopolymers and low quality bases were trimmed/filtered from the raw FASTQ data using Custom Perl script (Patel and Jain 2012). The reads having length less than 70 bp (after trimming the low quality bases/adapter sequences) were removed. Filtered reads were de novo assembled using Velvet-1.2.07 (Zerbino and Birney 2008). Velvet parameters used were: k-mer length of 41, auto coverage cut-off, minimum contig length: 100, expected coverage: 3. The script velvet estimate-exp_cov.pl in Velvet package was used to calculate this value, which is the minimum read depth for confirmation of a particular base in the contig, insert length: 152, Insert length standard deviation: 46.4806980845825.
After de novo assembly transcripts were assembled using Oases-0.2.08 assembler (Schulz et al. 2012). Oases parameters used were: the insert length obtained from Velvet results; the insert length standard deviation obtained from Velvet results, minimum transcript length: 200. In order to select a single transcript from a group of isoforms generated by Oases, the transcript with the largest number of exons, encoding longest ORF with highest Oases confidence score was chosen.
Gene, Pathways annotation and analysis and identification of transcription factors
The transcripts were subjected to the BLASTX analysis at an e-value cut-off of 10−5 against the databases including Aracyc, Swiss-Prot, TrEMBL. Best hit of each transcript with the highest sequence similarity from the above databases were chosen and the annotations were retrieved. KOG was used to analyse, predict and classify transcripts with probable functions whereas Aracyc database was used to assign pathways (Tatusov et al. 2001). Blast2GO program was used to get GO annotation according to molecular function, biological process and cellular component ontologies (Ashburner et al. 2000; Conesa and Gotz 2008). Transcription factors and transcriptional regulatory families were downloaded from PlnTFDB database (Perez-Rodriguez et al. 2010). The translated contigs were searched against this database using NCBI Blast-2.2.26+ .
Sequence similarity with other related organisms
To analyse the sequence similarity of Camelina with other related organisms, the transcripts were compared against the Arabidopsis thaliana, Arabidopsis lyrata, Glycine max, Oryza sativa, Ricinus communis and Sorghum bicolor proteome databases using BLASTX analysis at an e-value cut-off of 10−5.
Nucleotide content analysis and identification of SSR markers
The percentage compositions of the nucleotides A, T, G and C were calculated for each sequence and across the entire distribution of transcripts. Simple Sequence Repeats (SSRs) were detected using MIcroSAtellite tool. SSRs were detected by considering 100 bp flanking sequences on upstream and downstream of SSRs. Parameters used for development of SSRs were mentioned in Table 1.
Results and discussion
Earlier, bacterial cloning with DNA library construction and screening of the clones followed by Sanger sequencing was one of the preferred methods for sequencing which was tedious and cumbersome. Next generation sequencing technology has become an important molecular biology tool for sequencing and assembly of the genomes which has become convenient and cost effective (DiGuistini et al. 2009; Kudapa et al. 2012; Cheung et al. 2008; Lister et al. 2009; Thudi et al. 2012).
Transcriptome analysis is essential for understanding the fundamental activities of an organism which include developmental, cellular, molecular and biological processes which mainly depend on gene expression patterns. In order to acquire an overview of the transcriptome profiles, short reads with in-depth sequencing act as important tool (Garg et al. 2011; Troncoso-Ponce et al. 2011). In our present study, Illumina GAIIx platform was used for high throughput sequencing of C. sativa transcriptome to generate more number of short reads, capable of covering the complete transcriptome (Collins et al. 2008; Mizrachi et al. 2010).
Sequence assembly and data analysis
Recently, transcriptome analysis of Camelina has been performed by Liang et al. 2013 and Nguyen et al. 2013 where leaf and developing seeds respectively were used for the analysis (Liang et al. 2013; Nguyen et al. 2013). In the present study, in order to ensure maximum number of genes to be included in the transcriptome, RNA was pooled from two stages of plant development (3 days and 7-day-old seedlings), young and mature leaves, inflorescence (florets, immature and mature pods), stem, root and seeds. For Illumina sequencing, poly A RNA was purified, fragmented and reverse transcribed into cDNA. After adapter ligation to the synthesized cDNA, library was generated and this cDNA library was sequenced on Illumina GAIIX platform and paired end reads of 100 bp were obtained. A FASTQ format file size of 2.97 GB was generated; the total number of reads obtained were 10.83 million with mean read length of 101 bp. After adapter trimming, B-trimming and low-quality end-trimming, the number of high quality reads with >70 % of bases in a read having a phred score of ≥ 20 were 8.26 million (82.6 %), which were used for de novo assembly.
Due to the non-availability of a reference genome sequence, the assembly of the high quality reads was done with Velvet (1.2.07) followed by Oases (0.2.08) which resulted in 53,854 contigs with maximum and minimum read lengths of 10,836 and 200 bp respectively, with an average read length of 1,198.6 bp, indicating an increased coverage and depth of sequencing by generating longer fragment lengths (Table 2). With increase in the length of the contigs, the number of contigs decreased as is evident from the Fig. 2.

Number of transcripts versus transcript length: Size distribution of transcripts based on number of transcripts and transcript length
Gene and Pathway annotation and analysis
The sequence similarity search was done by subjecting the transcripts to BLAST X analysis (e-value ≤ 10−5) against the Aracyc, Swiss-Prot, TrEMBL, gene ontology (GO) and clusters of orthologous groups (KOG) databases. From a total of 50,341, 19,724 (39.18 %) sequences were aligned to Aracyc database with 9394 (47 %) having an e-value of 0 and 1e-05 (supplementary data 1); 31,178 hits were having similarity with Swiss-Prot database (supplementary data 2); 29,937 (59.46 %) were matched to KOG database with 13,900 (46.43 %) having an e-value of 0 and 1e-05 (supplementary data 3); 30,005 (59.6 %) transcripts showed a significant similarity to the sequences available in GO database (supplementary data 2); 50,022 (99.36 %) transcripts matched TrEMBL database with 12,085 (24.1 %) sequences having an e-value between 0 and 1e-05 (Supplementary data 4). Most of the transcripts did not match any of the databases; this could be due to the presence of novel transcripts, smaller size of the sequence or error in sequencing which needs to be curated manually.
In TrEMBL database, 51 % of the annotations were designated as putative, uncharacterized or hypothetical proteins. Experiments need to be conducted to show that these sequences encode that particular putative or uncharacterized protein. 48 % of the transcripts showed homology with that of Arabidopsis whereas only 0.6 % (333) sequences matched with Brassica, showing the close relatedness of Camelina with Arabidopsis (Nguyen et al. 2013; Liang et al. 2013). Further the transcripts were subjected to Aracyc, which is a comprehensive Arabidopsis metabolic pathway database. 2,378 transcripts were identified to be participating in various metabolic pathways which included glycolysis (190, 7.9 %), TCA cycle (160, 6.72 %), photorespiration (94, 3.95 %), gluconeogenesis (39, 1.64 %), Calvin-Benson-Bassham cycle (25, 1 %), Co enzyme A biosynthesis (11, 0.4 %) and Glyoxalate cycle (13, 0.54 %) (Supplementary data 5). Various other metabolic pathways that were identified were for the biosynthesis of amino acids, flavonoids, alkaloids, carotenoids, isoprenoids as well as biosynthesis and degradation of lipids which altogether accounted for 77.85 % (1,846).
The polyploid nature of Camelina was confirmed by southern blotting by analysing the fatty acid biosynthesis genes where three copies of FAD2, FAE1 and LFY were found to be functional, revealing the hexaploid nature of the plant (Hutcheon et al. 2010). In our study, we could also find this polyploidy nature of Camelina where one gene from Arabidopsis could be mapped to two or more transcripts, thus increasing the complexity of the genome (Supplementary data 6). To analyse the hexaploid nature of Camelina, we observed sequences encoding enzymes involved in lipid metabolism, where one gene of Arabidopsis was showing sequence homology with three transcripts of Camelina. For example, AT1G43800 gene which encodes for the enzyme, acyl-(ACP) desaturase from Arabidopsis was represented thrice at three different loci 10217, 14804 and 6438 in Camelina transcriptome. However, further experiments need to be performed to confirm the ploidy levels of Camelina.
Gene ontology (GO)
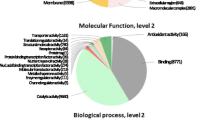
Blast2GO was used to classify the transcripts into different plant gene ontology categories such as molecular function, biological process and cellular components. The transcripts that show significant homology to the genes against swissprot database were selected for GO annotation (Fig. 3). Among the biological processes category, DNA dependent transcription and regulation were the main group, representing maximum number of transcripts (2,768, 5 and 2002, 3 % respectively), which were followed by response to salt stress (1,062, 3 %), cadmium stress (958, 2 %) and defence responses (904, 2 %) and further followed by the transcripts involved in translation (804, 1 %), proteolysis (757, 1 %), cold response (707, 1 %), transport (689, 1 %) and protein transport (687, 1 %).

GO classification: Gene ontology distribution of the transcripts into biological process, molecular function and cellular component. The number of transcripts encoded for each category is represented
Molecular function category with the decreasing order of their abundance included ATP binding (5,653, 10 %), protein binding (3,235, 5 %), DNA binding, zinc ion binding (2,277, 4 %), metal-ion binding (1,972, 4 %), protein serine/threonine kinase activity (1,939, 3 %), nucleotide binding (1,104, 2 %), RNA binding (1,070, 2 %), receptor activity (980, 2 %) and sequence-specific DNA binding transcription factor activity (971, 2 %).
Cellular responses category was represented by a large number of transcripts coding for proteins which belonged to integral to membranes (6,385, 12 %), nucleus (5,817, 11 %), plasma membrane (4,950, 10 %), cytosol (3,131, 6 %), chloroplast (2,326, 5 %), plasmodesma (2,043, 4 %), cytoplasm (1,957, 4 %), chloroplast stroma (1,615, 3 %) and mitochondria (1,380, 2 %) and extracellular region (1,059, 2 %).
The above data clearly demonstrates that the paired end Illumina sequencing has covered almost all the transcripts belonging to various categories, representing the depth and quality of Illumina sequencing.
KOG classification
KOG (Eukaryotic Orthologous Groups) is another form of COG (Clusters of Orthologous Groups) which is unique to eukaryotes (Natale et al. 2000). It aids in identifying the orthologous proteins and representing the phyletic classification of proteins coded in whole genome of almost 21 organisms including bacteria, algae and eukaryotes. The transcripts obtained in our study were compared with the KOG database and classified into 25 categories. Majority of transcripts belonged to general function prediction (17 %) which was followed by post translational modification (11 %), protein turnover (11 %) and chaperones (11 %) (Fig. 4).

KOG classification: Comparison of transcripts with the KOG database and classification into groups such as metabolism, information storage and processing, cellular processes and signalling resulting in 25 different categories
Discovery of lipid metabolism genes
As Camelina sativa is an upcoming biofuel crop, the transcripts belonging to lipid metabolism were analysed to understand the lipid formation, accumulation and degradation (Fig. 5). In our study, a total of 521 transcripts were found to be participating in lipid metabolism and 57 % (301) of the sequences were having an e-value of 0. As seen in Fig. 5, it is evident that 15 % of the transcripts are coding for the enzymes involved in the synthesis of phospholipases and linoleate biosynthesis, which were correlated with the earlier reports on Camelina fatty acids profiles and our study on GC analysis (data not presented), where the percentages of linoleic acid and linolenic acid were found to be high in seed oil (Moser 2010; Zubr and Matthaus 2002). The other abundant sequences observed were for TAG degradation (14 %), TAG biosynthesis (12 %), polyunsaturated fatty acid biosynthesis (11 %) and beta oxidation (9 %) (supplementary data 7), whereas the least number of transcripts were found for very long chain fatty acid biosynthesis and polyhydroxy fatty acid biosynthesis with 2 and 1 transcripts respectively (Table 3)(supplementary data 8). Thus, the transcriptome has covered all the genes encoding enzymes involved in fatty acid biosynthesis, initiation, termination, elongation and beta-oxidation along with TAG biosynthesis and degradation, indicating the depth of sequencing (Table 4) (Costa et al. 2010; Sato et al. 2011; Brown et al. 2012; Li et al. 2012).

Fatty acid biosynthesis pathway: Percentage distribution of transcripts involved in fatty acid biosynthesis pathway
Sequence similarity of Camelina with other plants
In our study, most of the transcripts obtained after annotation showed a significant homology with the Arabidopsis which is evident from the following data where C. sativa showed 93.3 % sequence similarity with the Arabidopsis thaliana and 93.1 % with Arabidopsis lyrata respectively, as it belongs to the family of Arabidopsis i.e., Brassicaceae. Since it is an oil yielding plant, the transcripts were compared with the oil yielding plants such as Glycine max and Ricinus communis which showed 86.8 % match for both. It shared 83 % similarities with Oryza sativa and 81.8 % with Sorghum bicolor (Fig. 6). In present study on Camelina transcriptome, only 0.6 % (333) of the transcripts showed sequence similarity with Brassica, most of them encoding for ribosomal proteins, transporters, defensins, disease resistance proteins (supplementary data 9) (Liang et al. 2013). We also found 16 transcripts that were showing sequence homology with Camelina microcarpa and C. sativa and not with Arabidopsis, which were coding for the following enzymes: fatty acid desaturases, acetolactate synthase, maturase k and phytochrome D (Supplementary data 10). Thus these sequences could be considered as Camelina specific, since no similarity/homology was found in any other plants including Arabidopsis.

Sequence similarity of Camelina sativa with other related organisms: Percentage homology of the Camelina proteins with other plant species from the protein database
Identification of transcription factors involved in lipid metabolism
Transcription factors are involved in the regulation of genes (Riano-Pachon et al. 2008), in order to understand these regulatory networks involved in various metabolic and signalling pathways of Camelina, the transcripts were subjected to Plant TFDB (Transcription factor database) and a total of 12,980 transcription factors were identified with a majority belonging to the family of C3H, MADS, FAR1, PHD, MYB-related, NAC while minimum number of transcripts for mTERF, MYB, TRAF, SBP (Fig. 7). To further comprehend the lipid metabolism pathway, transcription factors exclusive to this pathway were annotated and analysed (Fig. 8). The transcription factors involved in fatty acid biosynthesis were MYB, PLATZ, GRAS, MYB-related, bHLH, while MYB, PLATZ, MYB-related, bHLH were supposed to be regulating the pathway of fatty acid initiation. (Troncoso-Ponce et al. 2011; Venglat et al. 2011) The transcription factor MYB was known to be involved in palmitoleate biosynthesis, bHLH and GRAS in oleate biosynthesis and G2-like TF play a role in stearate biosynthesis. PHD and CCAAT are known to regulate the phosphlipases during fatty acid degradation.

Transcription factors distribution: Distribution of transcripts into transcription factor families

Transcription factors involved in fatty acid metabolism: Distribution of transcripts into transcription factor families involved in fatty acid metabolism
Nucleotide content analysis and Identification of SSRs
GC content is an important criterion for establishing the phylogenetic and evolutionary relationships among various species (Vinogradov 2003), and its percentage varies among different organisms ranging from 20 to 72 %. The percentage of GC content observed in Camelina sativa was around 49.419 % (Fig. 9).

Nucleotide content analysis: The percentage of A, T, G, C obtained for the transcripts
Molecular markers play an important role in the studies related to gene mapping and marker assisted molecular breeding to improve the plant varieties with desired traits (Kashi and King 2006). Various molecular markers used for studying these variations include RFLP, RAPD, SNPs and SSRs. SSRs are microsatellites with 2–6 nucleotides tandem repeats which are distributed randomly throughout the genome of all the eukaryotes (Kashi and King 2006; Cloutier et al. 2009; Wang et al. 2010; Dutta et al. 2011; Moe et al. 2011; Zhang et al. 2012).
Out of 53,854 sequences that were examined, a total of 19,379 SSRs were identified from Camelina. Statistical analysis of the identified SSRs was presented in Table 5. The number of mono, di, tri tetra, penta and hexa repeats were 6,166, 5,512, 7,405, 165, 63 and 68 respectively. Presence of more than one SSR was observed in the 3,793 sequences (Fig. 10).

Simple sequence Repeats: Distribution of SSR’s into di, tri, tetra, penta and hexa repeat types
This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GAFB00000000. The version described in this paper is the first version, GAFB01000000.
Conclusion
Our data on transcriptome analysis of Camelina sativa L. from different tissues using Illumina GAIIx platform has led to the identification of transcripts, transcription factors and SSR markers involved in various processes, functions, metabolic pathways along with the transcripts involved in lipid metabolism, which will not only add an insurmountable amount of information to the database for Camelina but will aid in the functional and comparative genomic studies, which could be used for the genetic improvement of this highly promising biofuel plant.
This is the first report on Camelina sativa L., where the transcriptome of the entire plant, including seedlings, seed, root, leaves and stem was done, thus increasing the probability of covering all the genes and providing an insight into the complex metabolic pathways and regulatory networks involved in different parts and at various developmental stages of the plant.
References
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29
Atabani AE, Silitonga AS, Badruddin Irfan Anjum et al (2012) A comprehensive review on biodiesel as an alternative energy resource and its characteristics. Renew Sust Energ Rev 16(4):211–245
Brown AP, Kroon JTM, Swarbreck D et al (2012) Tissue-specific whole transcriptome sequencing in castor, directed at understanding triacylglycerol lipid biosynthetic pathways. PLoS ONE 7(2):e30100
Cheung F, Win J, Lang JM, Hamilton J et al (2008) Analysis of the Pythium ultimum transcriptome using Sanger and Pyrosequencing approaches. BMC Genomics 9:542
Cloutier S, Niu Z, Datla R, Duguid S (2009) Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor Appl Genet 119(1):53–63
Collins LJ, Biggs PJ, Voelckel C, Joly S (2008) An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform 21:3–14
Conesa A, Gotz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832
Costa GG, Cardoso KC, Bem Del et al (2010) Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC Genomics 11(1):462
DiGuistini S, Liao N, Platt D et al (2009) De novo genome sequence assembly of a filamentous fungus using Sanger, 454 and Illumina sequence data. Genome Biol 10(9):R94
Dutta S, Kumawat G, Singh BP et al (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea (Cajanus cajan (L.) Millspaugh). BMC Plant Biol 11:17
Garg R, Patel RK, Tyagi AK, Jain M (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18(1):53–63
Grabherr MG, Haas BJ, Yassour M et al (2011) Full length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Hutcheon C, Ditt RF, Beilstein M et al (2010) Polyploid genome of Camelina sativa revealed by isolation of fatty acid synthesis genes. BMC Plant Biol 10:233
Illumina (2009) mRNA sequencing sample preparation guide. Illumina 24
Kashi Y, King DG (2006) Simple sequence repeats as advantageous mutators in evolution. Trends Genet 22(5):253–259
Kudapa H, Bharti AK, Cannon SB et al (2012) A comprehensive transcriptome assembly of pigeonpea (cajanus cajan l.) using sanger and second-generation sequencing platforms. Mol Plant 5(5):1020–1028
Li H, Dong Y, Yang J (2012) De novo transcriptome of safflower and the identification of putative genes for oleosin and the biosynthesis of flavonoids. PLoS ONE 7(2):e30987
Liang C, Liu X, Yiu SM, Lim BL (2013) De novo assembly and characterization of Camelina sativa transcriptome by paired end sequencing. BMC Genomics 14:146
Lister R, Gregory BD, Ecker JR (2009) Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond. Curr Opin Plant Biol 12(2):107–118
Mizrachi E, Hefer CA, Ranik M, Joubert F, Myburg AA (2010) De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics 11:681
Moe KT, Chung JW, Cho YI et al (2011) Sequence information on simple sequence repeats and single nucleotide polymorphisms through transcriptome analysis of Mungbean. J Integr Plant Biol 53(1):63–73
Morozova O, Hirst M, Marra MA (2009) Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet 10:135–151
Moser BR (2010) Camelina (Camelina sativa L.) oil as a biofuels feedstock: Golden opportunity or false hope? Lipid Technol 22(12):270–273
Natale DA, Shankavaram UT, Galperin MY, Wolf YI, Aravind L et al (2000) Towards understanding the first genome sequence of a crenarchaeon by genome annotation using clusters of orthologous groups of proteins (COGs). Genome Biol 1: research0009
Natarajan P, Parani M (2011) De novo assembly and transcriptome analysis of five major tissues of Jatropha curcas L. using GS FLX titanium platform of 454 pyrosequencing. BMC genomics 12:191
Nguyen HT, Silva JE, Podicheti R et al (2013) Camelina seed transcriptome: a tool for meal and oil improvement and translational research. Plant Biotech J 11(6):759–769
Patel RK, Jain M (2012) NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7(2):e30619
Perez-Rodriguez P, Riano-Pachon DM, Correa LG, Rensing SA, Kersten B, Mueller-Roeber B (2010) PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res 38(Database issue): D822–827
Riano-Pachon DM, Correa LGG, Trejos-Espinosa R, Mueller-Roeber B (2008) Green transcription factors: a Chlamydomonas overview. Genetics 179(1):31–39
Sato S, Hirakawa H, Isobe S et al (2011) Sequence analysis of the genome of an oil-bearing tree. Jatropha curcas L. DNA Res 18(1):65–76
Schulz MH, Zerbino DR, Vingron MA, Birney EC (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28(8):1086–1092
Soriano NU Jr, Narani A (2012) Evaluation of biodiesel derived from Camelina sativa oil. J Am Oil Chem Soc 89:917–923
Tatusov RL, Natale DA, Garkavtsev IV et al (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29(1):22–28
Thudi M, Li Y, Jackson SA, May GD, Varshney RK (2012) Current state-of-art of sequencing technologies for plant genomic research. Brief Funct Genomics 11(1):3–11
Troncoso-Ponce MA, Kilaru A, Cao X et al (2011) Comparative deep transcriptional profiling of four developing oilseeds. Plant J 68(6):1014–1027
Venglat P, Xiang D, Qiu S et al (2011) Gene expression analysis of flax seed development. BMC Plant Biol 11:74
Vera JC, Wheat CW, Fescemyer HW et al (2008) Rapid transcriptome characterization for a non-model organism using 454 pyrosequencing. Mol Ecol 17(7):1636–1647
Vinogradov AE (2003) DNA helix:the importance of being GC rich. Nucleic Acids Res 31(7):1838–1844
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1):57–63
Wang Z, Fang B, Chen J et al (2010) De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 11:726
Xia Z, Xu H, Zhai J et al (2011) RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol Biol 77(3):299–308
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18(5):821–829
Zhang J, Liang S, Duan J et al (2012) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.). BMC Genomics 13(1):90
Zubr J, Matthaus B (2002) Effects of growth conditions on fatty acids and tocopherols in Camelina sativa oil. Ind Crops Prod 15(2):155–162
Acknowledgments
The work was funded by DST grant number (DST/IS-STAC/CO2-SR-68/09) from Department of Science and Technology, Government of India. Thanks are due to Genotypic technology (P) Ltd., Bangalore, India, for library construction, sequencing and assembly. Shalini Mudalkar is thankful to UGC, New Delhi, India, for the fellowship. Ramesh Golla was supported by Dr. D.S. Kothari Postdoctoral fellowship from UGC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Shalini Mudalkar and Ramesh Golla contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Mudalkar, S., Golla, R., Ghatty, S. et al. De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol Biol 84, 159–171 (2014). https://doi.org/10.1007/s11103-013-0125-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0125-1