
Abstract
Populus euphratica, a typical hydro-halophyte, is ideal for studying salt stress responses in woody plants. MicroRNAs (miRNAs) are endogenous non-coding small RNAs that fulfilled an important post-transcriptional regulatory function. MiRNA may regulate tolerance to salt stress but this has not been widely studied in P. euphratica. In this investigation, the small RNAome, degradome and transcriptome were studied in salt stress treated P. euphratica by deep sequencing. Two hundred and eleven conserved miRNAs between Populus trichocarpa and P. euphratica have been found. In addition, 162 new miRNAs, belonging to 93 families, were identified in P. euphratica. Degradome sequencing experimentally verified 112 targets that belonged to 51 identified miRNAs, few of which were known previously in P. euphratica. Transcriptome profiling showed that expression of 15 miRNA-target pairs displayed reverse changing pattern under salt stress. Together, these results indicate that, in P. euphratica under salt stress, a large number of new miRNAs could be discovered, and both known and new miRNA were functionally cleaving to their target mRNA. Expression of miRNA and target were correspondingly induced by salt stress but that it was a complex process in P. euphratica.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Populus euphratica is a saline, desert living poplar species. It is an ideal model for salt tolerance research into woody plants (Chen and Polle 2010; Ottow et al. 2005a). A number of micrological discoveries were observed in this hydro-halophyte, such as leaf morphological plasticity, decreased vessel diameters and adjustment to osmotic changes (Chen and Polle 2010). These features mean that P. euphratica has substantial tolerance to salt stress compared with other poplar species. There have been many researches into why P. euphratica is able to withstand salt stress (Brinker et al. 2010), especially with regards to ion transport. When short- and long-term salt stress is applied to this hydro-halophyte, sodium and chloride ions are efficiently excreted from the cytosol into the intercellular space or deposited in a vacuole (Junghans et al. 2006). Potassium homeostasis is also well maintained (Ottow et al. 2005a). Furthermore, proton influx, calcium signaling, ABA signaling and reactive oxygen species homeostasis have been found to be involved in the regulation of salt stress tolerance in P. euphratica (Chen et al. 2001, 2009; Yang et al. 2007; Zhang et al. 2008; Wang et al. 2008).
Previous studies have revealed many regulation models for ion exchange and homeostasis, further understanding of the mechanism behind P. euphratica salt stress tolerance has been improved by the evidence on gene regulation for this hydro-halophyte. High throughput microarray technology has been used previously to profile transcript expression changes in P. euphratica under salt stress (Brinker et al. 2010; Gu et al. 2004; Brosche et al. 2005; Ottow et al. 2005a; Ding et al. 2010). Among these researches, a recently published work have combined physiology parameters with a microarray based transcriptome analysis (Janz et al. 2010), it concluded that P. euphratica exhibit high energy requirement in cellular metabolism and the loss of transcriptional regulation in saline environments. Meanwhile, another successful strategy was based on physiologically selecting and comparing poplar to Arabidopsis orthologs (Brinker et al. 2010). Many genes have been cloned and their possible functions, with regards to salt stress, have been described. These genes include two Na+/H+ antiporter genes (PeNhaD1 and PeSOS1), one Calcineurin B-like family (PeCBLs), two Shaker-like potassium channel families (PeKC1 and PeKC2) and one PeDREB (dehydration responsive element binding) gene family (Ottow et al. 2005b; Chen et al. 2009; Zhang et al. 2008; Wu et al. 2007). Genes related to salt tolerance regulation have been more extensively described in Arabidopsis, including genes involved in Na+ efflux, Na+ compartmentalization, K+ homeostasis and osmotic and oxidative stress response (Zhu 2002). Thus there are obvious potentials in discovering salt stress responsive genes and their regulators in P. euphratica.
MicroRNAs (miRNAs) are endogenous non-coding small RNAs, typically about 21 nucleotides in length that suppress gene translation or degrade target mRNAs, therefore miRNA have negative regulatory functions at the post-transcriptional level (Bartel 2004). In plants, a few miRNAs are conserved between species while the majority is species specific (Cuperus et al. 2011). In the newest Sanger miRNA database (miRBase 18.0 http://www.mirbase.org/), only 234 and 5 miRNAs are registered as Populus trichocarpa and Populus euphratica miRNAs, respectively. However, there are 581 rice miRNAs registered in this database, even though rice and poplar have almost the same size genome (~500 mega base pairs). This suggests that a large number of new or species specific miRNAs are waiting to be discovered in P. euphratica. A global search for miRNAs and their targets under salt stress will increase understanding of the post-transcriptional regulation networks involved in P. euphratica salt stress tolerance.
This study first applied short- and long-term salt stress to P. euphratica seedlings and then built sequencing libraries from the plant leaves. Using deep sequencing technology and by referring P. trichocarpa genome, 162 new P. euphratica miRNAs were found as well as 211 known P. trichocarpa miRNAs. MiRNA targets were validated using the Parallel Analysis of RNA Ends (PARE) method, also known as degradome sequencing. Overall, 112 targets were identified by the PARE when the plant was under short- or long- term salt stress. In addition, transcriptome sequencing was also used to profile expression changes of miRNA targets. These results finally found 15 miRNA-target pairs that showed reverse expression changes under salt stress. In summary, this study identified miRNA and their targets in P. euphratica under salt stress. These results increased the understanding of gene regulation in the hydro-halophyte, P. euphratica.
Methods and materials
Plant material and salt stress treatment
One year-old seedlings of P. euphratica, obtained from the Xinjiang Uygur Autonomous Region of China, were planted in individual pots containing a loam soil and placed in a greenhouse at Beijing Forestry University. Each pot contained three individual plants. The plants were irrigated according to evaporation demand and watered with 1 L of full strength Hoagland nutrient solution every 2 weeks for 2 months prior to the start of the salt stress treatment. The temperature in the greenhouse was maintained at 20–25 °C with a 16 h photo period (4 a.m.–8 p.m.). After establishment, l L of 200 mM NaCl solution was applied to each pot and every pot was placed on a tray to stop the solution flowing away. Mature leaves from the same position on each plant were collected and frozen immediately in liquid nitrogen for RNA extraction. Leaves collected 1 day after adding the NaCl solution were called short-term salt treatment samples and those collected 8 days after the NaCl treatment were called long-term salt treatment samples. Control leaf samples were also collected from pots that had not been treated with NaCl solution.
Ethics statement
Because all filed works were conducted on Beijing Forestry University owned fields, no specific permits were required for the described field studies. All field studies in this work did not involve endangered or protected species.
RNA extraction and deep sequencing
For each group of samples (control, short- and long-term), equal numbers of leaves from eight individual plants in their group were pooled together to represent the control or salt treatments. These combined samples were used for total RNA extraction using the standard CTAB method for plants (Chang et al. 2006). RNA quality and integrity were checked by Agilent 2100 Bioanalyzer with RNA 6000 Nano kit. All three methods of deep sequencing: small RNA (sRNA), degradome and transcriptome sequencing, used the total RNA from the different pooled samples. Transcriptome and sRNA sequencing libraries were prepared using an Illumina TruSeq RNA Sample Prep Kit and an Illumina Small RNA Sample Prep Kit, respectively. Both the sRNA and transcriptome sequencing used 10 μg of total RNA from the control, short- and long-term salt treatment samples. The degradome sequencing library was prepared using the previously published PARE protocol (German et al. 2009). Total RNA (270 μg) from each of the short- and long-term salt treatment samples was used to prepare the degradome sequencing libraries. After the libraries had been prepared, sRNA and transcriptome sequencing were undertaken using the Illumina Genome Analysis II platform. Standard pipeline degradome sequencing was performed using the sRNA sequencing pipeline as the degradome library fragments were about 20 nt in length.
Data processing for P. euphratica small RNA sequencing
Small RNA sequencing analysis
Raw reads from the small RNA sequencing process were first qualified by clipping adapters and trimming low quality reads using the fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). The left reads were then aligned against the P. trichocarpa genome (JGI Phytozome V 7.0: Populus trichocarpa) by bowtie software (http://bowtie-bio.sourceforge.net/index.shtml). Sequences without mismatches to genome were retained for further analysis. These sequences were then further aligned with other poplar genome databases. These databases included: Genebank (http://www.ncbi.nih.gov/Genbank/), Rfam (http://rfam.sanger.ac.uk/), miRBase (http://www.mirbase.org/) and P. trichocarpa sequencing annotation from Popgenie 2.0 (http://www.popgenie.org/). Following this process, the acquired small RNA sequences were classified into tRNA, snoRNA, snRNA, rRNA, miRNA, intron, exon, 3′UTR, 5′UTR, intergenic and unknown sequences.
SiRNA prediction
Small interfering RNA (siRNA) is a 20–25 nt long double-stranded RNA, each strand of which is 2 nt longer than the other at the 3′ end. According to this structural feature, tags from qualified reads were aligned with each other in order to find potential siRNA candidates that met this criterion. Bowtie was used as the alignment software combined with Perl scripts.
Identification of new miRNAs in P. euphratica
To identify new miRNA, all small RNA reads with non-annotation information in the genome, were first subjected to a secondary structure study. Genomic sequences, 100 nt in length and flanking each side of the sRNA sequences, were extracted and the secondary structures predicted using RNAfold (http://www.tbi.univie.ac.at/%7Eivo/RNA/RNAfold.html). Further identification of new miRNA in P. euphratica was conducted by Mireap (http://sourceforge.net/projects/mireap/) with adjusted parameters to meet the demands of plant miRNA identification: (1) the length range of the miRNA sequence was 20–23 bp; (2) the maximum free energy allowed for a miRNA precursor was −18 kcal mol−1; (3) the minimum number of common base pairs between miRNA and miRNA* was 16, with no more than four bulges and (4) the maximum asymmetry of the miRNA::miRNA* duplex was four bases. Out of these predicted new miRNA candidates, those that could be discovered in at least two different sequencing libraries or whose predicted miRNA* could be sequenced in the same library, were finally identified as true new miRNAs.
Profiling miRNA expression abundance
The high-throughput sequencing abundance profile analysis was based on the sequence reads in each library. The first step was to normalize the miRNA sequence reads to tags per million (TPM). The calculation of the p value for comparing the miRNA expression between salt treated samples with control sample was based on previously established methods (Audic and Claverie 1997; Man et al. 2000). Specifically, the log2 ratio formula was:
p value formulas were:
where N 1 is the total number of reads in the control sequencing library, N 2 is the total number of reads in the salt treatments sequencing library, x is the number of reads for a miRNA in the control library and y is the number of reads for a miRNA in the salt treatment library. All calculations were performed on a BGI Bio-Cloud Computing platform (http://cloud.genomics.org.cn). MiRNAs TPM of less than one were filtered out.
MiRNA target prediction and validation by degradome sequencing
New P. euphratica miRNA targets were predicted by Cleaveland 2.0 software (Addo-Quaye et al. 2009; Allen et al. 2005). A penalty score (alignment score) criterion was introduced according to the alignment between the miRNA and its potential target. The cut-off value for both the prediction and degradome sequencing data analysis was set to below 2.5, as used previously in poplar miRNA target prediction (Lu et al. 2008). The predicted targets of conserved miRNA for P. trichocarpa and P. euphratica were already available at the PopGenIE ftp site (http://aspnas.fysbot.umu.se/v1_archive/miRNA/). Raw reads from the degradome sequencing had adapters and low quality reads removed by fastx-toolkit. Then the reads were further analyzed by Cleaveland 2.0 software. Briefly, the reads were first mapped to the P. trichocarpa transcripts database from JGI Phytozome V 7.0. At this stage, a target plot was also conducted to distinguish a true miRNA cleavage site from background noise. Cleaveland 2.0 was run with default parameters using 100 randomized sequencing shuffles. The cleaved target transcripts were categorized into five categories using the following criteria: 0, the reads abundance at the cleavage site was the only maximum on the transcript; 1, the reads abundance at the cleavage site was the maximum but not unique to the transcript; 2, the reads abundance at the cleavage site was not the maximum but higher than the median for the transcript; 3 the reads abundance at the cleavage site was equal to or less than the median for the transcript and 4, there was only one raw read at the cleavage position. Only degradome sequencing verified targets were recorded in this research; predicted targets were not recorded.
Transcriptome sequencing analysis
After removing adapters and low quality reads, transcriptome sequencing reads from the control, short- and long-term salt treatments were aligned to the P. trichocarpa genome (JGI Phytozome V 7.0: Populus trichocarpa) by TopHat (http://tophat.cbcb.umd.edu/). Default parameters were used as well as those specific to the poplar genome: the minimum length of intron was set to 30 bp and the maximum was set to 4,000 bp. The results from TopHat were then inputted into Cufflink software (http://cufflinks.cbcb.umd.edu/) so that the sequencing reads could be assembled into known transcripts, according to P. trichocarpa transcript annotation (http://www.phytozome.net/poplar). At the same time, the abundance of assembled transcripts was also calculated in fragments per kilo-base of exon model per million mapped fragments (FPKM) (Ali Mortazavi et al. 2008). Following Cufflink, Cuffmerge software was used to merge the assembled transcripts from each sample into one file using a default parameter. Finally, transcript abundance profiling was undertaken by Cuffdiff software using a Poisson fragment distribution and a fault discovery rate (FDR) lower than 0.05 as the two default parameters. The Cufflink, Cuffmerge and Cuffdiff software were from the Cufflink software package (http://cufflinks.cbcb.umd.edu).
Real-time quantitative PCR validation of miRNA and target expression changes
Real-time quantitative PCR of mature miRNA was done following the high-stringency protocol in which a poly A polymerase was used to add a poly A tail (Li et al. 2009). MiRNA target real-time quantitative PCR were following protocol as published in Qin et al. (2011). The 3700 Fast Real-Time PCR System (ABI) and the Power SYBR Green PCR Master Mix (ABI) were used to carry out real-time quantitative PCR according to the standard protocol. Primers used in this research were listed in Table S7. P. euphratica action gene and 5.8s rRNA were used for reference gene for target and miRNA expression validation, respectively.
Pearson Correlation Coefficient analysis between miRNA and miRNA target
R software (www.r-project.org) was used for the calculation of Pearson Correlation Coefficient (PCC) between miRNA and miRNA target. Using results from small RNA sequencing and transcriptome sequencing, normalized expression of miRNA and target were directly input into R and standardized calculation of PCC were carried out by ‘cor’ command.
Results
Deep sequencing of P. euphratica small RNAs
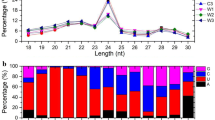
Small RNA deep sequencing of P. euphratica leaves acquired 13,864,803 sequences from the short-term salt stressed samples, 16,457,213 sequences from the long-term salt stressed samples and 14,286,843 sequences from the control samples (Table 1). Among these three data sets, approximately 65 % were 21 or 24 nt length sequences which accounted for the majority composition of P. euphratica sRNA (Figure S1). By comparing the acquired sRNA sequences to Genebank (http://www.ncbi.nih.gov/Genbank/), Rfam (http://rfam.sanger.ac.uk/), miRBase 18.0 (http://microrna.sanger.ac.uk/sequences) and the poplar genome (http://www.phytozome.net/poplar), P. euphratica small RNAs were classified into the following categories: tRNA, snoRNA, snRNA, rRNA, miRNA, intron, intergenic, exon, 5′UTR, 3′UTR and unknown sequences (Fig. 1; Table S1).

Summary of sequence classifications for all P. euphratica sRNA libraries. The sRNA sequences acquired by deep sequencing are classified into different categories by each available database. The ‘unknown’ category indicates the sRNA sequences that could not be mapped in any database. a Sequence classification of total reads. b Sequence classification of unique reads (total reads without redundancies). tRNA, transfer RNA; snoRNA, small nucleolar RNA; snRNA, small nuclear ribonucleic acid; rRNA, ribosomal RNA; miRNA, microRNA; 3′ or 5′UTR, 3′ or 5′ untranslated region
There was little variation in the different sRNA categories (SD = 2.1 %) among the three treatments. This indicated that the categories of P. euphratica sRNA remained relatively stable under salt stress. The deep sequencing generated sufficient data to cover most of the P. euphratica sRNA. Of all the sRNA categories, unknown sequences accounted for an average of 39.06 % in total acquired sequences and 90.33 % in non-redundant sequences (Fig. 1; Table S1). This suggested that a large number of the P. euphratica sRNA remained unidentified. Besides the unknown sRNA, miRNA made up 27.67 % of the total acquired sequences, which were represented in the main by 21 nt small RNAs (Fig. 1, Figure S1; Table S1).
When considering the classification of the P. euphratica sRNA, then the siRNA and miRNA proved the most interesting as their lengths (21–24 nt), were the most common lengths found in the our acquired sequences and, furthermore, they have been shown to have the most important post-transcription regulatory functions (Bartel 2004). Small interfering RNA is a kind of short (about 20–25 nt) double-stranded RNA, each strand of which is 2 nt longer than the other at the 3′ end. According to this structural feature, sRNA sequences were aligned with each other in order to identify siRNAs candidates that met this criterion. In total, the deep sequencing identified 151,217, 125,286 and 130,008 siRNA candidates in the control, short- and long-term salt stressed samples, respectively (Table S2).
Known miRNAs of P. trichocarpa discovered in P. euphratica
Among the 237 P. trichocarpa miRNAs registered in miRBase 18.0, 211 miRNAs could be found in P. euphratica sRNA sequencing. Specifically, 193, 190 and 208 known miRNAs could be found in the control, long- and short-term salt stress treated leaves, respectively (Table S2).
MiRNA* is the reverse strand of mature miRNA and degrades more easily in the nucleus (Bartel 2004). The identification of miRNA* can be further evidence of a new miRNA discovery (Meyers et al. 2008). Along with the 211 known and conserved miRNAs between P. euphratica and P. trichocarpa, 150 miRNA* were identified by alignment (Table 2, Table S2). Among them, ten miRNA*s (ptc-MIR160b*, ptc-MIR403c*, ptc-MIR408*, ptc-MIR475a*, ptc-MIR475b*, ptc-MIR1446b*, ptc-MIR1446e*, ptc-MIR478q*, ptc-MIR481e* and ptc-miR827*) had more sequencing reads than their mature miRNA in either one or two libraries. Based on previous studies, miRNA is more stable than miRNA* for the reason of joining into the RISC complex (Bartel 2004). The dominant sequence reads changed between miRNA and miRNA*, which implied that the choice of which strand of the miRNA::miRNA* complex entered the RISC complex may vary. Moreover, 20 miRNA*s had consistently higher reads than their mature sequences in all three libraries (Table 2, Table S2). These miRNA*s suggested different miRNA expression patterns between P. euphratica and P. trichocarpa. In most situations, more than one P. euphratica sRNA could completely match to a miRNA stem-loop sequence in miRBase 18.0. Among these small RNAs, the one with the most sequencing reads was named as the dominant sequence for that miRNA. Typically, the dominant sequence is supposed to be the mature sequence registered in miRBase 18.0. However, our study showed that 105 miRNAs in at least one library and 72 miRNAs overall three libraries had dominant sequences that were different from their registered mature sequences (Figure S2). The variation between the dominant sequences and the miRNA* sequences discovered in this study illustrated the complexity of miRNA generation in P. euphratica under salt stress.
Discovering new miRNAs in P. euphratica
Using an adjusted algorithm for miRNA identification in plants and Mireap software, 365 P. euphratica sRNA sequences met the algorithms and were identified as new miRNA candidates (Table S3). Some had more than one genomic location. According to previous research (Meyers et al. 2008), these were grouped and named by their genomic location and sequence homology. Finally, 473 miRNAs candidates, belonging to 300 families, were classified.
Research into plant miRNA discovery has been significantly improved by deep sequencing but stricter requirements are needed for identifying a miRNA candidate as a reliable new miRNA. There are two additional criteria regularly employed for identification of plant miRNA by deep sequencing (Meyers et al. 2008). These are: discovering miRNA and miRNA* in the same sequencing library or discovering miRNA candidates among multiple and independent libraries. Of the 473 P. euphratica miRNA candidates, 162 of them, belonging to 93 families, met at least one of these criteria, so they were identified as reliable new P. euphratica miRNAs. Furthermore, 49 miRNA*s, from these reliable new P. euphratica miRNAs, were also found. Among them, peu-MIR23*, peu-MIR115*, peu-MIR148bb*, peu-MIR199*, peu-MIR207ab* and peu-MIR309a* could be detected in all three sRNA libraries. Apart from the reliably identified new miRNAs, the remaining 311 miRNA candidates still have the potential to be qualified as true new miRNAs in future researches, so our study further analyzed both 162 newly identified miRNAs and the 311 potential new miRNA candidates.
To ascertain whether these new miRNAs or miRNA candidates were Populus species-specific, all their 365 mature sequences were aligned to all plant miRNA stem-loop sequences in miRBase 18.0. Results of the alignments showed that only peu-miR316* could be found in the stem-loop sequence of ptc-miR169b, 9 nt on the 5′ side of the mature ptc-miR169b sequence. Then mature sequences were further compared with all available plant genome sequences by Blast software (http://blast.ncbi.nlm.nih.gov/). The plant genome databases used in this study included 223 species assemblages that were deposited in PlantDB. More information can be accessed on the website: (http://www.plantgdb.org/prj/ESTCluster/progress.php ). In total, 224 of the 365 new miRNA or candidate sequences could be found in other plant genomes as well as in all Populus species EST database (Table S3). These conserved miRNAs or miRNA candidates could be helpful in discovering new miRNAs in future research into other plant genera. The remaining 141 new miRNA and miRNA candidate sequences, which had no homologies in other plant genera, could be Populus specific.
Salt responsive miRNAs in P. euphratica
For the purpose of discovering salt stress responsive miRNAs in P. euphratica, miRNA expression changes were profiled by statistical analysis using miRNA reads acquired by deep sequencing. Comparisons were made between the two salt stress treatments (short- and long-term) and the control treatment. At least one comparison showed significant changes at the level of p < 0.05. One hundred and thirty-two miRNAs in P. euphratica exhibited expression differentiations. For the purpose of discovering low differential expression changed miRNA, the log2 fold change threshold was set to 0.5, that’s about 1.5 fold change of the normalized miRNA reads number between salt treatment and control. Accordingly, 59 miRNAs showed differential expression between all salt treatment and control. Among them, 20 and 35 miRNAs showed consistent up or down-regulation, respectively, and 4 miRNAs were down-regulated under the short-term salt stress treatment but up-regulated under the long-term salt stress treatment, (Fig. 2; Table S4). These results showed that miRNA expression changes in P. euphratica under our short- or long-term salt stress were similar. Therefore, further analyses in our study no longer focused on the difference between these two conditions. Among the 59 miRNAs that showed expression change, 14 of them were newly identified P. euphratica miRNAs and the remaining 45 miRNAs were miRNAs that were conserved between P. euphratica and P. trichocarpa. To partly validate these miRNA expression changes, 20 miRNAs were randomly selected for real-time qPCR experiment. Results showed all miRNA showed consistence expression change trends comparing with sequencing results (Fig. 3). This indicated that the miRNA expression analysis based on small RNA sequencing data is reliable.

MiRNA expression profiling under short- and long-term salt stress treatment in P. euphratica leaves. miRNA expression scatter plot showing deep sequencing between the control, short- and long-term salt stress treated P. euphratica leaves. Figure is in three dimensions. The central axis represents no difference in expression. The more a miRNA spot leaves the central axis, the greater the expression differences of that miRNA; p value < 0.01. b Venn diagrams of the expression differences between miRNAs under short- or long-term salt stress. 59 miRNAs showed a significant (p value < 0.05) difference in expression under short- and long-term salt stress, only 4 and 19 miRNAs showed significant expression changes under short- or long-term salt stress, respectively. Most of these differently expressed miRNAs exhibited consistent up (20) or down (35) regulation under short- and long-term salt stress. +, −, consistent up (+) or down (−) expression under short- and long-term salt stress; ‘UD’, up then down or ‘DU’, down then up expression under short- and long-term salt stress

Real-time quantitative PCR validate expressions of 20 randomly selected miRNA. The amount of expression was normalized to the level of 5.8s rRNA. The normalized miRNA levels in control were arbitrarily set to 1
MiRNA targets verified by degradome sequencing
To further understand the role of miRNA in salt stress regulation in P. euphratica, degradome sequencing of P. euphratica under short- and long-term salt stress was used to identify miRNA targets (Figure S3). This study identified 112 miRNA targets, which were regulated by 51 miRNA families or subfamilies (Table 2, Table S5; Figure S4). Among them, 40 miRNAs targeted 103 of the 112 identified targets, which suggested that these miRNAs played various roles in the P. euphratica salt stress response. In contrast, 41 targets were regulated by multiple miRNAs, which was similar to previous degradome sequencing results in Arabidopsis and grapes (Addo-Quaye et al. 2008; Pantaleo et al. 2010).
Function annotations of the degradome sequencing verified targets were found to be diverse and included transcription factors, signal transduction factors and other proteins involved in various biological processes (Figure S5). The largest category of the targets (26 out of 112 qualified targets) were annotated as transcription factors, thus indicating their gene regulating functions in P. euphratica under salt stress. Another 17 targets were receptor like proteins and four targets were auxin signaling factors. These targets may be involved in the ion regulating ability of P. euphratica under salt stress. Further research needs to be undertaken into these signaling related miRNA targets in order to identify more P. euphratica specific salt stress regulation factors. The remaining targets had various other functions such as acting as glyoxylate reductase and polyketide cyclase (Table S5).
For the purpose of comparing with other miRNA target prediction methods, we further predicted targets of all new miRNAs using the psRNATarget web server (Table S5), among 61 degradome sequencing validated new miRNA and their target pairs, 40 pairs could be found in psRNATarget prediction results. Although the degradome analysis pipeline and psRNATarget use the same miRNA and target alignment scoring method, degradome analysis focuses on target cleavage sequence enrichments from high-throughput sequencing, while psRNATarget applies better developed bioinformatics criticisms in miRNA target prediction, like structure matching and energy calculating. For the reason that degradome sequencing and psRNATarget based on different algorithm, this comparing indicates that most our miRNA target results are highly reliable.
Transcriptome profiling in P. euphratica under salt stress
Three transcriptome sequencing procedures were used to profile expression changes of miRNA targets under salt stress. After genome mapping and statistical expression analysis by the TopHat and Cufflink software set, transcriptome sequencing results were further compared using sRNA and degradome sequencing. Specifically, transcriptome sequencing acquired around 40 million raw reads from the control, short- and long-term salt stressed P. euphratica leaves (Table 1). More than 29 million (>78 %) raw reads could be mapped to the P. trichocarpa genome. The mapped results were then integrally processed by Cufflink software set at the default significant level of p < 0.05 and with a false discovery rate qualification. Finally, 4,673 and 4,132 transcripts showed consistent up- and down-regulation, respectively, in both the short- and long-term salt stress samples. In contrast, only 477 transcripts exhibited reverse expression changes in two salt treatments. These results were similar to the miRNA profiling results, which indicated that a majority of the transcripts have same expression changing pattern under our short- and long-term salt stress treatment.
Although a large number of transcripts showed expression changes under salt stress, only 15 miRNAs-target pairs showed reverse expression changing pattern when the results from miRNA profiling, degradome sequencing and transcriptome profiling were compared (Table 3). The function annotations showed that three targets of these 15 pairs, POPTR_0007s08420, POPTR_0001s33030 and POPTR_0002s09050, were involved in auxin signaling, they are homologs of NAC1, AFB2 and ARF17 in Arabidopsis (Fig. 4) (Park et al. 2011; Navarro et al. 2006; Mallory et al. 2005). Another three targets were homologous to TOE1, HAM3 and TZP in Arabidopsis and responded to light or circadian rhythms (Hudson et al. 2003; Li et al. 2011b; Loudet et al. 2008). Nine targets, which were homologous to HAM3, NAC1, TOE1, AT1G27340.1, SPL4, AFB2, ARF17, TZP and APS3 in Arabidopsis, had functions related to tissue morphogenesis (Park et al. 2011; Hudson et al. 2003; Loudet et al. 2008; Engstrom et al. 2011; Wu and Poethig 2006; Parry et al. 2009; Liang et al. 2010). All 15 miRNA and target pairs were validated by Real-time qPCR, 14 of them showed consistent expression changes comparing with high throughput sequencing results (Figure S6). For the possible reason that no significant expression changes were observed in short-term salt treatment or the influence of homologies, target POPTR_0017s08860 of ptc-miR393a-d failed to pass the qPCR validation.

A combined view of reverse expressions between a miRNA and its target in P. euphratica under short- or long-term salt stress. Expression of P. euphratica miRNA (left side) was validated by small RNA sequencing of leaves from the control (C), under short-term salt stress treatment (SS) and under long-term salt stress treatment (LS). The miRNA targets (right side) were validated by degradome sequencing and their expressions were checked by transcriptome sequencing. Up (red) or down (green) regulation in expression were based on normalized data (color bar at the top) generated by Cluster3.0 software. Comparing to Arabidopsis shows homologous genes of miRNA targets in Arabidopsis. The table is combined with the expression hot spot figure and contains ‘Homology’, gene ID and gene ‘Name’ abbreviation and function ‘Annotation’
For all 132 differential expressed miRNA in salt treatment, 32 miRNA-target pairs were found in combining with degradome and transcriptome sequencing (Table S6). Among them, 9 miRNA-target pairs belonged to the 59 differentially expressed miRNAs with log2 fold change larger then 0.5, the other 23 pairs were then categorized as “Not correlated miRNA-target pairs in expression”. Previously published researches have evidenced that miRNA and target could be involved in diverse expression patterns and their co-regulation can beyond reverse changes in expression (Lopez-Gomollon et al. 2012; Kawashima et al. 2009; Voinnet 2009). According to the method developed by Lopez-Gomollon et al. in 2012, the gene expression PCC of miRNA and their targets were calculated. The PCC of all the 32 miRNA and target pairs was −0.041, which showed no correlation (Fig. 5). Similar results were concluded when we separately checked the PCC of short- and long-term salt treatments, they were 0.009 and −0.088, respectively. Finally, we calculated negative and positive miRNA-target pairs from the 9 ones with miRNA showing more than 0.5 of log2 fold change, and got the PCC −0.834 and PCC 0.927, respectively. This means that they were showing highly correlations. Specially, three pairs (ptc-miR395c::POPTR_0010s09140, ptc-miR1447::POPTR_0019s13730 and ptc-miR1447::POPTR_0063s00260) showed both high PCC positive and negative expression changes in different conditions, and they were categorized as mixed correlations. The calculation of expression PCC of all other 23 miRNA-target pairs got the value of 0.235. All these results indicated that complex regulation networks were existed between miRNA and target in P. euphratica under salt stress.

Pearson Correlation Coefficient between the expression of miRNA and miRNA target. Red circles represent miRNA and target pairs, PCC were presented by blue line slope and listed in blue numbers
Discussion
Complexities of miRNA generation and limitations of lacking P. euphratica genome
Comparing with previous discoveries in animals and plants, this study also found that ten miRNA* had larger sequencing read numbers than their mature miRNAs in one or two samples (Schwarz et al. 2003; Zhu et al. 2008). Additionally, 105 previously identified miRNAs have been found that their mature miRNA have dominant sequence changes in one or more libraries. Meanwhile, our published results identified 142 new miRNAs in P. euphratica under drought stress (Li et al. 2011a), a comparing between this drought study with our salt stress research showed that only 40 new miRNAs or miRNA* are the same (Table S3). In summary, the divergences discovered in this research showed the complexity of miRNA generation in P. euphratica under salt stress.
In the meanwhile, our study was inevitable restricted by lacking the P. euphratica genome as a reference. Firstly, we failed to discover new P. euphratica specific miRNAs which were not exit P. trichocarpa genome. Although many genome sequences of P. euphratica are available in NCBI database, like EST sequences, we did not find any new miRNA by referring these sequences. Another problem was whether our newly identified P. euphratica miRNAs are true new miRNAs or just fake signals which randomly mapping to P. euphratica genome. By avoiding this problem, we applied strict criterions in new miRNA identification, we first required non-mismatch mapping of sRNA to the P. trichocarpa genome, therefore our mapped sRNA were exist in both P. trichocarpa and P. euphratica genome. We further required that new miRNAs could be discovered in at least two samples or miRNA* sequences were also found. These requirements could remove fake miRNA sequences which randomly mapped to the P. trichocarpa genome.
Degradome sequencing verified previously predicted miRNA targets
In order to fully understand miRNA function, miRNA target prediction and validation are always accompanied to miRNA identification studies (Pantaleo et al. 2010; Li et al. 2011a; Sunkar and Zhu 2004). A modified 5′RACE method is widely used as a way to qualify the cleavage on predicted miRNA targets by experimental means (Lu et al. 2005). This method is limited due to its low efficiency in detecting the target cleavage sequences generated by miRNA splicing. This is because the cleavage sequences are single-stranded RNA and very short lived. The low abundance of cleavage sequences means that there are likely to be losses at the adaptor linkage step of the modified 5′RACE process. Furthermore, mRNA cleavage sequences generated randomly or as part of other procedures may interference the identification of miRNA cleavage sites. The newly developed method of miRNA target qualification by degradome sequencing uses the advantages of deep sequencing to discover low abundance cleavage sequences. Statistical analysis of abundance among all cleavage sequences in a transcript can help to distinguish the dominant miRNA cleavage signals from other random sequences. For these reasons, degradome sequencing is popular and has been shown to be highly efficient in recent studies into miRNA target discoveries (Pantaleo et al. 2010; Li et al. 2011a). Compared with previous results from experimental miRNA target verifications, the degradome sequencing in this study also confirmed the miRNA-target pairs that had been published previously for poplar or Arabidopsis. Among them, only two (ptc-miR475 targeted POPTR_0019s03770 and ptc-miR163a-e targeted POPTR_0007s08420) had previously been experimentally qualified in P. trichocarpa by 5′RACE (Gu et al. 2004). In contrast, a comparison between this study with two similar degradome sequencing studies in Arabidopsis, found that 35 and 48, respectively, of verified targets were consistence (Table S5) (Addo-Quaye et al. 2008; Lenz et al. 2011). The verification of previously published miRNA targets indicated the reliability of degradome sequencing in qualifying miRNA targets.
Reverse expressions between miRNA and its target in plants under salt stress
A large numbers of miRNA identification studies have been published for various plant species (Pantaleo et al. 2010; Li et al. 2011a; Sunkar and Zhu 2004), Furthermore, gradually improving predictive and experimental evidences form miRNA target cleavage have also been reported following the application of degradome sequencing (Addo-Quaye et al. 2009; Pantaleo et al. 2010). Although many of these miRNAs or targets responded to certain exogenous stimulants (Navarro et al. 2006; Liang et al. 2010), coordinated expression changes between miRNA and target were important as the evidence of function. Among the 15 miRNAs and target pairs that showed reverse expression changes in this study, 14 were newly verified in P. euphratica. These results also supplemented previously published research that had lacked evidence for corresponding expression changes between miRNAs and their targets. Navarro et al. (2006) verified that miR393 could repress TIR1, AFB2 and AFB3 at the post-transcription level during infection by a pathogen (Navarro et al. 2006). Other research extended the functions of these miRNA and target pairs to auxin-tolerance related root (Parry et al. 2009). Our results implied that the co-regulation between miR393 and AFB2 could be observed under salt stress. Similarly, in another example, miR395 was shown to target APS1, APS3 and APS4, which respond to sulfate limitation, while complementary expression could only be confirmed between miR395 and APS1 or APS4 (Liang et al. 2010; Liang and Yu 2010). Our results suggested the complementary expression pattern of miR395 and APS3 under salt stress.
Other salt stress regulation genes discovered by transcriptome profile sequencing
Research into how plants respond to salt stress has been reported for various plant species including Arabidopsis, rice and maize etc. (Xiong et al. 2002; Fukuda et al. 1999; Guan et al. 2000). These studies have shown that a large number of gene regulation factors were involved in cell processes, such as ion homeostasis and osmotic stress management. Our transcriptome profile sequencing results also discovered many this kinds of regulation factors. In the Na+ efflux pathway, homologs of SOS1, SOS2 and AVP1 in Arabidopsis were shown be to up-regulated under salt stress. SOS1 is a plasma membrane Na+/H+ antiporter that catalyze Na+ efflux. Under salt stress, SOS1 is up-regulated partly under the control of SOS2, which is a serine/threonine protein kinase (Zhu 2003). Overexpression of AVP1, a vacuolar H+ pyrophosphatase in Arabidopsis, could enhance salt tolerance (Gaxiola et al. 2001). In response to salt stress related osmotic stress, homologs of PEMAT and HK1 are also up-regulated in P. euphratica. These two genes are an osmotic response necessary methyltransferase gene and an osmo-sensing histidine kinase, respectively (Urao et al. 1999). With regards to ABA response and ABA metabolism, the homolog of ABA2 was also found to be up-regulated under salt stress in P. euphratica. The homolog of salt stress responsive gene RD22 was also found to be up-regulated in our results (Xiong et al. 2002). Besides the genes mentioned above, a large number of homologs of other genes, which have been reported to be involved in Arabidopsis salt regulation and response, were also found in this study’s transcriptome profiling sequencing results. Homologs Genes with statistical significant expression changes in P. euphratica were atrbohD, atrbohF, MEKK1, MPK3, MPK6, NDPK2, HB7, EIN2, P5PII, FRI/FIERY1, HSP70, ADH, PLD, CAT1, RAB18, RCI2A, ASK1, SPK1, SAD1, ARR2, CNGC1, RCD1, ENH1 and PLP (Table S6) (Xiong et al. 2002; Zhu 2003, 2001a, 2001b; Serrano and Rodriguez-Navarro 2001; Mahajan et al. 2008). Nevertheless, only two validated targets could be found amongst them. These two genes were a MYB transcription factor, targeted by the ptc-miR159 family, and EREBP (ethylene-responsive element binding proteins), targeted by the ptc-miR172 family.
Summary
In summary, for the purpose of identifying miRNA and their target in P. euphratica under salt stress, this study combined the advantages of three deep sequencing technologies: sRNA sequencing, degradome sequencing and transcriptome sequencing. The results illustrated many miRNA and their targets were responsive to salt stress in P. euphratica. The method and analysis protocols used in this study provided an efficient strategy for discovering miRNAs and their targets.
References
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ (2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18:758–762
Addo-Quaye C, Miller W, Axtell MJ (2009) CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25:130–131
Allen E, Xie Z, Gustafson AM, Carrington JC (2005) microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207–221
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7:986–995
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Brinker M, Brosche M, Vinocur B, Abo-Ogiala A, Fayyaz P et al (2010) Linking the salt transcriptome with physiological responses of a salt-resistant Populus species as a strategy to identify genes important for stress acclimation. Plant Physiol 154:1697–1709
Brosche M, Vinocur B, Alatalo ER, Lamminmaki A, Teichmann T et al (2005) Gene expression and metabolite profiling of Populus euphratica growing in the Negev desert. Genome Biol 6:R101
Chang Y, Chen S, Yin W, Wang RG, Liu YF et al (2006) Growth, gas exchange, abscisic acid, and calmodulin response to salt stress in three poplars. J Integr Plant Biol 48(3):286–293
Chen S, Polle A (2010) Salinity tolerance of Populus. Plant Biol (Stuttg) 12:317–333
Chen SL, Li JK, Wang SS, Huttermann A, Altman A (2001) Salt, nutrient uptake and transport, and ABA of Populus euphratica; a hybrid in response to increasing soil NaCl. Trees Struct Funct 15:186–194
Chen J, Xia X, Yin W (2009) Expression profiling and functional characterization of a DREB2-type gene from Populus euphratica. Biochem Biophys Res Commun 378:483–487
Cuperus JT, Fahlgren N, Carrington JC (2011) Evolution and functional diversification of MIRNA genes. Plant Cell 23:431–442
Ding M, Hou P, Shen X, Wang M, Deng S et al (2010) Salt-induced expression of genes related to Na(+)/K(+) and ROS homeostasis in leaves of salt-resistant and salt-sensitive poplar species. Plant Mol Biol 73:251–269
Engstrom EM, Andersen CM, Gumulak-Smith J, Hu J, Orlova E et al (2011) Arabidopsis homologs of the petunia hairy meristem gene are required for maintenance of shoot and root indeterminacy. Plant Physiol 155:735–750
Fukuda A, Nakamura A, Tanaka Y (1999) Molecular cloning and expression of the Na+/H+ exchanger gene in Oryza sativa. Biochim Biophys Acta 1446:149–155
Gaxiola RA, Li J, Undurraga S, Dang LM, Allen GJ et al (2001) Drought- and salt-tolerant plants result from overexpression of the AVP1 H+-pump. Proc Natl Acad Sci USA 98:11444–11449
German MA, Luo S, Schroth G, Meyers BC, Green PJ (2009) Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc 4:356–362
Gu R, Fonseca S, Puskas LG, Hackler L Jr, Zvara A et al (2004) Transcript identification and profiling during salt stress and recovery of Populus euphratica. Tree Physiol 24:265–276
Guan LM, Zhao J, Scandalios JG (2000) Cis-elements and trans-factors that regulate expression of the maize Cat1 antioxidant gene in response to ABA and osmotic stress: H2O2 is the likely intermediary signaling molecule for the response. Plant J 22:87–95
Hudson ME, Lisch DR, Quail PH (2003) The FHY3 and FAR1 genes encode transposase-related proteins involved in regulation of gene expression by the phytochrome A-signaling pathway. Plant J 34:453–471
Janz D, Behnke K, Schnitzler JP, Kanawati B, Schmitt-Kopplin P, et al (2010) Pathway analysis of the transcriptome and metabolome of salt sensitive and tolerant poplar species reveals evolutionary adaption of stress tolerance mechanisms. BMC Plant Biol 10:150
Junghans U, Polle A, Duchting P, Weiler E, Kuhlman B et al (2006) Adaptation to high salinity in poplar involves changes in xylem anatomy and auxin physiology. Plant Cell Environ 29:1519–1531
Kawashima CG, Yoshimoto N, Maruyama-Nakashita A et al (2009) Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J 57(2):313–321
Lenz D, May P, Walther D (2011) Comparative analysis of miRNAs and their targets across four plant species. BMC Res Notes 4:483
Li B, Yin W, Xia X (2009) Identification of microRNAs and their targets from Populus euphratica. Biochem Biophys Res Commun 388(2):272–277
Li B, Qin Y, Duan H, Yin W, Xia X (2011a) Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot 62:3765–3779
Li G, Siddiqui H, Teng Y, Lin R, Wan XY et al (2011b) Coordinated transcriptional regulation underlying the circadian clock in Arabidopsis. Nat Cell Biol 13:616–622
Liang G, Yu D (2010) Reciprocal regulation among miR395, APS and SULTR2;1 in Arabidopsis thaliana. Plant Signal Behav 5:1257–1259
Liang G, Yang F, Yu D (2010) MicroRNA395 mediates regulation of sulfate accumulation and allocation in Arabidopsis thaliana. Plant J 62:1046–1057
Lopez-Gomollon S, Mohorianu I, Szittya G, Moulton V, Dalmay T (2012) Diverse correlation patterns between microRNAs and their targets during tomato fruit development indicates different modes of microRNA actions. Planta 236(6):1875–1887
Loudet O, Michael TP, Burger BT, Le Mette C, Mockler TC et al (2008) A zinc knuckle protein that negatively controls morning-specific growth in Arabidopsis thaliana. Proc Natl Acad Sci USA 105:17193–17198
Lu S, Sun YH, Shi R, Clark C, Li L et al (2005) Novel and mechanical stress-responsive MicroRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 17:2186–2203
Lu S, Sun YH, Chiang VL (2008) Stress-responsive microRNAs in Populus. Plant J 55:131–151
Mahajan S, Pandey GK, Tuteja N (2008) Calcium- and salt-stress signaling in plants: shedding light on SOS pathway. Arch Biochem Biophys 471:146–158
Mallory AC, Bartel DP, Bartel B (2005) MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 17:1360–1375
Man MZ, Wang X, Wang Y (2000) POWER_SAGE: comparing statistical tests for SAGE experiments. Bioinformatics 16:953–959
Meyers BC, Axtell MJ, Bartel B et al (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 12:3186–3190
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N et al (2006) A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 312:436–439
Ottow EA, Brinker M, Teichmann T, Fritz E, Kaiser W et al (2005a) Populus euphratica displays apoplastic sodium accumulation, osmotic adjustment by decreases in calcium and soluble carbohydrates, and develops leaf succulence under salt stress. Plant Physiol 139:1762–1772
Ottow EA, Polle A, Brosche M, Kangasjarvi J, Dibrov P et al (2005b) Molecular characterization of PeNhaD1: the first member of the NhaD Na+/H+ antiporter family of plant origin. Plant Mol Biol 58:75–88
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V et al (2010) Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J 62:960–976
Park J, Kim YS, Kim SG, Jung JH, Woo JC et al (2011) Integration of auxin and salt signals by the NAC transcription factor NTM2 during seed germination in Arabidopsis. Plant Physiol 156:537–549
Parry G, Calderon-Villalobos LI, Prigge M, Peret B, Dharmasiri S et al (2009) Complex regulation of the TIR1/AFB family of auxin receptors. Proc Natl Acad Sci USA 106:22540–22545
Qin Y, Duan Z, Xia X, Yin W (2011) Expression profiles of precursor and mature microRNAs under dehydration and high salinity shock in Populus euphratica. Plant Cell Rep 30:1893–1907
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N et al (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199–208
Serrano R, Rodriguez-Navarro A (2001) Ion homeostasis during salt stress in plants. Curr Opin Cell Biol 13:399–404
Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019
Urao T, Yakubov B, Satoh R, Yamaguchi-Shinozaki K, Seki M et al (1999) A transmembrane hybrid-type histidine kinase in Arabidopsis functions as an osmosensor. Plant Cell 11:1743–1754
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136(4):669–687
Wang R, Chen S, Zhou X, Shen X, Deng L et al (2008) Ionic homeostasis and reactive oxygen species control in leaves and xylem sap of two poplars subjected to NaCl stress. Tree Physiol 28:947–957
Wu G, Poethig RS (2006) Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 133:3539–3547
Wu Y, Ding N, Zhao X, Zhao M, Chang Z et al (2007) Molecular characterization of PeSOS1: the putative Na(+)/H(+) antiporter of Populus euphratica. Plant Mol Biol 65:1–11
Xiong L, Schumaker KS, Zhu JK (2002) Cell signaling during cold, drought, and salt stress. Plant Cell 14(Suppl):S165–S183
Yang Y, Zhang F, Zhao M, An L, Zhang L et al (2007) Properties of plasma membrane H+-ATPase in salt-treated Populus euphratica callus. Plant Cell Rep 26:229–235
Zhang HC, Yin WL, Xia XL (2008) Calcineurin B-Like family in Populus: comparative genome analysis and expression pattern under cold, drought and salt stress treatment. Plant Growth Regul 56:129–140
Zhu JK (2001a) Cell signaling under salt, water and cold stresses. Curr Opin Plant Biol 4:401–406
Zhu JK (2001b) Plant salt tolerance. Trends Plant Sci 6:66–71
Zhu JK (2002) Salt and drought stress signal transduction in plants. Annu Rev Plant Biol 53:247–273
Zhu JK (2003) Regulation of ion homeostasis under salt stress. Curr Opin Plant Biol 6:441–445
Zhu QH, Spriggs A, Matthew L, Fan L, Kennedy G et al (2008) A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res 18:1456–1465
Acknowledgments
This work was supported by the National Natural Science Foundation of China (30972339, 31070597, 30730077), the Ministry of Science and Technology of China (2009CB119101), the Scientific Research and Graduate Training Joint Programs from BMEC (Regulation of Tree WUE) and National Science Foundation (NSF) Plant Genome Program (DBI0922604). B.L. was supported by Peking-Yale Joint Center Monsanto Fellowship, a fellowship from the China Scholarship Council and Beijing Forestry University Technology Innovation Program (BLYJ200902).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Li, B., Duan, H., Li, J. et al. Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol Biol 81, 525–539 (2013). https://doi.org/10.1007/s11103-013-0010-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0010-y