
Abstract
Human life and health are gravely threatened by brain diseases. The onset and progression of the illnesses are influenced by a variety of factors, including pathogenic causes, environmental factors, mental issues, etc. According to scientific studies, neuroinflammation and oxidative stress play a significant role in the development and incidence of brain diseases by producing pro-inflammatory cytokines and oxidative tissue damage to induce inflammation and apoptosis. Neuroinflammation, oxidative stress, and oxidative stress-related changes are inseparable factors in the etiology of several brain diseases. Numerous neurodegenerative diseases have undergone substantial research into the therapeutic alternatives that target oxidative stress, the function of oxidative stress, and the possible therapeutic use of antioxidants. Formerly, tBHQ is a synthetic phenolic antioxidant, which has been widely used as a food additive. According to recent researches, tBHQ can suppress the processes that lead to neuroinflammation and oxidative stress, which offers a fresh approach to treating brain diseases. In order to achieve the goal of decreasing inflammation and apoptosis, tBHQ is a specialized nuclear factor erythroid 2-related factor (Nrf2) activator that decreases oxidative stress and enhances antioxidant status by upregulating the Nrf2 gene and reducing nuclear factor kappa-B (NF-κB) activity. This article reviews the effects of tBHQ on neuroinflammation and oxidative stress in recent years and looks into how tBHQ inhibits neuroinflammation and oxidative stress through human, animal, and cell experiments to play a neuroprotective role in Alzheimer’s disease (AD), stroke, depression, and Parkinson’s disease (PD). It is anticipated that this article will be useful as a reference for upcoming research and the creation of drugs to treat brain diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain damage and behavioral abnormalities that characterize brain diseases impose a significant medical burden and degrade both the quality of life for patients and their families [1]. Neuroinflammation, oxidative stress, transcriptional alterations, abnormal protein deposition, and excitotoxicity are a few of the major causes of brain diseases [2]. Multiple brain diseases have been linked to the onset and progression of oxidative stress [3]. Oxidative stress and oxidative stress-associated neuroinflammation are widely acknowledged causative factors in many brain diseases, including AD, stroke, depression, PD, etc. [4]. Oxidative stress is caused by an imbalance between the production of radical species and antioxidant systems, which is linked to the development and progression of diseases. Emerging researches have confirmed that tBHQ has been shown to reduce oxidative stress to exert neuroprotection in a number of brain diseases [5,6,7].
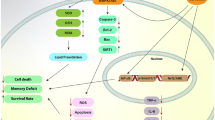
TBHQ is a very effective synthetic oil-soluble phenolic antioxidant with a significant antioxidant action, which has a low toxicity, low dosage, high-temperature resistance, antibacterial, low cost, among other properties (Fig. 1). Due to its excellent antioxidant activity and safety, tBHQ can also defend against oxidative stress and inflammation-induced dysfunction in animal cells and tissues [8,9,10]. TBHQ is an inducer of Nrf2 which is crucial for innate immune response regulation [11, 12], and when the body is stimulated by external stimuli, Nrf2 regulates the expression of related inflammatory factors by regulating the activity of the NF-κB signaling pathway [13]. TBHQ exerts antioxidant damage and neuroprotective effects on the central nervous system by increasing the stability of Nrf2 protein and activating Nrf2 transcription [14] (Fig. 2).

Characters of tBHQ

Antioxidative stress and neuroprotective mechanisms of tBHQ
Effect of tBHQ on Inflammation
Inflammation is the body’s natural defense mechanism and innate immunological reaction to outside stimuli, which is a protective response brought on by a damaging factor that affects the vascular system’s live tissues and has the potential to harm healthy tissues as well. The immune system, metabolism, and endocrine system will all be negatively impacted by an excessive inflammatory response, which is exceedingly damaging to health. Pro-inflammatory cytokines [15, 16], anti-inflammatory cytokines [17], and other nonspecific inflammatory cytokines [18] are among the inflammatory components that contribute to inflammatory reactions. Tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-2, IL-6, IL-8, IL-18, and interferon (IFN)-γ are the primary pro-inflammatory cytokines that play a key role in immune activation and causing inflammation. IL-4, IL-10, IL-13, transforming growth factor (TGF)-β, IL-1 receptor antagonist (IL-1Ra), and soluble receptors of various pro-inflammatory cytokines are anti-inflammatory cytokines that primarily suppress immunity and inflammation. Complex cytokine networks and immune cells are created by pro- and anti-inflammatory cytokines, and their dynamic equilibrium influences the course of inflammation and its outcome [19, 20].
TBHQ can inhibit inflammation and is crucial in the treatment of several illnesses. It is widely known that alterations in Nrf2-dependent redox homeostasis are closely correlated with inflammation [21]. As an illustration, it has been proven that tBHQ alleviates inflammation caused by fine particulate matter by promoting the transcriptional activity of Nrf2 [22]. NF-κB, pro-inflammatory cytokines, and intercellular adhesion molecule-1 (ICAM-1) are just a few of the inflammatory-related factors that tBHQ significantly reduces in the intestine [23, 24], which can prevent intestinal inflammation after traumatic brain injury (TBI) by activating the Nrf2 signaling pathway and lessen mucosal damage [23]. TBHQ alleviates oxidative stress during myocardial ischemia and reperfusion by inducing secretion of anti-inflammatory cytokines [25], which by increasing the activity of Nrf2 in macrophages and vascular smooth muscle cells in atherosclerotic lesions [26] inhibited the expression of cytokine induced pro-inflammatory and oxidative stress genes, changed the phenotype of macrophages, promoted autophagic activity, significantly reduced the size, extension and lipid content of atherosclerotic plaques, and reduced the size of macrophages and foam cells and the expression of chemokines to alleviate inflammation, so as to provide a method for the protection of atherosclerosis in diabetes. Moreover, in high salt-induced hypertension experiments [27], tBHQ increased the expression of Nrf2 in paraventricular nucleus (PVN), reduced oxidative stress, the expression of IL-1β and IL-6, and the neuronal activity and the plasma level of norepinephrine to reduce salt-induced hypertension and cardiac hypertrophy in hypertensive rats, inhibited the activation of NF-κB, that was demonstrated that tBHQ had a protective effect on high salt-induced hypertension by inhibiting oxidative stress and inflammation in PVN. The toll-like receptor 4 (TLR4)-NF-κB axis may be activated by oxidative stress, inflammation, and renal tubular cell apoptosis, which might aggravate renal ischemia/reperfusion (I/R) injury (RI/RI). However, by boosting antioxidant capacity and helpful inflammatory response modulation, tBHQ [28] might lessen the expression of pro-inflammatory cytokines and pro-apoptotic proteins, suppress the NF-κB signaling pathway, and lower the RI/RI of diabetic rats. On one hand, tBHQ alleviated oxidative abnormalities, reduced malondialdehyde (MDA) content, and increased superoxide dismutase (SOD) activity. On the other hand, the levels of TNF-α and IL-1β were decreased; concurrently, it also decreased the expression of pro-apoptotic proteins and increased the expression of beta cell lymphoma-2. TBHQ intervention dramatically reduced oxidative stress by up-regulating Nrf2 gene, suppressing inflammation, apoptosis, and promoting proliferation of testicular germ cells; more importantly, tBHQ could prevent probable long-term reproductive failure associated with medications [10]; simultaneously, in order to strengthen the body's antioxidant defense system both before and after cisplatin chemotherapy, tBHQ also raised steroidogenesis and enhanced sperm parameters.
Effect of tBHQ on Oxidative Stress
The idea of oxidative stress was initially put forth in 1985 as a redox biology and medicine concept and was derived from human understanding of aging. Highly reactive molecules including reactive oxygen species (ROS) and reactive nitrogen species (RNS) are overproduced in the organism when oxidative stress takes place [29,30,31,32]. When the production of intracellular ROS exceeds the ability of antioxidant system to scavenge ROS, excessive ROS not only attacks biological macromolecules such as proteins, lipids, and DNA to result in cell death or changes in organizational structure but also damages cells by causing mitochondrial dysfunction, which is one of the causes of neurodegenerative diseases, such as AD and PD. In order to create a dynamic physiological equilibrium, the body’s oxidation and antioxidant systems interact and restrain one another. The state of oxidative stress is demonstrated when the antioxidant system is compromised or when the oxidative system is strengthened. The oxidation system, which causes oxidative damage to cells, is mostly made up of reactive free radicals like ROS and RNS, including superoxide anion, hydroxyl radical, hydrogen peroxide, singlet oxygen, nitric oxide (NO), nitrogen dioxide, and nitrite peroxide [33,34,35], which causes oxidative damage to cells. Among them, mitochondria is susceptible to oxidative stress [36], which is the biggest cause of ROS production [37]. To scavenge ROS and guard cells from oxidative damage, the antioxidant system uses both enzymatic antioxidants like SOD, catalase (CAT), glutathione peroxidase (GPx), and peroxidase oxidoreductase as well as non-enzymatic antioxidants. As a result of the combination of internal and external stimuli, oxidative stress is currently thought to be one of the key mechanisms causing clinical damage in a number of diseases [38]. TBHQ significantly attenuated the production of oxidative products and inflammatory cytokines, increased GPx and SOD levels, decreased NF-κB, which protected against oxidative damage, and restored the antioxidant mechanism [39, 40]. Nrf2-Kelch-like ECH-associated protein 1 (Keap1)-antioxidant response elements (ARE) pathway has been linked to oxidative stress-related diseases [41], such as cancer, neurodegenerative diseases, diabetes and others [42, 43]. Nevertheless, tBHQ is one of the most effective activators of signaling pathways and plays a significant antioxidant role by activating the pathway [10, 44, 45].
According to research on ventilator-induced lung injury (VILI) [46], tBHQ increased pulmonary redox capacity by activating the Nrf2-ARE pathway, increased the expression of Nrf2-dependent genes like Nrf2 and SOD1, and the expression of antioxidant genes, all of which had a protective effect on VILI. For oxidative stress-induced osteoarthritis, tBHQ can improve the viability of chondrocytes, reduce excessive ROS production, raise SOD levels and lower MDA levels, reduce oxidative stress-induced mitochondrial damage and apoptosis, improve oxidative stress-induced matrix degradation, and activate Nrf2 signaling pathway, that indicated that tBHQ could prevent oxidative stress-induced chondrocyte apoptosis and extracellular matrix degradation in vitro, which had the potential to treat osteoarthritis [44]. Methamphetamine (MA) induces cardiotoxicity, neurotoxicity and pulmonary toxicity through oxidative stress, while tBHQ may inhibit oxidative stress-induced neurotoxicity through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system, astrocyte activation, and glutathione (GSH) pathway [47]; the up-regulation of Nrf2 expression to reverse the overexpression and phosphorylation of protein kinase-like ER kinase (PERK), alleviated MA-induced oxidative stress, and accelerated endoplasmic reticulum stress to initiate PERK-dependent apoptosis [48]. TBHQ may become a promising treatment medication for hyperlipidemia in chronic renal disease and lessen liver damage by inhibiting Nrf2/heme oxygenase-1 (HO-1) pathway-induced oxidative stress damage and lipid deposition and enhancing antioxidant defense [45, 49]. TBHQ inhibited lipopolysaccharide (LPS)-induced ROS production and macrophage repolarization, which significantly protected LPS-induced ROS accumulation in a dose-dependent manner; inhibited the activation of NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, and IL-1β-induced chondrocyte apoptosis, inflammation and differentiation defects, that experimental studies in vitro showed that tBHQ may prevent osteoarthritis from developing [9]. To protect pheochromocytoma cells from oxidative stress, inflammation, apoptosis, as well as oxidation-redox imbalance, tBHQ activated the Nrf2/ARE pathway, stabilized Nrf2, raised the expression of HO-1 and γ-glutamylcysteine synthetase, and enhanced the expression of HO-1 [50,51,52]. Simultaneously, tBHQ simultaneously decreased MDA and ROS levels, raised SOD and GPx activity, suppressed oxidative stress by raising Nrf2 levels, and activated Nrf2 to lower gestational diabetes mellitus [53]. Additionally, tBHQ may be employed as a potential neuroprotectant and adjuvant therapy for patients with traumatic brain damage (TBI) due to its ability to drastically attenuate NADPH oxidase protein expression, decrease MDA level, increase Nrf2 protein level, and activate the antioxidant enzyme SOD [54].
TBHQ in Brain Diseases
TBHQ, Neuroinflammation, Oxidative Stress, and AD
AD is one of the most prevalent neurodegenerative brain diseases characterized by progressive memory impairment, neuronal loss, tau hyperphosphorylation-induced neurofibrillary tangles, and extracellular amyloid β-protein (Aβ) accumulation-induced amyloid plaques [55, 56]. Millions of people all over the world are severely impacted by AD, and primarily affects the elderly and is characterized by progressive cognitive dysfunction and behavioral disorders, which is mainly manifested as systemic dementia such as cognitive decline, mental symptoms, and behavioral disorders, also causes a gradual loss of daily living skills [57]. Despite significant research efforts, there is still no effective therapy strategy for AD since the precise etiology and pathology of the disease are still unknown [58, 59]. Intriguingly, tBHQ's positive effects on AD were found in both in vivo and in vitro experiments [60]. In the brain and hippocampus of AβPP/PS1 mice, for instance, a 6-week tBHQ diet significantly decreased Aβ deposition. Notably, this effect was not due to the changes in AβPP expression/processing or Aβ production but the improved Aβ clearance, including increased Aβ degradation and Aβ efflux from the affected brain [60]. A further mechanistic investigation found that tBHQ could suppress the expression of plasminogen activator inhibitor-1 and enhance the activities of plasminogen activators, which significantly promote Aβ degradation in the brain of AβPP/PS1 mice [60]. Moreover, the increased expression of low-density lipoprotein receptor-related protein 1, a protein involved in Aβ efflux transport out of the brain, suggested that the improved Aβ efflux transport from the brain following tBHQ treatment contributes to the alleviation of Aβ load in AD mice. Furthermore, the antioxidant capacity of tBHQ also contributes to the improved AD pathology, as evidenced by increased concentration of GSH and decreased lipid peroxidation level in the transgenic AD mice, that indicates tBHQ has a neuroprotective effect on AD. NT2N is a well-documented cell line generating intracellular Aβ and is widely used in AD in vitro studies [61]. Intriguingly, NT2N neurons pretreated with tBHQ significantly reduced oxidative stress-induced Aβ production and markedly suppressed neuronal apoptosis. In fact, tBHQ’s neuroprotection depends on the Keap1-Nrf2 pathway, which is the main protective route in response to oxidative stress. Under normal conditions, Nrf2 binds to its inhibitor Keap1. However, in the presence of oxidative stress, Nrf2 dissociates from Keap1 and trans-locates into the nuclear, triggering an antioxidant response. The Keap1-Nrf2 pathway is stimulated by tBHQ, and the amount of Nrf2 in neuronal nuclei increases considerably, indicating an antioxidant response to tBHQ [61]. In line with these findings, tBHQ induced the elevated GSH levels and buffered the oxidative regents-induced redox status changes in NT2N neurons [61], indicating that the antioxidant effect of tBHQ contributes to the beneficial effects of tBHQ in AD. The neuroprotective effects of tBHQ are influenced by alterations in FoxO3a translocation as well as Nrf2 nuclear translocation. Following translocation from the cytosol to the nucleus, FoxO3a as one of the forkhead transcription factors to cause neuronal apoptosis [62]. Unlike promoting nuclear translocation of Nrf2, tBHQ suppressed nuclear translocation Nrf2 and activity by promoting phosphatidylinositol 3-kinase (PI3K)-AKT signaling activation-induced FoxO3a phosphorylation.
TBHQ, Neuroinflammation, Oxidative Stress, and Stroke
Stroke is the “number one killer” of people’s health and poses a major threat to human life. There are usually no obvious clinical symptoms prior to the onset of a stroke, but once the attack occurs, there is a high mortality rate, a high disability rate, and a high treatment cost, all of which pose a serious threat to public health [63]. Stroke is a type of acute cerebrovascular disease that includes both ischemic stroke and hemorrhagic stroke. Strokes are a collection of illnesses that result in hypoxia and ischemia of the brain tissue in the blood supply area. The damage to brain tissue is caused by sudden blood vessel rupture in the brain or by the inability of blood to flow into the brain owing to vascular blockage [64]. Ischemic stroke, which makes up around 70% of all strokes, is the most significant form of stroke among them [65]. Stroke with symptoms such as acute or sudden loss of balance or coordination, blurred vision, facial numbness or crooked eyes, limb weakness, trouble speaking, etc. [66]. Stroke is caused by a variety of reasons, both internal and external. Excitatory amino acid toxicity [67], free radical damage [68], inflammatory response [69], expression of related apoptosis genes [70], and immunosuppression [71] are the key contributors to the pathophysiology of ischemic stroke.
Both neuroinflammation and oxidative stress play significant roles in the development of stroke lesions, with neuroinflammation playing a complicated and multidimensional function in ischemic stroke [72]. Microglia are triggered by a variety of inflammatory signals to release pro-inflammatory cytokines like TNF-α, adhesion molecules, IL-18, and ROS, which exacerbate brain injury [73, 74]. Microglia and astrocytes can be rapidly activated within minutes after ischemic stroke [75] and produce a large number of pro-inflammatory mediators that worsen tissue damage [76] and lead to neutrophil infiltration in the brain [77]. The number and expression of astrocytes significantly increase in response to ischemic injury, release a variety of pro-inflammatory factors and induce the synthesis of NO synthase which the activation of astrocytes promoted the recovery process of the slow development of the whole brain and simultaneously microglia response was involved in local repair and cell debris removal [78]. However, the nerves were harmed by the neuronal degeneration brought on by a significant amount of pro-inflammatory substances generated by excessively active astrocytes [79]. JAK/STAT [80], MAPK [81], NF-κB [82], Toll-like receptors (TLRs)/myeloid differentiation primary response gene 88 (MyD88) [83], and other important signaling pathways in cells after stroke mediated inflammatory reactions. Additionally, cell adhesion molecules [84], chemokines [85], and blood–brain barrier [86] also play important roles in inflammation. One of the main causes of tissue damage in stroke is the excessive generation of ROS, which builds up in the body during a stroke, induces oxidative stress, and sets off a chain reaction of biological responses [87, 88]. Free radicals produced as a result of oxidative stress, particularly the high levels of ROS and RNS that caused protein malfunction, DNA damage, and lipid peroxidation, which ultimately led to cell death [68].
Nrf2, a protein that is biologically connected to the brain and has a role in tBHQ’s function, reduces the severity of local ischemic brain injury. The effects of tBHQ on GSH levels in the cortex and the basic and induced antioxidant/detoxifying enzyme activities in Nrf2(-/-) mice demonstrate that Nrf2 activation protects the brain from cerebral ischemia in the body, suggesting that activating Nrf2 may be a useful preventive treatment for patients who are at risk for stroke [89]. A study using a neonatal hypoxic-ischemic (HI) rat model was conducted to determine whether tBHQ offered protection against oxidative stress in neonatal HI brain injury. The findings [5] revealed that tBHQ activated the Nrf2-mediated antioxidant signaling pathway, reduced oxidative stress index, enhanced Nrf2 nuclear accumulation and DNA binding activity, and up-regulated the expression of Nrf2 downstream antioxidant genes, with the exception of the fact that it inhibited reactive. Through the activation of AKT [90] and the IL-10-mediated astrocytes pathway, tBHQ increased ischemic-induced angiogenesis, which may offer therapeutic guidance for the treatment intervention following oxidative stress injury such as ischemic stroke [91]. What is more, after cerebral ischemia, tBHQ increased angiogenesis and astrocyte activation by activating the Nrf2 pathway, increased HO-1 expression, and controlled the expression of vascular endothelial growth factor through the Nrf2/HO-1 pathway, in order to promote angiogenesis and enhance functional recovery [92]. Despite the fact that tBHQ accelerated the activation of the Nrf2-ARE signaling cascade, this activation would wane with continued exposure to tBHQ [93]. Intriguingly, tBHQ significantly reduced the mitochondrial respiration of cerebral cortical endothelial cells in vitro, and the induced mitochondrial inhibition could be enhanced in the presence of other mitochondrial toxins like LPS. The blood–brain barrier would be destroyed by LPS when the cerebral vascular endothelial cells’ mitochondrial function was compromised, aggravating the effects of a stroke and creating a negative consequence of tBHQ-enhanced mortality from permanent stroke [94, 95].
TBHQ, Neuroinflammation, Oxidative Stress, and Depression
One of the most prevalent psychological and mental diseases, depression is characterized by low mood, lack of interest in activities, poor focus, and slow thinking [96]. Depression is one of the most severe disease burdens of non-fatal neuropsychiatric disorders and is projected to be in the top three of all diseases burdens by 2030 [97], which seriously impairs the quality of life and brings a heavy burden to the affected patients and their families. Typical pathophysiological characteristics of depression include deficits in monoamine neurotransmission, resistance of glucocorticoid receptors, impaired neurotrophic factors, increased glutamate, corticosteroid-releasing hormone, and cortisol, and various hypotheses have been developed based on those pathophysiological changes, including the most widely accepted monoamine hypothesis [98]. Recently, growing evidence reveals that neuroinflammation is crucial for the emergence of depression, and anti-inflammatory treatments greatly reduced depressive-like symptoms [99, 100].
It is commonly acknowledged that neuroinflammation may play a role in the etiology of depression [101]. The neuro-inflammatory response mechanism is crucial to the development of depression, and it may alter the neurological function of the depression-related brain region through mechanisms including mitochondria and energy metabolism, resulting in aberrant emotional regulation [102, 103]. In some cases, microglia, astrocytes, and certain cytokines involved in the neuroinflammation process, which primarily manifests as the activation of microglia and astrocytes and the alteration of chemokine levels, may have abnormal effects that contribute to the development of depression [104,105,106]. Patients with depression had significantly higher levels of cytokines, macrophages, microglia, and astrocyte carbon monoxide synthase in their brains [107]. The prevalence and progression of depression are influenced by the balance of M1 type (classical/pro-inflammatory activation) and M2 type (alternative/anti-inflammatory activation) present in microglia [108], among which pro-inflammatory factors are involved in immune activation, and the occurrence of inflammation is positively correlated with the severity of depressive symptoms [109, 110]. The amount of TNF-α in the serum of depressive patients is markedly elevated [111], which may cause apoptosis through associated pathways [112] and influence specific chemicals that can affect depression by affecting those that can cross the blood–brain barrier [113]. The NF-κB signal transduction pathway and the expression of hippocampal neural progenitor cells are both impacted by IL-1β, which also reduces the proliferation of hippocampal cells [114], and participates in inflammatory responses that damage nerve cells in the brain [115]. IL-1β is a key mediator of depression. Fortunately, IL-4 therapy was able to block IL-1β-induced central nervous system glial activation and neurotransmitter changes, hence modulating IL-1β-induced depressive behavior [116]. Additionally, depressed patients had blood levels of IL-6 that were noticeably raised [117], which had neurotoxic effects on the brain by way of a variety of physiological stressors that led to structural and functional abnormalities. However, a research found that the levels of IL-1β and IL-6 in peripheral blood of the elderly with depression increased, while TNF-α levels did not rise [118]. The pathogenic process of depression involves oxidative stress, patients with depression experience an imbalance of oxidative stress, and changes in oxidative stress markers are linked to changes in the course and symptoms of depression [119, 120]. In the brains of depressed patients, lowered GSH levels fell [121], MDA and NO levels rose [122], which either directly or indirectly contributed to the development of depression. By controlling mitochondrial function to cause disorder, excessive autophagy to hasten the aging of brain neurons, and altering the function of the hippocampus, among other things, oxidative stress may have a role in the development of depression [123, 124]. Mitochondria are the susceptible sites of depression [103] and abnormal mitochondrial energy metabolism is involved in the occurrence of depression, mainly including the following aspects: the reduction of mitochondrial adenosine triphosphate (ATP) [125], the activity change of mitochondrial oxidative respiratory chain and its complex [126, 127], oxidative stress injury [128], abnormal mitochondrial morphological structure [129], mitochondrial related genes and molecular level abnormalities [130, 131] to result in a series of reactions such as mitochondrial dysfunction to affect brain function. A new ATP-sensitive potassium channel opener-Iptakalim could upregulate postsynaptic density 95 and synaptophysin, alleviate synaptic structural damage, reverse abnormal mitochondrial fission and fusion, and reduce mitochondrial ATP production and mitochondrial membrane potential collapse in depression models to alleviate abnormal mitochondrial dynamics and function dependent on mitochondrial ATP, which is helpful to improve synaptic plasticity and play an antidepressant role [132]. Moreover, mitochondrial transplantation was able to ameliorate LPS-induced depression-like behaviors, increase the expression of brain-derived neurotrophic factor and neurogenesis, and restore dysfunction of ATP production and oxidative stress in inflammation-induced depression, whose results suggest that mitochondrial transplantation may one day be used as a new treatment for major depressive disorder [133].
Studies have indicated that [6] Nrf2/PI3K may play a key role in the relationship between oxidative stress and apoptosis in MA-induced chronic neurotoxicity, which can result in the development of depressive-like behavior. However, tBHQ could amplify effect on the Nrf2/HO-1 pathway and protect dopamine (DA) neurons from MA-induced neurotoxicity, as result of reducing oxidative stress, protecting the normal signal transduction of PI3K/AKT pathway and the anti-apoptotic ability of PI3K/AKT; at the same time, PI3K/AKT pathway increased the immune content of Nrf2 protein and further enhanced antioxidant capacity through Nrf2/HO-1 pathway. Ghosh et al. [21] injected LPS into the abdominal cavity of Swiss albino mice to induce peripheral inflammation, which caused depression-like behavior in mice, increased the activation of microglia and the level of pro-inflammatory cytokines, and activated NF-κB-p65 pathway, after tBHQ is used, the changes of autophagy and cell death pathways in hippocampus were alleviated, and depression-like symptoms were reversed by activating Nrf2-dependent gene expression. Additionally, through activating the Nrf2/ARE pathway, tBHQ could reduce depressive and anxiety-like behaviors in diabetic rats [134].
TBHQ, Neuroinflammation, Oxidative Stress, and PD
PD, also referred to as “tremor paralysis,” is a prevalent neurodegenerative illness among the elderly and is the second most frequent neurodegenerative disease in this population. Clinically, the predominant symptoms are the static tremor, motor retardation, myotonia, postural and gait disorders, which are often accompanied by non-motor symptoms including depression, anxiety, constipation, autonomic nerve dysfunction, and so on [135]. The loss of the nigrostriatal pathway, which is primarily correlated with significantly decreased levels of DA and aggregation and misfolding of α-synuclein (α-syn) is the primary PD manifestation [136, 137]. In addition to factors including aging, environmental factors, familial inheritance, genetic mutations, and environmental factors, the etiology and pathophysiology of PD are extremely complicated and are yet unclear [138]. The abnormal regulation of α-syn, mitochondrial dysfunction, oxidative stress, excessive immune-inflammatory mechanisms, excessive accumulation of byproducts of DA oxidative metabolism, gastrointestinal related dysfunction, neurotoxicity, etc. are the main components of the pathogenesis of PD [139,140,141,142,143,144,145]. So far, there is no cure for PD but only to relieve its symptoms.
The process of neuroinflammation involves a wide number of enzymes and receptors, and the activation of microglia and astrocytes in the substantia nigra associated with neuroinflammation is thought to have a significant role in PD [146]. Numerous intracellular enzymes, including NADPH oxidase, cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), and myeloperoxidase (MPO), are activated to varying degrees when glial cells are stimulated, and a large number of inflammatory factors, including ROS, prostaglandin (PG), NO and hypochlorous acid are released, which can harm or even kill neurons. A huge quantity of oxygen free radicals and a range of pro-inflammatory cytokines can be produced by activated microglia, which can lead to the death and distortion of neurons [147,148,149]. What is even more remarkable is that dead neuron fragments can continue to promote microglial activation [150] to form a self-promoting vicious cycle to impel the malignant development of PD. A significant part of the pathophysiology of PD is attributed to astrocyte activation, which is connected to the PD pathogenesis. Under the influence of specific stimulating conditions, astrocytes release several pro-inflammatory substances that damage dopaminergic neurons and aid in the onset and progression of PD [151, 152]. The loss of DA neurons in the substantia nigra was finally caused by the oxidative stress and excitotoxicity of the astrocytes implicated in PD, and the expression of a number of proteins was important in the development of PD [153, 154]. Numerous studies have demonstrated the tight connection between oxidative stress and the onset and progression of PD [141]. Basic research and postmortem findings showed that oxidative stress, which was crucial in the degradation of PD DA neurons, was directly associated to the degeneration or death of dopaminergic neurons in the brain's substantia nigra [123, 155]. The etiology of PD is influenced by oxidative stress, which is correlated with a drop in GSH levels [156], a drop in SOD and CAT activity [157], a deficiency in mitochondrial complex I [158], and a significant amount of oxidized lipids, proteins, and DNA [156,157,158,159].
DA-induced oxidative stress may be directly associated to the pathophysiology of PD, and therapy with tBHQ may boost SH-SY5Y cells' intracellular antioxidant capacity to shield cells from 6-hydroxydopamine (6-OHDA)-induced oxidative stress, which plays a neuroprotective role, increases the intracellular GSH level and induces the expression of NADPH: quinone oxidoreductase (NQO1) mRNA [160]. Meanwhile, tBHQ had the ability to reverse oxidative stress such as cytoplasmic swelling, interstitial edema and neuronal loss induced by 1-methyl-4-(2′-methylphenyl)-1,2,3,6-tetrahydropyridine(2’CH3-MPTP) in mice; increased GSH brainstem content and reduce the MDA and SOD activity levels, while diminished histopathology and histochemical alterations may have beneficial benefits in exploring the genesis and pathogenic variables associated to dopaminergic damage and providing a direction for the research of PD treatment [161]. By activating the Nrf2/ARE pathway and Nrf2/ARE pathway to offer a direction for minimizing or preventing PD cell death, tBHQ was shown in another study to be able to protect 6-OHDA-induced damage in live mice [162]. Another experimental study revealed that cortical astrocytes from aged rats can respond to tBHQ pretreatment and stimulate the Nrf2-antioxidant response pathway to induce antioxidant strategies against MPP + (1-methyl-4-phenylpyridinium) toxicity, increase antioxidant enzymes and form cell protection, which is of great significance for the development and prevention or counteraction of diseases involving oxidative stress, such as AD, PD or other diseases [163].
Conclusion
In conclusion, tBHQ has neuroprotective effects on brain diseases through anti-inflammatory, anti-oxidative stress, and inhibition of apoptotic protein expression, among which, it is particularly important in inhibiting inflammation and anti-oxidative stress. In addition to up-regulating the Nrf2 gene and increasing the expression of Nrf2, tBHQ’s role as an agonist of Nrf2 also allows it to block the activity of NF-κB and pro-inflammatory factors while inducing the production of anti-inflammatory proteins. Although the antioxidant mechanism of tBHQ is not completely clear, it can also exert anti-oxidative stress through Nrf2. According to studies, tBHQ can prevent neurotoxicity brought on by oxidative stress by activating the Nrf2-ARE and Keap1-Nrf2-ARE signaling pathways [164, 165], regulating NADPH oxidase system, regulating the activation of astrocytes and regulating GSH pathway [166]. In addition, tBHQ effectively prevents the production of related oxidation products, increases the level of antioxidant substances, and has a protective effect on the oxidative damage generated by the body, and at the same time promotes the recovery of the anti-oxidative mechanism of the damaged body. The preceding sections of this research go into great length into the neuroprotective mechanisms of tBHQ against AD, stroke, depression, PD and other brain diseases; they are summarized in Table 1.
In-depth study on tBHQ has drawn a lot of attention to its safety, even though certain research reports have indicated that tBHQ has a great deal of potential in the treatment of brain diseases [167]. Although tBHQ is frequently utilized as an antioxidant [168], researches have revealed that it also has certain harmful and carcinogenic properties [169,170,171]. The ability of tBHQ to generate ROS during the redox cycle can lead to cytotoxicity at high concentrations, which is strongly tied to this ability [172]. High-dose tBHQ can promote apoptosis and carcinogenicity in food additives, which slows the rate of growth of normal cells by causing apoptosis through chromatin and DNA fragmentation and causes cytotoxicity in a dose- and time-dependent manner [173]. Under in vitro conditions, the threshold concentration of tBHQ (30 M or less) induction of possible adverse effects is 30 M, and the range of tBHQ concentrations from beneficial to toxic may be 10 to 30 M, and tBHQ exhibits antioxidant effects below the concentration that causes adverse cellular reactions, so the blood concentration of tBHQ should not exceed 1.66 mg/L [174]. What is even more surprising is that studies have shown that a new type of antioxidant and prebiotic material can inhibit the toxicity of tBHQ by grafting tBHQ onto chitosan and further cross-linking to agavin, and the experimental results of acute oral toxicity in mice show that edible synthetic materials do not have adverse short-term effects [175]. This positive research data offers a point of reference and a specific research and development path for how to decrease the toxicity of tBHQ and enhance the safe use of clinical in the future.
As a result, even though tBHQ’s application has some flaws, this does not diminish the drug’s effectiveness. On the contrary, by examining its toxicity, tBHQ offers a safety assurance for the use of medications and paves the way for new avenues of investigation into ways to lessen tBHQ’s toxicity in upcoming research projects.
Data Availability
Not applicable.
References
Wu C, Yang L, Tucker D et al (2018) Beneficial effects of exercise pretreatment in a sporadic Alzheimer’s rat model. Med Sci Sports Exerc 50. https://doi.org/10.1249/MSS.0000000000001519
Yang L, Youngblood H, Wu C, Zhang Q (2020) Mitochondria as a target for neuroprotection: role of methylene blue and photobiomodulation. Transl Neurodegener 9. https://doi.org/10.1186/s40035-020-00197-z
Zuo L, Prather ER, Stetskiv M et al (2019) Inflammaging and oxidative stress in human diseases: from molecular mechanisms to novel treatments. Int J Mol Sci 20. https://doi.org/10.3390/ijms20184472
Spaas J, van Veggel L, Schepers M et al (2021) Oxidative stress and impaired oligodendrocyte precursor cell differentiation in neurological disorders. Cell Mol Life Sci CMLS 78. https://doi.org/10.1007/s00018-021-03802-0
Zhang J, Tucker LD, Lu Y et al (2018) Tert-butylhydroquinone post-treatment attenuates neonatal hypoxic-ischemic brain damage in rats. Neurochem Int 116. https://doi.org/10.1016/j.neuint.2018.03.004
Meng X, Zhang C, Guo Y et al (2020) TBHQ attenuates neurotoxicity induced by methamphetamine in the VTA through the Nrf2/HO-1 and PI3K/AKT signaling pathways. Oxidative Med Cell Longev 2020. https://doi.org/10.1155/2020/8787156
Zhang ZW, Liang J, Yan JX et al (2020) TBHQ improved neurological recovery after traumatic brain injury by inhibiting the overactivation of astrocytes. Brain Res 1739. https://doi.org/10.1016/j.brainres.2020.146818
Boss AP, Freeborn RA, Duriancik DM et al (2018) The Nrf2 activator tBHQ inhibits the activation of primary murine natural killer cells. Food Chem Toxicol:an Int J Published British Ind Biol Res Assoc 121. https://doi.org/10.1016/j.fct.2018.08.067
Zhang H, Li J, Xiang X et al (2021) Tert-butylhydroquinone attenuates osteoarthritis by protecting chondrocytes and inhibiting macrophage polarization. Bone Joint Res 10. https://doi.org/10.1302/2046-3758.1011.BJR-2020-0242.R4
Nna VU, Ujah GA, Suleiman JB et al (2020) Tert-butylhydroquinone preserve testicular steroidogenesis and spermatogenesis in cisplatin-intoxicated rats by targeting oxidative stress, inflammation and apoptosis. Toxicology 441. https://doi.org/10.1016/j.tox.2020.152528
Han G, Cao C, Yang X et al (2022) Nrf2 expands the intracellular pool of the chaperone AHSP in a cellular model of β-thalassemia. Redox Biol 50. https://doi.org/10.1016/j.redox.2022.102239
Ying YT, Yang J, Tan X et al (2021) Escherichia coli and Staphylococcus aureus differentially regulate Nrf2 pathway in bovine mammary epithelial cells: relation to distinct innate immune response. Cells 10. https://doi.org/10.3390/cells10123426
Ren J, Li L, Wang Y et al (2019) Gambogic acid induces heme oxygenase-1 through Nrf2 signaling pathway and inhibits NF-κB and MAPK activation to reduce inflammation in LPS-activated RAW264.7 cells. Biomed Pharmacother=Biomed Pharmacotherapie 109. https://doi.org/10.1016/j.biopha.2018.10.112
An Y, Li H, Wang M et al (2022) Nuclear factor erythroid 2-related factor 2 agonist protects retinal ganglion cells in glutamate excitotoxicity retinas. Biomed Pharmacother 153:113378. https://doi.org/10.1016/j.biopha.2022.113378
Ding Z, Pothineni NVK, Goel A et al (2020) PCSK9 and inflammation: role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc Res 116:908–915. https://doi.org/10.1093/cvr/cvz313
Olkowska-Truchanowicz J, Białoszewska A, Zwierzchowska A et al (2021) Peritoneal fluid from patients with ovarian endometriosis displays immunosuppressive potential and stimulates Th2 response. Int J Mol Sci 22:8134. https://doi.org/10.3390/ijms22158134
Chen Z, Bozec A, Ramming A, Schett G (2019) Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol 15:9–17. https://doi.org/10.1038/s41584-018-0109-2
Vallée A, Lecarpentier Y (2018) Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical WNT/β-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol 9:745. https://doi.org/10.3389/fimmu.2018.00745
Huggard D, Kelly L, Ryan E et al (2020) Increased systemic inflammation in children with Down syndrome. Cytokine 127. https://doi.org/10.1016/j.cyto.2019.154938
Sundd P, Gladwin MT, Novelli EM (2019) Pathophysiology of sickle cell disease. Ann Rev Pathol 14. https://doi.org/10.1146/annurev-pathmechdis-012418-012838
Ghosh S, Choudhury S, Chowdhury O et al (2020) Inflammation-induced behavioral changes is driven by alterations in Nrf2-dependent apoptosis and autophagy in mouse hippocampus: Role of fluoxetine. Cell Signal 68. https://doi.org/10.1016/j.cellsig.2019.109521
Zhu Z, Chen X, Sun J et al (2019) Inhibition of nuclear thioredoxin aggregation attenuates PM 2.5-induced NF-κB activation and pro-inflammatory responses. Free Radic Biol Med 130. https://doi.org/10.1016/j.freeradbiomed.2018.10.438
Jin W, Ni H, Dai Y et al (2010) Effects of tert-butylhydroquinone on intestinal inflammatory response and apoptosis following traumatic brain injury in mice. Mediat Inflamm 2010. https://doi.org/10.1155/2010/502564
Jin W, Ni H, Hou X et al (2014) Tert-butylhydroquinone protects the spinal cord against inflammatory response produced by spinal cord injury. Ann Clin Lab Sci 44
Huang Z, Li X, Zhou T et al (2021) Phosphorylated nuclear factor erythroid 2-related factor 2 promotes the secretion of C-C motif chemokine ligand 2 and the recruitment of M2 macrophages. Ann Transl Med 9. https://doi.org/10.21037/atm-21-2947
Lazaro I, Lopez-Sanz L, Bernal S et al (2018) Nrf2 activation provides atheroprotection in diabetic mice through concerted upregulation of antioxidant, anti-inflammatory, and autophagy mechanisms. Front Pharmacol 9. https://doi.org/10.3389/fphar.2018.00819
Bai J, Yu XJ, Liu KL et al (2017) Tert-butylhydroquinone attenuates oxidative stress and inflammation in hypothalamic paraventricular nucleus in high salt-induced hypertension. Toxicol Lett 281. https://doi.org/10.1016/j.toxlet.2017.08.018
Gong D-J, Wang L, Yang Y-Y et al (2019) Diabetes aggravates renal ischemia and reperfusion injury in rats by exacerbating oxidative stress, inflammation, and apoptosis. Ren Fail 41:750–761. https://doi.org/10.1080/0886022X.2019.1643737
Merelli A, Repetto M, Lazarowski A, Auzmendi J (2021) Hypoxia, oxidative stress, and inflammation: three faces of neurodegenerative diseases. J Alzheimers Dis:JAD 82. https://doi.org/10.3233/JAD-201074
Marcovecchio GE, Ferrua F, Fontana E et al (2021) Premature senescence and increased oxidative stress in the thymus of Down syndrome patients. Front Immunol 12. https://doi.org/10.3389/fimmu.2021.669893
Minguzzi M, Cetrullo S, D’Adamo S et al (2018) Emerging players at the intersection of chondrocyte loss of maturational arrest, oxidative stress, senescence and low-grade inflammation in osteoarthritis. Oxidative Med Cell Longev 2018. https://doi.org/10.1155/2018/3075293
Ghezzi P, Jaquet V, Marcucci F, Schmidt HH (2017) The oxidative stress theory of disease: levels of evidence and epistemological aspects. Br J Pharmacol 174. https://doi.org/10.1111/bph.13544
Bellmaine S, Schnellbaecher A, Zimmer A (2020) Reactivity and degradation products of tryptophan in solution and proteins. Free Radic Biol Med 160:696–718. https://doi.org/10.1016/j.freeradbiomed.2020.09.002
Bresolí-Obach R, Frattini M, Abbruzzetti S et al (2020) Tetramethylbenzidine: an acoustogenic photoacoustic probe for reactive oxygen species detection. Sensors (Basel) 20:E5952. https://doi.org/10.3390/s20205952
Chauvin J, Judée F, Yousfi M et al (2017) Analysis of reactive oxygen and nitrogen species generated in three liquid media by low temperature helium plasma jet. Sci Rep 7:4562. https://doi.org/10.1038/s41598-017-04650-4
Emami NK, Jung U, Voy B, Dridi S (2020) Radical response: effects of heat stress-induced oxidative stress on lipid metabolism in the avian liver. Antioxidants (Basel, Switzerland) 10. https://doi.org/10.3390/antiox10010035
Ray PD, Huang B-W, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24:981–990. https://doi.org/10.1016/j.cellsig.2012.01.008
Farzaei MH, Zobeiri M, Parvizi F et al (2018) Curcumin in liver diseases: a systematic review of the cellular mechanisms of oxidative stress and clinical perspective. Nutrients 10. https://doi.org/10.3390/nu10070855
Gavia-García G, Rosas-Trejo MDLÁ, García-Mendoza E et al (2018) t-BHQ protects against oxidative damage and maintains the antioxidant response in malnourished rats. Dose-Response : Publ Int Hormesis Soc 16. https://doi.org/10.1177/1559325818796304
Zhang H, Zhou L, Zhou Y et al (2021) Intermittent hypoxia aggravates non-alcoholic fatty liver disease via RIPK3-dependent necroptosis-modulated Nrf2/NFκB signaling pathway. Life Sci 285. https://doi.org/10.1016/j.lfs.2021.119963
García-Aguilar A, Palomino O, Benito M, Guillén C (2021) Dietary polyphenols in metabolic and neurodegenerative diseases: molecular targets in autophagy and biological effects. Antioxidants 10. https://doi.org/10.3390/antiox10020142
Camiña N, Penning TM (2022) Genetic and epigenetic regulation of the NRF2-KEAP1 pathway in human lung cancer. Br J Cancer 126. https://doi.org/10.1038/s41416-021-01642-0
Tu W, Wang H, Li S et al (2019) The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis 10:637–651. https://doi.org/10.14336/AD.2018.0513
Yang B, Huang H, He Q et al (2021) Tert-butylhydroquinone prevents oxidative stress-mediated apoptosis and extracellular matrix degradation in rat chondrocytes. Evidence-based complementary and alternative medicine : eCAM 2021. https://doi.org/10.1155/2021/1905995
Li R, Zhang P, Li C et al (2020) Tert-butylhydroquinone mitigates carbon tetrachloride induced hepatic injury in mice. Int J Med Sci 17. https://doi.org/10.7150/ijms.45842
Veskemaa L, Graw JA, Pickerodt PA et al (2021) Tert-butylhydroquinone augments Nrf2-dependent resilience against oxidative stress and improves survival of ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 320. https://doi.org/10.1152/ajplung.00131.2020
Zhao YL, Zhao W, Liu M et al (2020) TBHQ-overview of multiple mechanisms against oxidative stress for attenuating methamphetamine-induced neurotoxicity. Oxidative Med Cell Longev 2020. https://doi.org/10.1155/2020/8874304
Wang Y, Gu YH, Liu M et al (2017) TBHQ alleviated endoplasmic reticulum stress-apoptosis and oxidative stress by PERK-Nrf2 crosstalk in methamphetamine-induced chronic pulmonary toxicity. Oxidative Med Cell Longev 2017. https://doi.org/10.1155/2017/4310475
Hu L, Tian K, Zhang T et al (2019) Cyanate induces oxidative stress injury and abnormal lipid metabolism in liver through Nrf2/HO-1. Molecules (Basel, Switzerland) 24. https://doi.org/10.3390/molecules24183231
Li HY, Zhong YF, Wu SY, Shi N (2007) NF-E2 related factor 2 activation and heme oxygenase-1 induction by tert-butylhydroquinone protect against deltamethrin-mediated oxidative stress in PC12 cells. Chem Res Toxicol 20. https://doi.org/10.1021/tx700076q
Wu J, Cheng M, Liu Q et al (2015) Protective role of tert-butylhydroquinone against sodium fluoride-induced oxidative stress and apoptosis in PC12 cells. Cell Mol Neurobiol 35. https://doi.org/10.1007/s10571-015-0196-4
Xu W, Li F, Xu Z et al (2017) Tert-butylhydroquinone protects PC12 cells against ferrous sulfate-induced oxidative and inflammatory injury via the Nrf2/ARE pathway. Chem-Biol Interact 273. https://doi.org/10.1016/j.cbi.2017.05.021
Song H, Xu Y, Yang X et al (2019) Tertiary butylhydroquinone alleviates gestational diabetes mellitus in C57BL/KsJ-Lep db/+ mice by suppression of oxidative stress. J Cell Biochem 120. https://doi.org/10.1002/jcb.28798
Lu XY, Wang HD, Xu JG et al (2014) Pretreatment with tert-butylhydroquinone attenuates cerebral oxidative stress in mice after traumatic brain injury. J Surg Res 188. https://doi.org/10.1016/j.jss.2013.11.1106
Misrani A, Tabassum S, Yang L (2021) Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front Aging Neurosci 13:617588. https://doi.org/10.3389/fnagi.2021.617588
Zhang D-F, Xu M, Bi R, Yao Y-G (2019) Genetic analyses of Alzheimer’s disease in China: achievements and perspectives. ACS Chem Neurosci 10:890–901. https://doi.org/10.1021/acschemneuro.8b00435
Andrieu S, Coley N, Lovestone S et al (2015) Prevention of sporadic Alzheimer’s disease: lessons learned from clinical trials and future directions. Lancet Neurol 14:926–944. https://doi.org/10.1016/S1474-4422(15)00153-2
Peng Y, Chang X, Lang M (2021) Iron homeostasis disorder and Alzheimer’s disease. Int J Mol Sci 22:12442. https://doi.org/10.3390/ijms222212442
Schott JM, Aisen PS, Cummings JL et al (2019) Unsuccessful trials of therapies for Alzheimer’s disease. Lancet (London, England) 393. https://doi.org/10.1016/S0140-6736(18)31896-8
Akhter H, Katre A, Li L et al (2011) Therapeutic potential and anti-amyloidosis mechanisms of tert-butylhydroquinone for Alzheimer’s disease. J Alzheimers Dis:JAD 26. https://doi.org/10.3233/JAD-2011-110512
Eftekharzadeh B, Maghsoudi N, Khodagholi F (2010) Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 92. https://doi.org/10.1016/j.biochi.2009.12.001
Bahia PK, Pugh V, Hoyland K et al (2012) Neuroprotective effects of phenolic antioxidant tBHQ associate with inhibition of FoxO3a nuclear translocation and activity. J Neurochem 123. https://doi.org/10.1111/j.1471-4159.2012.07877.x
Ferriero DM, Fullerton HJ, Bernard TJ et al (2019) Management of stroke in neonates and children: a scientific statement from the American Heart Association/American Stroke Association. Stroke 50:e51–e96. https://doi.org/10.1161/STR.0000000000000183
Gaire BP (2018) Herbal medicine in ischemic stroke: challenges and prospective. Chin J Integr Med 24. https://doi.org/10.1007/s11655-018-2828-2
Wang W, Jiang B, Sun H et al (2017) Prevalence, incidence, and mortality of stroke in China: results from a nationwide population-based survey of 480 687 adults. Circulation 135. https://doi.org/10.1161/CIRCULATIONAHA.116.025250
Phipps MS, Cronin CA (2020) Management of acute ischemic stroke. BMJ 368:l6983. https://doi.org/10.1136/bmj.l6983
Liu Y, Chu S, Hu Y et al (2021) Exogenous adenosine antagonizes excitatory amino acid toxicity in primary astrocytes. Cell Mol Neurobiol 41. https://doi.org/10.1007/s10571-020-00876-5
Sun MS, Jin H, Sun X et al (2018) Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxidative Med Cell Longev 2018. https://doi.org/10.1155/2018/3804979
Bourhy L, Mazeraud A, Bozza FA et al (2022) Neuro-inflammatory response and brain-peripheral crosstalk in sepsis and stroke. Front Immunol 13. https://doi.org/10.3389/fimmu.2022.834649
Sehara Y, Inaba T, Urabe T et al (2018) Survivin overexpression via adeno-associated virus vector Rh10 ameliorates ischemic damage after middle cerebral artery occlusion in rats. Eur J Neurosci 48. https://doi.org/10.1111/ejn.14169
Faura J, Bustamante A, Miró-Mur F, Montaner J (2021) Stroke-induced immunosuppression: implications for the prevention and prediction of post-stroke infections. J Neuroinflammation 18. https://doi.org/10.1186/s12974-021-02177-0
Zhang W, Tian T, Gong SX et al (2021) Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke. Neural Regen Res 16. https://doi.org/10.4103/1673-5374.286954
Nguyen KD, Qiu Y, Cui X et al (2011) Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480. https://doi.org/10.1038/nature10653
Liu L, Liu J, Bao J et al (2020) Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol 11. https://doi.org/10.3389/fimmu.2020.01024
Bylicky MA, Mueller GP, Day RM (2018) Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxidative Med Cell Longev 2018. https://doi.org/10.1155/2018/6501031
Taylor RA, Chang CF, Goods BA et al (2017) TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Investig 127. https://doi.org/10.1172/JCI88647
Beuker C, Strecker JK, Rawal R et al (2021) Immune cell infiltration into the brain after ischemic stroke in humans compared to mice and rats: a systematic review and meta-analysis. Transl Stroke Res 12. https://doi.org/10.1007/s12975-021-00887-4
Nowicka D, Rogozinska K, Aleksy M et al (2008) Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol Exp 68
Appel SH, Zhao W, Beers DR, Henkel JS (2011) The microglial-motoneuron dialogue in ALS. Acta myologica : myopathies and cardiomyopathies : Off J Mediterr Soc Myology 30
Zhou M, Guo C, Li X et al (2020) JAK/STAT signaling controls the fate of CD8 + CD103 + tissue-resident memory T cell in lupus nephritis. J Autoimmun 109. https://doi.org/10.1016/j.jaut.2020.102424
Ge J-W, Deng S-J, Xue Z-W et al (2022) Imperatorin inhibits mitogen-activated protein kinase and nuclear factor kappa-B signaling pathways and alleviates neuroinflammation in ischemic stroke. CNS Neurosci Ther 28:116–125. https://doi.org/10.1111/cns.13748
Won JS, Kim J, Annamalai B et al (2013) Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J Alzheimers Dis:JAD 34. https://doi.org/10.3233/JAD-121786
Kim MS, Bang JH, Lee J et al (2015) Salvia miltiorrhiza extract protects white matter and the hippocampus from damage induced by chronic cerebral hypoperfusion in rats. BMC Complement Alternat Med 15. https://doi.org/10.1186/s12906-015-0943-6
Winneberger J, Schöls S, Lessmann K et al (2021) Platelet endothelial cell adhesion molecule-1 is a gatekeeper of neutrophil transendothelial migration in ischemic stroke. Brain, Behav, Immun 93. https://doi.org/10.1016/j.bbi.2020.12.026
Farris BY, Monaghan KL, Zheng W et al (2019) Ischemic stroke alters immune cell niche and chemokine profile in mice independent of spontaneous bacterial infection. Immun, Inflamm Dis 7. https://doi.org/10.1002/iid3.277
Yang C, Hawkins KE, Doré S, Candelario-Jalil E (2019) Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol 316. https://doi.org/10.1152/ajpcell.00136.2018
Cheng YC, Sheen JM, Hu WL, Hung YC (2017) Polyphenols and oxidative stress in atherosclerosis-related ischemic heart disease and stroke. Oxidative Med Cell Longev 2017. https://doi.org/10.1155/2017/8526438
Kahles T, Brandes RP (2012) NADPH oxidases as therapeutic targets in ischemic stroke. Cell Mol Life Sci 69:2345–2363. https://doi.org/10.1007/s00018-012-1011-8
Shih AY, Li P, Murphy TH (2005) A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci: Off J Soc Neurosci 25. https://doi.org/10.1523/JNEUROSCI.4014-05.2005
Zhou NQ, Liu N, Li P et al (2017) Tert-butylhydroquinone promotes angiogenesis and improves heart functions in rats after myocardial infarction. Clin Exp Hypertens (New York, NY : 1993) 39. https://doi.org/10.1080/10641963.2016.1259322
Segev-Amzaleg N, Trudler D, Frenkel D (2013) Preconditioning to mild oxidative stress mediates astroglial neuroprotection in an IL-10-dependent manner. Brain, Behav, Immunity 30. https://doi.org/10.1016/j.bbi.2012.12.016
Chen Y, Zhang X, Yang Y et al (2019) Tert-butylhydroquinone enhanced angiogenesis and astrocyte activation by activating nuclear factor-E2-related factor 2/heme oxygenase-1 after focal cerebral ischemia in mice. Microvasc Res 126. https://doi.org/10.1016/j.mvr.2019.103891
Sun J, Ren X, Simpkins JW (2015) Sequential upregulation of superoxide dismutase 2 and heme oxygenase 1 by tert-butylhydroquinone protects mitochondria during oxidative stress. Mol Pharmacol 88. https://doi.org/10.1124/mol.115.098269
Doll DN, Hu H, Sun J et al (2015) Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 46. https://doi.org/10.1161/STROKEAHA.115.009099
Sun J, Hu H, Ren X, Simpkins JW (2016) Tert-butylhydroquinone compromises survival in murine experimental stroke. Neurotoxicol Teratol 54. https://doi.org/10.1016/j.ntt.2016.01.004
Donovan NJ, Wu Q, Rentz DM et al (2017) Loneliness, depression and cognitive function in older U.S. adults. Int J Geriatr Psychiatr 32. https://doi.org/10.1002/gps.4495
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3:e442. https://doi.org/10.1371/journal.pmed.0030442
Zhou S, Chen R, She Y et al (2022) A new perspective on depression and neuroinflammation: non-coding RNA. J Psychiatr Res 148. https://doi.org/10.1016/j.jpsychires.2022.02.007
Yang L, Wu C, Tucker L et al (2021) Photobiomodulation therapy attenuates anxious-depressive-like behavior in the TgF344 rat model. J Alzheimers Dis:JAD 83. https://doi.org/10.3233/JAD-201616
Wang YL, Wu HR, Zhang SS et al (2021) Catalpol ameliorates depressive-like behaviors in CUMS mice via oxidative stress-mediated NLRP3 inflammasome and neuroinflammation. Transl Psychiatr 11. https://doi.org/10.1038/s41398-021-01468-7
Li W, Ali T, He K et al (2021) Ibrutinib alleviates LPS-induced neuroinflammation and synaptic defects in a mouse model of depression. Brain Behav Immun 92:10–24. https://doi.org/10.1016/j.bbi.2020.11.008
Allen J, Romay-Tallon R, Brymer KJ et al (2018) Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Front Neurosci 12. https://doi.org/10.3389/fnins.2018.00386
Zhang Z, Ni J, Zhang J et al (2016) A haplotype in the 5’-upstream region of the NDUFV2 gene is associated with major depressive disorder in Han Chinese. J Affect Disord 190. https://doi.org/10.1016/j.jad.2015.10.034
Hayley S, Hakim AM, Albert PR (2021) Depression, dementia and immune dysregulation. Brain 144:746–760. https://doi.org/10.1093/brain/awaa405
Salvador AF, de Lima KA, Kipnis J (2021) Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol 21:526–541. https://doi.org/10.1038/s41577-021-00508-z
Cazareth J, Guyon A, Heurteaux CH et al (2014) Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. J Neuroinflammation 11. https://doi.org/10.1186/1742-2094-11-132
Torres-Platas SG, Cruceanu C, Chen GG et al (2014) Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain, Behavior, Immunity 42. https://doi.org/10.1016/j.bbi.2014.05.007
Xia W, Xu Y, Gong Y et al (2022) Microglia involves in the immune inflammatory response of poststroke depression: a review of evidence. Oxid Med Cell Longev 2022:2049371. https://doi.org/10.1155/2022/2049371
Passos IC, Vasconcelos-Moreno MP, Costa LG et al (2015) Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2. https://doi.org/10.1016/S2215-0366(15)00309-0
Manigault AW, Ganz PA, Irwin MR et al (2021) Moderators of inflammation-related depression: a prospective study of breast cancer survivors. Transl Psychiatry 11:615. https://doi.org/10.1038/s41398-021-01744-6
Schmidt FM, Lichtblau N, Minkwitz J et al (2014) Cytokine levels in depressed and non-depressed subjects, and masking effects of obesity. J Psychiatric Res 55. https://doi.org/10.1016/j.jpsychires.2014.04.021
Varfolomeev EE, Ashkenazi A (2004) Tumor necrosis factor: an apoptosis JuNKie? Cell 116. https://doi.org/10.1016/s0092-8674(04)00166-7
Karson A, Demirtaş T, Bayramgürler D et al (2013) Chronic administration of infliximab (TNF-α inhibitor) decreases depression and anxiety-like behaviour in rat model of chronic mild stress. Basic Clin Pharmacol Toxicol 112. https://doi.org/10.1111/bcpt.12037
JW DK (2008) IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S Am 105. https://doi.org/10.1073/pnas.0708092105
Kosuge A, Kunisawa K, Arai S et al (2021) Heat-sterilized Bifidobacterium breve prevents depression-like behavior and interleukin-1β expression in mice exposed to chronic social defeat stress. Brain Behav Immun 96:200–211. https://doi.org/10.1016/j.bbi.2021.05.028
Park HJ, Shim HS, An K et al (2015) IL-4 inhibits IL-1β-induced depressive-like behavior and central neurotransmitter alterations. Mediat Inflamm 2015. https://doi.org/10.1155/2015/941413
Dahl J, Ormstad H, Aass HCD et al (2014) The plasma levels of various cytokines are increased during ongoing depression and are reduced to normal levels after recovery. Psychoneuroendocrinology 45. https://doi.org/10.1016/j.psyneuen.2014.03.019
Ng A, Tam WW, Zhang MW et al (2018) IL-1β, IL-6, TNF- α and CRP in elderly patients with depression or Alzheimer’s disease: systematic review and meta-analysis. Sci Rep 8. https://doi.org/10.1038/s41598-018-30487-6
Black CN, Bot M, Scheffer PG et al (2015) Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 51. https://doi.org/10.1016/j.psyneuen.2014.09.025
Black CN, Bot M, Scheffer PG, Penninx BWJH (2017) Oxidative stress in major depressive and anxiety disorders, and the association with antidepressant use; results from a large adult cohort. Psychol Med 47. https://doi.org/10.1017/S0033291716002828
Aydın EP, Genc A, Dalkıran M et al (2018) Thioredoxin is not a marker for treatment-resistance depression but associated with cognitive function: An rTMS study. Progress Neuro-Psychopharmacol Biol Psychiatry 80. https://doi.org/10.1016/j.pnpbp.2017.04.025
Talarowska M, Gałecki P, Maes M et al (2012) Malondialdehyde plasma concentration correlates with declarative and working memory in patients with recurrent depressive disorder. Mol Biol Rep 39. https://doi.org/10.1007/s11033-011-1335-8
Anderson G (2018) Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Progress Neuro-psychopharmacol Biol Psychiatry 80. https://doi.org/10.1016/j.pnpbp.2017.04.022
Gałecki P, Talarowska M, Anderson G et al (2015) Mechanisms underlying neurocognitive dysfunctions in recurrent major depression. Med Sci Monitor : Int Med J Exp Clin Res 21. https://doi.org/10.12659/MSM.893176
Kuffner K, Triebelhorn J, Meindl K et al (2020) Major depressive disorder is associated with impaired mitochondrial function in skin fibroblasts. Cells 9. https://doi.org/10.3390/cells9040884
Holper L, Ben-Shachar D, Mann JJ (2019) Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacol: Off Publ Am Coll Neuropsychopharmacol 44. https://doi.org/10.1038/s41386-018-0090-0
Emmerzaal TL, Preston G, Geenen B et al (2020) Impaired mitochondrial complex I function as a candidate driver in the biological stress response and a concomitant stress-induced brain metabolic reprogramming in male mice. Transl Psychiatr 10. https://doi.org/10.1038/s41398-020-0858-y
Islam MR, Islam MR, Ahmed I et al (2018) Elevated serum levels of malondialdehyde and cortisol are associated with major depressive disorder: a case-control study. SAGE Open Med 6. https://doi.org/10.1177/2050312118773953
Yuan Q, Li Y, Deng X et al (2019) Effects of Xingpi Kaiyu Fang on ATP, Na/K-ATPase, and respiratory chain complexes of hippocampus and gastrocnemius muscle in depressed rats. Evid-Based Complement Alternat Med: eCAM 2019. https://doi.org/10.1155/2019/6054926
Consortium C (2015) Sparse whole genome sequencing identifies two loci for major depressive disorder. Nature 523:588. https://doi.org/10.1038/nature14659
Gebara E, Zanoletti O, Ghosal S et al (2021) Mitofusin-2 in the nucleus accumbens regulates anxiety and depression-like behaviors through mitochondrial and neuronal actions. Biol Psychiatr 89. https://doi.org/10.1016/j.biopsych.2020.12.003
Guo W, Tang ZY, Cai ZY et al (2021) Iptakalim alleviates synaptic damages via targeting mitochondrial ATP-sensitive potassium channel in depression. FASEB J: Off Publ Fed Am Soc Exp Biol 35. https://doi.org/10.1096/fj.202100124RR
Wang Y, Ni J, Gao C et al (2019) Mitochondrial transplantation attenuates lipopolysaccharide- induced depression-like behaviors. Progress Neuro-Psychopharmacol Biol Psychiatr 93. https://doi.org/10.1016/j.pnpbp.2019.04.010
Zhu X, Liu H, Liu Y et al (2020) The antidepressant-like effects of hesperidin in streptozotocin‐induced diabetic rats by activating Nrf2/ARE/glyoxalase 1 pathway. Front Pharmacol 11. https://doi.org/10.3389/fphar.2020.01325
Tolosa E, Garrido A, Scholz SW, Poewe W (2021) Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol 20. https://doi.org/10.1016/S1474-4422(21)00030-2
Cardinale A, Calabrese V, de Iure A, Picconi B (2021) Alpha-synuclein as a prominent actor in the inflammatory synaptopathy of Parkinson’s disease. Int J Mol Sci 22. https://doi.org/10.3390/ijms22126517
Picconi B, Hernández LF, Obeso JA, Calabresi P (2018) Motor complications in Parkinson’s disease: striatal molecular and electrophysiological mechanisms of dyskinesias. Mov Disord: Off J Mov Disord Soc 33. https://doi.org/10.1002/mds.27261
Jankovic J, Tan EK (2020) Parkinson’s disease: etiopathogenesis and treatment. J Neurol, Neurosurg, Psychiatr 91. https://doi.org/10.1136/jnnp-2019-322338
Ingelsson M (2016) Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front Neurosci 10. https://doi.org/10.3389/fnins.2016.00408
Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy - PubMed. https://pubmed.ncbi.nlm.nih.gov/33323315/. Accessed 1 Dec 2022
Dionísio PA, Amaral JD, Rodrigues CMP (2021) Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res Rev 67. https://doi.org/10.1016/j.arr.2021.101263
Nalls MA, Blauwendraat C, Vallerga CL et al (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18. https://doi.org/10.1016/S1474-4422(19)30320-5
Carballo-Carbajal I, Laguna A, Romero-Giménez J, et al (2019) Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nature Commun 10. https://doi.org/10.1038/s41467-019-08858-y
Chai M, Kohyama J (2019) Non-cell-autonomous neurotoxicity in Parkinson’s disease mediated by astroglial α-synuclein. Stem Cell Reports 12:183–185. https://doi.org/10.1016/j.stemcr.2019.01.011
Fasano A, Visanji NP, Liu L et al (2015) Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 14. https://doi.org/10.1016/S1474-4422(15)00007-1
Kam T-I, Hinkle JT, Dawson TM, Dawson VL (2020) Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol Dis 144:105028. https://doi.org/10.1016/j.nbd.2020.105028
Hiebert NM, Seergobin KN, Vo A et al (2014) Dopaminergic therapy affects learning and impulsivity in Parkinson’s disease. Ann Clin Transl Neurol 1. https://doi.org/10.1002/acn3.128
Niranjan R, Mishra KP, Thakur AK (2018) Inhibition of cyclooxygenase-2 (COX-2) Initiates autophagy and potentiates MPTP-induced autophagic cell death of human neuroblastoma cells, SH-SY5Y: an inside in the pathology of Parkinson’s disease. Mol Neurobiol 55. https://doi.org/10.1007/s12035-018-0950-y
Maki RA, Holzer M, Motamedchaboki K et al (2019) Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radical Biol Med 141. https://doi.org/10.1016/j.freeradbiomed.2019.05.033
Gao HM, Hong JS (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29. https://doi.org/10.1016/j.it.2008.05.002
Kwon HS, Koh S-H (2020) Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener 9:42. https://doi.org/10.1186/s40035-020-00221-2
Russ K, Teku G, Bousset L et al (2021) TNF-α and α-synuclein fibrils differently regulate human astrocyte immune reactivity and impair mitochondrial respiration. Cell Rep 34:108895. https://doi.org/10.1016/j.celrep.2021.108895
Angelopoulou E, Paudel YN, Piperi C (2021) Emerging role of S100B protein implication in Parkinson’s disease pathogenesis. Cell Mol Life Sci: CMLS 78. https://doi.org/10.1007/s00018-020-03673-x
Xu J, Xiao C, Song W et al (2021) Elevated heme oxygenase-1 correlates with increased brain iron deposition measured by quantitative susceptibility mapping and decreased hemoglobin in patients with Parkinson’s disease. Front Aging Neurosci 13. https://doi.org/10.3389/fnagi.2021.656626
Singh B, Pandey S, Yadav SK et al (2017) Role of ethanolic extract of Bacopa monnieri against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced mice model via inhibition of apoptotic pathways of dopaminergic neurons. Brain Res Bull 135. https://doi.org/10.1016/j.brainresbull.2017.10.007
Bjørklund G, Peana M, Maes M et al (2021) The glutathione system in Parkinson’s disease and its progression. Neurosci Biobehavioral Rev 120. https://doi.org/10.1016/j.neubiorev.2020.10.004
Liu CB, Wang R, Pan HB et al (2013) [Effect of lycopene on oxidative stress and behavioral deficits in rotenone induced model of Parkinson’s disease]. Zhongguo ying yong sheng li xue za zhi = Zhongguo yingyong shenglixue zazhi = Chin J Appl Physiol 29
Jiménez-Delgado A, Ortiz GG, Delgado-Lara DL et al (2021) Effect of melatonin administration on mitochondrial activity and oxidative stress markers in patients with Parkinson’s disease. Oxidative Med Cell Longev 2021. https://doi.org/10.1155/2021/5577541
Romano A, Serviddio G, Calcagnini S et al (2017) Linking lipid peroxidation and neuropsychiatric disorders: focus on 4-hydroxy-2-nonenal. Free Radical Biol Med 111. https://doi.org/10.1016/j.freeradbiomed.2016.12.046
Hara H, Ohta M, Ohta K et al (2003) Increase of antioxidative potential by tert-butylhydroquinone protects against cell death associated with 6-hydroxydopamine-induced oxidative stress in neuroblastoma SH-SY5Y cells. Brain Res Mol Brain Res 119. https://doi.org/10.1016/j.molbrainres.2003.08.021
Abdel‐Wahab MH (2005) Potential neuroprotective effect of t-butylhydroquinone against neurotoxicity-induced by 1-methyl-4-(2’-methylphenyl)-1,2,3,6-tetrahydropyridine (2’-methyl-MPTP) in mice. J Biochem Mol Toxicol 19. https://doi.org/10.1002/jbt.20053
Jakel RJ, Townsend JA, Kraft AD, Johnson JA (2007) NRF2-mediated protection against 6-hydroxydopamine. Brain Res 1144:192. https://doi.org/10.1016/j.brainres.2007.01.131
Alarcón-Aguilar A, Luna-López A, Ventura-Gallegos JL et al (2014) Primary cultured astrocytes from old rats are capable to activate the Nrf2 response against MPP+ toxicity after tBHQ pretreatment. Neurobiol Aging 35:1901–1912. https://doi.org/10.1016/j.neurobiolaging.2014.01.143
Veskemaa L, Graw JA, Pickerodt PA et al (2021) Tert-butylhydroquinone augments Nrf2-dependent resilience against oxidative stress and improves survival of ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 320:L17–L28. https://doi.org/10.1152/ajplung.00131.2020
Tkachev VO, Menshchikova EB, Zenkov NK (2011) Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Mosc) 76:407–422. https://doi.org/10.1134/s0006297911040031
Yadav E, Yadav P, Khan MMU et al (2022) Resveratrol: a potential therapeutic natural polyphenol for neurodegenerative diseases associated with mitochondrial dysfunction. Front Pharmacol 13:922232. https://doi.org/10.3389/fphar.2022.922232
Xi M, Shen D, Dai P et al (2022) TBHQ alleviates pyroptosis and necroptosis in chicken alveolar epithelial cells induced by fine particulate matter from broiler houses. Poult Sci 101:101593. https://doi.org/10.1016/j.psj.2021.101593
Townsend BE, Chen Y-J, Jeffery EH, Johnson RW (2014) Dietary broccoli mildly improves neuroinflammation in aged mice but does not reduce lipopolysaccharide-induced sickness behavior. Nutr Res 34:990–999. https://doi.org/10.1016/j.nutres.2014.10.001
Huang W, Gu Y, Niu H (2008) Determination of tertiary-butylhydroquinone and its metabolites in rat serum by liquid chromatography-ion trap mass spectrometry. Lipids 43. https://doi.org/10.1007/s11745-007-3135-4
Yang X, Sun Z, Wang W et al (2018) Developmental toxicity of synthetic phenolic antioxidants to the early life stage of zebrafish. Sci Total Environ 643. https://doi.org/10.1016/j.scitotenv.2018.06.213
Xu X, Liu A, Hu S et al (2021) Synthetic phenolic antioxidants: metabolism, hazards and mechanism of action. Food Chem 353. https://doi.org/10.1016/j.foodchem.2021.129488
Braeuning A, Vetter S, Orsetti S, Schwarz M (2012) Paradoxical cytotoxicity of tert-butylhydroquinone in vitro: what kills the untreated cells? Arch Toxicol 86. https://doi.org/10.1007/s00204-012-0841-3
Eskandani M, Hamishehkar H, Dolatabadi JEN (2014) Cytotoxicity and DNA damage properties of tert-butylhydroquinone (TBHQ) food additive. Food Chem 153. https://doi.org/10.1016/j.foodchem.2013.12.087
Kamemura N, Oyama K, Kanemaru K et al (2017) Diverse cellular actions of tert-butylhydroquinone, a food additive, on rat thymocytes. Toxicol Res 6. https://doi.org/10.1039/c7tx00183e
Hernández-Valdepeña MA, Pedraza-Chaverri J, Gracia-Mora I et al (2016) Suppression of the tert-butylhydroquinone toxicity by its grafting onto chitosan and further cross-linking to agavin toward a novel antioxidant and prebiotic material. Food Chem 199:485–491. https://doi.org/10.1016/j.foodchem.2015.12.042
Funding
The work was funded by Key Research and Development Program of Shandong Province (Grant numbers 2022TZXD0033 and 2019GSF107003) and National Natural Science Foundation of China (Grant numbers 21877075).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Preparation of materials and collection of literature were performed by Xiaojin Liu and Luodan Yang. The first draft of the manuscript was written by Xiaojin Liu, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Consent for Publication
The authors confirmed that all participants had consented to publication and that the study had not been published before.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, X., Yang, L., Zhang, G. et al. Neuroprotective Effects of Phenolic Antioxidant Tert-butylhydroquinone (tBHQ) in Brain Diseases. Mol Neurobiol 60, 4909–4923 (2023). https://doi.org/10.1007/s12035-023-03370-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03370-3