
Abstract
The link between mitochondrial dysfunction, redox impairment, and inflammation leads to increased rates of brain cells loss in neurodegenerative diseases and in affective disorders. Carvacrol (CAR) is a component of essential oils found in Labiatae. CAR exerts antioxidant and anti-inflammatory effects in different cell types, as assessed in both in vitro and in vivo experimental designs. Nonetheless, it was not previously investigated whether and how CAR would prevent mitochondrial impairment in human cells exposed to a pro-oxidant challenge. Therefore, we analyzed here whether a pretreatment (for 4 h) with CAR (10–1000 µM) would promote mitochondrial protection in the human neuroblastoma cells SH-SY5Y exposed to hydrogen peroxide (H2O2). We found that CAR at 100 µM prevented the H2O2-induced decline in the activity of the complexes I and V, as well as on the levels of adenosine triphosphate (ATP). CAR also prevented the H2O2-elicited decrease in the activity of the mitochondrial enzymes aconitase, α-ketoglutarate dehydrogenase, and succinate dehydrogenase. Moreover, CAR induced an antioxidant action by decreasing the levels of lipid peroxidation, protein carbonylation, and protein nitration in the mitochondrial membranes. Interestingly, CAR prevented the pro-inflammatory action of H2O2 by downregulating the transcription factor nuclear factor-κB (NF-κB). The inhibition of the heme oxygenase-1 (HO-1) enzyme by zinc protoporphyrin IX (ZnPP IX, 10 µM) suppressed the preventive effects caused by CAR regarding mitochondrial function and inflammation. Thus, it is suggested that CAR caused cytoprotective effects by an HO-1-dependent manner in SH-SY5Y cells exposed to H2O2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases present several common aspects, such as impairment in the redox environment, neuroinflammation, and disruption in the bioenergetics state, mainly affecting mitochondrial function and dynamics [1,2,3]. Moreover, alterations in signaling pathways mediating cell survival lead to increase death rates of brain cells (both neuron and glia) [4]. Similar routes leading to neuronal loss are also observed in the brain of patients suffering from major depression, bipolar disorder, and schizophrenia, which are affective disorders [5,6,7]. Since there is not a cure for those brain diseases, prevention of neuronal and glial dysfunction in the sporadic cases is an interesting strategy to decrease the number of individuals affected by such maladies [8]. In that context, mitochondria are an interesting pharmacological target when aiming to ameliorate brain cells function in the case of neurodegeneration [9].
The mitochondria present the molecular apparatus necessary to produce more than 90% of the adenosine triphosphate (ATP) in the nucleated human cells [10]. The electron transfer chain (ETC) contains the complexes I (NADH dehydrogenase), II (succinate dehydrogenase, SDH), III (coenzyme Q:cytochrome c—oxidoreductase), and IV (cytochrome c oxidase), as well as the electron transfer components ubiquinone (the so-called coenzyme Q10) and cytochrome c (a heme protein) [11, 12]. The flux of electrons in the ETC is utilized by the complexes to generate a proton (H+) gradient across the inner mitochondrial membrane, which is measured as the mitochondrial membrane potential (MMP) experimentally [13]. The H+ gradient is utilized by the complex V (ATP synthase/ATPase) protein to produce ATP [11]. Mitochondrial damage leads to decreased ability to sustain the production of ATP in the organelles, as well as enhances the production of reactive oxygen and nitrogen species (ROS and RNS, respectively) by the mitochondria [14]. Actually, the mitochondria are the main site of ROS production in the mammalian cells [14]. The ETC generates radical anion superoxide (O2−⋅) due to electron leakage in the complexes I–III and IV [14]. Then, O2−⋅ is converted into hydrogen peroxide (H2O2) by the manganese-dependent superoxide dismutase (Mn-SOD), the mitochondrial form of SOD [15]. Catalase (CAT) or glutathione peroxidase (GPx) generates water by consuming H2O2 in the mitochondria or in the cytosol [15]. H2O2 is not a free radical, but it can react with iron or cupper ions leading to the formation of the hydroxyl radical (⋅OH), the most powerful free radical generated in human cells [15]. It would particularly important in the mitochondria because these organelles contain high concentrations of iron and cupper due to the work of ETC [15]. Therefore, mitochondria are susceptible to a pro-oxidant impairment (i.e., lipid peroxidation and the consequences of this deleterious action) generated by H2O2.
In this regard, several natural compounds have been described as potential neuroprotective agents, as evaluated in both in vitro and in vivo experimental models, by promoting mitochondrial protection. We have previously demonstrated that carnosic acid, pinocembrin, naringenin, and tanshinone I, for example, attenuated the effects of different chemical stressors on both redox and functional parameters related to mitochondria in the dopaminergic cell line SH-SY5Y [16,17,18,19,20,21]. At least in part, the benefits resulting from the pretreatment of SH-SY5Y cells with such bioactive molecules are dependent on the cytoprotective enzyme heme oxygenase-1 (HO-1) [21,22,23]. HO-1 generates free iron ions, carbon monoxide (CO), and biliverdin during the degradation of heme [24]. The enzyme biliverdin reductase (BVR) consumes biliverdin producing bilirubin, a potent antioxidant [25]. On the other hand, CO has been associated with an anti-inflammatory effect in animal cells due to the ability in inhibiting the transcription factor nuclear factor-κB (NF-κB), the master regulator in the immune response [26, 27]. Nonetheless, some studies have indicated a pro-oxidant and pro-apoptotic role for HO-1 in some cell types [28, 29]. The mechanisms underlying the pro-survival and cytotoxic actions of HO-1 are focus of intense research [29].
In this context, carvacrol (CAR; 5-isopropyl-2-methylphenol; C10H14O) has been demonstrated to be an antioxidant, anti-inflammatory, and anti-apoptotic agent experimentally [30]. CAR is found in the essential oil of some plants such as Origanum vulgare L. and Rosmarinus officinalis L. [31]. It was previously shown that CAR protected SH-SY5Y cells exposed to iron ions by a mechanism involving the inhibition of NF-κB [32]. Moreover, CAR alleviated the effects of cisplatin in the mice kidney by a mechanism involving HO-1 modulation [33]. It was shown that CAR caused neuroprotection in mice subjected to focal cerebral ischemia and reperfusion, indicating that CAR presents the ability to cross the blood–brain barrier (BBB) [34]. Nonetheless, it was not previously reported whether CAR would be able to promote mitochondrial protection in experimental models of neuronal dysfunction. Thus, we investigated here whether would exert mitochondrial protection in SH-SY5Y cells exposed to H2O2, a pro-oxidant stressor that is produced by brain cells at high rates and that induces mitochondrial dysfunction by several ways [35].
Materials and Methods
Materials
The plastic materials utilized in the maintenance of cell culture were acquired from Corning, Inc. (NY, USA) and Beckton Dickson (NJ, USA). The chemicals and other materials necessary to cell culture have been obtained from Sigma (MO, USA). Other reagents and assay kits utilized in this work were purchased from different manufacturers, as indicated whenever necessary.
Chemical Assays
Epinephrine Autoxidation
The autoxidation of epinephrine was assayed in alkaline pH (7.4) according to a protocol previously published [36]. The autoxidation of epinephrine was detected at 480 nm in a spectrophotometer.
DPPH Assay
The assay using the 2,2-diphenyl-1-picryl-hydrazyl-hydrate (DPPH) free-radical has been performed based on the protocol published by others [37]. CAR at different concentrations was incubated with DPPH and the absorbance was read at 518 nm in a spectrophotometer.
Biological Assays
Cell Culture and Treatments
We utilized the human neuroblastoma SH-SY5Y cell line as an in vitro experimental model of dopaminergic cells. The SH-SY5Y cell line was acquired from the American Type Culture Collection (Manassas, VA, USA) and was cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 HAM nutrient medium (1:1 mixture) containing fetal bovine serum (FBS, 10%), L-glutamine (2 mM), penicillin (1000 units/mL), streptomycin (1000 µg/mL), and amphotericin B (2.5 µg/mL) in a 5% CO2 humidified incubator (37 °C).
H2O2 at 300 µM was used as a pro-oxidant stressor for 3 h or 24 h according to each specific assay. CAR (dissolved in dimethyl sulfoxide, DMSO) at 10–1000 µM was administrated for 4 h prior exposing the cells to H2O2 (pretreatment experimental model). The specific inhibitor of HO-1, zinc protoporphyrin IX (ZnPP IX, 10 µM), was administrated to the cells for 1 h before the treatment with CAR. Additional information is described in the figure legends. The data are shown as the mean ± S.E.M. of three or five independent experiments each done in triplicate.
Cell Viability and Cytotoxicity Assays
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to analyze the effect of specific treatments on the viability of the SH-SY5Y cells [38]. At the end of each experiment, the cells were exposed to MTT for 1 h at 37 °C. After this period, the culture medium was removed and the wells were washed twice with PBS (pH 7.4). The insoluble formazan formed intracellularly was dissolved by the administration of 100 µL DMSO to each well and incubated for 30 min. The absorbance was read at 570 nm in a spectrophotometer.
The leakage of the cytoplasmic lactate dehydrogenase (LDH) enzyme was quantified in the culture medium as an index of membrane integrity based on the protocol indicated by the manufacturer (CytoTox 96-NonRadioactive Cytotoxicity Assay, Promega).
Isolation of Mitochondria
Mitochondria were isolated from the SH-SY5Y cells by washing the and re-suspending in a buffer with 250 mM sucrose, 10 mM KCl, 1 mM EGTA, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulphonyl floride (PMSF), 1 mM benzamidine, 1 mM pepstatin A, 10 mg/mL leupeptin, 2 mg/mL aprotonin, and 20 mM HEPES (pH 7.4). The samples were centrifuged at 1000×g for 10 min at 4 °C in order to remove cell debris and nuclei, as well as unbroken cells. The mitochondrial fraction was obtained after centrifuging the resulting supernatant at 11,000×g for 20 min at 4 °C [39].
Isolation of Submitochondrial Particles
We obtained submitochondrial particles (SMP) after isolating mitochondria from the SH-SY5Y cells. The solution presenting mitochondria was frozen and thawed (three times), leading to the rupture of mitochondrial membranes and to the release of the components of the mitochondrial matrix. This solution was washed (twice) with a buffer containing 140 mM KCl, 20 mM Tris–HCl (pH 7.4) in order to generate SMP without the enzyme Mn-SOD (which would interfere in the quantification of reactive species by the organelles, as described below). We have also utilized this protocol to determine the production of O2−⋅ and to evaluate the effects of chemical stressors (H2O2, in this work) and/or CAR on the levels of markers of redox impairment in the membranes of mitochondria, since SMP are mitochondrial membranes without any component of the matrix of these organelles [36].
Quantification of the Production of O2 −⋅ and NO⋅
We quantified the generation of O2−⋅ by using the SMP obtained from the SH-SY5Y cells in a reaction medium containing 230 mM mannitol, 70 mM sucrose, 10 mM HEPES-KOH (pH 7.4), 4.2 mM succinate, 0.5 mM KH2PO4, 0.1 µM catalase, and 1 mM epinephrine, as previously published [36]. The levels of NO⋅ were quantified in the cellular level following the protocol of the manufacturer of a commercial kit (Abcam, MA, USA).
Examination of the Mitochondria-Related Apoptotic Factors and Cell Death-Associated Parameters
The immunocontents of cytochrome c (mitochondrial and cytosolic) and of the cleaved form of PARP were examined through the utilization of ELISA assay kits, based on the instructions of the manufacturer (Abcam, MA, USA). Caspase-9 and caspase-3 enzyme activities were measured by using fluorimetric assay kits following the instructions of the manufacturer (Abcam, MA, USA) [16].
Quantification of Enzyme Activities
The enzymatic activity of aconitase, α-ketoglutarate dehydrogenase, succinate dehydrogenase, complex I, and complex V were quantified by using commercial kits according to the instructions of the manufacturer (Abcam, MA, USA).
Evaluation of the Levels of ATP
The levels of ATP were evaluated by following the protocol of a commercial kit (Abcam, MA, USA). After the deproteinization of the samples, it was centrifuged and the levels of ATP were determined in the supernatants [20].
Measurement of the Mitochondrial Membrane Potential (MMP)
The MMP was measured by the use of a commercial kit utilizing tetraethylbenzimidazolylcarbocyanide iodine (JC-1) as a lipophilic cationic dye that can cross mitochondrial membranes, accumulating in the organelles according to the changes in the membrane potential (Abcam, MA, USA).
Examination of the Levels of Malondialdehyde (MDA), Protein Carbonyl, and 8-Oxo-2′-Deoxyguanosine (8-Oxo-dG)
We quantified the levels of MDA, protein carbonyl, and 8-oxo-dG by following the instructions of the manufacturer of commercial kits (Abcam, MA, USA), as previously described [20]. MDA and protein carbonyl levels were quantified in both mitochondrial and total samples.
Determination of the Levels of 3-Nitrotyrosine
We quantified the levels of 3-nitrotyrosine in the membranes of the mitochondria through the utilization of a polyclonal antibody (Calbiochem, Germany), which was diluted 1:2000 in phosphate-buffered saline (PBS containing albumin at 5%, pH 7.4), as previously published [40].
Isolation of the Cell Nucleus
The cell nucleus was isolated by using the Nuclear Extraction Kit, following the instructions of the manufacturer of a commercial assay kit (Cayman Chemical, MI, USA) and as previously published by us [16]. The protein determination was performed through the Bradford method.
Measurement of the Levels of Interleukin-1β (IL-1β) and Tumor Necrosis Factor-α (TNF-α)
The levels of the pro-inflammatory cytokines IL-1β and TNF-α were measured based on the instructions of the manufacturer of a commercial ELISA assay kit (Abcam, MA, USA).
Quantification of the Activity of the Nuclear Factor-κB (NF-κB)
We quantified the activity of the p65 subunit of NF-κB according to the protocol of the manufacturer of a commercial assay kit (Abcam, MA, USA) [21].
Statistical Analyses
We performed the statistical analyses by using the GraphPad 5.0 software. Data are shown as the mean ± standard error of the mean (S.E.M.) of three or five independent experiments each done in triplicate; p values were considered significant when p < 0.05. The differences between the experimental groups were examined by one-way ANOVA, followed by the post hoc Tukey’s test.
Results
CAR Attenuated the Effects of H2O2 on the Viability of SH-SY5Y Cells by an HO-1-Dependent Fashion
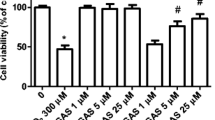
According to Fig. 1, CAR at 10 and 100 µM did not affect the viability of SH-SY5Y cells. However, CAR at 500 and 1000 µM induced a significant decrease in the cell viability in this experimental model (p < 0.05). In this regard, only CAR at 100 µM attenuated the H2O2-induced decline in the viability of SH-SY5Y cells (p < 0.05). Therefore, this CAR concentration was utilized in the other experiments that were performed in this work. In order to evaluate whether the HO-1 enzyme would be involved in the cytoprotection caused by CAR in H2O2-treated SH-SY5Y cells, ZnPP IX was administrated to the cells before the treatment with CAR. Thus, as depicted in Fig. 2A, the inhibition of HO-1 by ZnPP IX abolished the protection mediated by CAR in H2O2-treated cells (p < 0.05). Moreover, CAR attenuated the H2O2-induced cytotoxicity (as assessed through the measurement of LDH leakage from the cells) by an HO-1-associated manner (Fig. 2B, p < 0.05).

The effects of carvacrol (CAR) on the viability of SH-SY5Y cells exposed to H2O2. The cells were treated with CAR at 10–1000 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the viability (A) and leakage of lactate dehydrogenase (LDH) (B) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group
CAR also alleviated the pro-apoptotic effects induced by H2O2 in SH-SY5Y cells by a mechanism dependent on HO-1. CAR significantly blocked the release of cytochrome c to the cytosol (Fig. 3A, p < 0.05). Consequently, CAR prevented the loss of cytochrome c in the mitochondria of H2O2-treated cells (Fig. 3B, p < 0.05). The activation of the pro-apoptotic caspases-9 and -3 was decreased by CAR in the cells exposed to H2O2 (Fig. 4A, B, respectively; p < 0.05). Also, the cleavage of PARP, a target of caspase-3 during apoptotic cell death, was attenuated by CAR in H2O2-treated cells (Fig. 4C, p < 0.05). The anti-apoptotic effects induced by CAR were suppressed by the inhibition of HO-1, showing a role for this enzyme in the CAR-induced mitochondria-related blockade of cell death during the exposure to H2O2.

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the cytosolic (A) and mitochondrial (B) contents of cytochrome c (cyt c) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the activity of the pro-apoptotic enzymes caspase-3 (A) and caspase-9 (B), and on the cleavage of poly [ADP-ribose] polymerase (PARP) (C) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group
CAR Promoted Mitochondrial Protection by a HO-1-Dependent Mechanism in SH-SY5Y Cells Exposed to the Pro-oxidant Agent H2O2
CAR significantly attenuated the H2O2-induced impairment in the function of the complex I by a mechanism dependent on HO-1, since the inhibition of this enzyme by ZnPP IX blocked this effect in SH-SY5Y cells (Fig. 5A, p < 0.05). Similarly, ZnPP IX abrogated the mitochondrial protection mediated by CAR regarding the function of complex V in the mitochondria of the cells exposed to H2O2 (Fig. 5B, p < 0.05). As expected, CAR prevented the decline in the ATP content in the mitochondria of H2O2-treated cells by an HO-1-dependent manner (Fig. 5C, p < 0.05). Moreover, CAR efficiently prevented the H2O2-induced loss of MMP in SH-SY5Y cells by a mechanism involving the HO-1 enzyme (Fig. 6; p < 0.05). The mitochondria-related protection caused by CAR was also observed regarding the function of the TCA cycle in SH-SY5Y cells exposed to H2O2. As depicted in Fig. 7A, CAR significantly attenuated the H2O2-induced impairment in the activity of aconitase (p < 0.05). Similar effects were seen regarding the function of α-KGDH and SDH (Fig. 7B, C, respectively; p < 0.05). Nonetheless, the inhibition of HO-1 by ZnPP IX suppressed the TCA cycle-related protection caused by CAR in cells challenged by H2O2 (p < 0.05).

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the activity of the complexes I (A) and V (B) and on the levels of ATP (C) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the mitochondrial membrane potential (MMP) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the activity of the tricarboxylic acid cycle (TCA) enzymes aconitase (A), α-ketoglutarate dehydrogenase (B), and succinate dehydrogenase (C) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group
CAR also prevented both cellular and mitochondrial redox impairment elicited by H2O2 in SH-SY5Y cells. CAR pretreatment reduced the levels of lipid peroxidation and protein carbonylation in the SH-SY5Y cells (Fig. 8A, B, respectively; p < 0.05). Moreover, CAR efficiently attenuated the levels of oxidative stress in the DNA of SH-SY5Y cells, as assessed through the formation of 8-oxo-dG (Fig. 8C, p < 0.05). In spite of this, the inhibition of HO-1 blocked the antioxidant effects caused by CAR in the cells exposed to H2O2 (p < 0.05). Similar antioxidant effects were seen in the mitochondria obtained from the SH-SY5Y cells. CAR decreased the lipid peroxidation levels in the membranes of mitochondria isolated from the H2O2-treated cells (Fig. 9A, p < 0.05). Also, protein carbonylation in mitochondrial membranes was reduced by CAR in the H2O2-treated cells (Fig. 9B, p < 0.05). CAR was also effective in decreasing the levels of protein nitration in the membranes of mitochondria obtained from SH-SY5Y cells facing H2O2 (Fig. 9C, p < 0.05). The antioxidant effects caused by CAR in the mitochondrial membranes obtained from the H2O2-challenged cells were suppressed by ZnPP IX, indicating a role for HO-1 in mediating this protective effects elicited by CAR (p < 0.05).

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the total levels of malondialdehyde (MDA) (A), protein carbonyl (B), and 8-oxo-2′-deoxyguanosine (8-oxo-dG) (C) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the levels of malondialdehyde (MDA) (A), protein carbonyl (B), and 3-nitrotyrosine (C) in the membranes of mitochondria obtained from SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group
The production of free radicals was also assessed in this work in SH-SY5Y cells treated with CAR and/or H2O2. As demonstrated in Fig. 10A, CAR reduced the production of O2−⋅ in the SMP obtained from the H2O2-treated cells by a mechanism involving the enzyme HO-1 (p < 0.05). Similarly, HO-1 played a role in mediating the CAR-induced decrease in the cellular production of NO⋅ in the H2O2-treated SH-SY5Y cells (Fig. 10B, p < 0.05). Interestingly, CAR was not effective in inhibiting the autoxidation of epinephrine, as may be observed in Fig. 11. On the other hand, CAR presented a concentration-dependent antioxidant effect by inhibiting the oxidation of DPPH, as depicted in Fig. 12 (p < 0.05). These data indicate that CAR may fail as a direct antioxidant depending on the type of redox stressor present in a particular environment.

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the production of radical anion superoxide (O2−⋅) (A) and nitric oxide (NO⋅) (B) by SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. O2−⋅ production was determined by using submitochondrial particles (SMP) obtained from SH-SY5Y cells, as described in the “Materials and Methods”. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group

The effects of carvacrol (CAR) at different concentrations on the autoxidation of adrenaline. Adrenaline was incubated with CAR at 10–1000 µM and the autoxidation of the catecholamine was assessed as described in “Materials and Methods”. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test

The effects of carvacrol (CAR) at different concentrations on the oxidation of 2,2-diphenyl-1-picrylhydrazyl (DPPH). DPPH was incubated with CAR at 10–1000 µM and the oxidation of this free radical was assessed as described in “Materials and Methods”. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test
CAR Induced Anti-inflammatory Effects in H2O2-treated SH-SY5Y Cells by a Mechanism Associated with HO-1
Since pro-oxidant agents are able to cause redox impairment, mitochondrial dysfunction, and inflammation, we tested here whether CAR would be able to modulate the pro-inflammatory state elicited by H2O2 in SH-SY5Y cells. As depicted in Fig. 13A, CAR prevented the increase in the levels of IL-1β in the SH-SY5Y cells exposed to H2O2 (p < 0.05). Similarly, CAR attenuated the H2O2-induced increase in the levels of TNF-α in SH-SY5Y (Fig. 13B, p < 0.05). CAR pretreatment was also effective in inhibiting the activation of the transcription factor NF-κB in the cells treated with H2O2 (Fig. 13C, p < 0.05). The anti-inflammatory effects elicited by CAR were blocked by ZnPP IX (p < 0.05).

The effects of heme oxygenase-1 (HO-1) inhibition by zinc protoporphyrin IX (ZnPP IX) on the production of the pro-inflammatory cytokines interleukin-1β (IL-1β) (A) and tumor necrosis factor-α (TNF-α) (B), and on the activity of the transcription factor nuclear factor-κB (NF-κB) (C) in SH-SY5Y cells treated with carvacrol (CAR) and/or H2O2. ZnPP IX at 10 µM was administrated to the cells for 1 h before the exposure to CAR. The cells were treated with CAR at 100 µM during 4 h prior to the challenge with H2O2 at 300 µM for further 24 h. Data are shown as the mean ± SEM of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 different from the control group; ap < 0.05 different from H2O2-treated group; bp < 0.05 different from the CAR + H2O2-treated group
Discussion
In the present work, CAR pretreatment attenuated the deleterious effects induced by the pro-oxidant agent H2O2 by a mechanism involving the enzyme HO-1. The cytoprotective actions caused by HO-1 have been reported by several research groups, showing that this enzyme mediates antioxidant, anti-apoptotic, and anti-inflammatory effects in mammalian cells [41,42,43,44]. Nonetheless, it was not previously demonstrated whether HO-1 would mediate the beneficial effects induced by CAR experimentally. The role of HO-1 in promoting mitochondrial protection is still on debate, since it was found that HO-1 also works as a pro-oxidant in some cases, causing cellular injury and altering physiological parameters [28]. In this regard, the modulation of HO-1 may serve as a therapeutic target in the case of neurodegenerative diseases, as recently reviewed [29]. We have demonstrated that several natural compounds can modulate HO-1 through the activation of signal pathways related to cell survival, leading to the mitochondrial protection, which is crucial to suppress the pro-apoptotic actions of different chemical stressors [18, 20, 45].
HO-1 generates CO, biliverdin, and free iron as products of the heme degradation [46]. Biliverdin is converted into bilirubin, a potent antioxidant, by BVR [47]. CO, on the other hand, has been viewed as an inhibitor of the master regulator of inflammation, the transcription factor NF-κB [26, 48]. Therefore, it is expected that the combination of direct antioxidant actions exerted by bilirubin and the attenuation of inflammation by CO would cause mitochondrial protection by decreasing the pro-oxidant signal of pro-inflammatory cytokines on the mitochondria. During inflammation, the production of ROS by the mitochondria is enhanced [49]. Also, depending on the duration of the pro-inflammatory stimulus, the intrinsic apoptotic pathway, which presents the mitochondria as central players, is activated during inflammation, causing increased rates of cell death [50]. This is particularly important in brain cells, which are very sensitive to redox impairment and mitochondrial dysfunction [51]. Thus, the modulation of the HO-1/CO/NF-κB signaling pathway by CAR is expected to act both directly and indirectly on the maintenance of mitochondrial function and dynamics. HO-1 is an inducible enzyme whose expression is controlled by the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), among others [52]. Nrf2 is upregulated by certain natural compounds in mammalian cells, and the activation of this transcription factor leads to the expression of several cytoprotective proteins [53]. These effects have been associated with prevention of cellular dysfunction in different experimental models using chemical stressors [22, 23, 42]. Even though we have not analyzed whether the link between HO-1 and Nrf2 in the present work, it may be suggested that such regulator would be associated with the cytoprotection seen here. Further research would be useful in investigating the role of Nrf2 in the HO-1-mediated preventive effects induced by CAR in mammalian cells.
Prevention of mitochondrial dysfunction is central when considering neurodegenerative diseases, since alterations in the activity of these organelles appear in Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD), as well as in affective disorders [54, 55]. Indeed, the use of anti-depressants by patients suffering from major depression or bipolar disorder may not be sufficient to downregulate the pro-inflammatory status observed in different brain areas [56,57,58,59]. Sustained pro-inflammatory signal leads to neuronal loss, an event that would amplify the impact of major depression of the life quality of the patients [60]. On the other hand, some environmental factors may cause mitochondrial dysfunction, as is the case of agrochemicals that have been associated with the induction of PD, for example [61, 62]. By impairing mitochondrial function, these neurotoxic agents enhance the production of ROS by the organelles, leading to posterior inflammation [63]. A similar mechanism has been seen in the pathophysiology of some diseases in in vivo experimental models [64, 65]. Thus, the mitochondrial impaired function may be a cause or a consequence of inflammation. Therefore, targeting mitochondria during neuroinflammation may be an interesting pharmacological strategy to decrease the loss of brain cells in the case of neurodegeneration and/or affective disorders, among others [1, 66].
Overall, CAR promoted mitochondrial protection by a mechanism related to HO-1 in SH-SY5Y cells exposed to H2O2. The antioxidant effects CAR exerted in mitochondrial membranes are crucial to maintain both mitochondrial function (mainly the OXPHOS system) and dynamics (during events such as mitochondrial fusion and fission, as well as mitochondrial biogenesis). In spite of the results seen here, future research is necessary to investigate whether CAR would exert similar effects in in vivo experimental models, since the concentration of this natural compound that induced beneficial effects here is considered high when analyzing its bioavailability [67]. Furthermore, CAR at high concentrations induced toxic effects in the SH-SY5Y cell line, as assessed here through the cell viability assay. Thus, it should be studied in the future to avoid intoxication when using CAR therapeutically.
References
Cadonic C, Sabbir MG, Albensi BC (2016) Mechanisms of mitochondrial dysfunction in Alzheimer’s disease. Mol Neurobiol 53:6078–6090. https://doi.org/10.1007/s12035-015-9515-5
Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, Filip M (2016) Oxidative stress in neurodegenerative diseases. Mol Neurobiol 53:4094–4125. https://doi.org/10.1007/s12035-015-9337-5
Giorgi C, Marchi S, Simoes ICM, Ren Z, Morciano G, Perrone M et al (2018) Mitochondria and reactive oxygen species in aging and age-related diseases. Int Rev Cell Mol Biol 340:209–344. https://doi.org/10.1016/bs.ircmb.2018.05.006
Erpapazoglou Z, Mouton-Liger F, Corti O (2017) From dysfunctional endoplasmic reticulum–mitochondria coupling to neurodegeneration. Neurochem Int 109:171–183. https://doi.org/10.1016/j.neuint.2017.03.021
Kato T (2017) Neurobiological basis of bipolar disorder: mitochondrial dysfunction hypothesis and beyond. Schizophr Res 187:62–66. https://doi.org/10.1016/j.schres.2016.10.037
Liu CS, Adibfar A, Herrmann N, Gallagher D, Lanctôt KL (2017) Evidence for inflammation-associated depression. Curr Top Behav Neurosci 31:3–30. https://doi.org/10.1007/7854_2016_2
Morris G, Walder K, McGee SL, Dean OM, Tye SJ, Maes M, Berk M (2017) A model of the mitochondrial basis of bipolar disorder. Neurosci Biobehav Rev 74:1–20. https://doi.org/10.1016/j.neubiorev.2017.01.014
Sarrafchi A, Bahmani M, Shirzad H, Rafieian-Kopaei M (2016) Oxidative stress and Parkinson’s disease: new hopes in treatment with herbal antioxidants. Curr Pharm Des 22:238–246
Picard M, Wallace DC, Burelle Y (2016) The rise of mitochondria in medicine. Mitochondrion 30:105–116. https://doi.org/10.1016/j.mito.2016.07.003
Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505:335–343. https://doi.org/10.1038/nature12985
Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D, Signorile A, Petruzzella V (2012) The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol 942:3–37. https://doi.org/10.1007/978-94-007-2869-1_1
Genova ML, Bianchi C, Lenaz G (2005) Supercomplex organization of the mitochondrial respiratory chain and the role of the Coenzyme Q pool: pathophysiological implications. Biofactors 25:5–20
Solaini G, Sgarbi G, Lenaz G, Baracca A (2007) Evaluating mitochondrial membrane potential in cells. Biosci Rep 27:11–21
Naoi M, Maruyama W, Shamoto-Nagai M, Yi H, Akao Y, Tanaka M (2005) Oxidative stress in mitochondria: decision to survival and death of neurons in neurodegenerative disorders. Mol Neurobiol 31:81–93
Sies H, Berndt C, Jones DP (2017) Oxidative stress. Annu Rev Biochem 86:715–748. https://doi.org/10.1146/annurev-biochem-061516-045037
de Oliveira MR, Ferreira GC, Schuck PF, Dal Bosco SM (2015) Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem Biol Interact 242:396–406. https://doi.org/10.1016/j.cbi.2015.11.003
de Oliveira MR, Brasil FB, Andrade CMB (2017) Naringenin attenuates H2O2-induced mitochondrial dysfunction by an Nrf2-dependent mechanism in SH-SY5Y cells. Neurochem Res 42:3341–3350. https://doi.org/10.1007/s11064-017-2376-8
de Oliveira MR, Fürstenau CR, de Souza ICC, da Costa Ferreira G (2017) Tanshinone I attenuates the effects of a challenge with H2O2 on the functions of tricarboxylic acid cycle and respiratory chain in SH-SY5Y cells. Mol Neurobiol 54:7858–7868. https://doi.org/10.1007/s12035-016-0267-7
de Oliveira MR, Peres A, Ferreira GC (2017) Pinocembrin attenuates mitochondrial dysfunction in human neuroblastoma SH-SY5Y cells exposed to methylglyoxal: role for the Erk1/2-Nrf2 signaling pathway. Neurochem Res 42:1057–1072. https://doi.org/10.1007/s11064-016-2140-5
de Oliveira MR, da Costa Ferreira G, Peres A, Bosco SMD (2018) Carnosic acid suppresses the H2O2-induced mitochondria-related bioenergetics disturbances and redox impairment in SH-SY5Y cells: role for Nrf2. Mol Neurobiol 55:968–979. https://doi.org/10.1007/s12035-016-0372-7
de Oliveira MR, da Costa Ferreira G, Brasil FB, Peres A (2018) Pinocembrin suppresses H2O2-induced mitochondrial dysfunction by a mechanism dependent on the Nrf2/HO-1 axis in SH-SY5Y Cells. Mol Neurobiol 55:989–1003. https://doi.org/10.1007/s12035-016-0380-7
de Oliveira MR, Peres A, Ferreira GC, Schuck PF, Gama CS, Bosco SMD (2017) Carnosic acid protects mitochondria of human neuroblastoma SH-SY5Y cells exposed to paraquat through activation of the Nrf2/HO-1 Axis. Mol Neurobiol 54:5961–5972. https://doi.org/10.1007/s12035-016-0100-3
de Oliveira MR, de Bittencourt Brasil F, Fürstenau CR (2018) Inhibition of the Nrf2/HO-1 axis suppresses the mitochondria-related protection promoted by gastrodin in human neuroblastoma cells exposed to paraquat. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1222-6
Chung HT, Ryter SW, Kim HP (2013) Heme oxygenase-1 as a novel metabolic player. Oxid Med Cell Longev 2013:814058. https://doi.org/10.1155/2013/814058
Ollinger R, Wang H, Yamashita K, Wegiel B, Thomas M, Margreiter R, Bach FH (2007) Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid Redox Signal 9:2175–2185
Bach FH (2006) Carbon monoxide: from the origin of life to molecular medicine. Trends Mol Med 12:348–350
Hoesel B, Schmid JA (2013) The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer 12:86. https://doi.org/10.1186/1476-4598-12-86
Chen S, Khan ZA, Barbin Y, Chakrabarti S (2004) Pro-oxidant role of heme oxygenase in mediating glucose-induced endothelial cell damage. Free Radic Res 38:1301–1310
Schipper HM, Song W, Tavitian A, Cressatti M (2018) The sinister face of heme oxygenase-1 in brain aging and disease. Prog Neurobiol. https://doi.org/10.1016/j.pneurobio.2018.06.008
Suntres ZE, Coccimiglio J, Alipour M (2015) The bioactivity and toxicological actions of carvacrol. Crit Rev Food Sci Nutr 55:304–318. https://doi.org/10.1080/10408398.2011.653458
Friedman M (2014) Chemistry and multibeneficial bioactivities of carvacrol (4-isopropyl-2-methylphenol), a component of essential oils produced by aromatic plants and spices. J Agric Food Chem 62:7652–7670. https://doi.org/10.1021/jf5023862
Cui ZW, Xie ZX, Wang BF, Zhong ZH, Chen XY, Sun YH, Sun QF, Yang GY, Bian LG (2015) Carvacrol protects neuroblastoma SH-SY5Y cells against Fe(2+)-induced apoptosis by suppressing activation of MAPK/JNK-NF-κB signaling pathway. Acta Pharmacol Sin 36:1426–1436. https://doi.org/10.1038/aps.2015.90
Potočnjak I, Domitrović R (2016) Carvacrol attenuates acute kidney injury induced by cisplatin through suppression of ERK and PI3K/Akt activation. Food Chem Toxicol 98:251–261. https://doi.org/10.1016/j.fct.2016.11.004
Yu H, Zhang ZL, Chen J, Pei A, Hua F, Qian X, He J, Liu CF, Xu X (2012) Carvacrol, a food-additive, provides neuroprotection on focal cerebral ischemia/reperfusion injury in mice. PLoS ONE 7:e33584. https://doi.org/10.1371/journal.pone.0033584
Scandroglio F, Tórtora V, Radi R, Castro L (2014) Metabolic control analysis of mitochondrial aconitase: influence over respiration and mitochondrial superoxide and hydrogen peroxide production. Free Radic Res 48:684–693. https://doi.org/10.3109/10715762.2014.900175
Poderoso JJ, Carreras MC, Lisdero C, Riobó N, Schöpfer F, Boveris A (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92
Mensor LL, Menezes FS, Leitão GG, Reis AS, dos Santos TC, Coube CS, Leitão SG (2001) Screening of Brazilian plant extracts for antioxidant activity by the use of DPPH free radical method. Phytother Res 15:127–130
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Wang K, Zhu L, Zhu X, Zhang K, Huang B, Zhang J, Zhang Y, Zhu L, Zhou B, Zhou F (2014) Protective effect of paeoniflorin on Aβ25-35-induced SH-SY5Y cell injury by preventing mitochondrial dysfunction. Cell Mol Neurobiol 34:227–234. https://doi.org/10.1007/s10571-013-0006-9
de Oliveira MR, da Rocha RF, Pasquali MA, Moreira JC (2012) The effects of vitamin A supplementation for 3 months on adult rat nigrostriatal axis: increased monoamine oxidase enzyme activity, mitochondrial redox dysfunction, increased β-amyloid(1–40) peptide and TNF-α contents, and susceptibility of mitochondria to an in vitro H2O2 challenge. Brain Res Bull 87:432–444. https://doi.org/10.1016/j.brainresbull.2012.01.005
Wang Y, Miao Y, Mir AZ, Cheng L, Wang L, Zhao L, Cui Q, Zhao W, Wang H (2016) Inhibition of beta-amyloid-induced neurotoxicity by pinocembrin through Nrf2/HO-1 pathway in SH-SY5Y cells. J Neurol Sci 368:223–230. https://doi.org/10.1016/j.jns.2016.07.010
de Oliveira MR, Brasil FB, Fürstenau CR (2018) Sulforaphane attenuated the pro-inflammatory state induced by hydrogen peroxide in SH-SY5Y cells through the Nrf2/HO-1 signaling pathway. Neurotox Res 34:241–249. https://doi.org/10.1007/s12640-018-9881-7
de Oliveira MR (2018) Carnosic acid as a promising agent in protecting mitochondria of brain cells. Mol Neurobiol 55:6687–6699. https://doi.org/10.1007/s12035-017-0842-6
Jo MG, Ikram M, Jo MH, Yoo L, Chung KC, Nah SY, Hwang H, Rhim H, Kim MO (2018) Gintonin mitigates MPTP-induced loss of nigrostriatal dopaminergic neurons and accumulation of α-synuclein via the Nrf2/HO-1 pathway. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1020-1
de Oliveira MR, de Bittencourt Brasil F, Fürstenau CR (2018) Sulforaphane promotes mitochondrial protection in SH-SY5Y cells exposed to hydrogen peroxide by an Nrf2-dependent mechanism. Mol Neurobiol 55:4777–4787. https://doi.org/10.1007/s12035-017-0684-2
Rochette L, Zeller M, Cottin Y, Vergely C (2018) Redox functions of heme oxygenase-1 and biliverdin reductase in diabetes. Trends Endocrinol Metab 29:74–85. https://doi.org/10.1016/j.tem.2017.11.005
O’Brien L, Hosick PA, John K, Stec DE, Hinds TD Jr (2015) Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab 26:212–220. https://doi.org/10.1016/j.tem.2015.02.001
Qin W, Zhang J, Lv W, Wang X, Sun B (2013) Effect of carbon monoxide-releasing molecules II-liberated CO on suppressing inflammatory response in sepsis by interfering with nuclear factor kappa B activation. PLoS One 8:e75840. https://doi.org/10.1371/journal.pone.0075840
Tschopp J (2011) Mitochondria: sovereign of inflammation? Eur J Immunol 41:1196–1202. https://doi.org/10.1002/eji.201141436
Green DR, Galluzzi L, Kroemer G (2014) Metabolic control of cell death. Science 345:1250256. https://doi.org/10.1126/science.1250256
Cobley JN, Fiorello ML, Bailey DM (2018) 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 15:490–503. https://doi.org/10.1016/j.redox.2018.01.008
Alam J, Cook JL (2003) Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des 9:2499–2511
Niture SK, Khatri R, Jaiswal AK (2014) Regulation of Nrf2-an update. Free Radic Biol Med 66:36–44. https://doi.org/10.1016/j.freeradbiomed.2013.02.008
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34:13–25
Allen J, Romay-Tallon R, Brymer KJ, Caruncho HJ, Kalynchuk LE (2018) Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Front Neurosci 12:386. https://doi.org/10.3389/fnins.2018.00386
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741. https://doi.org/10.1016/j.biopsych.2008.11.029
Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15:384–392. https://doi.org/10.1038/mp.2009.47
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C, Brisch R, Bielau H, Meyer zu Schwabedissen L, Bogerts B, Myint AM (2011) Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation 8:94. https://doi.org/10.1186/1742-2094-8-94
Alcocer-Gómez E, de Miguel M, Casas-Barquero N, Núñez-Vasco J, Sánchez-Alcazar JA, Fernández-Rodríguez A, Cordero MD (2014) NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun 36:111–117
Liu YZ, Wang YX, Jiang CL (2017) Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci 11:316. https://doi.org/10.3389/fnhum.2017.00316
Blanco-Ayala T, Andérica-Romero AC, Pedraza-Chaverri J (2014) New insights into antioxidant strategies against paraquat toxicity. Free Radic Res 48:623–640. https://doi.org/10.3109/10715762.2014.899694
Stojkovska I, Wagner BM, Morrison BE (2015) Parkinson’s disease and enhanced inflammatory response. Exp Biol Med (Maywood) 240:1387–1395. https://doi.org/10.1177/1535370215576313
Jang Y, Lee AY, Jeong SH, Park KH, Paik MK, Cho NJ, Kim JE, Cho MH (2015) Chlorpyrifos induces NLRP3 inflammasome and pyroptosis/apoptosis via mitochondrial oxidative stress in human keratinocyte HaCaT cells. Toxicology 338:37–46. https://doi.org/10.1016/j.tox.2015.09.006
Naik E, Dixit VM (2011) Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 208:417–420. https://doi.org/10.1084/jem.20110367
Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, Zang QS (2015) Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE 10:e0139416. https://doi.org/10.1371/journal.pone.0139416
Wilkins HM, Swerdlow RH (2016) Relationships between mitochondria and neuroinflammation: implications for Alzheimer’s disease. Curr Top Med Chem 16:849–857
Salehi B, Mishra AP, Shukla I, Sharifi-Rad M, Contreras MDM, Segura-Carretero A, Fathi H, Nasrabadi NN, Kobarfard F, Sharifi-Rad J (2018) Thymol, thyme, and other plant sources: health and potential uses. Phytother Res 32:1688–1706. https://doi.org/10.1002/ptr.6109
Acknowledgements
This work was supported by the Conselho Nacional de Pesquisa e Desenvolvimento Tecnológico (CNPq; Edital Universal 2016; 400216/2016-7) and by the Fundação de Apoio à Pesquisa do Estado de Mato Grosso (FAPEMAT; Edital Universal; 222541/2015). ALC receives a CAPES fellow (Bolsa de Mestrado).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11064_2019_2724_MOESM1_ESM.pdf
Fig. S1 The mitochondrial and nuclear fractions were checked in order to confirm that the samples do not contain cytoplasmic enzymes. (A) The activity of the cytoplasmic enzyme lactate dehydrogenase (LDH) was analyzed in the mitochondrial fraction obtained after isolation of the organelles. (B) The activity of the cytoplasmic enzyme lactate dehydrogenase (LDH) was analyzed in the nuclear fraction obtained after isolation of the cell nucleus. Supplementary material 1 (PDF 84 KB)
Rights and permissions
About this article

Cite this article
Chenet, A.L., Duarte, A.R., de Almeida, F.J.S. et al. Carvacrol Depends on Heme Oxygenase-1 (HO-1) to Exert Antioxidant, Anti-inflammatory, and Mitochondria-Related Protection in the Human Neuroblastoma SH-SY5Y Cells Line Exposed to Hydrogen Peroxide. Neurochem Res 44, 884–896 (2019). https://doi.org/10.1007/s11064-019-02724-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02724-5