
Abstract
The phenolic diterpene carnosic acid (CA, C20H28O4) exerts antioxidant, anti-inflammatory, anti-apoptotic, and anti-cancer effects in mammalian cells. CA activates the nuclear factor erythroid 2-related factor 2 (Nrf2), among other signaling pathways, and restores cell viability in several in vitro and in vivo experimental models. We have previously reported that CA affords mitochondrial protection against various chemical challenges. However, it was not clear yet whether CA would prevent chemically induced impairment of the tricarboxylic acid cycle (TCA) function in mammalian cells. In the present work, we found that a pretreatment of human neuroblastoma SH-SY5Y cells with CA at 1 μM for 12 h prevented the hydrogen peroxide (H2O2)-induced impairment of the TCA enzymes (aconitase, α-ketoglutarate dehydrogenase (α-KGDH), succinate dehydrogenase (SDH)) and abolished the inhibition of the complexes I and V and restored the levels of ATP by a mechanism associated with Nrf2. CA also exhibited antioxidant abilities by enhancing the levels of reduced glutathione (GSH) and decreasing the content oxidative stress markers (cellular 8-oxo-2′-deoxyguanosine (8-oxo-dG), and mitochondrial malondialdehyde (MDA), protein carbonyl, and 3-nitrotyrosine). Silencing of Nrf2 by small interfering RNA (siRNA) abrogated the protective effects elicited by CA in mitochondria of SH-SY5Y cells. Therefore, CA prevented the H2O2-triggered mitochondrial impairment by an Nrf2-dependent mechanism. The specific role of Nrf2 in ameliorating the function of TCA enzymes function needs further research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondria, the major organelles involved in ATP synthesis, take a central role in the redox biology of mammalian cells. These double-membrane organelles are both a source and a target of reactive species [1–3]. The respiratory chain (complexes I–IV) generates the electrochemical potential necessary to the production of ATP by the complex V/ATP synthase [4, 5]. Disruption in the electron transport and consequent electron leakage from the respiratory chain leads to the production of the radical anion superoxide (O2 −•) [6–11], which undergoes dismutation after reacting with the mitochondrial form of superoxide dismutase (Mn-SOD) generating hydrogen peroxide (H2O2) [12–15]. H2O2 is a membrane-diffusible molecule, meaning that it may spread the pro-oxidant from one mitochondrion to another, as well as to other cellular compartments [16]. Indeed, H2O2 is utilized by mammalian cells as a signaling agent [17–19]. H2O2, which is not a free radical, is converted in water by catalase (CAT) or glutathione peroxidase (GPx) [20]. There is consumption of reduced glutathione (GSH) when GPx consumes H2O2 [21]. GSH is the major nonenzymatic antioxidant within mammalian cells and may be found in both cytosol and mitochondria [22]. In this regard, investigating strategies that lead to improvement of the mitochondria-located antioxidant defenses is of pharmacological interest due to the role of this intrinsic antioxidant in rescuing mammalian cells in cases of redox impairment [15, 23–29]. Actually, redox disturbances have been seen in several human diseases, such as neurodegeneration, cardiovascular dysfunction, diabetes mellitus, and inborn errors of metabolism, among others [30–34]. Furthermore, redox impairment may lead to disruption of the bioenergetics-related reactions in mammalian mitochondria, compromising cellular function and dynamics due to decreased access to ATP [35–37].
Carnosic acid (CA; C20H28O4; MW 332.43392 g/mol) is a diterpene found in rosemary (Rosmarinus officinalis L.) and exhibits antioxidant, anti-inflammatory, and anti-apoptotic capacities, as has been studied in several experimental models [38–44]. Moreover, CA is an anti-tumor agent [45–47]. We have recently demonstrated that CA protects mitochondria of SH-SY5Y cells challenged with different chemical stressors by a mechanism associated with the activation of the master regulator of the redox biology in mammalian cells, nuclear factor erythroid 2-related factor 2 (Nrf2) [48–50]. Nrf2 upregulation induced by CA caused an increase in the expression of antioxidant and phase II detoxification enzymes in SH-SY5Y cells, such as heme oxygenase-1 (HO-1), glutathione reductase (GR), GPx, and Mn-SOD, decreasing the susceptibility of mitochondria to pro-oxidant stressors. Other research groups also demonstrated that CA exhibited a protective role against different stimuli in neuronal cells in both in vitro and in vivo experimental designs [51–55]. Nonetheless, this is the first work reporting the ability of CA in preventing the H2O2-triggered mitochondria-related redox disruption in SH-SY5Y cells.
In the present work, we investigated whether CA would be effective in preventing mitochondrial impairment in SH-SY5Y cells exposed to the pro-oxidant agent H2O2.
Materials and Methods
Materials
Plastic materials utilized in cell culture were obtained from Corning, Inc. (NY, USA) and Beckton Dickson (NJ, USA). Culture analytical grade reagents were acquired from Sigma (MO, USA). Other chemicals and assay kits were obtained as described here.
Cell Culture and Chemical Treatment
Human dopaminergic neuroblastoma SH-SY5Y cells (obtained from the American Type Culture Collection (Manassas, VA, USA)) were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 HAM nutrient medium (1:1 mixture; supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin (1000 units/mL), streptomycin (1000 μg/mL), and amphotericin B (2.5 μg/mL)) in a 5% CO2 humidified incubator at 37 °C. SH-SY5Y cells were cultured until a confluence of 80–90% was achieved and then trypsinized.
H2O2 was utilized at 300 μM for different periods of incubation according to each specific assay. A pretreatment with CA (dissolved in DMSO) at 1 μM for 12 h was performed in order to test the ability of this diterpene in preventing the deleterious effects triggered by H2O2 in SH-SY5Y cells. More detailed information (specific concentrations and periods of treatment) may be achieved in the figure legends. The data are exhibited as the mean ± S.E.M. of three or five independent experiments each done in triplicate.
Analysis of Cell Viability
The viability of SH-SY5Y cells was analyzed by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, as previously described [56].
Quantification of Mitochondria-Related Apoptotic Factors and Cell Death-Associated Parameters
The immunocontents of Bcl-2, Bax, cytochrome c (mitochondrial and cytosolic), and cleaved PARP were quantified by using ELISA assay kits according to the instructions of the manufacturer (Abcam, MA, USA). The activities of the apoptotic enzymes caspase-9 and caspase-3 were assessed through the utilization of fluorimetric assay kits according to the instructions of the manufacturer (Abcam, MA, USA), as previously described [48–50, 57]. DNA fragmentation in cell lysates was measured by the utilization of an ELISA kit following the manufacturer’s instructions (Roche, Germany).
Measurement of Intracellular Reactive Oxygen Species Production
We utilized the nonpolar compound 2′-7′-dichlorodihydrofluorescein diacetate (DCFH-DA) to assess the intracellular production of ROS in this experimental model, as previously described [58]. The cells were loaded with 100 μM DCFH-DA dissolved in medium and were incubated for 30 min at 37 °C to allow cellular incorporation. After this incubation, the medium was changed to fresh medium and the oxidation of DCFH was registered during 30 min (1-min intervals) at 37 °C in a fluorescence (Em/Ex = 535/485 nm) plate reader (Molecular Devices, USA).
Quantification of MDA, Protein Carbonyl, and 8-Oxo-dG Levels
The levels of MDA, protein carbonyl, and 8-oxo-dG were determined by utilizing commercial kits (Abcam, MA, USA), as previously described [48–50, 57].
Analysis of 3-Nitrotyrosine Levels
The levels of 3-nitrotyrosine in mitochondrial membranes were examined by using a polyclonal antibody to 3-nitrotyrosine (Calbiochem, Germany) diluted to 1:2000 in phosphate-buffered saline (PBS) pH 7.4 with 5% albumin in an indirect ELISA assay, as previously reported [59–62].
Quantification of Total and Mitochondrial GSH Contents
The SH-SY5Y neuroblastoma cells were washed and collected, and GSH was measured according to the protocol of a commercial kit (Abcam, MA, USA), as described elsewhere [48–50, 57].
Isolation of Mitochondria
In order to obtain mitochondria from cultured cells, SH-SY5Y cells were washed and re-suspended in a buffer (250 mM sucrose, 10 mM KCl, 1 mM EGTA, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 1 mM pepstatin A, 10 mg/mL leupeptin, 2 mg/mL aprotonin, and 20 mM HEPES—pH 7.4), and differential centrifugations were performed, as previously reported [63].
Extraction of Submitochondrial Particles
Submitochondrial particles (SMPs) were obtained from SH-SY5Y cells according to a protocol previously published [64, 65]. Briefly, after the end of the incubations, the cells were homogenized in a buffer (230 mM mannitol, 70 mM sucrose, 10 mM Tris-HCl, and 1 mM EDTA—pH 7.4). The mitochondrial solution was frozen and thawed (three times) originating superoxide dismutase-free SMP. The solution containing SMP was washed (twice) using a buffer (140 mM KCl, 20 mM Tris-HCl—pH 7.4) in order to ensure Mn-superoxide dismutase leakage from the organelle. This protocol was used to quantify the production of superoxide anion radical (O2 −•) and to examine the effects of H2O2 and/or CA on the levels of malondialdehyde (MDA), protein carbonyl, and 3-nitrotyrosine in mitochondrial membranes [59].
Enzyme Activities
The activities of aconitase, α-ketoglutarate dehydrogenase (α-KGDH), succinate dehydrogenase (SDH), complex I, and complex V enzymes were measured through the utilization of commercial kits following the instructions of the manufacturer (Abcam, MA, USA).
Quantification of ATP Levels
ATP levels were measured by using a commercial kit (Abcam, MA, USA). Briefly, the cells were re-suspended and homogenized in a specific ATP assay buffer. After centrifugation, the samples rendered supernatants, which were collected and transferred to another tube. The samples were then deproteinizated and centrifuged, and the supernatants were utilized to determine the ATP levels. The reaction was read in a fluorescence (Ex/Em = 535/590 nm) plate reader (Molecular Devices, USA).
Determination of Mitochondrial Membrane Potential
Mitochondrial membrane potential (MMP) was studied by utilizing a kit that applies tetraethylbenzimidazolylcarbocyanide iodine (JC-1) as a lipophilic cationic dye that penetrates and accumulates in mitochondria according to variations in the membrane potential, as previously described [48–50, 57].
Nrf2 Knockdown by siRNA Transfection
Briefly, Nrf2 knockdown was performed through transient transfection of SH-SY5Y neuroblastoma cells by utilizing Nrf2 siRNA duplex, following the instructions of the manufacturer (Santa Cruz, CA, USA) [66, 67].
Statistical Analyses
Statistical analyses were performed with GraphPad 5.0 software. Data are exhibited as the mean ± standard error of the mean (S.E.M.) of three or five independent experiments each done in triplicate; p values were considered significant when p < 0.05. Differences between the experimental groups were determined by one-way ANOVA followed by the post hoc Tukey’s test.
Results
CA Prevented Loss of Cell Viability and Inhibited Apoptosis in SH-SY5Y Cells Exposed to H2O2
We first examined the effect of various H2O2 concentrations on the viability of SH-SY5Y neuroblastoma cells. As depicted in Fig. 1a, H2O2 at 25–400 μM for 24 h decreased cell viability in a dose-dependent manner. We also investigated the CA concentrations that efficiently suppressed the H2O2-induced loss of cell viability. According to Fig. 1b, CA pretreatment for 12 h at 1–2 μM efficiently prevented loss of viability in H2O2-treated SH-SY5Y cells. We have previously demonstrated that CA at 1 μM exerted cytoprotective effects in SH-SY5Y cells exposed to different chemical stressors. Therefore, we decided to use pretreatment with CA at 1 μM for 12 h in further assays in the herein presented experimental model.

The effect of H2O2 at different concentrations for 24 h on the viability of SH-SY5Y cells. CA pretreatment at different concentrations (0.1–2 μM) for 12 h on the viability of SH-SY5Y cells exposed to 300 μM H2O2 for additional 24 h. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells, #p < 0.05 vs CA-treated cells exposed to H2O2
H2O2 at 300 μM for 24 h increased the levels of pro-apoptotic markers, as may be viewed in Fig. 2. Otherwise, CA (1 μM) pretreatment (12 h) suppressed apoptosis in H2O2-treated cells by blocking the H2O2-induced decrease in Bcl-2 (Fig. 2a) and the increase in Bax (Fig. 2b) levels. CA also abrogated the release of cytochrome c to the cytosol (Fig. 2c), consequently maintaining the mitochondrial content of cytochrome c near the values observed in the control group (Fig. 2d). In this context, CA prevented the increase observed in the activities of the pro-apoptotic enzymes caspase-9 (Fig. 2e) and caspase-3 (Fig. 2f) in the cells exposed to H2O2.

The effects of a pretreatment with CA at 1 μM for 12 h on Bcl-2 immunocontent (a), Bax immunocontent (b), cytosolic cytochrome c content (c), mitochondrial cytochrome c content (d), caspase-9 activity (e), and caspase-3 activity (f). H2O2 was utilized at 300 μM for further 24 h. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells; #p < 0.05 vs H2O2-treated cells
In order to confirm the anti-apoptotic effect of CA in H2O2-treated SH-SY5Y cells, we examined the levels of hallmarks of apoptosis (Fig. 3). CA pretreatment significantly prevented the H2O2-elicited increase in the cleavage of PARP (Fig. 3a) and in the fragmentation of DNA (Fig. 3b).

The effects of a pretreatment with CA at 1 μM for 12 h on the levels of cleaved PARP (a) and DNA fragmentation (b). H2O2 was utilized at 300 μM for further 24 h. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells; #p < 0.05 vs H2O2-treated cells
CA Exerted Antioxidant Effects in H2O2-Treated SH-SY5Y Cells
We next examined whether CA would prevent redox impairment in cells exposed to H2O2 (Fig. 4). The reactions performed by reactive species occur at very high rates [20, 68]; therefore, we decided to investigate the levels of markers of redox disturbances 3 h after administration of H2O2 to the cells. H2O2 increased the cellular production of reactive species (Fig. 4a), causing a decrease in the levels of cellular GSH (Fig. 4b) and oxidizing total DNA, as assessed through the quantification of 8-oxo-2′-deoxyguanosine (8-oxodG) (Fig. 4c). Pretreatment with CA prevented the redox disturbances caused by H2O2 also in mitochondria by reducing the levels of lipid peroxidation (Fig. 5a), protein carbonylation (5b), and protein nitration (Fig. 5c) in mitochondrial membranes.

The effects of a pretreatment with CA at 1 μM for 12 h on the cellular levels of reactive species generation (a), GSH (b), and 8-oxo-dG (c). H2O2 was utilized at 300 μM for further 3 h (DCF assay) or 24 h (GSH and 8-oxo-dG). The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells; a p < 0.05 vs control cells; #p < 0.05 vs H2O2-treated cells

The effects of a pretreatment with CA at 1 μM for 12 h on the levels of lipid peroxidation (a), protein carbonylation (b), and protein nitration (c) in mitochondrial membranes. H2O2 was utilized at 300 μM for further 24 h. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells; #p < 0.05 vs H2O2-treated cells
CA Affords Mitochondrial Protection in Cells Exposed to H2O2
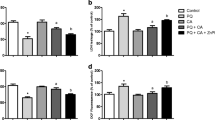
According to Figs. 6 and 7, H2O2 impaired mitochondrial function. However, pretreatment with CA efficiently abrogated the effects of H2O2 on the activities of aconitase (Fig. 6a), α-KGDH (Fig. 6b), and SDH (Fig. 6c). CA suppressed the H2O2-induced inhibition of complex I (Fig. 7a) and complex V (Fig. 7b) enzymes and prevented the decrease in ATP levels elicited by H2O2 (Fig. 7c). CA also protected mitochondria by inhibiting the H2O2-induced loss of MMP (Fig. 7d). CA attenuated the effect of H2O2 on the production of O2 −• by mitochondria (Fig. 7e). The levels of GSH in mitochondria were also modulated by CA and H2O2, as may be seen in Fig. 7f.

The effects of a pretreatment with CA at 1 μM for 12 h on the activities of aconitase (a), α-ketoglutarate dehydrogenase (b), and succinate dehydrogenase (c) enzymes. H2O2 was utilized at 300 μM for further 24 h. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells; #p < 0.05 vs H2O2-treated cells

The effects of a pretreatment with CA at 1 μM for 12 h on the activities of the complexes I (a) and V (b), on the levels of ATP (c), on MMP (d), on the production of O2 −• by SMP (e), and on the levels of GSH in mitochondria (f). H2O2 was utilized at 300 μM for further 3 h (measurement of O2 −•) or 24 h (other analyses). The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs control cells; a p < 0.05 vs control cells; #p < 0.05 vs H2O2-treated cells
CA Protected Mitochondrial Function by an Nrf2-Dependent Manner in SH-SY5Y Cells Challenged with H2O2
In order to investigate by which mechanism CA protected mitochondria in SH-SY5Y cells exposed to H2O2, we examined the consequences of silencing the Nrf2 transcription factor in this experimental model. CA failed to restore mitochondrial function in cells in which Nrf2 was knocked down, as may be observed in relation to the activities of complexes I (Fig. 8a) and V (Fig. 8b). CA did not enhance the levels of GSH efficiently in SH-SY5Y cells in which Nrf2 was silenced (Fig. 9a) and failed to suppress loss of MMP in cells exposed to H2O2 (Fig. 9b).

The effects of Nrf2 siRNA (48 h) on the activities of the complexes I (a) and V (b) of SH-SY5Y cells treated or not with CA and/or H2O2. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs the CA and H2O2-treated cells transfected with negative control (NC) siRNA

The effects of Nrf2 siRNA (48 h) on the activities of the mitochondrial content of GSH (a) and MMP (b) of SH-SY5Y cells treated or not with CA and/or H2O2. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs the CA and H2O2-treated cells transfected with negative control (NC) siRNA
CA Protected SH-SY5Y Cells Through an Nrf2-Dependent Manner
Finally, we analyzed whether Nrf2 would present a role in the CA-induced cytoprotective effects in H2O2-treated SH-SY5Y cells. According to Fig. 10, CA failed to abolish the cellular impairment in Nrf2-silenced SH-SY5Y cells.

The effects of Nrf2 siRNA (48 h) on the activities of the viability of SH-SY5Y cells treated or not with CA and/or H2O2. The results are presented as the mean ± S.E.M. of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 vs the carnosic acid and H2O2-treated cells transfected with negative control (NC) siRNA
Discussion
Redox impairment causes mitochondrial dysfunction by inhibiting mitochondria-located enzymes that maintain bioenergetics-related reactions associated with ATP production [69, 70]. Disruption of the tricarboxylic acid cycle (TCA), for example, affects directly the flux of electrons in the respiratory chain, causing a decrease in the electrochemical gradient across the inner mitochondrial membrane [71–74]. This may be accompanied by electron leakage of the oxidative phosphorylation system, enhancing the production of reactive species, such as O2 −•, among others [10, 11, 75]. Therefore, this is a vicious cycle that gradually amplifies mitochondrial dysfunction, leading to the activation of the intrinsic apoptotic pathway and cell death [76]. Actually, disruption of the mitochondrial function, increased production of reactive species by these organelles, and enhanced rates of cell death have been observed in several human pathologies, including neurodegeneration and neurotoxicity [37, 77–81].
In the present work, we have found that a pretreatment with CA prevented the H2O2-induced mitochondrial dysfunction, loss of cell viability, and apoptosis in SH-SY5Y neuroblastoma cells. CA suppressed the H2O2-mediated inhibition of components of the TCA and of the oxidative phosphorylation system and decreased the production of O2 −• by mitochondria exposed to H2O2. CA also upregulated the levels of GSH in the mitochondria, probably causing mitochondrial protection, as previously demonstrated by our research group [82–84]. However, we have also demonstrated previously that CA increased the levels of Mn-SOD in SH-SY5Y cells [48, 49]. This enzyme is located in the mitochondrial matrix and takes a crucial role in converting O2 −• in H2O2 [20]. Therefore, the participation of enzymatic antioxidant defenses against exposure to H2O2 and the consequences this reactive species causes should not be discarded in this experimental model. It has been demonstrated that oxidants, including H2O2, inhibit mitochondrial enzymes, such as aconitase, α-KGDH, and SDH, in different experimental models [85, 86]. Additionally, impaired function and/or content of the TCA enzymes in the brain of patients with Parkinson’s disease [35, 87–89], Alzheimer’s disease [90, 91], or Huntington’s disease [92] has been published. In this regard, there is increasing interest in investigating natural compounds that may prevent mitochondrial dysfunction in case of neurologic disorders, among other diseases [15, 23, 24, 26–29, 93, 94].
We found that Nrf2 knockdown abrogated the beneficial effects elicited by CA in SH-SY5Y cells exposed to H2O2. Indeed, a close relation between mitochondrial function and Nrf2-dependent signaling has been reported [95]. Even though Nrf2 is a master regulator of the redox environment in mammalian cells, this transcription factor may be associated with other mitochondria-related parameters, such as oxidation of fatty acids [96] and synthesis of ATP [97]. Indeed, Nrf2 silencing abolished the CA-induced upregulation of mitochondrial enzymes of the oxidative phosphorylation system related to the maintenance of the energetic status in SH-SY5Y cells in this work. In spite of this, it remains to be determined exactly how Nrf2 knockdown impacted the effects mediated by CA, since the regulation of Nrf2 is not dependent only on redox reactions [98]. We [48, 49] and others [99] have previously shown that CA activates the PI3K/Akt signaling pathway, leading to an upregulation of Nrf2 and a consequent increase in the expression of antioxidant and phase II detoxification enzymes. The activation of the PI3K/Akt axis is not associated only with redox disturbances and takes a crucial role in mediating metabolic effects, for example, in mammalian cells [100]. Nonetheless, further research is necessary in order to investigate whether there is a role for this signaling pathway in modulating the CA-induced Nrf2-dependent metabolic effects in cultured cells.
Overall, CA prevented the H2O2-triggered mitochondrial redox impairment and dysfunction (i.e., TCA impairment) and cell death in SH-SY5Y cells by a mechanism involving the transcription factor Nrf2. Further research is necessary to investigate whether CA would be able to trigger mitochondrial biogenesis in mammalian cells, among other mitochondria-related effects.
References
Lushchak VI (2014) Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact 224:164–175. doi:10.1016/j.cbi.2014.10.016
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13. doi:10.1042/BJ20081386
Phaniendra A, Jestadi DB, Periyasamy L (2015) Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem 30:11–26. doi:10.1007/s12291-014-0446-0
Bornhövd C, Vogel F, Neupert W, Reichert AS (2006) Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J Biol Chem 281:13990–13998
McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse. Curr Biol 16:R551–R560
Butler J, Jayson GG, Swallow AJ (1975) The reaction between the superoxide anion radical and cytochrome c. Biochim Biophys Acta 408:215–222
Cadenas E, Boveris A, Ragan CI, Stoppani AO (1977) Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys 180:248–257
Beyer RE (1990) The participation of coenzyme Q in free radical production and antioxidation. Free Radic Biol Med 8:545–565
Barja G (1999) Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr 31:347–366
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–244
Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD (2010) Mitochondrial proton and electron leaks. Essays Biochem 47:53–67. doi:10.1042/bse0470053
Li C, Zhou HM (2011) The role of manganese superoxide dismutase in inflammation defense. Enzyme Res 2011:387176. doi:10.4061/2011/387176
Bakthavatchalu V, Dey S, Xu Y, Noel T, Jungsuwadee P, Holley AK, Dhar SK, Batinic-Haberle I et al (2012) Manganese superoxide dismutase is a mitochondrial fidelity protein that protects Polγ against UV-induced inactivation. Oncogene 31:2129–2139. doi:10.1038/onc.2011.407
Candas D, Li JJ (2014) MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid Redox Signal 20:1599–1617. doi:10.1089/ars.2013.5305
de Oliveira MR, Nabavi SF, Habtemariam S, Erdogan Orhan I, Daglia M, Nabavi SM (2015) The effects of baicalein and baicalin on mitochondrial function and dynamics: a review. Pharmacol Res 100:296–308. doi:10.1016/j.phrs.2015.08.021
Paravicini TM, Drummond GR, Sobey CG (2004) Reactive oxygen species in the cerebral circulation: physiological roles and therapeutic implications for hypertension and stroke. Drugs 64:2143–2157
Kamsler A, Segal M (2004) Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol 29:167–178
Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ (2005) Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J 19:854–856
Gough DR, Cotter TG (2011) Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis 2:e213. doi:10.1038/cddis.2011.96
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Deponte M (2013) Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim Biophys Acta 1830:3217–3266. doi:10.1016/j.bbagen.2012.09.018
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830:3143–3153. doi:10.1016/j.bbagen.2012.09.008
Atamna H, Mackey J, Dhahbi JM (2012) Mitochondrial pharmacology: electron transport chain bypass as strategies to treat mitochondrial dysfunction. Biofactors 38:158–166. doi:10.1002/biof.197
Gruber J, Fong S, Chen CB, Yoong S, Pastorin G, Schaffer S, Cheah I, Halliwell B (2013) Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnol Adv 31:563–592. doi:10.1016/j.biotechadv.2012.09.005
de Oliveira MR (2016) Evidence for genistein as a mitochondriotropic molecule. Mitochondrion 29:35–44. doi:10.1016/j.mito.2016.05.005
de Oliveira MR, Jardim FR, Setzer WN, Nabavi SM, Nabavi SF (2016) Curcumin, mitochondrial biogenesis, and mitophagy: exploring recent data and indicating future needs. Biotechnol Adv 34:813–826. doi:10.1016/j.biotechadv.2016.04.004
de Oliveira MR, Nabavi SF, Manayi A, Daglia M, Hajheydari Z, Nabavi SM (2016) Resveratrol and the mitochondria: from triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim Biophys Acta 1860:727–745. doi:10.1016/j.bbagen.2016.01.017
de Oliveira MR, Nabavi SM, Braidy N, Setzer WN, Ahmed T, Nabavi SF (2016) Quercetin and the mitochondria: a mechanistic view. Biotechnol Adv 34:532–549. doi:10.1016/j.biotechadv.2015.12.014
Oliveira MR, Nabavi SF, Daglia M, Rastrelli L, Nabavi SM (2016) Epigallocatechin gallate and mitochondria—a story of life and death. Pharmacol Res 104:70–85. doi:10.1016/j.phrs.2015.12.027
Wiernsperger NF (2003) Oxidative stress: the special case of diabetes. Biofactors 19:11–18
Pérez-Neri I, Ramírez-Bermúdez J, Montes S, Ríos C (2006) Possible mechanisms of neurodegeneration in schizophrenia. Neurochem Res 31:1279–1294
Mc Guire PJ, Parikh A, Diaz GA (2009) Profiling of oxidative stress in patients with inborn errors of metabolism. Mol Genet Metab 98:173–180. doi:10.1016/j.ymgme.2009.06.007
Nunomura A, Moreira PI, Castellani RJ, Lee HG, Zhu X, Smith MA, Perry G (2012) Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res 22:231–248. doi:10.1007/s12640-012-9331-x
Anderson G, Maes M (2014) Neurodegeneration in Parkinson’s disease: interactions of oxidative stress, tryptophan catabolites and depression with mitochondria and sirtuins. Mol Neurobiol 49:771–783. doi:10.1007/s12035-013-8554-z
Kasote DM, Hegde MV, Katyare SS (2013) Mitochondrial dysfunction in psychiatric and neurological diseases: cause(s), consequence(s), and implications of antioxidant therapy. Biofactors 39:392–406. doi:10.1002/biof.1093
de Oliveira MR (2015) Vitamin A and retinoids as mitochondrial toxicants. Oxidative Med Cell Longev 2015:140267. doi:10.1155/2015/140267
de Oliveira MR, Jardim FR (2016) Cocaine and mitochondria-related signaling in the brain: a mechanistic view and future directions. Neurochem Int 92:58–66. doi:10.1016/j.neuint.2015.12.006
Foresti R, Bains SK, Pitchumony TS, de Castro Brás LE, Drago F, Dubois-Randé JL, Bucolo C, Motterlini R (2013) Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 76:132–148. doi:10.1016/j.phrs.2013.07.010
de Oliveira MR (2015) The dietary components carnosic acid and carnosol as neuroprotective agents: a mechanistic view. Mol Neurobiol IN PRESS doi. doi:10.1007/s12035-015-9519-1
Wu CR, Tsai CW, Chang SW, Lin CY, Huang LC, Tsai CW (2015) Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: involvement of antioxidative enzymes induction. Chem Biol Interact 225:40–46. doi:10.1016/j.cbi.2014.11.011
Chen SD, Ji BB, Yan YX, He X, Han KY, Dai QX, Zhang MX, Mo YC et al (2016) Carnosic acid attenuates neuropathic pain in rat through the activation of spinal sirtuin1 and down-regulation of p66shc expression. Neurochem Int 93:95–102. doi:10.1016/j.neuint.2016.01.004
Jung KJ, Min KJ, Park JW, Park KM, Kwon TK (2016) Carnosic acid attenuates unilateral ureteral obstruction-induced kidney fibrosis via inhibition of Akt-mediated Nox4 expression. Free Radic Biol Med 97:50–57. doi:10.1016/j.freeradbiomed.2016.05.020
Su K, Wang CF, Zhang Y, Cai YJ, Zhang YY, Zhao Q (2016) The inhibitory effects of carnosic acid on cervical cancer cells growth by promoting apoptosis via ROS-regulated signaling pathway. Biomed Pharmacother 82:180–191. doi:10.1016/j.biopha.2016.04.056
Tian X, Hu Y, Li M, Xia K, Yin J, Chen J, Liu Z (2016) Carnosic acid attenuates acute ethanol-induced liver injury via a SIRT1/p66Shc-mediated mitochondrial pathway. Can J Physiol Pharmacol 94:416–425. doi:10.1139/cjpp-2015-0276
Kapoor S (2013) Carnosic acid and its inhibitory effect on tumor growth in systemic malignancies. Oral Dis 19:427. doi:10.1111/odi.12055
Gao Q, Liu H, Yao Y, Geng L, Zhang X, Jiang L, Shi B, Yang F (2015) Carnosic acid induces autophagic cell death through inhibition of the Akt/mTOR pathway in human hepatoma cells. J Appl Toxicol 35:485–492. doi:10.1002/jat.3049
Hao L, Ran W, Xiang-Xin L, Lu-Qun W, Xiao-Ning Y (2016) Carnosic acid-combined arsenic trioxide antileukaemia cells in the establishment of NB4/SCID mouse model. Basic Clin Pharmacol Toxicol 119:259–266. doi:10.1111/bcpt.12580
de Oliveira MR, Ferreira GC, Schuck PF, Dal Bosco SM (2015) Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem Biol Interact 242:396–406. doi:10.1016/j.cbi.2015.11.003
de Oliveira MR, Ferreira GC, Schuck PF (2016) Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in SH-SY5Y cells: role for PI3K/Akt/Nrf2 pathway. Toxicol in Vitro 32:41–54. doi:10.1016/j.tiv.2015.12.005
de Oliveira MR, Peres A, Ferreira GC, Schuck PF, Bosco SM (2016) Carnosic acid affords mitochondrial protection in chlorpyrifos-treated Sh-Sy5y cells. Neurotox Res IN PRESS doi. doi:10.1007/s12640-016-9620-x
Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J et al (2008) Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 104:1116–1131
Vaka SR, Shivakumar HN, Repka MA, Murthy SN (2013) Formulation and evaluation of carnosic acid nanoparticulate system for upregulation of neurotrophins in the brain upon intranasal administration. J Drug Target 21:44–53. doi:10.3109/1061186X.2012.725405
Meng P, Yoshida H, Tanji K, Matsumiya T, Xing F, Hayakari R, Wang L, Tsuruga K et al (2015) Carnosic acid attenuates apoptosis induced by amyloid-β 1-42 or 1-43 in SH-SY5Y human neuroblastoma cells. Neurosci Res 94:1–9. doi:10.1016/j.neures.2014.12.003
Miller DM, Singh IN, Wang JA, Hall ED (2015) Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol 264:103–110. doi:10.1016/j.expneurol.2014.11.008
Zhang D, Lee B, Nutter A, Song P, Dolatabadi N, Parker J, Sanz-Blasco S, Newmeyer T et al (2015) Protection from cyanide-induced brain injury by the Nrf2 transcriptional activator carnosic acid. J Neurochem 133:898–908. doi:10.1111/jnc.13074
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
de Oliveira MR, Schuck PF, Bosco SM (2016) Tanshinone I induces mitochondrial protection through an Nrf2-dependent mechanism in paraquat-treated human neuroblastoma SH-SY5Y cells. Mol Neurobiol IN PRESS. doi:10.1007/s12035-016-0009-x
LeBel CP, Ischiropoulos H, Bondy SC (1992) Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5:227–231
De Oliveira MR, Oliveira MW, Da Rocha RF, Moreira JC (2009) Vitamin A supplementation at pharmacological doses induces nitrosative stress on the hypothalamus of adult Wistar rats. Chem Biol Interact 180:407–413. doi:10.1016/j.cbi.2009.02.006
de Oliveira MR, Lorenzi R, Schnorr CE, Morrone M, Moreira JC (2011) Increased 3-nitrotyrosine levels in mitochondrial membranes and impaired respiratory chain activity in brain regions of adult female rats submitted to daily vitamin A supplementation for 2 months. Brain Res Bull 86:246–253. doi:10.1016/j.brainresbull.2011.08.006
de Oliveira MR, da Rocha RF, Moreira JC (2012) Increased susceptibility of mitochondria isolated from frontal cortex and hippocampus of vitamin A-treated rats to non-aggregated amyloid-β peptides 1-40 and 1-42. Acta Neuropsychiatr 24:101–108. doi:10.1111/j.1601-5215.2011.00588.x
de Oliveira MR, da Rocha RF, Schnorr CE, Moreira JC (2012) L-NAME cotreatment did prevent neither mitochondrial impairment nor behavioral abnormalities in adult Wistar rats treated with vitamin A supplementation. Fundam Clin Pharmacol 26:513–529. doi:10.1111/j.1472-8206.2011.00943.x
Wang K, Zhu L, Zhu X, Zhang K, Huang B, Zhang J, Zhang Y, Zhu L et al (2014) Protective effect of paeoniflorin on Aβ25-35-induced SH-SY5Y cell injury by preventing mitochondrial dysfunction. Cell Mol Neurobiol 34:227–234. doi:10.1007/s10571-013-0006-9
Poderoso JJ, Carreras MC, Lisdero C, Riobó N, Schöpfer F, Boveris A (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92
de Oliveira MR, da Rocha RF, Stertz L, Fries GR, de Oliveira DL, Kapczinski F, Moreira JC (2011) Total and mitochondrial nitrosative stress, decreased brain-derived neurotrophic factor (BDNF) levels and glutamate uptake, and evidence of endoplasmic reticulum stress in the hippocampus of vitamin A-treated rats. Neurochem Res 36:506–517. doi:10.1007/s11064-010-0372-3
Quesada A, Ogi J, Schultz J, Handforth A (2011) C-terminal mechano-growth factor induces heme oxygenase-1-mediated neuroprotection of SH-SY5Y cells via the protein kinase Cϵ/Nrf2 pathway. J Neurosci Res 89:394–405. doi:10.1002/jnr.22543
Jin X, Liu Q, Jia L, Li M, Wang X (2015) Pinocembrin attenuates 6-OHDA-induced neuronal cell death through Nrf2/ARE pathway in SH-SY5Y cells. Cell Mol Neurobiol 35:323–333. doi:10.1007/s10571-014-0128-8
D’Autréaux, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8:813–824
Chinopoulos C, Tretter L, Adam-Vizi V (1999) Depolarization of in situ mitochondria due to hydrogen peroxide-induced oxidative stress in nerve terminals: inhibition of alpha-ketoglutarate dehydrogenase. J Neurochem 73:220–228
Nulton-Persson AC, Szweda LI (2001) Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem 276:23357–23361. doi:10.1074/jbc.M100320200
Chernyak BV, Bernardi P (1996) The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem 238:623–630
Costantini P, Chernyak BV, Petronilli V, Bernardi P (1996) Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem 271:6746–6751
Chernyak BV (1997) Redox regulation of the mitochondrial permeability transition pore. Biosci Rep 17:293–302
Lieven CJ, Vrabec JP, Levin LA (2003) The effects of oxidative stress on mitochondrial transmembrane potential in retinal ganglion cells. Antioxid Redox Signal 5:641–646
Fato R, Bergamini C, Leoni S, Lenaz G (2008) Mitochondrial production of reactive oxygen species: role of complex I and quinone analogues. Biofactors 32:31–39
Wang CH, Wu SB, Wu YT, Wei YH (2013) Oxidative stress response elicited by mitochondrial dysfunction: implication in the pathophysiology of aging. Exp Biol Med (Maywood) 238:450–460. doi:10.1177/1535370213493069
Schinder AF, Olson EC, Spitzer NC, Montal M (1996) Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 16:6125–6133
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Deschepper M, Hoogendoorn B, Brooks S, Dunnett SB, Jones L (2012) Proteomic changes in the brains of Huntington’s disease mouse models reflect pathology and implicate mitochondrial changes. Brain Res Bull 88:210–222. doi:10.1016/j.brainresbull.2011.01.012
Lezi E, Swerdlow RH (2012) Mitochondria in neurodegeneration. Adv Exp Med Biol 942:269–286. doi:10.1007/978-94-007-2869-1_12
de Oliveira MR (2016) Fluoxetine and the mitochondria: a review of the toxicological aspects. Toxicol Lett 258:185–191. doi:10.1016/j.toxlet.2016.07.001
Fernández-Checa JC, Kaplowitz N, García-Ruiz C, Colell A, Miranda M, Marí M, Ardite E, Morales A (1997) GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Phys 273:G7–G17
García-Ruiz C, Colell A, Marí M, Morales A, Fernández-Checa JC (1997) Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione J Biol Chem 272:11369–11377
Fernández-Checa JC, García-Ruiz C, Colell A, Morales A, Marí M, Miranda M, Ardite E (1998) Oxidative stress: role of mitochondria and protection by glutathione. Biofactors 8:7–11
Tretter L, Adam-Vizi V (2000) Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci 20:8972–8979
Kil IS, Park JW (2005) Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem 280:10846–10854
Hattori N, Tanaka M, Ozawa T, Mizuno Y (1991) Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann Neurol 30:563–571
Kingsbury AE, Cooper M, Schapira AH, Foster OJ (2001) Metabolic enzyme expression in dopaminergic neurons in Parkinson’s disease: an in situ hybridization study. Ann Neurol 50:142–149
Gibson GE, Kingsbury AE, Xu H, Lindsay JG, Daniel S, Foster OJ, Lees AJ, Blass JP (2003) Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem Int 43:129–135
Mastrogiacomo F, Bergeron C, Kish SJ (1993) Brain alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. J Neurochem 61:2007–2014
Sheu KF, Cooper AJ, Koike K, Koike M, Lindsay JG, Blass JP (1994) Abnormality of the alpha-ketoglutarate dehydrogenase complex in fibroblasts from familial Alzheimer’s disease. Ann Neurol 35:312–318
Browne SE, Beal MF (2004) The energetics of Huntington’s disease. Neurochem Res 29:531–546
Armstrong JS (2007) Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol 151:1154–1165
Heller A, Brockhoff G, Goepferich A (2012) Targeting drugs to mitochondria. Eur J Pharm Biopharm 82:1–18. doi:10.1016/j.ejpb.2012.05.014
Hayes JD, Dinkova-Kostova AT (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39:199–218. doi:10.1016/j.tibs.2014.02.002
Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT (2014) Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J 457:415–424. doi:10.1042/BJ20130863
Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D, Lee YM, Ku SK, Jung Y et al (2011) NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res 71:2260–2275. doi:10.1158/0008-5472.CAN-10-3007
Cornejo P, Vargas R, Videla LA (2013) Nrf2-regulated phase-II detoxification enzymes and phase-III transporters are induced by thyroid hormone in rat liver. Biofactors 39:514–521. doi:10.1002/biof.1094
Lin CY, Chen JH, Fu RH, Tsai CW (2014) Induction of Pi form of glutathione S-transferase by carnosic acid is mediated through PI3K/Akt/NF-κB pathway and protects against neurotoxicity. Chem Res Toxicol 27:1958–1966. doi:10.1021/tx5003063
Rohlenova K, Neuzil J, Rohlena J (2016) The role of Her2 and other oncogenes of the PI3K/AKT pathway in mitochondria. Biol Chem 397:607–615. doi:10.1515/hsz-2016-0130
Acknowledgements
GCF is supported by Edital APQ1/FAPERJ and receives a “Produtividade em Pesquisa do CNPq - Nível 2” fellow. This work was supported by CNPq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
de Oliveira, M.R., da Costa Ferreira, G., Peres, A. et al. Carnosic Acid Suppresses the H2O2-Induced Mitochondria-Related Bioenergetics Disturbances and Redox Impairment in SH-SY5Y Cells: Role for Nrf2. Mol Neurobiol 55, 968–979 (2018). https://doi.org/10.1007/s12035-016-0372-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0372-7