
Abstract
Cognitive impairment is one of the most salient non-motor symptoms of Parkinson disease (PD) that poses a significant burden on the patients and carers as well as being a risk factor for early mortality. People with PD show a wide spectrum of cognitive dysfunctions ranging from subjective cognitive decline and mild cognitive impairment (MCI) to frank dementia. The mean frequency of PD with MCI (PD-MCI) is 25.8% and the pooled dementia frequency is 26.3% increasing up to 83% 20 years after diagnosis. A better understanding of the underlying pathological processes will aid in directing disease-specific treatment. Modern neuroimaging studies revealed considerable changes in gray and white matter in PD patients with cognitive impairment, cortical atrophy, hypometabolism, dopamine/cholinergic or other neurotransmitter dysfunction and increased amyloid burden, but multiple mechanism are likely involved. Combined analysis of imaging and fluid markers is the most promising method for identifying PD-MCI and Parkinson disease dementia (PDD). Morphological substrates are a combination of Lewy- and Alzheimer-associated and other concomitant pathologies with aggregation of α-synuclein, amyloid, tau and other pathological proteins in cortical and subcortical regions causing destruction of essential neuronal networks. Significant pathological heterogeneity within PD-MCI reflects deficits in various cognitive domains. This review highlights the essential neuroimaging data and neuropathological changes in PD with cognitive impairment, the amount and topographical distribution of pathological protein aggregates and their pathophysiological relevance. Large-scale clinicopathological correlative studies are warranted to further elucidate the exact neuropathological correlates of cognitive impairment in PD and related synucleinopathies as a basis for early diagnosis and future disease-modifying therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
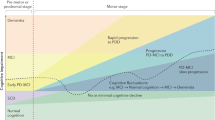
Parkinson disease (PD), the most common movement disorder and the second most common neurodegenerative disorder after Alzheimer disease (AD), is characterized by progressive degeneration not only of the dopaminergic striatonigral system but also by involvement of many other neurological systems and organs, due to widespread intraneuronal and neuritic deposition of abnormal phosphorylated α-synuclein (αSyn), forming intracytoplasmic Lewy bodies (LBs) and Lewy neurites, the morphological hallmarks of PD and related LB disorders. However, multiple mechanisms and pathways play a role in the pathogenesis of PD including oxidative stress, mitochondrial dysfunction, calcium imbalance, neuroinflammation, and multiple neurotransmitter deficits (Jellinger 2012a; Zaman et al. 2021): The resulting biochemical deficits cause a heterogeneous spectrum of motor and non-motor symptoms that contribute greatly to the overall disease burden of this multisystem/organ disorder (Dickson et al. 2009a; Jellinger 2012b). Cognitive impairment (CI) that has been recognized as an important part of PD since the historical description of Charcot (1877), shows a full spectrum ranging from subjective cognitive decline (SCD) and mild cognitive impairment (MCI) to full-blown dementia (PDD). It severely affects the quality of life, is a risk factor for early mortality (Oosterveld et al. 2015; Schrag et al. 2000), and has been shown to have substantial consequences over and above the motor symptoms, even at early stages of PD (Chandler et al. 2021; Leroi et al. 2012). SCD is a self-perceived decline in cognitive ability with normal age-, sex- and education-adjusted performance on standardized cognitive tests (Jessen et al. 2014); PD with MCI (PD-MCI) is a gradual decline in cognitive ability affecting single or multiple cognitive domains on complex functional tasks, including amnestic (aMCI) and non-amnestic (naMCI) phenotypes (Litvan et al. 2012; Petersen et al. 2009). It is a risk factor for PDD (Hoogland et al. 2017), which is defined as CI in PD patients with deficits in at least four cognitive domains (memory, attention, executive and visuospatial abilities) being severe enough to significantly affect routine functions of life (Emre et al. 2007; Goetz et al. 2008; Kiesmann et al. 2013). PDD can be denoted as mild, moderate and severe (inability for independent living). Cognitive decline may occur in presymptomatic stages (Fengler et al. 2017), at the time of diagnosis or a few years or decades after diagnosis of PD and has a high variability in its severity, rate of progression and involved cognitive domains (Aarsland et al. 2021). Mild neurocognitive deficits can occur even in the presymptomatic phase of PD (Bougea et al. 2019) and may precede the onset of dementia by up to 20 years. This is suggested to affect 19–30% of newly diagnosed, untreated (de novo) PD patients and may be associated with subtle changes of cognitive function that are not apparent to patients, families or clinicians. The most frequent phenotypes of MCI in prodromal PD are executive dysfunction and multidomain amnestic phenotypes (Pan et al. 2022), but not memory or attention (Speelberg et al. 2022). Although the estimated frequency of cognitive dysfunction in nondemented PD varies between 19 and 55%, it is underrecognized in practice (Barone et al. 2011). The cognitive symptoms experienced in PD are highly variable and may reflect both molecular, neurochemical, and morphological changes, such as αSyn- and Alzheimer-related and other pathologies, which will be critically reviewed. The relations between PDD and dementia with Lewy bodies (DLB) will not be discussed, since they have been reviewed recently (Jellinger 2018; Jellinger and Korczyn 2018).
Epidemiology
PD patients have a 2.5–6 times higher risk of developing dementia than people without PD of similar age (Aarsland et al. 2021; Perez et al. 2012). However, the epidemiology of CI in PD is not entirely clear, since population-based studies rarely include PD-MCI and PDD, and most studies assess the prevalence and incidence of CI in established PD cohorts. MCI is often described as a transitory stage between normal condition and dementia; conversion rates for PDD are markedly increased in those with MCI, and were reported to be almost 60% at 5 years of follow-up (Pedersen et al. 2017). Early onset PD patients exhibit a poorer cognitive performance than those with late onset PD (Kim et al. 2017). The frequency of PD-MCI ranges from about 21 to 70%, with a mean of 25.8% (Aarsland et al. 2021; Monastero et al. 2018; Nicoletti et al. 2019). A recent meta-analysis reported a pooled prevalence of 40% in a sample of 7053 PD patients (Baiano et al. 2020). Its estimated point prevalence is 30%, the cumulative prevalence is > 75% for PD patients surviving more than 10 years (Hely et al. 2008). The cumulative incidence of PD-MCI is 9.9% after 1 year, 23.2% after 3 years, and 28.9% after 5 years follow-up (Pedersen et al. 2017). Within 3 years, in PD with normal cognition (PD-NC), 25% (95% CI 20–30%) converted to PD-MCI and 2% (95% CI 1–7%) converted to PDD, whereas 28% (95% CI 20–37%) reverted back to normal cognitive function (Saredakis et al. 2019). A comprehensive meta-analysis of PD-MCI cognitive outcome and predictors in its conversion to PDD was published recently (Wallace et al. 2022). Approximately 20–30% have at least mild cognitive changes even at the time of diagnosis of PD (Poletti et al. 2012), increasing to 40–50% after 5 years’ follow-up (Domellöf et al. 2015; Pedersen et al. 2017). By contrast, the estimated prevalence of MCI in the general population (age 60–90 years) ranges between 16 and 20% (Roberts and Knopman 2013). 59.1% of patients with persistent PD-MCI within 1 year develop PDD (Pedersen et al. 2017). Importantly, the value of MCI for the development of PDD is influenced by the diagnostic criteria chosen for MCI (Wood et al. 2016). About 30.3% of de novo PD patients complained of memory issues and were more likely to develop MCI within 2 years’ follow-up compared to those who did not complain of memory issues (Purri et al. 2020), although other factors, such as affective symptoms, may contribute to progression of MCI (Chua et al. 2021). Cognitive deficits have been recently defined as a prodromal marker and have been included in the last research criteria of prodromal PD (Heinzel et al. 2019).
The global pooled frequency of PDD is 26.3% with variations according to the methodologies (14–55%) (Severiano et al. 2022), the estimated prevalence is 24–31% (Aarsland et al. 2005b), the cumulated prevalence in patients with a mean age of 54–70 years is 17% at 5 years after diagnosis, 83% at 20 years after diagnosis (Hely et al. 2008), and up to 95% by age 90 years (Rongve and Aarsland 2013). PDD has a relative risk of 2.47 (1.55–3.95) (Perez et al. 2012), a prevalence of 31.3% (95% CI 20.1–40.1) and incidence rates from 42.6 to 112.5/100,000 person-years (Marder 2010), indicating that around 10% of a PD population develop dementia per year (Hall and Lewis 2019). Systemic reviews suggest that 3–4% of the dementia in the general population would be due to PDD; its estimated prevalence in the population older than 65 years is 0.2–0.5% (Aarsland et al. 2005b).
Cognitive dysfunction/dementia in genetic forms of PD is variable, depending on the affected gene or genetic risk factors, e.g., DNA mutations, LRRK2, GBA1, Parkin/PINK1, APOE ε4, MAPT/H1, or other unknown factors, like additional genetic modifiers and environmental factors, which have been reviewed recently (Aarsland et al. 2021; Fan et al. 2021; Koros et al. 2022; Szwedo et al. 2022; Wise and Alcalay 2022). AD tau has been shown to be a prominent pathology in LRRK2 PD (Henderson et al. 2019).
Neuroimaging findings in cognitive impairment in PD
Unlike clinical behavioral research and fluid biomarkers, brain imaging studies offer a unique opportunity to relate changes in brain structure and function, changes in cerebral blood flow, neuronal activation and neurochemical changes in the brain to cognition and cognitive impairment. Neuroimaging approaches to cognition in PD have been reviewed recently (Hall and Lewis 2019; Hou and Shang 2022; Montaser-Kouhsari et al. 2022; Weil et al. 2019). Although there is a continuum from PD-NC to SCD, PD-MCI and PDD, the major neuroimaging changes in the progression of normal to impaired cognition have been described separately.
Gray matter changes in early PD
While in noncomplicated PD, structural neuroimaging may be normal or shows only mild diffuse brain atrophy or temporal lobe changes in early PD (Martin et al. 2009; Pereira et al. 2014), voxel-based morphometry in PD patients with subjective memory complaints revealed reduced gray matter (GM) intensities in anterior cingulate and right parietal lobe than in uncomplicated ones (Hong et al. 2012). Earlier studies showed reduced gray matter volume (GMV) in frontal lobe in patients with PD and no dementia (PDND) compared with control subjects, while there was significant GM atrophy in the occipital lobe in PDD patients which extended from frontal areas to temporal, occipital and subcortical areas (Burton et al. 2004). Measurement of cortical thickness revealed distinct limbic and subtle GM atrophy in anterior cingulate, precuneus and temporal neocortex in PD-NC compared to healthy controls (Kunst et al. 2019).
Recent studies indicated that reduction of GM density in superior frontal gyrus and cerebellum were related with cognitive performance in early PD-MCI (Donzuso et al. 2021), while right entorhinal cortex atrophy was seen in early, drug-naive PD-MCI, which provided new evidence in differentiating the neuroanatomical states between PD-MCI and PD-NC (Jia et al. 2019).
Magentic resonance imaging (MRI) findings in PD-MCI (Table 1)
At baseline, compared with stable PD-NC cases, those with conversion to MCI showed cortical atrophy in the parietal and occipital lobes, similar to PD with stable MCI, while those with CI from the study entry showed additional involvement of the frontotemporal cortices (Weintraub et al. 2011). MCI is linked with a faster rate of cortical thinning in patients with PD longitudinally, as well as with significant diminishment of limbic subcortical structures (Hanganu et al. 2014). PD-MCI subjects revealed significant enlargement of bilateral temporal, occipital and left frontal lateral ventricles relative to PDND ones (Apostolova et al. 2012). GMV loss in MCI is characterized by prefrontal and occipital GM atrophy (Weintraub et al. 2011). A study using voxel-based morphometry, showed atrophy of the right entorhinal cortex in PD-MCI patients compared to PD-NC ones (Jia et al. 2019), while a resting-state functional MRI study documented hyperactivity (reflecting a compensatory mechanism) in the right inferior frontal gyrus and hypoactivity in the occipital area in early PD with MCI (Wang et al. 2018). PD-MCI showed greater GM atrophy than PD-NC in orbitofrontal regions, left superior parietal lobule, more wide-spread limbic and fronto-parietal-occipital neocortical atrophy (Kunst et al. 2019). While frontostriatal atrophy may be a predictor for dementia in PD-MCI (Lee et al. 2010), other reduced GMV regions, including temporal and parietal cortices, amygdala, putamen and hippocampus have also been implicated (Melzer et al. 2012), the latter particularly associated with memory impairment (Chen et al. 2016; Weintraub et al. 2011).
A meta-analysis of around 1400 PD patients reported a significantly higher GM atrophy in bilateral prefrontal cortex, left angular gyrus, right supramarginal gyrus, left insula, and midcingulate cortex in the PD-MCI group, but atrophy of bilateral insula and right hippocampus in the PDD group (Mihaescu et al. 2019), while another meta-analysis reported severe GM atrophy in the left anterior insula, inferior and orbital-frontal gyrus (Zheng et al. 2019). Smaller cornu ammonis (CA) 1 region and hippocampal-amygdaloid transition area volumes have been observed in PD-MCI compared to PDND (Becker et al. 2021). Early PD-MCI showed reduction of GM density in superior frontal gyrus and cerebellum (Donzuso et al. 2021).
Longitudinal studies have shown a significantly greater progression of cortical thinning in posterior brain region in PD-MCI compared to PDND (Garcia-Diaz et al. 2018), while another 4-year follow-up study showed that both PDND and PD-MCI patients have a more severe decline in anterior and posterior hippocampus related to memory dysfunction (Uribe et al. 2018). Significant correlations were found between global cognitive status and lateral hippocampus volume, with significant reduction of bilateral CA4, and other subfields and right presubiculum, indicating selective regional vulnerability of the hippocampus in the progression of PD (Foo et al. 2016; Xu et al. 2020).
MRI findings in PD-MCI and PDD converters (Table 2)
Relative to PD-MCI patients who did not convert to PDD, the converters showed lower GM densities in prefrontal areas, insular caudate nucleus and lesser cortical thickness extending from the posterior cortical area into the frontal region and frontotemporal cortices (Chung et al. 2019; Filippi et al. 2020). PD-MCI is associated with a faster rate of GM thinning in temporal and medial occipital lobes as well as limbic subcortical structures (Hanganu et al. 2014); others observed early atrophy in temporal lobes and progressive atrophy in frontal lobes in patients who converted to PD-MCI (Zhou et al. 2020).
Few studies using longitudinal MRI metrics to predict MCI or dementia conversion in PD patients suggested that atrophy of fronto-temporal areas, hippocampus, thalamus and accumbens play a role in this process. Stratifying patients according to disease severity findings appeared partially controversial, although showing progressive atrophy of basal ganglia over one year of follow-up and a widespread cortical thinning over 3–6 years in patients with mild to moderate CI (Sarasso et al. 2021).
A longitudinal analysis showed that PD patients with stable MCI and those with no conversion to dementia accumulated the least cortical damage, while those with conversion to dementia showed progressive volume loss of right thalamus and hippocampus. PD patients with conversion to MCI had cortical thinning in the medial and superior frontal gyri, inferior temporal, precuneus, cingulate and supramarginal gyri bilaterally, whereas those with stable normal cognition showed cortical thinning progression mainly in parietal and occipital regions bilaterally. In general, cortical thinning was more prominent in the initial stage of PD cognitive decline, whereas involvement of the frontotemporoparietal regions, hippocampus and thalamus is associated with conversion to a more severe stage of CI (Filippi et al. 2020).
MRI in PDD (Table 3)
One study investigating whole brain atrophy in PDD showed a rate of atrophy of 1.12% in PDD patients, compared to 0.31% in non-demented ones and 0.34% in healthy age-matched controls. Rather surprisingly, it found no correlations between atrophy rate and dementia severity, which might be attributed to an insensitive scale used (Burton et al. 2005). One of the first identified predictive markers for cognitive decline in PD was temporo-parietal atrophy, which is indicative of AD pathology (Weintraub et al. 2012), confirmed by many subsequent studies (Hall and Lewis 2019). In addition, basal forebrain atrophy is also associated with CI in PD (Pereira et al. 2020; Ray et al. 2018). Memory impairment is correlated with frontal and hippocampal diffusivity impairments (Carlesimo et al. 2012; Gargouri et al. 2019; Melzer et al. 2013). Dorsomedial thalamus free water (FW) correlates with cognitive decline in early PD, while baseline hippocampal FW was associated with CI at 3 years, and baseline dorsomedial thalamic FW distinguished PD-NC from PD with cognitive impairment (Guttuso et al. 2022).
Cluster analysis of multimodal imaging data identified three PD subtypes, with prominent GM patterns and little white matter (WM) involvement: One group with widespread cortical and subcortical GMV and WM fractional anisotropy (FA) reductions and pronounced cognitive deficits; a second group with only cortical atrophy limited to orbitofrontal and temporal regions and more specific neuropsychological impairment, and a third one without detectable atrophy or CI and earlier disease onset (Inguanzo et al. 2021). Early onset PDD patients exhibit more severe atrophy in the left anterior cingulate and right inferior temporal gyrus with significantly decreased substantia innominata volume (Kim et al. 2017). These results are in line with recent results showing structural connectivity differences in PD subtypes (Abbasi et al. 2020).
A meta-analysis showed consistent GM loss bilaterally in the medial temporal lobes and the striatum (Pan et al. 2013). A discrimination analysis demonstrated that the volume of hippocampus, in combination with cortical thickness could identify PDD patients with an 80% accuracy (Zarei et al. 2013). PPD patients have GMV reduction in the superior temporal, inferior frontal lobe, insula and anterior cingulate cortex (Xu et al. 2016).
White matter (WM) changes in PD-MCI and PDD
Prominent WM changes are observed in both PD-MCI and PDD patients. Early changes in WM in PD-MCI patients with intact GMV have been reported (Agosta et al. 2014; Rektor et al. 2018). White matter hyperintensities (WMH) burden in PD-MCI patients was significantly different from that in PD-NC (Liu et al. 2021). WMH volume changed over time and was associated with impairment in global cognition, executive functions and language, whereas WM microstructural changes did not vary significantly with those clinical parameters (Scamarcia et al. 2022). However, significant reductions in WM volume have not been consistently found with PD-MCI compared with healthy controls (Butt et al. 2021; Hanning et al. 2019; Hattori et al. 2012; Yarnall et al. 2014). This suggests that the heterogeneous phenotypes seen in PD-MCI may impact on these distinctions and that either brain atrophy may not be as prominent in the early stages of PD-MCI (Hall and Lewis 2019). Moreover, microstructural damage in the main motor and associative WM tracts are present and rapidly progress, even in early phases of PD (Sarasso et al. 2021). PDD patients had a significantly higher burden of WMH, especially deep WMH, which might be an imaging marker for CI in PDD but not in PD-MCI (Liu et al. 2021). Whole brain studies revealed the involvement of the corpus callosum, cingulum and major association tracts in PD-MC patients, but not in PD-NC (Agosta et al. 2014; Chen et al. 2016; Hattori et al. 2012). PD-MCI shows increased hyperintensity in frontal and interhemispheric WM (genu and body of corpus callosum) (Agosta et al. 2014; Deng et al. 2013; Melzer et al. 2013). Thinning of corpus callosum in PDD compared to PD-MCI and PD-NC correlated with thickness of left orbitofrontal cortex in PD-MCI, while changes in corpus callosum in PDD occur in line with changes in the cortex in advanced disease stage (Owens-Walton et al. 2022). The corpus callosum, the cingulum bundle, and the corticospinal tract showed the same trend in the decline of cognitive function (Sang et al. 2022). In addition, the PDD group showed FA decrease and/or mean diffusivity increase in the bilateral cingulate tract (Kamagata et al. 2012; Matsui et al. 2007), in genu of corpus callosum (Chondrogiorgi et al. 2019; Kamagata et al. 2013), and hippocampus (Chen et al. 2015).
Correlation analyses between memory and voxel-based WM measures showed that PD-aMCI had smaller FA values than PD-NC in diffuse WM areas (Chen et al. 2019). Overall, WM abnormalities in PD patients with CI seem to be widespread (Hall and Lewis 2019), involving multiple brain regions with a heterogeneous pattern, abnormal diffusivity variables being widely distributed in WM adjacent to cortices and limbic subcortices (Zhang and Burock 2020). PDD patients show a significantly higher burden of periventricular and deep WMHs compared to PD-NC (Beyer et al. 2006; Lee et al. 2010), which might be an imaging marker for CI in PDD but not in PD-MCI (Liu et al. 2021).
In summary, GM changes in PDD predominantly involve the temporal regions including the hippocampus, frontal and parietal areas as well as subcortical areas including thalamus and nucleus basalis of Meynert (NBM), while WM lesions are most typically observed in the corpus callosum and cingulate gyrus, inducing dysfunctions of cortico-cortical and cortico-subcortical networks, while local network analysis showed reduced efficiency predominantly in the frontal and parietal regions with the PD-MCI group (Colon-Perez et al. 2018). However, the clinical heterogeneity of MCI in PD is reflected in the variability of structural imaging findings and identifying a unique structural signature of PD-MCI remains challenging (Hall and Lewis 2019).
Degeneration of neurotransmitter systems
Dopaminergic system
Cognitive deficits in early PD are associated with impaired striatal and extrastriatal dopaminergic dysfunction (Siepel et al. 2014), which results in abnormal processing in the cortico-basal ganglia circuit with reduced prefrontal and parietal metabolism in PD-MCI (Bohnen et al. 2011; Ekman et al. 2012), in the salience network (SAN), and in the medial temporal lobe (Christopher et al. 2015), which contribute to memory impairment in PD, whereas mesocortical dopamine transmission appears to be preserved (Huang et al. 2008). Lower presynaptic dopamine uptake in striatum correlated with under-recruitment of anterior cingulate cortex suggesting frontostriatal dysfunction (Ekman et al. 2012). Functional MRI studies have shown frontostriatal and temporal lobe deficits in some PDD patients suggesting an involvement of both the nigrostriatal and the mesocortical dopaminergic pathways. Resting-state functional MRI studies that provide evidence of functional connectivity changes are consistent with the concept of two distinct cognitive syndromes in PD, which include dopaminergically mediated frontostriatal executive impairments and a "posterior cortical syndrome" more frequently associated with the later development of dementia (Baggio et al. 2015; Lebedev et al. 2014; Olde Dubbelink et al. 2014). Striatal dopamine transporter availability mediates the association between WMHs and CI in the visuospatial and memory domains (Jeong et al. 2022).
All patients with PD have a moderate to severe loss of dopaminergic neurons in the nigrostriatal pathway. More widespread degeneration of dopamine terminals in the striatum, particularly in the dorsal caudate nucleus, occurs in patients with PD-MCI than in those without CI. However, in PD-MCI patients there is relative preservation of the other dopaminergic systems in the brain, while those with PDD have a considerable loss of the lateral dopaminergic systems in frontal, parietal and temporal cortical regions (Sasikumar and Strafella 2020). Dysfunction of subcortical-cortical networks is the result of neuronal loss in the brainstem and limbic areas; cholinergic deficits in the cortex, thalamus, and NBM; striatal dopamine loss, decreased nicotinic acetylcholine receptors, and degeneration of the medial substantia nigra (SN) and striatofrontal and mesocorticolimbic loops. Dopaminergic differences in the SAN and the medial temporal lobes also contribute to memory impairment in PD (Christopher et al. 2015).
Forebrain cholinergic system
In vivo cholinergic forebrain atrophy predicts cognitive decline in de novo PD (Grothe et al. 2021; Ray et al. 2018). Microstructural alterations within the cholinergic NBM, detected by diffusion tensor imaging, have been identified as a strong predictor for development of CI in PD, and precede structural GM volume loss (Wilson et al. 2021). Volume loss of the NBM is specific to PD and progressive supranuclear palsy but not to multiple system atrophy (Rogozinski et al. 2022).
WM lesions were found in the cholinergic pathway projecting from the NBM to the cortex, associated with severe memory impairment (Park et al. 2015); these lesions were increased in PDD compared to PD-MCI and PD-NC, supporting the notion that memory dysfunction is related to cholinergic impairment (Schulz et al. 2018). Patients with smaller volumes of the NBM had a 3.5-fold greater risk of developing PD-MCI over about 5 years (Ray et al. 2018).
PDD is associated with selective destruction of corticostriatal resting functional MRI correlations (Seibert et al. 2012), while acetylcholinesterase-PET (positron emission tomography) demonstrated that posterior brain areas are related to cognitive decline in PD (Hirano et al. 2012). PD patients showed a reduction in volume and density of the forebrain cholinergic region and their projections to neocortex, hippocampus and amygdala, which was associated with CI over a 2-year period and predicted CI in those with PD-NC over 5 years (Bohnen et al. 2015; Ray et al. 2018; Schulz et al. 2018). The loss of the basal forebrain cholinergic projections to the hippocampus correlates with memory deficits and conversion to PDD (Gargouri et al. 2019; Pereira et al. 2020). Loss of hippocampal cholinergic fibers is seen in patients with PD-MCI, whereas those with PDD show a subsequent increase in αSyn deposition and dysfunction in both hippocampal and basal forebrain cholinergic systems (Hall et al. 2014; Liu et al. 2018). Significant subcortical degeneration with neuronal loss and LBs in NBM may precede the onset of PDD due to cortical cholinergic denervation and αSyn pathology (Jellinger 2007a). Cortical cholinergic denervation and early posterior cortical atrophy induced by caudate dopaminergic denervation contribute to CI in PD (Bohnen et al. 2015; Sampedro et al. 2019). Reduction of cholinergic markers in PDD is due to early degeneration of the corticopetal basal forebrain projection involving both the NBM and the nucleus of the diagonal band of Broca (Liu et al. 2018; Ray et al. 2018; Schulz et al. 2018).
The noradrenergic locus ceruleus, serotonergic dorsal raphe nucleus and ventral tegmental area are also involved (Del Tredici and Braak 2013; Espay et al. 2014; Halliday et al. 2014; Tilley et al. 2021; Vermeiren and De Deyn 2017; Ye et al. 2022). PD-MCI patients showed a reduction in the neuromelanin-sensitive MRI signal of the locus ceruleus (Li et al. 2019; Prasuhn et al. 2021). MRI techniques sensitive to brain iron content found higher brain tissue iron content in cerebral cortices, hippocampus, thalamus, and putamen related to lower Montreal Cognitive Assessment scores in early and mid-stage PD (Thomas et al. 2020).
Connectivity and network degradation
Multimodal imaging studies showed a loss of functional connectivity and topological features without structural damage in the SAN in PD-MCI (Aracil-Bolaños et al. 2019), while recent studies revealed disrupted myelin networks in the cingulate cortex of PD (Xie et al. 2022).
Comparison of corticostriatal connectivity in PD-MCI showed decreased function between the striatal network and both the default mode (DMN), central executive and saliency (SAN) networks compared to PD/nonMCI and age-matched control subjects. This was explained partly by increased atrophy within the SAN in PD-MCI. The seed analysis revealed a relationship between higher MCI scores and lower connectivity of the left caudate head to the dorsal anterior cingulate and left middle frontal cortex, as well as to decreased connectivity of the right caudate head with the anterior cingulate cortex, precuneus, and left supramarginal gyrus, and increased connectivity to the left hippocampus and right cerebellar hemisphere. These results suggest that PD-MCI is associated with both global behavioral and cognitive symptoms in PD (Lang et al. 2020). Disrupted WM connectivity in frontal and posterior cortical regions, which correlates with frontal/executive dysfunction, are associated with early dementia conversion in PD-MCI (Chung et al. 2022). Furthermore, PD-MCI is associated with reduced connectivity of the mediodorsal thalamus with the paracingulate cortex, while also demonstrating increased functional connectivity of the mediodorsal thalamus with posterior cingulate cortex, compared to PDD. Structures with basal ganglia-thalamo-cortical circuits are implicated in CI and dementia in PD, which are associated with a breakdown in the connectivity of mediodorsal thalamus with para- and posterior cingulate regions, respectively (Owens-Walton et al. 2021). The brain regions involved in PD-MCI are associated with the somatosensory and executive processing networks (Mihaescu et al. 2019), and specific change in resting-state functional connections in frontostriatal and posterior cortical subtypes of PD-MCI (Devignes et al. 2022).
Reduced cognitive performance in PD patients was also associated with functional connectivity of the dorsal insular cortex with the DMN, highlighting the relevance of the insula in cognitive dysfunction in PD (Fathy et al. 2020). Tracts between dorsal anterior insular cortex and anterior cingulate cortex showed lower fractional anisotrophy and higher mean diffusivity in PD patients with lower working memory and executive functions, indicating a structural damage in the dorsal limbs of the SAN in PD, possibly due to loss of interconnecting anterior insular cortex subregions and anterior cingulate cortex. This provided evidence for clinically relevant structural damage to the cortical limbs of the SAN in PD due to extensive neuropathology and loss of interconnecting anterior insular and anterior cingulate cortex (Jonkman et al. 2021).
Studies of the connectivity within two distinct DMN systems—left-to-right hippocampal (LHC-RHC) and medial prefrontal cortex to posterior cingulate cortex (mPFC-PCC)—showed that LHC-RHC connectivity was significantly associated with global and domain-specific cognitive impairments, while the mPFC-PCC was associated with future global and episodic memory impairment. This suggests that there is a functionally distinct role of the hippocampal subsystems within the DMN resting state network and that intrinsic connectivity between the hippocampus is related to a broad range of cognitive functions in PD (Zarifkar et al. 2021). Reduced hippocampal FA correlating with global cortical decline in PD (Chen et al. 2015) is associated with disruption of cortex functional connectivity (Rektorova et al. 2012; Seibert et al. 2012) with predominant frontal cortical disruption, while others showed altered temporal properties in dynamic connectivity in PDD (Fiorenzato et al. 2019). Examination of altered (dynamic) functional interactions between brain networks relating to cognitive dysfunctions in PD patients showed that the severity of executive dysfunction was correlated with higher static and lower dynamic functional connectivity between deep GM regions and the frontoparietal network (DGM-FPN). Declining executive function was related to increasing static DGM-FPN connectivity, together with changes of connectivity involving the dorsal attention network. These findings demonstrate that in PD patients, dysfunctional connections between subcortical fronto-parietal and attention networks mostly underlie worsening in executive functioning (Boon et al. 2020). In general, CI in PD is associated with reduced connectivity in networks relevant to cognition, most prominently to the DMN (Gratwicke et al. 2015; Wolters et al. 2019).
Brain positron emission tomography studies in PDD
18FFluorodeoxyglucose positron emission tomography (FDG-PET) studies showed hypometabolism in parietal, precuneus, hippocampus, and occipital lobes in PD with incident dementia (Bohnen et al. 2011), while hypometabolism in medial frontal and parietal regions was associated with decline in memory and executive functions (Huang et al. 2007), and reduced metabolism in posterior cortical regions was observed in PD-MCI patients (Schrag et al. 2017). Aβ PET studies showed higher rates of tracer retention in PDD but the degree of uptake was less than that seen in AD (Foster et al. 2010; Mashima et al. 2017; Oh et al. 2021; Villemagne et al. 2011), Patients who show higher degree of Aβ uptake are at higher risk of developing CI (Petrou et al. 2012; Shah et al. 2016). 18FFlorbetapir PET showed that severe Aβ deposition is common in PDD patients (52.4%), contributing to memory impairment and driving a faster rate of cognitive decline (Palermo et al. 2019). In other PET studies, prevalence of Aβ-positive cases was 0.34 (95% CI 0.13–0.56) in the PDD group and 0.05 (95% CI − 0.07 to 0.17) in the PD-MCI group (Petrou et al. 2015). Other groups did not find an association between Aβ deposition and CI in PD (Ko et al. 2017; Melzer et al. 2019). Frequency of positive Aβ PET scans in PD-MCI (5–11%) was not different from age-matched controls (Melzer et al. 2019; Petrou et al. 2015; Winer et al. 2018). The patterns of cortical Aβ and tau did not significantly differ between people with PD-NC, those with PD-MCI and healthy older adults. Thus, age, Aβ and tau did not differentiate patients with PD-NC and PD-MCI (Winer et al. 2018). A recent study showed that the Aβ-positive PD group had higher frequency of MCI, especially amnestic type, and lower dopaminergic activities in the left ventral striatum, suggesting that PD patients with Aβ positivity have AD-related cognitive changes (Na et al. 2020; Oh et al. 2021). In general, PDD patients have a lower incidence of Aβ deposition than DLB patients (Akhtar et al. 2016; Frey and Petrou 2015). No significant increase of tau-PET in SN or cortex brain flortaucipir uptake was seen across a 2-year follow-up in PD patients (Hansen et al. 2020). Preliminary tau-PET studies using 18Fflortaucipir (formerly called AV-1451) indicated a gradient of tau binding from PD-NC (none to minimal) via PD-MCI (minimal), PDD (low/modest) to DLB (intermediate/strong) to AD (highest) (Bohnen et al. 2017), uptake in PDD being intermediate between PDND and AD (Coughlin et al. 2020; Gomperts et al. 2016; Kantarci et al. 2017). Similar to postmortem data for tau pathology, increased flortaucipir uptake antemortem is also associated with dementia in PD (Smith et al. 2018). The recently described binding of 18Fflortaucipir uptake by neuromelanin (Marquie et al. 2017) and the relevance of radioiodinated benzimidazole derivates for selective imaging of αSyn aggregates (Alzghool et al. 2022; Roshanbin et al. 2022; Watanabe et al. 2017) deserve further confirmation.
Neuropathology of PD-MCI
Although the heterogeneous pathology of PDD and PD-MCI are well documented (Halliday et al. 2014; Molano et al. 2010; Sabbagh et al. 2009), there are few neuropathological studies of PD-MCI. Two neuropathological studies described 16 PD-MCI cases: among 365 autopsy-proven PD, eight (2.2%) met the criteria for PD-MCI (mean age 82.2, mean disease duration 11.4 years). Four patients had aMCI memory, three naMCI with frontal executive and one with executive and visuospatial dysfunction. Three cases were brainstem-dominant and brainstem-limbic-dominant, and two neocortical LB stage (Beach et al. 2009). Two patients with naMCI and one with aMCI showed multiple brain infarcts, emphasizing the role of co-existent cerebrovascular pathology (Adler et al. 2010). In addition, there was severe amyloid plaque intensity in the cortex; four with moderate to severe cerebral amyloid angiopathy (CAA), while one case each had moderate to severe CAA (Adler and Beach 2010). Among 233 autopsy-proven cases of PD (54.6% cognitively unimpaired), eight (3.4%) met the criteria for PD-MCI (mean 76.7, disease duration 13.4 years). Four patients were aMCI memory only; three naMCI with frontal dysexecution, and one multiple-domain aMCI. Two were brainstem, 5 brainstem-limbic, and one neocortical LB stage (Jellinger 2010a). Neuritic Braak stages ranged from I to III (mean 1.3); a few neuritic plaques and mild generalized CAA were detected in only two brains, while no diffuse plaques were seen in the basal ganglia. In the case of multidomain MCI, there was a correlation between amyloid and neuritic plaques and CAA (Jellinger 2010b), confirming the contribution of both Aβ plaque load and CAA to CI (Jellinger and Attems 2008). The neuropathological data in these 16 PD-MCI cases (8 aMCI-PD, 7 naMCI-PD, one amnestic multiple domain, mean age 78 years) can be summarized as follows: 50% were brainstem dominant LB disease, 31% brainstem-limbic forms, 19% neocortical type. Neuritic Braak stage in aMCI was slightly higher than in naMCI (mean 2.7 vs 2.1); mild neuritic plaques were seen in 12%, moderate ones in 31%; mild CAA in 11%, lacunar state in 25%, and old cerebral infarcts in 12.5%. These data indicated a heterogeneous neuropathology in PD-MCI (Jellinger 2013). Recently the neuropathological findings of 49 cases (15 with the clinico-pathological diagnosis of PD) with amnestic aMCI and naMCI were compared, reporting the propensity of increased neurofibrillary tangles (NFT) in the aMCI group and increased LBs in the naMCI group (Dugger et al. 2015). In a recent study of 159 autopsy-confirmed PD cases, 25 had PD-MCI and 102 PDD. In the PD-MCI group 56% met criteria for aMCI and 44% of naMCI, showing no significant differences in age, gender, PD duration, etc. In the naMCI group, all were brainstem-limbic stage (III), which was significantly different from the aMCI group in which only 22% were at neocortical stage (IV). Concomitant non-AD tauopathy was present in 9 PD-MCI cases (42% aMCI and 18% naMCI). Both aging-related tau astrogliopathy (ARTAG) and argyrophilic grains were seen in 5 cases with no significant differences between both groups. Two aMCI cases also met neuropathological criteria of progressive supranuclear palsy. No differences were found in neuritic plaque density, total plaque density score, WM rarefaction, cerebral infarct volume. CAA score or APOE carrier frequency were similar between both groups (Knox et al. 2020). This study also confirmed a clear morphological heterogeneity in PD-MCI similar to that in MCI without PD (Markesbery 2010). In this cohort, 56% of the PD-MCI cases had aMCI with no preponderance of naMCI as reported in other series (Litvan et al. 2012). The aMCI cases had slightly higher Braak NFT stages, while a previous study of autopsy-proven PDD cases showed that 54.9% of them had concomitant AD, although there was little difference in their clinical dementia presentation (Sabbagh et al. 2009), while another recent study revealed an increase in LB pathology in naMCI (Knox et al. 2020). Furthermore, the presence of non-AD pathology in this PD-MCI cohort suggests that the role of tauopathies in PD-MCI and PDD should be further explored.
Neuropathology of PDD
There is a large number of extensive reports about the neuropathological substrates of PDD, most of them discussing the convergence and interactions of αSyn, tau and Aβ pathologies and their contribution to dementia pathogenesis, the relations between PD and AD, associated dysfunctions of various neurotransmitter systems, metabolic disorders (Compta et al. 2011; Coughlin and Irwin 2022; Hall et al. 2014; Halliday et al. 2014; Irwin et al. 2012, 2013; Jellinger 2012b; Kalaitzakis and Pearce 2009; Liu et al. 2019; Smith et al. 2019; Wills et al. 2010), the influence of co-pathologies on cognition in PD (Coughlin and Irwin 2022; Daida et al. 2018; Homma et al. 2015; Smith et al. 2019) or discussing specific changes, like protein pathology (Kouli et al. 2020; Tu et al. 2022), neuroinflammation (Kouli et al. 2020) or mitochondrial disorders in PDD (Garcia-Esparcia et al. 2018; Gatt et al. 2016).
Lewy/αSyn pathology
Although few cortical LBs are found in virtually all cases of sporadic PD, there is no consensus on the structural basis of CI in PD (Jellinger 2009; Sonnen et al. 2010). The function of αSyn remains under investigation, but it is localized in presynaptic neuronal membranes and regulated endocytosis and trafficking (Bendor et al. 2013; Vargas et al. 2014). Due to the ubiquitous deposition of αSyn in the central nervous system with high enrichment in presynaptic terminals, PD is denoted a synucleinopathy (Uversky 2009), showing specific synaptic pathology of αSyn aggregation (Schulz-Schaeffer 2010). The morphological substrate of PDD is heterogeneous and includes (1) Lewy/αSyn pathology in cortical, limbic, and subcortical structures, (2) AD-related neuropathological changes (ADNC) (diffuse and neuritic plaques, neurofibrillary tangles and CAA), and (3) a combination of these pathologies that has been shown to most robustly correlate with the severity of CI (Compta et al. 2011; Halliday et al. 2014; Irwin et al. 2012; Jellinger 2012b; Smith et al. 2019). Based on a large autopsy series of PD patients, a stereotypical pattern of spread of Lewy body pathology (LBP) from brainstem regions and olfactory bulb via limbic areas to neocortical areas was suggested (Braak et al. 2003), and later modified (Beach et al. 2009). The reasons for this selective vulnerability to accumulate LBP of these regions remains unclear but may be due to the fact that the longer, poorly myelinated axons or functionally connected networks may be prone to develop pathology and favor transsynaptic neuronal spread of pathogenic αSyn (Braak et al. 2004; Surmeier et al. 2017), suggesting a prion-like mechanism of αSyn pathology in PD.
Limbic and neocortical LBP are approximately 10 times higher in PDD cases than in PDND ones (Apaydin et al. 2002). CI in PD is often correlated with the density of LBs in frontal cortex and Lewy neurites and neuritic degeneration in hippocampus and periamygdaloid cortex, causing a disruption of the limbic loop similar to that described in AD (Mattila et al. 1999). The severity of LBP in the CA2/3 region of the hippocampus has been shown to correlate with episodic memory loss (Adamowicz et al. 2017; Harding et al. 2002), although hippocampal atrophy and cell loss are not necessarily involved in memory impairment in PD (Joelving et al. 2006). The severity of CI correlates strongly with Braak PD stage (Braak et al. 2005). In a large autopsy series, 50% of PDD patients showed Braak Lewy neurite stages 4–6, particularly when cases with coexistent ADNC were excluded (Mattila et al. 2000). PDD cases also showed higher LBP in subcortical regions compared to PDND cases. In the striatum, insoluble αSyn levels were twice as high (Wills et al. 2010), while in the amygdala and hippocampus, LB density correlated with dementia severity (Apaydin et al. 2002; Churchyard and Lees 1997; Halliday et al. 2011; Mattila et al. 2000). Parahippocampal αSyn scores showed excellent sensitivity (91–93%) and specificity (84–88%) for separating PD cases with and without dementia (Harding and Halliday 2001). PDD cases usually showed higher LBP in subcortical regions relative to PDND ones. In a large study, the severity of cortical LBP was the factor that best correlated with dementia (Irwin et al. 2012), and in a community-based study of 872 autopsies, 103 showed neocortical LBP associated with increased odds of dementia and more rapid decline in all cognitive domains, whereas a limbic distribution was specifically associated with more rapid decline in visuospatial skills, which was not modified by coexistent AD pathology (Schneider et al. 2012). It should be considered, however, that not all patients with cortical LBP may develop dementia (Colosimo et al. 2003; Irwin et al. 2012; Kempster et al. 2010), although LB densities in temporal lobe were significantly higher in PDD compared to PDND cases, which was not observed in frontal or limbic cortical regions (Harding and Halliday 2001). The more severe increase of αSyn in inferior frontal gyrus in PDD patients compared to those without dementia (Wills et al. 2010), enhances the discussion in defining the underlying substrate of CI in PD, in particular with regard to the impact of neocortical LB burden (Jellinger 2007b; Kalaitzakis and Pearce 2009; Selikhova et al. 2009). However, the findings of more increased αSyn burden in the inferior frontal cortex in PDD subjects appear to favor increased LBP in neocortex contributing to dementia.
Insoluble αSyn in the striata being substantially higher than soluble levels in normal controls showed significant increase in both PDND and PDD, with much higher increase in PDD (176% vs. 141%) and in inferior frontal cortex (41- vs. 20-fold; p < 0.019), suggesting that there is a substantial increase of αSyn in both regions, being significantly greater in PDD (Wills et al. 2010). Striatal αSyn pathology in PDD was associated with Braak LB stage 3, and only mild striatal αSyn burden in PD brains scored LB Braak stages 3–5 (Jellinger and Attems 2006).
The strong association between extensive αSyn pathology and dementia was challenged by some studies that reported that 15–44.7% of cognitive intact PD patients were associated with severe neocortical LBP (Compta et al. 2011; Horvath et al. 2013; Irwin et al. 2012; Kempster et al. 2010), while a small study described PD cases without dementia despite limbic and neocortical LB pathology and concluded that no clear threshold of LB burden can distinguish PD cases with and without dementia (Colosimo et al. 2003). On the other side, few cases with dementia were described without LBs outside the brainstem and only mild or absent concomitant AD or cerebrovascular pathology (Libow et al. 2009), and dementia cases with αSyn pathology confined to the brainstem were observed in 14.7% of 109 PDD cases (Horvath et al. 2013), while other studies reported much lower figures (Aarsland et al. 2005a; Colosimo et al. 2003; Compta et al. 2011; Harding and Halliday 2001; Irwin et al. 2012; Kotzbauer et al. 2012; Sierra et al. 2016; Walker et al. 2015).
Role of AD pathology
Coexisting tau and Aβ pathology of varying severity is common in PD with CI and relates to a faster onset of dementia (Compta et al. 2011; Halliday et al. 2014; Howlett et al. 2015; Irwin et al. 2012, 2017; Jellinger et al. 2002). AD-related changes, severe enough for a secondary contribution to CI, were present in about 10% of PDND and in about 35% of PDD patients in various autopsy series (Irwin et al. 2012; Jellinger 2008; Smith et al. 2019). In general, both LBP and ADNC may occur and act synergistically (Colom-Cadena et al. 2017; Halliday et al. 2014; Hepp et al. 2016; Irwin et al. 2012, 2013; Jellinger 2009; Kotzbauer et al. 2012; Lashley et al. 2008; Nelson et al. 2010). In large autopsy series around 50% of PDD patients showed Braak LB stages 4–6 together with severe ADNC (Braak neuritic stages 5 and 6) (Irwin et al. 2013; Jellinger 2007a), while others suggested a significant positive relationship between cortical αSyn deposition and CI (Biundo et al. 2016; Petrou et al. 2012). ADNC has been considered by some to be a more specific correlate of dementia than cortical LBP, since the majority of PDD cases with sufficient numbers of cortical NFTs could be assigned a diagnosis of PD plus AD (Compta et al. 2011; Irwin et al. 2012). However, the proportion of PD with comorbid AD varies considerably. The four largest studies (n = 88 to n = 200) that defined AD as intermediate or high probability by NIA/AA (National Institute on Aging/Alzheimer’s Association) criteria, showed reasonably consistent results: comorbid AD was diagnosed in 19.3–31.5% of total PD cases, while the rate of comorbid AD in PDD cases showed much higher variation between 21.5 and 89.4% (Braak et al. 2005; Irwin et al. 2012; Jellinger et al. 2002), tau pathologies in PDD cases affected the prefrontal cortex more severe than the temporal cortex, while the occipital cortex was rarely affected (Vermersch et al. 1993). Two reports related advanced ADNC with severe dementia and concluded that PDD was particularly related to comorbid AD (Bancher et al. 1993; Jellinger et al. 1991), which was confirmed by later studies from the same research group (Jellinger et al. 2002; Jellinger and Attems 2008). The patterns of Aβ pathology and spread of NFTs in PDD are similar to that seen in typical AD, although in some cases the medial temporal lobe was relatively spared and there were some differences in neocortical tau burden (Coughlin et al. 2019b; Walker et al. 2015). The presence of co-existing ADNC relates to faster onset of dementia in PD (Compta et al. 2011; Halliday et al. 2014; Irwin et al. 2012, 2017; Jellinger et al. 2002). Moreover, AD co-pathology is related to older age at disease onset and decreased survival (Irwin et al. 2017; Kotzbauer et al. 2012; Sabbagh et al. 2009); some reports suggest that ADNC has a greater influence on dementia onset than αSyn pathology (Compta et al. 2014; Howlett et al. 2015). Co-existent ADNC has been shown to produce greater deficits in episodic memory (Coughlin et al. 2019a; Kraybill et al. 2005; Peavy et al. 2016).
A clinicopathological study identified three subgroups of PDD: (1) predominant synucleinopathy (LB Braak stages 5–6; 38%), (2) synucleinopathy with Aβ deposition but minimal or no tau pathology (59%), and (3) synucleinopathy with considerable to severe neocortical tau pathology (Braak neuritic stages 5–6; 3%). Patients in group II showed significantly shorter survival than those with pure synucleinopathy (Kotzbauer et al. 2012). Another study showed three groups with different LBP distributions: PD patients without comorbid AD, PD with AD (PD-AD) and DLB with AD (DLB-AD). The PD-AD group had ADNC with increased LBP; the DLB-AD group showed relative preservation of SN, while coincident ADNC was associated with increased LBP suggesting interaction of both. These cluster-defined groups were associated with different rate of progression to dementia (Toledo et al. 2016). LBP has typically been considered the most significant predictor of dementia in PD (Horvath et al. 2013; Irwin et al. 2012; Kövari et al. 2003; Ruffmann et al. 2016), while in some studies Aβ and tau pathologies were suggested to be independent predictors of dementia (Compta et al. 2011; Horvath et al. 2013). However, the additive or synergistic effect of αSyn on AD pathologies may influence clinical features of PDD, like shorter disease duration or more malignant course (Compta et al. 2011, 2014; Halliday et al. 2014; Irwin et al. 2017).
Contribution of αSyn, Aβ and tau to PDD
There is increasing evidence that abnormal αSyn, Aβ and tau are significant predictors of dementia in PD (Horvath et al. 2013; Irwin et al. 2012; Ruffmann et al. 2016). One study found that the variance in cognitive scores was related to LBP in entorhinal, anterior cingulate and temporal cortices, with smaller contributions from entorhinal and temporal Aβ (Kövari et al. 2003). Braak NFT stage remained independently associated with CI, while LBP was consistently the best predictor for dementia (Horvath et al. 2013). Another study of 104 PD cases found that the LB score alone was the best predictor for dementia (Ruffmann et al. 2016), while another study indicated that diagnostic accuracy was improved by addition of indicators of Aβ and tau pathology (Compta et al. 2011). A multivariate regression analysis examining dementia severity found that anterior cingulate and entorhinal LB burden together accounted for about 60%, while values for Aβ and tau were not significant (Kövari et al. 2003). A small study found that cognitive scores in PD patients were unrelated to any measure of Aβ, tau and αSyn, though the LB score predicted the annual rate of cognitive decline causing dementia in PD (Aarsland et al. 2005a, 2005b), whereas a study using multiple backward regressions showed that the best predictor of annual decline was a summated score incorporating both LB and AD pathologies that are both common, particularly in PDD cases in the prefrontal cortex (Howlett et al. 2015). There is convincing evidence that coexistence of limbic and neocortical αSyn pathology and notable ADNC contribute to dementia in PD, and we can reliably conclude that both tau and Aβ pathologies are common particularly in PDD cases. While one research group found advanced ADNC in most PDD cases (Bancher et al. 1993; Jellinger et al. 2002), in other studies ADNC was less frequent and less severe; while tau indices independently predicted dementia in PD cases in one study (Horvath et al. 2013), two other studies found no such association (Irwin et al. 2012; Ruffmann et al. 2016). In spite of some differences between study groups, the majority of results indicates that tau pathology contributes to dementia in a majority of PD cases, whereas Aβ was found not to be independently related to dementia in most studies. Thus, tau has a closer relationship with CI in PD than Aβ, which is consistent with observations in AD (Nelson et al. 2012). While Aβ deposition was not associated with dementia in PD, severe changes were linked with more rapid cognitive deterioration and earlier mortality (Compta et al. 2014; Halliday et al. 2011; Jellinger et al. 2002; Kotzbauer et al. 2012; Ruffmann et al. 2016; Sabbagh et al. 2009).
The relationship between αSyn deposition and dementia is strong despite some variations between studies. Global cortical αSyn burden was the best predictor of dementia (Horvath et al. 2013; Irwin et al. 2012; Kövari et al. 2003; Ruffmann et al. 2016), although the addition of tau and Aβ scores improved predicative accuracy for dementia (Compta et al. 2011). On the other hand, significant αSyn burden in limbic and neocortical areas were found in 15–45% of PD cases without CI (Compta et al. 2011; Irwin et al. 2012; Kempster et al. 2010) and other studies found severe αSyn as well as Aβ and tau pathologies in elderly PD cases without CI (Parkkinen et al. 2005), which probably might be explained by higher cognitive reserve in these patients (Hindle et al. 2014). Human brain autopsy findings and both cell and animal model data provide evidence for a synergistic interaction of αSyn, tau and Aβ pathologies inducing each other and their spreading in the brain (Bassil et al. 2020, 2021).
In conclusion, whereas there has been a discussion about the role of individual pathologies causing dementia in PD, there is increasing evidence from multiple clinicopathological studies for a synergistic effect between αSyn pathology, age and ADNC (both tau and Aβ) as the main drive of cognitive decline in PD, suggesting a triad of neurodegeneration, the molecular pathogenesis remains to be further elucidated (Dickson et al. 2009b; Halliday et al. 2014; Jellinger 2011; Pletnikova et al. 2005; Wills et al. 2010). A recent study on the disease-specific patterns of αSyn multimer destabilization in PD, based on local regional neuronal vulnerability and "prion-like" aggregation transmission enabled by destabilization of local endogenous αSyn protein, revealed differences of the cytosolic unfolded, monomeric form of αSyn (αSU) and helically folded multimeric form (αSH) equilibrium comparing demented and cognitively intact PD patients (de Boni et al. 2022). These data suggest that different brain region-specific susceptibility of LBP might be important for development of cognitive impairment in PD.
Impact of other co-pathologies on cognition in PD
Other common neuropathologies associated with age can influence the course of PD. Cerebrovascular disease and WMHs have been demonstrated to be associated with cognitive dysfunction in PD (Chahine et al. 2019; Mak et al. 2015; Malek et al. 2016; Rektor et al. 2009), while other studies did not find such an association (González-Redondo et al. 2012; Haugarvoll et al. 2005). Among the different subtypes of cerebrovascular disease, cerebral small vessel disease has been associated with cortical thinning in the frontoparietal regions with concomitant decline in memory (Foo and Kandiah 2016). A meta-analysis of the influence of cerebral small vessel disease showed different effects on cognitive function in PD, most effective on executive ability, memory and overall cognitive function (Wan et al. 2022). Higher perivascular space in the basal ganglia and WMH severity are independent positive predictors of future cognitive decline in PD (Chen et al. 2022).
Cerebral microbleeds (CMB) related to hypertension also have been associated with cognitive decline (Qin et al. 2022), while others did not, but they were seen more frequently in PDD than in PDND patients (Daida et al. 2018; Ham et al. 2014). A regression analysis showed that the presence of lobar CMBs was strongly associated with PDD (Daida et al. 2018). Other recent studies showed that amyloid-related CMBs and reduced hippocampal volume are associated with PDD (Tsai et al. 2021); earlier studies also showed association of severe CAA with PDD (Compta et al. 2011; Irwin et al. 2012). While according to some authors, cerebrovascular and TDP 43 pathologies do not generally contribute to PDD (Smith et al. 2019), one study found hippocampal and entorhinal TDP-43 inclusions more often in subjects with PDD than in those with PDND and healthy controls. Furthermore, significant association between co-morbid ADNC and TDP-43 was observed (Nakashima-Yasuda et al. 2007). Argyrophilic grain disease, another form of age-related tauopathy largely related to medial temporal lobe (Ferrer et al. 2008), appears to be rare in PD, but has been reported as an important factor affecting dementia in PD (Homma et al. 2015), while according to others, it was not associated with worse cognitive outcome (Aarsland et al. 2021; Irwin et al. 2012). Many of these pathologies can occur in advanced age and make it difficult to disentangle their individual contribution to cognitive decline (Compta et al. 2011; Coughlin et al. 2019b; Irwin et al. 2017). In general, there is likely a complex interaction of various neuropathologies in the expression of cognitive and other clinical features in PD (Buchman et al. 2019; Coughlin and Irwin 2022), which, however deserves further elucidation.
Conclusion and outlook
PD is a common and heterogeneous neurodegenerative disorder; it is much more than a movement disorder, and a wide range of nonmotor symptoms has been recognized. Among them, cognitive decline, in a wide range of severity and involved domains, is particularly important, due to its enormous impact on the quality of life of patients and caregivers, as well as the economic burden brought about by this severe condition. The morphological and molecular/biochemical basis of CI is heterogeneous, and modern neuroimaging studies revealed widespread changes in cerebral GM and WM, involving multiple brain areas and causing loss of functional connectivity between critical neuronal networks involved in cognitive and behavioral functions due to neurodegenerative changes. PD patients who exhibit ‘AD-like’ patterns of brain atrophy are at a greater risk for future cognitive decline. SPARE-AD (Spatial Pattern of Abnormality for Recognition of Early Alzheimer’s disease), an MRI index capturing AD-related atrophy, has been shown to be higher in PD-MCI and PDD patients than in PD-NC and healthy controls (Charissé et al. 2022).
The majority of autopsy-based studies to date support the strong association of limbic and neocortical LBP with CI in PD, while AD co-pathology is often observed as well and may play a synergistic role in the development of dementia with some unique cognitive features (episodic memory deficits and others). The global number of individuals who live with dementia has been expected to increase to 100 million by 2050 (Nichols and Collaborators 2019), and research challenges are increasingly being recognized for both PD and dementia, and further data on the prevalence of PD-associated CI are urgently warranted. The proposal that dementia prior to or simultaneous with or after development of motor symptoms might be included in the diagnosis of PD (Berg et al. 2014; Postuma et al. 2015) has reopened the discussion on whether PDD and DLB should be considered the same disease or phenotypes of a spectrum of LB diseases (Friedman 2018; Jellinger 2018; Jellinger and Korczyn 2018). A deeper understanding of the pathophysiological processes underlying these two synucleinopathies, such as the relative contribution of Aβ and tau pathologies in cortex and striatum, the extent of cortical and entorhinal LBP, the severity of neuronal loss in SN and other subcortical nuclei and the involvement of various neurotransmitter systems is required to better understanding the relationship between the different forms of CI in PD and related LB diseases. The prospective assessment and validation of CI in PD will be improved by combined assessment of neuroimaging and biomarker signatures, making decisions more homogenous. There is an urgent need for quantitative in vivo biomarkers and multicentered autopsy studies of well-characterized longitudinally followed patients to further elucidate the pathobiological contributions of different neuropathologies to CI and domain-specific features in PDD. These and other interdisciplinary efforts are critical to the development of meaningful disease-modifying therapies and preventive measures to slow or halt progression of PD and resultant cognitive deterioration.
Abbreviations
- AD:
-
Alzheimer disease
- ADNC:
-
Alzheimer disease-related neuropathological changes
- aMCI:
-
Amnestic mild cognitive impairment
- Aβ:
-
β-amyloid
- αSyn:
-
α-synuclein
- CA:
-
Cornu ammonis
- CAA:
-
Cerebral amyloid angiopathy
- CI:
-
Cognitive impairment
- CMBs:
-
Cerebral microbleeds
- DLB:
-
Dementia with lewy bodies
- DMN:
-
Default mode network
- FA:
-
Fractional anisotropy
- FW:
-
Free water
- GM:
-
Gray matter
- GMV:
-
Gray matter volume
- LB:
-
Lewy body
- LBP:
-
Lewy body pathology
- MCI:
-
Mild cognitive impairment
- MMSE:
-
Mini mental state examination
- MRI:
-
Magentic resonance imaging
- naMCI:
-
Non-amnestic mild cognitive impairment
- NBM:
-
Nucleus basalis of Meynert
- NFT:
-
Neurofibrillary tangle
- PD:
-
Parkinson disease
- PDD:
-
Parkinson disease dementia
- PD-MCI:
-
Parkinson disease with mild cognitive impairment
- PD-NC:
-
Parkinson disease with normal cognition
- PDND:
-
Parkinson disease-no dementia
- PET:
-
Positron emission tomography
- SAN:
-
Salience network
- SCD:
-
Subjective cognitive decline
- SN:
-
Substantia nigra
- WM:
-
White matter
- WMH:
-
White matter hyperintensity
- WMV:
-
White matter volume
References
Aarsland D, Perry R, Brown A, Larsen JP, Ballard C (2005a) Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann Neurol 58:773–776
Aarsland D, Zaccai J, Brayne C (2005b) A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov Disord 20:1255–1263
Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Ray Chaudhuri K, Weintraub D (2021) Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers 7:47
Abbasi N, Fereshtehnejad SM, Zeighami Y, Larcher KM, Postuma RB, Dagher A (2020) Predicting severity and prognosis in Parkinson’s disease from brain microstructure and connectivity. Neuroimage Clin 25:102111
Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, Gage FH (2017) Hippocampal alpha-synuclein in dementia with lewy bodies contributes to memory impairment and is consistent with spread of pathology. J Neurosci 37:1675–1684
Adler CH, Beach TG (2010) Variability of diffuse plaques and amyloid angiopathy in Parkinson’s disease with mild cognitive impairment. Acta Neuropathol 120:831–832
Adler CH, Caviness JN, Sabbagh MN, Shill HA, Connor DJ, Sue L, Evidente VG, Driver-Dunckley E, Beach TG (2010) Heterogeneous neuropathological findings in Parkinson’s disease with mild cognitive impairment. Acta Neuropathol 120:827–828
Agosta F, Canu E, Stefanova E, Sarro L, Tomic A, Špica V, Comi G, Kostic VS, Filippi M (2014) Mild cognitive impairment in Parkinson’s disease is associated with a distributed pattern of brain white matter damage. Hum Brain Mapp 35:1921–1929
Akhtar RS, Xie SX, Brennan L, Pontecorvo MJ, Hurtig HI, Trojanowski JQ, Weintraub D, Siderowf AD (2016) Amyloid-beta positron emission tomography imaging of Alzheimer’s pathology in Parkinson’s disease dementia. Mov Disord Clin Pract 3:367–375
Alzghool OM, van Dongen G, van de Giessen E, Schoonmade L, Beaino W (2022) Alpha-synuclein radiotracer development and in vivo imaging: recent advancements and new perspectives. Mov Disord 37:936–948
Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW (2002) Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol 59:102–112
Apostolova L, Alves G, Hwang KS, Babakchanian S, Bronnick KS, Larsen JP, Thompson PM, Chou YY, Tysnes OB, Vefring HK, Beyer MK (2012) Hippocampal and ventricular changes in Parkinson’s disease mild cognitive impairment. Neurobiol Aging 33:2113–2124
Aracil-Bolaños I, Sampedro F, Marín-Lahoz J, Horta-Barba A, Martínez-Horta S, Botí M, Pérez-Pérez J, Bejr-Kasem H, Pascual-Sedano B, Campolongo A, Izquierdo C, Gironell A, Gómez-Ansón B, Kulisevsky J, Pagonabarraga J (2019) A divergent breakdown of neurocognitive networks in Parkinson’s disease mild cognitive impairment. Hum Brain Mapp 40:3233–3242
Baggio HC, Segura B, Sala-Llonch R, Marti MJ, Valldeoriola F, Compta Y, Tolosa E, Junque C (2015) Cognitive impairment and resting-state network connectivity in Parkinson’s disease. Hum Brain Mapp 36:199–212
Baiano C, Barone P, Trojano L, Santangelo G (2020) Prevalence and clinical aspects of mild cognitive impairment in Parkinson’s disease: a meta-analysis. Mov Disord 35:45–54
Bancher C, Braak H, Fischer P, Jellinger KA (1993) Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci Lett 162:179–182
Barone P, Aarsland D, Burn D, Emre M, Kulisevsky J, Weintraub D (2011) Cognitive impairment in nondemented Parkinson’s disease. Mov Disord 26:2483–2495
Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, Cox TO, Riddle DM, Zhang B, Trojanowski JQ, Lee VM (2020) Amyloid-beta (aBeta) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of lewy body disorders with aBeta pathology. Neuron 105(260–275):e266
Bassil F, Meymand ES, Brown HJ, Xu H, Cox TO, Pattabhiraman S, Maghames CM, Wu Q, Zhang B, Trojanowski JQ, Lee VM. (2021) Alpha-synuclein modulates tau spreading in mouse brains. J Exp Med 218:e20192193
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634
Becker S, Granert O, Timmers M, Pilotto A, Van Nueten L, Roeben B, Salvadore G, Galpern WR, Streffer J, Scheffler K, Maetzler W, Berg D, Liepelt-Scarfone I (2021) Association of hippocampal subfields, CSF biomarkers, and cognition in patients with Parkinson disease without dementia. Neurology 96:e904–e915
Bendor JT, Logan TP, Edwards RH (2013) The function of alpha-synuclein. Neuron 79:1044–1066
Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T, Goetz CG, Halliday GM, Hardy J, Lang AE, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G (2014) Time to redefine PD? Introductory statement of the MDS task force on the definition of Parkinson’s disease. Mov Disord 29:454–462
Beyer MK, Aarsland D, Greve OJ, Larsen JP (2006) Visual rating of white matter hyperintensities in Parkinson’s disease. Mov Disord 21:223–229
Biundo R, Weis L, Antonini A (2016) Cognitive decline in Parkinson’s disease: the complex picture. NPJ Parkinsons Dis 2:16018
Bohnen NI, Koeppe RA, Minoshima S, Giordani B, Albin RL, Frey KA, Kuhl DE (2011) Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med 52:848–855
Bohnen NI, Albin RL, Muller ML, Petrou M, Kotagal V, Koeppe RA, Scott PJ, Frey KA (2015) Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol 72:194–200
Bohnen NI, Muller M, Frey KA (2017) Molecular imaging and updated diagnostic criteria in lewy body dementias. Curr Neurol Neurosci Rep 17:73
Boon LI, Hepp DH, Douw L, van Geenen N, Broeders TAA, Geurts JJG, Berendse HW, Schoonheim MM (2020) Functional connectivity between resting-state networks reflects decline in executive function in Parkinson’s disease: a longitudinal fMRI study. Neuroimage Clin 28:102468
Bougea A, Maraki MI, Yannakoulia M, Stamelou M, Xiromerisiou G, Kosmidis MH, Ntanasi E, Dardiotis E, Hadjigeorgiou GM, Sakka P, Anastasiou CA, Stefanis L, Scarmeas N (2019) Higher probability of prodromal Parkinson disease is related to lower cognitive performance. Neurology 92:e2261–e2272
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134
Braak H, Rüb U, Jansen Steur EN, Del Tredici K, de Vos RA (2005) Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64:1404–1410
Buchman AS, Yu L, Wilson RS, Leurgans SE, Nag S, Shulman JM, Barnes LL, Schneider JA, Bennett DA (2019) Progressive parkinsonism in older adults is related to the burden of mixed brain pathologies. Neurology 92:e1821–e1830
Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT (2004) Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with lewy bodies and controls. Brain 127:791–800
Burton EJ, McKeith IG, Burn DJ, O’Brien JT (2005) Brain atrophy rates in Parkinson’s disease with and without dementia using serial magnetic resonance imaging. Mov Disord 20:1571–1576
Butt A, Kamtchum-Tatuene J, Khan K, Shuaib A, Jickling GC, Miyasaki JM, Smith EE, Camicioli R (2021) White matter hyperintensities in patients with Parkinson’s disease: a systematic review and meta-analysis. J Neurol Sci 426:117481
Carlesimo GA, Piras F, Assogna F, Pontieri FE, Caltagirone C, Spalletta G (2012) Hippocampal abnormalities and memory deficits in Parkinson disease: a multimodal imaging study. Neurology 78:1939–1945
Chahine LM, Dos Santos C, Fullard M, Scordia C, Weintraub D, Erus G, Rosenthal L, Davatzikos C, McMillan CT (2019) Modifiable vascular risk factors, white matter disease and cognition in early Parkinson’s disease. Eur J Neurol 26:246-e218
Chandler JM, Nair R, Biglan K, Ferries EA, Munsie LM, Changamire T, Patel N (2021) Characteristics of Parkinson’s disease in patients with and without cognitive impairment. J Parkinsons Dis 11:1381–1392
Charcot J-M. (1877) De la paralysie agitante. Oeuvres Complétes: Leçons sur les maladies du systéme nerveux. Vol 1. Paris: Bureaux du Progrés Mèdical, 1872; On Parkinson’s disease. Lectures on the diseases of the nervous system. G. Sigerson, trans. London: New Sydenham Society
Charissé D, Erus G, Pomponio R, Gorges M, Schmidt N, Schneider C, Liepelt-Scarfone I, Riedel O, Reetz K, Schulz JB, Berg D, Storch A, Witt K, Dodel R, Kalbe E, Kassubek J, Hilker-Roggendorf R, Baudrexel S (2022) Brain age and Alzheimer’s-like atrophy are domain-specific predictors of cognitive impairment in Parkinson’s disease. Neurobiol Aging 109:31–42
Chen B, Fan GG, Liu H, Wang S (2015) Changes in anatomical and functional connectivity of Parkinson’s disease patients according to cognitive status. Eur J Radiol 84:1318–1324
Chen F, Wu T, Luo Y, Li Z, Guan Q, Meng X, Tao W, Zhang H (2019) Amnestic mild cognitive impairment in Parkinson’s disease: white matter structural changes and mechanisms. PLoS One 14:e0226175
Chen FX, Kang DZ, Chen FY, Liu Y, Wu G, Li X, Yu LH, Lin YX, Lin ZY (2016) Gray matter atrophy associated with mild cognitive impairment in Parkinson’s disease. Neurosci Lett 617:160–165
Chen H, Wan H, Zhang M, Wardlaw JM, Feng T, Wang Y (2022) Perivascular space in Parkinson's disease: Association with CSF amyloid/tau and cognitive decline. Parkinsonism Relat Disord 95:70-76
Chondrogiorgi M, Astrakas LG, Zikou AK, Weis L, Xydis VG, Antonini A, Argyropoulou MI, Konitsiotis S (2019) Multifocal alterations of white matter accompany the transition from normal cognition to dementia in Parkinson’s disease patients. Brain Imaging Behav 13:232–240
Christopher L, Duff-Canning S, Koshimori Y, Segura B, Boileau I, Chen R, Lang AE, Houle S, Rusjan P, Strafella AP (2015) Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann Neurol 77:269–280
Chua CY, Koh MRE, Chia NS, Ng SY, Saffari SE, Wen MC, Chen RY, Choi X, Heng DL, Neo SX, Tay KY, Au WL, Tan EK, Tan LC, Xu Z (2021) Subjective cognitive complaints in early Parkinson’s disease patients with normal cognition are associated with affective symptoms. Parkinsonism Relat Disord 82:24–28
Chung SJ, Yoo HS, Lee YH, Lee HS, Ye BS, Sohn YH, Kwon H, Lee PH (2019) Frontal atrophy as a marker for dementia conversion in Parkinson’s disease with mild cognitive impairment. Hum Brain Mapp 40:3784–3794
Chung SJ, Kim YJ, Jung JH, Lee HS, Ye BS, Sohn YH, Jeong Y, Lee PH (2022) Association between white matter connectivity and early dementia in patients with Parkinson disease. Neurology. https://doi.org/10.1212/WNL.0000000000200152
Churchyard A, Lees AJ (1997) The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson’s disease. Neurology 49:1570–1576
Colom-Cadena M, Grau-Rivera O, Planellas L, Cerquera C, Morenas E, Helgueta S, Munoz L, Kulisevsky J, Marti MJ, Tolosa E, Clarimon J, Lleo A, Gelpi E (2017) Regional overlap of pathologies in lewy body disorders. J Neuropathol Exp Neurol 76:216–224
Colon-Perez LM, Tanner JJ, Couret M, Goicochea S, Mareci TH, Price CC (2018) Cognition and connectomes in nondementia idiopathic Parkinson’s disease. Netw Neurosci 2:106–124
Colosimo C, Hughes AJ, Kilford L, Lees AJ (2003) Lewy body cortical involvement may not always predict dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 74:852–856
Compta Y, Parkkinen L, O’Sullivan SS, Vandrovcova J, Holton JL, Collins C, Lashley T, Kallis C, Williams DR, de Silva R, Lees AJ, Revesz T (2011) Lewy- and alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 134:1493–1505
Compta Y, Parkkinen L, Kempster P, Selikhova M, Lashley T, Holton JL, Lees AJ, Revesz T (2014) The significance of alpha-synuclein, amyloid-beta and tau pathologies in Parkinson’s disease progression and related dementia. Neurodegener Dis 13:154–156
Coughlin D, Phillips J, Roll E, Wolk D, Das S, Nasrallah I, Vaishnavi S, Siderowf A, Weintraub D, Shaw L, Trojanowski JQ, Grossman M, Irwin DJ, McMillan CT (2019a) Cerebrospinal fluid AD biomarkers and regional [18F]-flortaucipir uptake in lewy body disorders (abstr). Neurology 92(15 Suppl):S10.009
Coughlin D, Xie SX, Liang M, Williams A, Peterson C, Weintraub D, McMillan CT, Wolk DA, Akhtar RS, Hurtig HI, Branch Coslett H, Hamilton RH, Siderowf AD, Duda JE, Rascovsky K, Lee EB, Lee VM, Grossman M, Trojanowski JQ, Irwin DJ (2019b) Cognitive and pathological influences of tau pathology in lewy body disorders. Ann Neurol 85:259–271
Coughlin DG, Phillips JS, Roll E, Peterson C, Lobrovich R, Rascovsky K, Ungrady M, Wolk DA, Das S, Weintraub D, Lee EB, Trojanowski JQ, Shaw LM, Vaishnavi S, Siderowf A, Nasrallah IM, Irwin DJ, McMillan CT (2020) Multimodal in vivo and postmortem assessments of tau in lewy body disorders. Neurobiol Aging 96:137–147
Coughlin DG, Irwin DJ (2022) Neuropathological substrates of cognition in Parkinson’s disease. Prog Brain Res 269:177–193
Daida K, Tanaka R, Yamashiro K, Ogawa T, Oyama G, Nishioka K, Shimo Y, Umemura A, Hattori N (2018) The presence of cerebral microbleeds is associated with cognitive impairment in Parkinson’s disease. J Neurol Sci 393:39–44
de Boni L, Watson AH, Zaccagnini L, Wallis A, Zhelcheska K, Kim N, Sanderson J, Jiang H, Martin E, Cantlon A, Rovere M, Liu L, Sylvester M, Lashley T, Dettmer U, Jaunmuktane Z, Bartels T (2022) Brain region-specific susceptibility of lewy body pathology in synucleinopathies is governed by alpha-synuclein conformations. Acta Neuropathol 143:453–469
Del Tredici K, Braak H (2013) Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson’s disease-related dementia. J Neurol Neurosurg Psychiatry 84:774–783
Deng B, Zhang Y, Wang L, Peng K, Han L, Nie K, Yang H, Zhang L, Wang J (2013) Diffusion tensor imaging reveals white matter changes associated with cognitive status in patients with Parkinson’s disease. Am J Alzheimers Dis Other Demen 28:154–164
Devignes Q, Bordier C, Viard R, Defebvre L, Kuchcinski G, Leentjens AFG, Lopes R, Dujardin K (2022) Resting-state functional connectivity in frontostriatal and posterior cortical subtypes in Parkinson's disease-mild cognitive impairment. Mov Disord 37:502-512
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009a) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157
Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA, Frigerio R, Burnett M, Parisi JE, Klos KJ, Ahlskog JE (2009b) Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord 15(Suppl 3):S1-5
Domellöf ME, Ekman U, Forsgren L, Elgh E (2015) Cognitive function in the early phase of Parkinson’s disease, a five-year follow-up. Acta Neurol Scand 132:79–88
Donzuso G, Monastero R, Cicero CE, Luca A, Mostile G, Giuliano L, Baschi R, Caccamo M, Gagliardo C, Palmucci S, Zappia M, Nicoletti A (2021) Neuroanatomical changes in early Parkinson’s disease with mild cognitive impairment: a VBM study; the Parkinson’s disease cognitive impairment study (PaCoS). Neurol Sci 42:3723–3731
Dugger BN, Davis K, Malek-Ahmadi M, Hentz JG, Sandhu S, Beach TG, Adler CH, Caselli RJ, Johnson TA, Serrano GE, Shill HA, Belden C, Driver-Dunckley E, Caviness JN, Sue LI, Jacobson S, Powell J, Sabbagh MN (2015) Neuropathological comparisons of amnestic and nonamnestic mild cognitive impairment. BMC Neurol 15:146
Ekman U, Eriksson J, Forsgren L, Mo SJ, Riklund K, Nyberg L (2012) Functional brain activity and presynaptic dopamine uptake in patients with Parkinson’s disease and mild cognitive impairment: a cross-sectional study. Lancet Neurol 11:679–687
Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S, Goldman J, Goetz C, Korczyn A, Lees A, Levy R, Litvan I, McKeith I, Olanow W, Poewe W, Quinn N, Sampaio C, Tolosa E, Dubois B (2007) Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 22:1689–1707 (quiz 1837)
Espay AJ, LeWitt PA, Kaufmann H (2014) Norepinephrine deficiency in Parkinson’s disease: the case for noradrenergic enhancement. Mov Disord 29:1710–1719
Fan TS, Liu SC, Wu RM (2021) Alpha-synuclein and cognitive decline in Parkinson disease. Life (Basel) 11:1239
Fathy YY, Hepp DH, de Jong FJ, Geurts JJG, Foncke EMJ, Berendse HW, van de Berg WDJ, Schoonheim MM (2020) Anterior insular network disconnection and cognitive impairment in Parkinson’s disease. Neuroimage Clin 28:102364
Fengler S, Liepelt-Scarfone I, Brockmann K, Schäffer E, Berg D, Kalbe E (2017) Cognitive changes in prodromal Parkinson’s disease: a review. Mov Disord 32:1655–1666
Ferrer I, Santpere G, van Leeuwen FW (2008) Argyrophilic grain disease. Brain 131:1416–1432
Filippi M, Canu E, Donzuso G, Stojkovic T, Basaia S, Stankovic I, Tomic A, Markovic V, Petrovic I, Stefanova E, Kostic VS, Agosta F (2020) Tracking cortical changes throughout cognitive decline in Parkinson’s disease. Mov Disord 35:1987–1998
Fiorenzato E, Strafella AP, Kim J, Schifano R, Weis L, Antonini A, Biundo R (2019) Dynamic functional connectivity changes associated with dementia in Parkinson’s disease. Brain 142:2860–2872
Foo H, Kandiah N. (2016) The role of cerebrovascular disease in Parkinson's disease related cognitive impairment. J Parkinsons Dis Alzheimer Dis 3:7 - https://www.avensonline.org/fulltextarticles/jpa-2376-2922x-2303-0017.html
Foo H, Mak E, Chander RJ, Ng A, Au WL, Sitoh YY, Tan LC, Kandiah N (2016) Associations of hippocampal subfields in the progression of cognitive decline related to Parkinson’s disease. Neuroimage Clin 14:37–42
Foster ER, Campbell MC, Burack MA, Hartlein J, Flores HP, Cairns NJ, Hershey T, Perlmutter JS (2010) Amyloid imaging of lewy body-associated disorders. Mov Disord 25:2516–2523
Frey KA, Petrou M (2015) Imaging amyloidopathy in Parkinson disease and parkinsonian dementia syndromes. Clin Transl Imaging 3:57–64
Friedman JH (2018) Dementia with Lewy bodies and Parkinson disease dementia: it is the same disease! Parkinsonism Relat Disord 46(Suppl 1):S6–S9
Garcia-Diaz AI, Segura B, Baggio HC, Uribe C, Campabadal A, Abos A, Marti MJ, Valldeoriola F, Compta Y, Bargallo N, Junque C (2018) Cortical thinning correlates of changes in visuospatial and visuoperceptual performance in Parkinson’s disease: A 4-year follow-up. Parkinsonism Relat Disord 46:62–68
Garcia-Esparcia P, Koneti A, Rodríguez-Oroz MC, Gago B, Del Rio JA, Ferrer I (2018) Mitochondrial activity in the frontal cortex area 8 and angular gyrus in Parkinson’s disease and Parkinson’s disease with dementia. Brain Pathol 28:43–57
Gargouri F, Gallea C, Mongin M, Pyatigorskaya N, Valabregue R, Ewenczyk C, Sarazin M, Yahia-Cherif L, Vidailhet M, Lehéricy S (2019) Multimodal magnetic resonance imaging investigation of basal forebrain damage and cognitive deficits in Parkinson’s disease. Mov Disord 34:516–525
Gatt AP, Duncan OF, Attems J, Francis PT, Ballard CG, Bateman JM (2016) Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov Disord 31:352–359
Goetz CG, Emre M, Dubois B (2008) Parkinson’s disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol 64(Suppl 2):S81-92
Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N, Sperling R, Growdon JH, Dickerson BC, Johnson K (2016) Tau positron emission tomographic imaging in the lewy body diseases. JAMA Neurol 73:1334–1341
González-Redondo R, Toledo J, Clavero P, Lamet I, García-García D, García-Eulate R, Martínez-Lage P, Rodríguez-Oroz MC (2012) The impact of silent vascular brain burden in cognitive impairment in Parkinson’s disease. Eur J Neurol 19:1100–1107
Gratwicke J, Jahanshahi M, Foltynie T (2015) Parkinson’s disease dementia: a neural networks perspective. Brain 138:1454–1476
Grothe MJ, Labrador-Espinosa MA, Jesús S, Macías-García D, Adarmes-Gómez A, Carrillo F, Camacho EI, Franco-Rosado P, Lora FR, Martín-Rodríguez JF, Barberá MA, Pastor P, Arroyo SE, Vila BS, Foraster AC, Martínez JR, Padilla FC, Morlans MP, Aramburu IG, Ceberio JI, Vara JH, de Fábregues-Boixar O, de Deus FT, Pascual-Sedano B, Kulisevsky J, Martínez-Martín P, Santos-García D, Mir P (2021) In vivo cholinergic basal forebrain degeneration and cognition in Parkinson’s disease: Imaging results from the COPPADIS study. Parkinsonism Relat Disord 88:68–75
Guttuso T Jr, Sirica D, Tosun D, Zivadinov R, Pasternak O, Weintraub D, Baglio F, Bergsland N (2022) Thalamic dorsomedial nucleus free water correlates with cognitive decline in Parkinson’s disease. Mov Disord 37:490–501
Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D (2014) Hippocampal lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 137:2493–2508
Hall JM, Lewis SJG (2019) Neural correlates of cognitive impairment in Parkinson’s disease: a review of structural MRI findings. Int Rev Neurobiol 144:1–28
Halliday GM, Song YJ, Harding AJ (2011) Striatal beta-amyloid in dementia with lewy bodies but not Parkinson’s disease. J Neural Transm (vienna) 118:713–719
Halliday GM, Leverenz JB, Schneider JS, Adler CH (2014) The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov Disord 29:634–650
Ham JH, Yi H, Sunwoo MK, Hong JY, Sohn YH, Lee PH (2014) Cerebral microbleeds in patients with Parkinson’s disease. J Neurol 261:1628–1635
Hanganu A, Bedetti C, Degroot C, Mejia-Constain B, Lafontaine AL, Soland V, Chouinard S, Bruneau MA, Mellah S, Belleville S, Monchi O (2014) Mild cognitive impairment is linked with faster rate of cortical thinning in patients with Parkinson’s disease longitudinally. Brain 137:1120–1129
Hanning U, Teuber A, Lang E, Trenkwalder C, Mollenhauer B, Minnerup H (2019) White matter hyperintensities are not associated with cognitive decline in early Parkinson’s disease—the DeNopa cohort. Parkinsonism Relat Disord 69:61–67
Hansen AK, Parbo P, Ismail R, Østergaard K, Brooks DJ, Borghammer P (2020) Tau tangles in Parkinson’s disease: a 2-year follow-up flortaucipir PET study. J Parkinsons Dis 10:161–171
Harding AJ, Halliday GM (2001) Cortical lewy body pathology in the diagnosis of dementia. Acta Neuropathol 102:355–363
Harding AJ, Broe GA, Halliday GM (2002) Visual hallucinations in lewy body disease relate to lewy bodies in the temporal lobe. Brain 125:391–403
Hattori T, Orimo S, Aoki S, Ito K, Abe O, Amano A, Sato R, Sakai K, Mizusawa H (2012) Cognitive status correlates with white matter alteration in Parkinson’s disease. Hum Brain Mapp 33:727–739
Haugarvoll K, Aarsland D, Wentzel-Larsen T, Larsen JP (2005) The influence of cerebrovascular risk factors on incident dementia in patients with Parkinson’s disease. Acta Neurol Scand 112:386–390
Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB (2019) Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord 34:1464–1470
Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG (2008) The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord 23:837–844
Henderson MX, Sengupta M, Trojanowski JQ, Lee VMY (2019) Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun 7:183
Hepp DH, Vergoossen DL, Huisman E, Lemstra AW, Berendse HW, Rozemuller AJ, Foncke EM, van de Berg WD (2016) Distribution and load of amyloid-beta pathology in Parkinson disease and dementia with Lewy bodies. J Neuropathol Exp Neurol 75:936–945
Hindle JV, Martyr A, Clare L (2014) Cognitive reserve in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord 20:1–7
Hirano S, Shinotoh H, Eidelberg D (2012) Functional brain imaging of cognitive dysfunction in Parkinson’s disease. J Neurol Neurosurg Psychiatry 83:963–969
Homma T, Mochizuki Y, Takahashi K, Komori T (2015) Medial temporal regional argyrophilic grain as a possible important factor affecting dementia in Parkinson’s disease. Neuropathology 35:441–451
Hong JY, Lee JE, Sohn YH, Lee PH (2012) Neurocognitive and atrophic patterns in Parkinson’s disease based on subjective memory complaints. J Neurol 259:1706–1712
Hoogland J, Boel JA, de Bie RMA, Geskus RB, Schmand BA, Dalrymple-Alford JC, Marras C, Adler CH, Goldman JG, Tröster AI, Burn DJ, Litvan I, Geurtsen GJ (2017) Mild cognitive impairment as a risk factor for Parkinson’s disease dementia. Mov Disord 32:1056–1065
Horvath J, Herrmann FR, Burkhard PR, Bouras C, Kövari E (2013) Neuropathology of dementia in a large cohort of patients with Parkinson’s disease. Parkinsonism Relat Disord 19:864–868 (Discussion 864)
Hou Y, Shang H (2022) Magnetic resonance imaging markers for cognitive impairment in Parkinson’s disease: current view. Front Aging Neurosci 14:788846
Howlett DR, Whitfield D, Johnson M, Attems J, O’Brien JT, Aarsland D, Lai MK, Lee JH, Chen C, Ballard C, Hortobagyi T, Francis PT (2015) Regional multiple pathology scores are associated with cognitive decline in lewy body dementias. Brain Pathol 25:401–408
Huang C, Mattis P, Tang C, Perrine K, Carbon M, Eidelberg D (2007) Metabolic brain networks associated with cognitive function in Parkinson’s disease. Neuroimage 34:714–723
Huang C, Mattis P, Perrine K, Brown N, Dhawan V, Eidelberg D (2008) Metabolic abnormalities associated with mild cognitive impairment in Parkinson disease. Neurology 70:1470–1477
Inguanzo A, Sala-Llonch R, Segura B, Erostarbe H, Abos A, Campabadal A, Uribe C, Baggio HC, Compta Y, Marti MJ, Valldeoriola F, Bargallo N, Junque C (2021) Hierarchical cluster analysis of multimodal imaging data identifies brain atrophy and cognitive patterns in Parkinson’s disease. Parkinsonism Relat Disord 82:16–23
Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, Lee VM, Leverenz JB, Montine TJ, Duda JE, Hurtig HI, Trojanowski JQ (2012) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72:587–598
Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinsons disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14:626–636
Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, Keene CD, Montine TJ, Zabetian CP, Trojanowski JQ (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16:55–65
Jellinger K, Braak H, Braak E, Fischer P (1991) Alzheimer lesions in the entorhinal region and isocortex in Parkinson’s and Alzheimer’s diseases. Ann NY Acad Sci 640:203–209
Jellinger KA, Seppi K, Wenning GK, Poewe W (2002) Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm 109:329–339
Jellinger KA, Attems J (2006) Does striatal pathology distinguish Parkinson disease with dementia and dementia with lewy bodies? Acta Neuropathol 112:253–260
Jellinger KA (2007) Lewy body disorders. In: Youdim MBH, Riederer P, Mandel SA, Battistin L, Lajtha A (eds) Degenerative diseases of the nervous system. Springer Science, New York, pp 270–343
Jellinger KA. (2007b) Morphological substrates of parkinsonism with and without dementia: a retrospective clinico-pathological study. J Neural Transm Suppl 72:91–104
Jellinger KA (2008) A critical reappraisal of current staging of lewy-related pathology in human brain. Acta Neuropathol 116:1–16
Jellinger KA, Attems J (2008) Prevalence and impact of vascular and Alzheimer pathologies in lewy body disease. Acta Neuropathol 115:427–436
Jellinger KA (2009) Significance of brain lesions in Parkinson disease dementia and lewy body dementia. Front Neurol Neurosci 24:114–125
Jellinger KA (2010) Neuropathology in Parkinson’s disease with mild cognitive impairment. Acta Neuropathol 120:829–830 (Author reply 831)
Jellinger KA (2010b) Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neurodegener Dis 7:112–115
Jellinger KA (2011) Interaction between alpha-synuclein and tau in Parkinson’s disease comment on Wills et al.: elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol 227:13–18 (Exp Neurol 2010; 225: 210-218)
Jellinger KA (2012a) Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord 27:8–30
Jellinger KA (2012b) Neurobiology of cognitive impairment in Parkinson’s disease. Expert Rev Neurother 12:1451–1466
Jellinger KA (2013) Mild cognitive impairment in Parkinson disease: heterogenous mechanisms. J Neural Transm (Vienna) 120:157–167
Jellinger KA (2018) Dementia with lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J Neural Transm (Vienna) 125:615–650
Jellinger KA, Korczyn AD (2018) Are dementia with lewy bodies and Parkinson’s disease dementia the same disease? BMC Med 16:34
Jeong SH, Lee HS, Jung JH, Baik K, Sohn YH, Chung SJ, Lee PH (2022) Associations between white matter hyperintensities, striatal dopamine loss, and cognition in drug-naive Parkinson's disease. Parkinsonism Relat Disord 97:1-7
Jessen F, Amariglio RE, van Boxtel M, Breteler M, Ceccaldi M, Chételat G, Dubois B, Dufouil C, Ellis KA, van der Flier WM, Glodzik L, van Harten AC, de Leon MJ, McHugh P, Mielke MM, Molinuevo JL, Mosconi L, Osorio RS, Perrotin A, Petersen RC, Rabin LA, Rami L, Reisberg B, Rentz DM, Sachdev PS, de la Sayette V, Saykin AJ, Scheltens P, Shulman MB, Slavin MJ, Sperling RA, Stewart R, Uspenskaya O, Vellas B, Visser PJ, Wagner M (2014) A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 10:844–852
Jia X, Wang Z, Yang T, Li Y, Gao S, Wu G, Jiang T, Liang P (2019) Entorhinal cortex atrophy in early, drug-naive Parkinson’s disease with mild cognitive impairment. Aging Dis 10:1221–1232
Joelving FC, Billeskov R, Christensen JR, West M, Pakkenberg B (2006) Hippocampal neuron and glial cell numbers in Parkinson’s disease—a stereological study. Hippocampus 16:826–833
Jonkman LE, Fathy YY, Berendse HW, Schoonheim MM, van de Berg WDJ (2021) Structural network topology and microstructural alterations of the anterior insula associate with cognitive and affective impairment in Parkinson’s disease. Sci Rep 11:16021
Kalaitzakis ME, Pearce RK (2009) The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol 118:587–598
Kamagata K, Motoi Y, Abe O, Shimoji K, Hori M, Nakanishi A, Sano T, Kuwatsuru R, Aoki S, Hattori N (2012) White matter alteration of the cingulum in Parkinson disease with and without dementia: evaluation by diffusion tensor tract-specific analysis. AJNR Am J Neuroradiol 33:890–895
Kamagata K, Motoi Y, Tomiyama H, Abe O, Ito K, Shimoji K, Suzuki M, Hori M, Nakanishi A, Sano T, Kuwatsuru R, Sasai K, Aoki S, Hattori N (2013) Relationship between cognitive impairment and white-matter alteration in Parkinson’s disease with dementia: tract-based spatial statistics and tract-specific analysis. Eur Radiol 23:1946–1955
Kantarci K, Lowe VJ, Boeve BF, Senjem ML, Tosakulwong N, Lesnick TG, Spychalla AJ, Gunter JL, Fields JA, Graff-Radford J, Ferman TJ, Jones DT, Murray ME, Knopman DS, Jack CR Jr, Petersen RC (2017) AV-1451 tau and beta-amyloid positron emission tomography imaging in dementia with lewy bodies. Ann Neurol 81:58–67
Kempster PA, O’Sullivan SS, Holton JL, Revesz T, Lees AJ (2010) Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain 133:1755–1762
Kiesmann M, Chanson JB, Godet J, Vogel T, Schweiger L, Chayer S, Kaltenbach G (2013) The movement disorders society criteria for the diagnosis of Parkinson’s disease dementia: their usefulness and limitations in elderly patients. J Neurol 260:2569–2579
Kim Y, Lee D, Cho KH, Lee JJ, Ham JH, Ye BS, Lee SK, Lee JM, Sohn YH, Lee PH (2017) Cognitive and neuroanatomical correlates in early versus late onset Parkinson’s disease dementia. J Alzheimers Dis 55:485–495
Knox MG, Adler CH, Shill HA, Driver-Dunckley E, Mehta SA, Belden C, Zamrini E, Serrano G, Sabbagh MN, Caviness JN, Sue LI, Davis KJ, Dugger BN, Beach TG (2020) Neuropathological findings in Parkinson’s disease with mild cognitive impairment. Mov Disord 35:845–850
Ko JH, Katako A, Aljuaid M, Goertzen AL, Borys A, Hobson DE, Kim SM, Lee CS (2017) Distinct brain metabolic patterns separately associated with cognition, motor function, and aging in Parkinson’s disease dementia. Neurobiol Aging 60:81–91
Koros C, Stefanis L, Scarmeas N (2022) Parkinsonism and dementia. J Neurol Sci 433:120015
Kotzbauer PT, Cairns NJ, Campbell MC, Willis AW, Racette BA, Tabbal SD, Perlmutter JS (2012) Pathologic accumulation of alpha-synuclein and abeta in Parkinson disease patients with dementia. Arch Neurol 69:1326–1331
Kouli A, Camacho M, Allinson K, Williams-Gray CH (2020) Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun 8:211
Kövari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, Giannakopoulos P (2003) Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol 106:83–88
Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD, Kukull WA, Leverenz JB, Cherrier MM (2005) Cognitive differences in dementia patients with autopsy-verified AD, lewy body pathology, or both. Neurology 64:2069–2073
Kunst J, Marecek R, Klobusiakova P, Balazova Z, Anderkova L, Nemcova-Elfmarkova N, Rektorova I (2019) Patterns of grey matter atrophy at different stages of Parkinson’s and Alzheimer’s diseases and relation to cognition. Brain Topogr 32:142–160
Lang S, Yoon EJ, Kibreab M, Kathol I, Cheetham J, Hammer T, Sarna J, Ismail Z, Monchi O (2020) Mild behavioral impairment in Parkinson’s disease is associated with altered corticostriatal connectivity. Neuroimage Clin 26:102252
Lashley T, Holton JL, Gray E, Kirkham K, O’Sullivan SS, Hilbig A, Wood NW, Lees AJ, Revesz T (2008) Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol 115:417–425
Lebedev AV, Westman E, Simmons A, Lebedeva A, Siepel FJ, Pereira JB, Aarsland D (2014) Large-scale resting state network correlates of cognitive impairment in Parkinson’s disease and related dopaminergic deficits. Front Syst Neurosci 8:45
Lee SJ, Kim JS, Yoo JY, Song IU, Kim BS, Jung SL, Yang DW, Kim YI, Jeong DS, Lee KS (2010) Influence of white matter hyperintensities on the cognition of patients with Parkinson disease. Alzheimer Dis Assoc Disord 24:227–233
Leroi I, McDonald K, Pantula H, Harbishettar V (2012) Cognitive impairment in Parkinson disease: impact on quality of life, disability, and caregiver burden. J Geriatr Psychiatry Neurol 25:208–214
Li Y, Wang C, Wang J, Zhou Y, Ye F, Zhang Y, Cheng X, Huang Z, Liu K, Fei G, Zhong C, Zeng M, Jin L (2019) Mild cognitive impairment in de novo Parkinson’s disease: a neuromelanin MRI study in locus coeruleus. Mov Disord 34:884–892
Libow LS, Frisina PG, Haroutunian V, Perl DP, Purohit DP (2009) Parkinson’s disease dementia: a diminished role for the lewy body. Parkinsonism Relat Disord 15:572–575
Litvan I, Goldman JG, Tröster AI, Schmand BA, Weintraub D, Petersen RC, Mollenhauer B, Adler CH, Marder K, Williams-Gray CH, Aarsland D, Kulisevsky J, Rodriguez-Oroz MC, Burn DJ, Barker RA, Emre M (2012) Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: movement disorder society task force guidelines. Mov Disord 27:349–356
Liu AKL, Lim EJ, Ahmed I, Chang RC, Pearce RKB, Gentleman SM (2018) Review: revisiting the human cholinergic nucleus of the diagonal band of broca. Neuropathol Appl Neurobiol 44:647–662
Liu AKL, Chau TW, Lim EJ, Ahmed I, Chang RC, Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RKB (2019) Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol Commun 7:61
Liu H, Deng B, Xie F, Yang X, Xie Z, Chen Y, Yang Z, Huang X, Zhu S, Wang Q (2021) The influence of white matter hyperintensity on cognitive impairment in Parkinson’s disease. Ann Clin Transl Neurol 8:1917–1934
Mak E, Dwyer MG, Ramasamy DP, Au WL, Tan LC, Zivadinov R, Kandiah N (2015) White matter hyperintensities and mild cognitive impairment in Parkinson’s disease. J Neuroimaging 25:754–760
Malek N, Lawton MA, Swallow DM, Grosset KA, Marrinan SL, Bajaj N, Barker RA, Burn DJ, Hardy J, Morris HR, Williams NM, Wood N, Ben-Shlomo Y, Grosset DG (2016) Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson’s disease. Mov Disord 31:1518–1526
Marder K (2010) Cognitive impairment and dementia in Parkinson’s disease. Mov Disord 25(Suppl 1):S110-116
Markesbery WR (2010) Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimers Dis 19:221–228
Marquie M, Verwer EE, Meltzer AC, Kim SJW, Aguero C, Gonzalez J, Makaretz SJ, Siao Tick Chong M, Ramanan P, Amaral AC, Normandin MD, Vanderburg CR, Gomperts SN, Johnson KA, Frosch MP, Gomez-Isla T (2017) Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson’s case. Acta Neuropathol Commun 5:75
Martin WR, Wieler M, Gee M, Camicioli R (2009) Temporal lobe changes in early, untreated Parkinson’s disease. Mov Disord 24:1949–1954
Mashima K, Ito D, Kameyama M, Osada T, Tabuchi H, Nihei Y, Yoshizaki T, Noguchi E, Tanikawa M, Iizuka T, Date Y, Ogata Y, Nakahara T, Iwabuchi Y, Jinzaki M, Murakami K, Suzuki N (2017) Extremely low prevalence of amyloid positron emission tomography positivity in Parkinson’s disease without dementia. Eur Neurol 77:231–237
Matsui H, Nishinaka K, Oda M, Niikawa H, Kubori T, Udaka F (2007) Dementia in Parkinson’s disease: diffusion tensor imaging. Acta Neurol Scand 116:177–181
Mattila PM, Rinne JO, Helenius H, Roytta M (1999) Neuritic degeneration in the hippocampus and amygdala in Parkinson’s disease in relation to Alzheimer pathology. Acta Neuropathol 98:157–164
Mattila PM, Rinne JO, Helenius H, Dickson DW, Röyttä M (2000) Alpha-synuclein-immunoreactive cortical lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol 100:285–290
Melzer TR, Watts R, MacAskill MR, Pitcher TL, Livingston L, Keenan RJ, Dalrymple-Alford JC, Anderson TJ (2012) Grey matter atrophy in cognitively impaired Parkinson’s disease. J Neurol Neurosurg Psychiatry 83:188–194
Melzer TR, Watts R, MacAskill MR, Pitcher TL, Livingston L, Keenan RJ, Dalrymple-Alford JC, Anderson TJ (2013) White matter microstructure deteriorates across cognitive stages in Parkinson disease. Neurology 80:1841–1849
Melzer TR, Stark MR, Keenan RJ, Myall DJ, MacAskill MR, Pitcher TL, Livingston L, Grenfell S, Horne KL, Young BN, Pascoe MJ, Almuqbel MM, Wang J, Marsh SH, Miller DH, Dalrymple-Alford JC, Anderson TJ (2019) Beta amyloid deposition is not associated with cognitive impairment in Parkinson’s disease. Front Neurol 10:391
Mihaescu AS, Masellis M, Graff-Guerrero A, Kim J, Criaud M, Cho SS, Ghadery C, Valli M, Strafella AP (2019) Brain degeneration in Parkinson’s disease patients with cognitive decline: a coordinate-based meta-analysis. Brain Imaging Behav 13:1021–1034
Molano J, Boeve B, Ferman T, Smith G, Parisi J, Dickson D, Knopman D, Graff-Radford N, Geda Y, Lucas J, Kantarci K, Shiung M, Jack C, Silber M, Pankratz VS, Petersen R (2010) Mild cognitive impairment associated with limbic and neocortical lewy body disease: a clinicopathological study. Brain 133:540–556
Monastero R, Cicero CE, Baschi R, Davì M, Luca A, Restivo V, Zangara C, Fierro B, Zappia M, Nicoletti A (2018) Mild cognitive impairment in Parkinson’s disease: the Parkinson’s disease cognitive study (PACOS). J Neurol 265:1050–1058
Montaser-Kouhsari L, Young CB, Poston KL (2022) Neuroimaging approaches to cognition in Parkinson’s disease. Prog Brain Res 269:257–286
Na S, Jeong H, Park JS, Chung YA, Song IU (2020) The impact of amyloid-beta positivity with 18F-florbetaben PET on neuropsychological aspects in Parkinson’s disease dementia. Metabolites 10:380
Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ (2007) Co-morbidity of TDP-43 proteinopathy in lewy body related diseases. Acta Neuropathol 114:221–229
Nelson PT, Jicha GA, Kryscio RJ, Abner EL, Schmitt FA, Cooper G, Xu LO, Smith CD, Markesbery WR (2010) Low sensitivity in clinical diagnoses of dementia with lewy bodies. J Neurol 257:359–366
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381
Nichols E, Collaborators GD (2019) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol 18:88–106
Nicoletti A, Luca A, Baschi R, Cicero CE, Mostile G, Davì M, Pilati L, Restivo V, Zappia M, Monastero R (2019) Incidence of mild cognitive impairment and dementia in Parkinson’s disease: the Parkinson’s disease cognitive impairment study. Front Aging Neurosci 11:21
Oh YS, Yoo SW, Lyoo CH, Yoo JY, Yoon H, Ha S, Lee KS, Kim JS (2021) The association of beta-amyloid with cognition and striatal dopamine in early, non-demented Parkinson’s disease. J Parkinsons Dis 11:605–613
Olde Dubbelink KT, Schoonheim MM, Deijen JB, Twisk JW, Barkhof F, Berendse HW (2014) Functional connectivity and cognitive decline over 3 years in Parkinson disease. Neurology 83:2046–2053
Oosterveld LP, Allen JC Jr, Reinoso G, Seah SH, Tay KY, Au WL, Tan LC (2015) Prognostic factors for early mortality in Parkinson’s disease. Parkinsonism Relat Disord 21:226–230
Owens-Walton C, Jakabek D, Power BD, Walterfang M, Hall S, van Westen D, Looi JCL, Shaw M, Hansson O (2021) Structural and functional neuroimaging changes associated with cognitive impairment and dementia in Parkinson’s disease. Psychiatry Res Neuroimaging 312:111273
Owens-Walton C, Adamson C, Walterfang M, Hall S, van Westen D, Hansson O, Shaw M, Looi JCL (2022) Midsagittal corpus callosal thickness and cognitive impairment in Parkinson’s disease. Eur J Neurosci 55:1859–1872
Palermo G, Tommasini L, Aghakhanyan G, Frosini D, Giuntini M, Tognoni G, Bonuccelli U, Volterrani D, Ceravolo R (2019) Clinical correlates of cerebral amyloid deposition in Parkinson’s disease dementia: evidence from a PET study. J Alzheimers Dis 70:597–609
Pan C, Li Y, Ren J, Li L, Huang P, Xu P, Zhang L, Zhang W, Zhang MM, Chen J, Liu W (2022) Characterizing mild cognitive impairment in prodromal Parkinson's disease: A community-based study in China. CNS Neurosci Ther 28:259-268
Pan PL, Shi HC, Zhong JG, Xiao PR, Shen Y, Wu LJ, Song YY, He GX, Li HL (2013) Gray matter atrophy in Parkinson’s disease with dementia: evidence from meta-analysis of voxel-based morphometry studies. Neurol Sci 34:613–619
Park HE, Park IS, Oh YS, Yang DW, Lee KS, Choi HS, Ahn KJ, Kim JS (2015) Subcortical whiter matter hyperintensities within the cholinergic pathways of patients with dementia and parkinsonism. J Neurol Sci 353:44–48
Parkkinen L, Kauppinen T, Pirttilã T, Autere JM, Alafuzoff I (2005) Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 57:82–91
Peavy GM, Edland SD, Toole BM, Hansen LA, Galasko DR, Mayo AM (2016) Phenotypic differences based on staging of Alzheimer’s neuropathology in autopsy-confirmed dementia with lewy bodies. Parkinsonism Relat Disord 31:72–78
Pedersen KF, Larsen JP, Tysnes OB, Alves G (2017) Natural course of mild cognitive impairment in Parkinson disease: a 5-year population-based study. Neurology 88:767–774
Pereira JB, Svenningsson P, Weintraub D, Brønnick K, Lebedev A, Westman E, Aarsland D (2014) Initial cognitive decline is associated with cortical thinning in early Parkinson disease. Neurology 82:2017–2025
Pereira JB, Hall S, Jalakas M, Grothe MJ, Strandberg O, Stomrud E, Westman E, van Westen D, Hansson O (2020) Longitudinal degeneration of the basal forebrain predicts subsequent dementia in Parkinson’s disease. Neurobiol Dis 139:104831
Perez F, Helmer C, Foubert-Samier A, Auriacombe S, Dartigues JF, Tison F (2012) Risk of dementia in an elderly population of Parkinson’s disease patients: a 15-year population-based study. Alzheimers Dement 8:463–469
Petersen RC, Roberts RO, Knopman DS, Boeve BF, Geda YE, Ivnik RJ, Smith GE, Jack CR Jr (2009) Mild cognitive impairment: ten years later. Arch Neurol 66:1447–1455
Petrou M, Bohnen NI, Muller ML, Koeppe RA, Albin RL, Frey KA (2012) Abeta-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology 79:1161–1167
Petrou M, Dwamena BA, Foerster BR, MacEachern MP, Bohnen NI, Muller ML, Albin RL, Frey KA (2015) Amyloid deposition in Parkinson’s disease and cognitive impairment: a systematic review. Mov Disord 30:928–935
Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC (2005) Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in lewy body diseases. Neurobiol Aging 26:1183–1192
Poletti M, Frosini D, Pagni C, Baldacci F, Nicoletti V, Tognoni G, Lucetti C, Del Dotto P, Ceravolo R, Bonuccelli U (2012) Mild cognitive impairment and cognitive-motor relationships in newly diagnosed drug-naive patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 83:601–606
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30:1591–1601
Prasuhn J, Prasuhn M, Fellbrich A, Strautz R, Lemmer F, Dreischmeier S, Kasten M, Münte TF, Hanssen H, Heldmann M, Brüggemann N (2021) Association of locus coeruleus and substantia nigra pathology with cognitive and motor functions in patients with Parkinson disease. Neurology 97:e1007–e1016
Purri R, Brennan L, Rick J, Xie SX, Deck BL, Chahine LM, Dahodwala N, Chen-Plotkin A, Duda JE, Morley JF, Akhtar RS, Trojanowski JQ, Siderowf A, Weintraub D (2020) Subjective cognitive complaint in Parkinson’s disease patients with normal cognition: canary in the coal mine? Mov Disord 35:1618–1625
Qin Q, Wan H, Wang D, Li J, Yang Q, Zhao J, Xue Z (2022) Effect of cerebral microbleeds on cognitive function and quality of life in Parkinson disease. Med Sci Monit 28:e935026
Ray NJ, Bradburn S, Murgatroyd C, Toseeb U, Mir P, Kountouriotis GK, Teipel SJ, Grothe MJ (2018) In vivo cholinergic basal forebrain atrophy predicts cognitive decline in de novo Parkinson’s disease. Brain 141:165–176
Rektor I, Goldemund D, Sheardovã K, Rektorovã I, Michãlkovã Z, Dufek M (2009) Vascular pathology in patients with idiopathic Parkinson’s disease. Parkinsonism Relat Disord 15:24–29
Rektor I, Svátková A, Vojtíšek L, Zikmundová I, Vanícek J, Király A, Szabó N (2018) White matter alterations in Parkinson’s disease with normal cognition precede grey matter atrophy. PLoS One 13:e0187939
Rektorova I, Krajcovicova L, Marecek R, Mikl M (2012) Default mode network and extrastriate visual resting state network in patients with Parkinson’s disease dementia. Neurodegener Dis 10:232–237
Roberts R, Knopman DS (2013) Classification and epidemiology of MCI. Clin Geriatr Med 29:753–772
Rogozinski S, Klietz M, Respondek G, Oertel WH, Grothe MJ, Pereira JB, Höglinger GU (2022) Reduction in volume of nucleus basalis of Meynert is specific to Parkinson’s disease and progressive supranuclear palsy but not to multiple system atrophy. Front Aging Neurosci 14:851788
Rongve A, Aarsland D (2013) Dementia in Parkinson’s disease and dementia with lewy bodies. In: Dening T, Thomas A (eds) Oxford textbook of old age psychiatry 2e. Oxford Univ. Press, Oxford, pp 469–478
Roshanbin S, Xiong M, Hultqvist G, Söderberg L, Zachrisson O, Meier S, Ekmark-Lewén S, Bergström J, Ingelsson M, Sehlin D, Syvänen S (2022) In vivo imaging of alpha-synuclein with antibody-based PET. Neuropharmacology 208:108985
Ruffmann C, Calboli FC, Bravi I, Gveric D, Curry LK, de Smith A, Pavlou S, Buxton JL, Blakemore AI, Takousis P, Molloy S, Piccini P, Dexter DT, Roncaroli F, Gentleman SM, Middleton LT (2016) Cortical lewy bodies and abeta burden are associated with prevalence and timing of dementia in lewy body diseases. Neuropathol Appl Neurobiol 42:436–450
Sabbagh MN, Adler CH, Lahti TJ, Connor DJ, Vedders L, Peterson LK, Caviness JN, Shill HA, Sue LI, Ziabreva I, Perry E, Ballard CG, Aarsland D, Walker DG, Beach TG (2009) Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord 23:295–297
Sampedro F, Marin-Lahoz J, Martinez-Horta S, Pagonabarraga J, Kulisevsky J (2019) Dopaminergic degeneration induces early posterior cortical thinning in Parkinson’s disease. Neurobiol Dis 124:29–35
Sang T, He J, Wang J, Zhang C, Zhou W, Zeng Q, Yuan Y, Yu L, Feng Y (2022) Alterations in white matter fiber in Parkinson disease across different cognitive stages. Neurosci Lett 769:136424
Sarasso E, Agosta F, Piramide N, Filippi M (2021) Progression of grey and white matter brain damage in Parkinson’s disease: a critical review of structural MRI literature. J Neurol 268:3144–3179
Saredakis D, Collins-Praino LE, Gutteridge DS, Stephan BCM, Keage HAD (2019) Conversion to MCI and dementia in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord 65:20–31
Sasikumar S, Strafella AP (2020) Imaging mild cognitive impairment and dementia in Parkinson’s disease. Front Neurol 11:47
Scamarcia PG, Agosta F, Spinelli EG, Basaia S, Stojkovic T, Stankovic I, Sarasso E, Canu E, Markovic V, Petrovic I, Stefanova E, Pagani E, Kostic VS, Filippi M (2022) Longitudinal white matter damage evolution in Parkinson’s disease. Mov Disord 37:315–324
Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, Bennett DA (2012) Cognitive impairment, decline and fluctuations in older community-dwelling subjects with lewy bodies. Brain 135:3005–3014
Schrag A, Jahanshahi M, Quinn N (2000) What contributes to quality of life in patients with Parkinson’s disease? J Neurol Neurosurg Psychiatry 69:308–312
Schrag A, Siddiqui UF, Anastasiou Z, Weintraub D, Schott JM (2017) Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: a cohort study. Lancet Neurol 16:66–75
Schulz J, Pagano G, Fernández Bonfante JA, Wilson H, Politis M (2018) Nucleus basalis of Meynert degeneration precedes and predicts cognitive impairment in Parkinson’s disease. Brain 141:1501–1516
Schulz-Schaeffer WJ (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143
Seibert TM, Murphy EA, Kaestner EJ, Brewer JB (2012) Interregional correlations in Parkinson disease and Parkinson-related dementia with resting functional MR imaging. Radiology 263:226–234
Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ (2009) A clinico-pathological study of subtypes in Parkinson’s disease. Brain 132:2947–2957
Severiano ESC, Alarcão J, Pavão Martins I, Ferreira JJ (2022) Frequency of dementia in Parkinson’s disease: a systematic review and meta-analysis. J Neurol Sci 432:120077
Shah N, Frey KA, Muller ML, Petrou M, Kotagal V, Koeppe RA, Scott PJ, Albin RL, Bohnen NI (2016) Striatal and cortical beta-amyloidopathy and cognition in Parkinson’s disease. Mov Disord 31:111–117
Siepel FJ, Bronnick KS, Booij J, Ravina BM, Lebedev AV, Pereira JB, Gruner R, Aarsland D (2014) Cognitive executive impairment and dopaminergic deficits in de novo Parkinson’s disease. Mov Disord 29:1802–1808
Sierra M, Gelpi E, Marti MJ, Compta Y (2016) Lewy- and Alzheimer-type pathologies in midbrain and cerebellum across the lewy body disorders spectrum. Neuropathol Appl Neurobiol 42:451–462
Smith C, Malek N, Grosset K, Cullen B, Gentleman S, Grosset DG (2019) Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry 90:1234–1243
Smith R, Schöll M, Londos E, Ohlsson T, Hansson O (2018) (18)F-AV-1451 in Parkinson’s disease with and without dementia and in dementia with lewy bodies. Sci Rep 8:4717
Sonnen JA, Postupna N, Larson EB, Crane PK, Rose SE, Montine KS, Leverenz JB, Montine TJ (2010) Pathologic correlates of dementia in individuals with lewy body disease. Brain Pathol 20:654–659
Speelberg DHB, Janssen Daalen JM, Bloem BR, Gagnon JF, Post B, Darweesh SKL (2022) Prodromal cognitive deficits and the risk of subsequent Parkinson's disease. Brain Sci 12:199
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113
Szwedo AA, Dalen I, Pedersen KF, Camacho M, Bäckström D, Forsgren L, Tzoulis C, Winder-Rhodes S, Hudson G, Liu G, Scherzer CR, Lawson RA, Yarnall AJ, Williams-Gray CH, Macleod AD, Counsell CE, Tysnes OB, Alves G, Maple-Grødem J (2022) GBA and APOE impact cognitive decline in Parkinson’s disease: a 10-year population-based study. Mov Disord. https://doi.org/10.1002/mds.28932
Thomas GEC, Leyland LA, Schrag AE, Lees AJ, Acosta-Cabronero J, Weil RS (2020) Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J Neurol Neurosurg Psychiatry 91:418–425
Tilley BS, Patel SR, Goldfinger MH, Pearce RKB, Gentleman SM (2021) Locus coeruleus pathology indicates a continuum of lewy body dementia. J Parkinsons Dis 11:1641–1650
Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, Lee EB, Arnold SE, Duda JE, Hurtig H, Lee VM, Adler CH, Beach TG, Trojanowski JQ (2016) Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and lewy body pathology. Acta Neuropathol 131:393–409
Tsai HH, Tsai LK, Lo YL, Lin CH (2021) Amyloid related cerebral microbleed and plasma Abeta40 are associated with cognitive decline in Parkinson’s disease. Sci Rep 11:7115
Tu H, Zhang ZW, Qiu L, Lin Y, Jiang M, Chia SY, Wei Y, Ng ASL, Reynolds R, Tan EK, Zeng L (2022) Increased expression of pathological markers in Parkinson’s disease dementia post-mortem brains compared to dementia with lewy bodies. BMC Neurosci 23:3
Uribe C, Segura B, Baggio HC, Campabadal A, Abos A, Compta Y, Marti MJ, Valldeoriola F, Bargallo N, Junque C (2018) Differential progression of regional hippocampal atrophy in aging and Parkinson’s disease. Front Aging Neurosci 10:325
Uversky VN (2009) Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci (Landmark Ed) 14:5188–5238
Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS (2014) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34:9364–9376
Vermeiren Y, De Deyn PP (2017) Targeting the norepinephrinergic system in Parkinson’s disease and related disorders: the locus coeruleus story. Neurochem Int 102:22–32
Vermersch P, Delacourte A, Javoy-Agid F, Hauw JJ, Agid Y (1993) Dementia in Parkinson’s disease: biochemical evidence for cortical involvement using the immunodetection of abnormal tau proteins. Ann Neurol 33:445–450
Villemagne VL, Ong K, Mulligan RS, Holl G, Pejoska S, Jones G, O’Keefe G, Ackerman U, Tochon-Danguy H, Chan JG, Reininger CB, Fels L, Putz B, Rohde B, Masters CL, Rowe CC (2011) Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J Nucl Med 52:1210–1217
Walker Z, Moreno E, Thomas A, Inglis F, Tabet N, Rainer M, Pizzolato G, Padovani A (2015) Clinical usefulness of dopamine transporter SPECT imaging with 123I-FP-CIT in patients with possible dementia with lewy bodies: randomised study. Br J Psychiatry 206:145–152
Wallace ER, Segerstrom SC, van Horne CG, Schmitt FA, Koehl LM (2022) Meta-analysis of cognition in Parkinson's disease mild cognitive impairment and dementia progression. Neuropsychol Rev 32:149-160
Wan H, Wang G, Liu Q, Wang Y (2022) Effect of cerebral small vessel disease on cognitive impairment in Parkinson's disease: a systematic review and meta-analysis. Ann Transl Med 10:288
Wang Z, Jia X, Chen H, Feng T, Wang H (2018) Abnormal spontaneous brain activity in early Parkinson’s disease with mild cognitive impairment: a resting-state fMRI study. Front Physiol 9:1093
Watanabe H, Ariyoshi T, Ozaki A, Ihara M, Ono M, Saji H (2017) Synthesis and biological evaluation of novel radioiodinated benzimidazole derivatives for imaging a-synuclein aggregates. Bioorg Med Chem 25:6398–6403
Weil RS, Hsu JK, Darby RR, Soussand L, Fox MD (2019) Neuroimaging in Parkinson’s disease dementia: connecting the dots. Brain Commun 1:fcz006
Weintraub D, Doshi J, Koka D, Davatzikos C, Siderowf AD, Duda JE, Wolk DA, Moberg PJ, Xie SX, Clark CM (2011) Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch Neurol 68:1562–1568
Weintraub D, Dietz N, Duda JE, Wolk DA, Doshi J, Xie SX, Davatzikos C, Clark CM, Siderowf A (2012) Alzheimer’s disease pattern of brain atrophy predicts cognitive decline in Parkinson’s disease. Brain 135:170–180
Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A (2010) Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol 225:210–218
Wilson H, de Natale ER, Politis M (2021) Nucleus basalis of Meynert degeneration predicts cognitive impairment in Parkinson’s disease. Handb Clin Neurol 179:189–205
Winer JR, Maass A, Pressman P, Stiver J, Schonhaut DR, Baker SL, Kramer J, Rabinovici GD, Jagust WJ (2018) Associations between tau, beta-amyloid, and cognition in Parkinson disease. JAMA Neurol 75:227–235
Wise AH, Alcalay RN (2022) Genetics of cognitive dysfunction in Parkinson’s disease. Prog Brain Res 269:195–226
Wolters AF, van de Weijer SCF, Leentjens AFG, Duits AA, Jacobs HIL, Kuijf ML (2019) Resting-state fMRI in Parkinson’s disease patients with cognitive impairment: a meta-analysis. Parkinsonism Relat Disord 62:16–27
Wood KL, Myall DJ, Livingston L, Melzer TR, Pitcher TL, MacAskill MR, Geurtsen GJ, Anderson TJ, Dalrymple-Alford JC (2016) Different PD-MCI criteria and risk of dementia in Parkinson’s disease: 4-year longitudinal study. NPJ Parkinsons Dis 2:15027
Xie S, Yang J, Huang S, Fan Y, Xu T, He J, Guo J, Ji X, Wang Z, Li P, Chen J, Zhang Y (2022) Disrupted myelination network in the cingulate cortex of Parkinson’s disease. IET Syst Biol. https://doi.org/10.1049/syb2.12043
Xu R, Hu X, Jiang X, Zhang Y, Wang J, Zeng X (2020) Longitudinal volume changes of hippocampal subfields and cognitive decline in Parkinson’s disease. Quant Imaging Med Surg 10:220–232
Xu Y, Yang J, Hu X, Shang H (2016) Voxel-based meta-analysis of gray matter volume reductions associated with cognitive impairment in Parkinson’s disease. J Neurol 263:1178–1187
Yarnall AJ, Breen DP, Duncan GW, Khoo TK, Coleman SY, Firbank MJ, Nombela C, Winder-Rhodes S, Evans JR, Rowe JB, Mollenhauer B, Kruse N, Hudson G, Chinnery PF, O’Brien JT, Robbins TW, Wesnes K, Brooks DJ, Barker RA, Burn DJ (2014) Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology 82:308–316
Ye R, O'Callaghan C, Rua C, Hezemans FH, Holland N, Malpetti M, Jones PS, Barker RA, Williams-Gray CH, Robbins TW, Passamonti L, Rowe J (2022) Locus coeruleus integrity from 7 T MRI relates to apathy and cognition in parkinsonian disorders. Mov Disord online May 16: https://doi.org/10.1002/mds.29072
Zaman V, Shields DC, Shams R, Drasites KP, Matzelle D, Haque A, Banik NL (2021) Cellular and molecular pathophysiology in the progression of Parkinson’s disease. Metab Brain Dis 36:815–827
Zarei M, Ibarretxe-Bilbao N, Compta Y, Hough M, Junque C, Bargallo N, Tolosa E, Martí MJ (2013) Cortical thinning is associated with disease stages and dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 84:875–881
Zarifkar P, Kim J, La C, Zhang K, YorkWilliams S, Levine TF, Tian L, Borghammer P, Poston KL (2021) Cognitive impairment in Parkinson’s disease is associated with default mode network subsystem connectivity and cerebrospinal fluid Aß. Parkinsonism Relat Disord 83:71–78
Zhang Y, Burock MA (2020) Diffusion tensor imaging in Parkinson’s disease and parkinsonian syndrome: a systematic review. Front Neurol 11:531993
Zheng D, Chen C, Song W, Yi Z, Zhao P, Zhong J, Dai Z, Shi H, Pan P (2019) Regional gray matter reductions associated with mild cognitive impairment in Parkinson’s disease: a meta-analysis of voxel-based morphometry studies. Behav Brain Res 371:111973
Zhou C, Guan XJ, Guo T, Zeng QL, Gao T, Huang PY, Xuan M, Gu QQ, Xu XJ, Zhang MM (2020) Progressive brain atrophy in Parkinson’s disease patients who convert to mild cognitive impairment. CNS Neurosci Ther 26:117–125
Acknowledgements
The author thanks Mr. E. Mitter-Ferstl for secretarial and editorial work.
Funding
The study was funded by the Society for the Promotion of Research in Experimental Neurology, Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jellinger, K.A. Morphological basis of Parkinson disease-associated cognitive impairment: an update. J Neural Transm 129, 977–999 (2022). https://doi.org/10.1007/s00702-022-02522-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02522-4