
Abstract
Lipases from Pseudomonas species are particularly useful due to their broader biocatalytic applications and temperature activity. In this study, we amplified the gene encoding wild-type cold-active lipase from the genome of psychrotrophic bacterium isolated from the Himalayan glacier. The isolated CRBC14 strain was identified as Pseudomonas sp. based on the 16S rRNA gene sequence. Lipase activity was determined by observing the hydrolysis zone on nutrient agar containing tributyrin (1%, v/v). The sequence analysis of cold-active lipase revealed a protein of 611 amino acids with a calculated molecular mass of 63.71 kDa. The three-dimensional structure of this lipase was generated through template-supported modeling. Distinct techniques stamped the model quality, following which the binding free energies of tributyrin and oleic acid in the complex state with this enzymatic protein were predicted through molecular mechanics generalized born surface area (MMGBSA). A relative comparison of binding free energy values of these substrates indicated tributyrin’s comparatively higher binding propensity towards the lipase. Using molecular docking, we evaluated the binding activity of cold-active lipase against tributyrin and oleic acid. Our docking analysis revealed that the lipase had a higher affinity for tributyrin than oleic acid, as evidenced by our measurement of the hydrolysis zone on two media plates. This study will help to understand the bacterial diversity of unexplored Himalayan glaciers and the possible application of their cold-adapted enzymes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lipases are the potential bioresources, mainly responsible for the hydrolysis of acylglycerides, while some are more suitable for synthesis (Kumar et al. 2020). Among all the lipases, cold-active lipases found naturally in psychrotrophic and psychrophilic bacterial species have gained a lot of attention in structural investigations and industrial applications due to their remarkable stability at low temperatures (Bhatia et al. 2020). These lipases have presented great activity in biofuel production (Ribeiro et al. 2011), as detergent additives (Al-Ghanayem and Joseph 2020), in environmental bioremediations and food industries (Chandra et al. 2020), leather processing and suppressing the formation of inclusion bodies in protein expression studies at low temperatures (Sathish Yadav et al. 2011; Joseph et al. 2008). Moreover, the global market for lipases is expected to reach $ 0.79 billion by 2025 (Fatima et al. 2020). Cold-active lipases are in growing demand (Mhetras et al. 2021) because they are active at low temperatures and low water concentrations due to improved flexibility compared to their mesophilic and thermophilic counterparts (Kumar et al. 2020).
Furthermore, microbial lipases have a lower cost of manufacture, a more selective activity, lower energy usage (Kavitha 2016), easy handling and transportation in powdered form in the enzyme market in comparison to lipases from plant and animal sources (Chandra et al. 2020). Pseudomonas lipases, in particular, have received special attention among bacterial lipases because of their thermo-resistance, active at alkaline pHs and higher production rate (Chandra et al. 2020; Ramnath et al. 2016). According to their molecular properties and the need for correct folding and secretion of helper proteins, these lipases have been categorized into three groups viz., the groups I, II and III. Group III lipases do not require a lipase-specific foldase to obtain enzymatically active lipases and have a molecular mass between 50 and 68 kDa (Karakaş and Arslanoğlu 2020).
The common methods of identifying the new lipases isolated from natural sources such as plants, animals and microorganisms used established protocols or selected methods. These methods include fermentation, precipitation and purification of enzymes, which are usually time and resource consuming (Bharathi and Rajalakshmi 2019). Alternatively, the amplification of the target gene can be carried out using degenerate primers (Abd. Jalil et al. 2018). In addition, the in silico characterization of these enzymes also offers a higher success rate, increased discoverability and lower consumption of time and resources (Kamble et al. 2018). The discovery of new lipases through the combined molecular and in silico approach for industrial use has become a valuable tool due to the increasing availability of whole-genome sequences (Kamble et al. 2018). Thus, several lipase-encoding genes have either been amplified from the wild-type bacterial species or cloned into various other species (Baweja et al. 2016; Perfumo et al. 2020).
In light of this, there is greater interest in exploring cold habitats to isolate such enzymes from psychrotrophic bacteria for commercial purposes. Kashmir Himalaya, which lies to the north-western extremity of the Himalayan biodiversity hotspot, has been less explored for lipase-producing psychrotrophic bacteria (Yadav et al. 2016; Joseph et al. 2012). In this study, we used a combinatorial approach to characterize the cold-active lipase isolated from a psychrotrophic bacterium, CRBC14 of the Himalayan Thajwas glacier. Further, we explored the binding free energy of this lipase towards tributyrin and oleic acid through an extra-precision molecular docking approach in flexible mode and next-generation solvation model-based molecular mechanics generalized born surface area (MMGBSA) approach.
Materials and methods
Sample collection
The soil sample was collected at an altitude of 2944 m (34°16′30′′N; 75°17′10′′E) from the Himalayan Thajwas glacier. The soil sample was collected in 100 ml sterile plastic vials and transported to the laboratory in ice packs for analysis.
Isolation of the psychrotrophic bacteria
Isolation of bacterial colonies was carried on Luria–Bertani (LB) agar plates as per Srinivas et al. (2011). Soil sample (1 g) was dissolved in 100 ml of NaCl solution (0.9%, w/v) and kept in an orbital shaker incubator for 2 h at 150 rpm and 15 °C. Following that, 0.1 ml of the soil sample solution was inoculated on pre-prepared LB agar plates. The inoculated LB plates were then incubated at 4, 15, 20 and 30 °C for 2–15 days, colony counts were taken and distinct morphotypes were purified and kept on LB agar medium.
Screening for lipolytic activity
The isolated, pure colonies were screened for their lipolytic activity (clear zone) on nutrient agar (NA) plates containing tributyrin (1%, v/v). The strain CRBC14 with a maximum lipolytic activity was chosen for further analysis. The pure isolate was grown at five different temperatures (4, 10, 15, 20 and 30 °C) and pHs (6.0, 7.0, 8.0, 9.0 and 10.0) on two sets of NA plates. One set contained tributyrin (1%, v/v) and the other olive oil (1%, v/v) for the determination of lipolytic activity by analyzing the zone of hydrolysis (Joseph et al. 2012).
Determination of optimal growth temperature
The purified isolate was inoculated on pre-prepared LB plates and incubated at 7 different temperatures (0, 5, 10, 15, 20, 30 and 37 °C) to determine the optimal growth temperature. Growth was observed every 24 h for 2–30 days (Zhang et al. 2013).
Genomic DNA extraction and 16S rRNA gene amplification
The extraction of genomic DNA from psychrotrophic bacterial isolate CRBC14 was done by DNA purification kit (HiPura bacterial genomic DNA), as per the manufacturer’s instructions. The extracted DNA was used as a template for 16S rRNA gene amplification through PCR using 27F and 1429R primers (Shivaji et al. 2004). The PCR product purification and sequencing were done at Agrigenome labs, Kerala, India. Identification of isolate (CRBC14) was done by BLASTn (Nucleotide BLAST) search at NCBI to check the similarity with our sequence. Further, the sequence alignment of CRBC14 with similar species (downloaded at NCBI) was done with ClustalW (https://www.genome.jp/tools-bin/clustalw). MEGA 7 was used to create the phylogenetic tree (Kumar et al. 2016) by the Neighbor-joining method (Bootstrapping at 1000 replicates).
Amplification of lipase gene
Lipase gene was amplified by PCR using the primers based on known Pseudomonas lipase sequences: (LF) 5′-ATGGCTGTGTAGGACAAAAGAAC-3′ and (LR) 5′-TCAGGCGATTACAATGCCATCAGC-3′ (Zhang et al. 2007). Protein sequences of cold-active lipase from CRBC14 strain and of other similar lipases were aligned together in Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and Fig. 2 was developed with ESPript 3.0.0 (Robert and Gouet 2014).
Model generation and assuring its validity
Using the amino acid sequence of cold-active lipase as input, the three-dimensional (3D) structure of this enzyme was generated using the sophisticated homology modeling tool, namely SWISS-MODEL (Bordoli et al. 2009). Among the various templates recognized by the search algorithm, the best template was chosen, taking various aspects like sequence identity, sequence coverage and best resolution into consideration. 2Z8X, the PDB ID of the extracellular enzyme (lipase), from Pseudomonas sp. MIS38 was used as template for model building (Angkawidjaja et al. 2007). The model quality was tested using various methods, including PROCHECK, VERIFY3D, PROSA and by estimating RMSD between model and template (Laskowski et al. 1993; Lüthy et al. 1992; Wiederstein and Sippl 2007).
Preparing protein (lipase) and substrate (tributyrin and oleic acid) coordinates
As a rule of thumb, one has to perform protein preparation before molecular docking. The needful was done by taking advantage of the protein preparation wizard, the component of the Schrödinger suite. During the preparation of protein, the standard protocol was followed (Madhavi Sastry et al. 2013; Mir et al. 2020). After fixing missing side chains and other parameters, the protein was optimized and then refined. The active site was selected by specifying the residues that were confirmed to be active site residues by various tools, including ScanProsite and Computed Atlas of Surface Topography of proteins (CASTp) (De Castro et al. 2006; Tian et al. 2018). Coordinates of tributyrin and oleic acid were retrieved from the huge database, PubChem. The PubChem CID of these molecules is 6050 and 445,639, respectively (Kim et al. 2020). Tributyrin and oleic acid structures were prepared for molecular docking with the help of LigPrep software (LigPrep, Schrödinger, LLC, New York, 2021). Substrates, as usual, were minimized, desalted, metal-binding states and tautomers were generated and default parameter for chirality was opted (Shankaran et al. 2016).
Flexible molecular docking and binding free energy quantification
The prepared tributyrin and oleic acid were docked into the predefined active site of cold-active lipase. As it is well proven that flexible docking in extra-precision mode has better accuracy than standard-precision mode, thus we executed molecular docking in the former mode. Docking was executed using the pandemically authenticated Glide tool. The best pose, in either case, was picked based on docking score criteria (Friesner et al. 2006; Ganai 2021). The individual tributyrin–lipase and oleic acid–lipase docked complexes were used as input for estimating the values of binding free energy. Calculations were done with the popular implicit solvation reliant MMGBSA method. MMGBSA of Prime module performs multiple energy estimations and from those, the binding free energy value is finally deduced using the standard equation;
E_complex (minimized) – E_ligand (minimized) − E_receptor (minimized) = ΔG (bind).
Default parameters, including the novel energy model, namely VSGB 2.0 were maintained during the calculations in both cases (Ganai 2021; Li et al. 2011).
Results
Identification of psychrotrophic bacterium
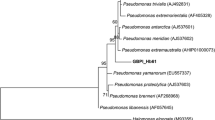
The taxonomical study conducted on the lipase-producing psychrotrophic bacterial strain, CRBC14 showed that the strain was rod-shaped, Gram-negative and aerobic with an optimum growth temperature of 20 °C. Besides, the isolate could grow at temperatures between 4 and 30 °C, but could not grow at 37 °C. This behavior towards temperature indicates that it is a psychrotrophic bacterium. The CRBC14 strain was closely associated (100–98%) with Pseudomonas sp. based on the 16S rRNA gene sequence review. A high similarity rate was observed with Pseudomonas sp. ICMP 13 603 (Fig. 1) after the nucleotide sequence was BLAST searched at NCBI. The sequence of 16S rRNA gene was deposited in NCBI GenBank under the accession number MT478141.

Phylogenetic tree of strain CRBC14 (indicated by bold letters) based on the sequence of 16S rRNA genes. Using MEGA 7.0 software, the phylogenetic tree was developed from the neighbor-joining approach, and 1000 bootstrap analysis trials were performed
Screening for lipase activity
The lipolytic activity was seen at temperatures between 4 and 30 °C on 1% (v/v) tributyrin, with a maximum at 20 °C and pH 8.0 in comparison to olive oil (Fig. S1). Lipase activity increased from 4 to 20 °C, after which the enzymatic activity started decreasing, indicating its cold-active nature.
Amplification of lipase gene and sequence analysis
Sequence analysis of our lipase (GenBank accession no. MW417497) revealed an open reading frame of 1,835 nucleotides with 46% G + C content. The polypeptide sequence of the cold-active lipase showed 99% identity with that of Pseudomonas sp. 7323 lipase (GenBank: CAJ76166), 79% with LipA of Pseudomonas fluorescens F113 (GenBank: G8Q328) (Fig. 2). The nucleotide sequence encoded a protein comprising 611 amino acids with a calculated molecular mass of 63.71 kDa.

Similarity analysis of multiple amino acids sequence. The red backdrop highlights exclusively conserved residues, and boxed are the conservatively replaced residues. The vertical row displays the signal peptide’s cleavage site, and the horizontal rows show the N-terminal residues
Template guided modeling and validation
The extracellular lipase from Pseudomonas sp. MIS38 (PDB ID: 2Z8X) was utilized as template to build the 3D structure of the cold-active lipase. This refined model (red) on superimposing with template (yellow) showed a very low RMSD (0.055 Å), suggesting its structural closeness with the latter (Fig. 3). Additionally, this model qualified stereochemical quality check as its Ramachandran plot showed localization of 92.5% residues in the most preferred region (acceptable quality), 6.5% in the allowed region and 0.2% residues in the disallowed region (Fig. S2). Besides, the structure-sequence compatibility of this model was found to be highly acceptable as the percentage of residues scoring over or equal to 0.2 was found to be 98.52%, the threshold being 80% (Fig. 4). PROSA analysis also supported the acceptable model quality as its z-score aligned with similar-sized proteins possessing experimentally determined structures (Fig. 5).

RMSD between the target (model) and template structure. On superimposing template and target RMSD value of 0.055 Å was estimated. This indicates that the target is very close to the experimentally solved template. For easy understanding, model has been shown in red and the template in yellow

Measurement of model compatibility with its primary structure (sequence). 98.52% of residues scored beyond or equal to 0.2 (80% lowest limit), thereby confirming the model exactitude

Testing model quality by estimating z-score. The cold-active lipase model showed z-score in agreement with the z-score displayed by similar-sized proteins whose structures have been solved by empirical methods. The cold lipase model demonstrated a z-score of − 10.7 that falls well within the range
Flexible molecular docking and binding free energy estimation
It was observed from the binding free energy estimates for two docked complexes that tributyrin has a higher affinity for cold-active lipase than oleic acid. While oleic acid’s value was estimated to be -29.3826 kcal/mol, tributyrin’s value was found to be -33.3136 kcal/mol (Figs. 6 and 7). From the binding free energy estimations on two docked complexes, it can be inferred that tributyrin has more affinity towards cold-active lipase when compared to oleic acid. This crux is taken as tributyrin manifested more negative binding free energy value in comparison to oleic acid. While this value for oleic acid was found to be − 29.3826 kcal/mol, a value of − 33.3136 kcal/mol was demonstrated by tributyrin (Figs. 6 and 7). More negative binding free energy infers more binding inclination.

Tributyrin and oleic acid in the docked state with the lipase. As in both cases, protein is lipase thus, the same color has been used for protein. The ligands being different are presented in two distinct colors. Tributyrin has been shown in red and oleic acid in yellow

The binding proclivity of tributyrin and oleic acid towards lipase. From the binding free energy values, it is perceptible that tributyrin–lipase stability is relatively more compared to oleic acid–lipase stability. In other words, tributyrin has higher binding strength with lipase when compared to oleic acid
Discussion
Most of the Himalayan glaciers of north-western side are still untouched for their bacterial diversity, which can be a goldmine of potential bacterial enzymes. Isolation of such microbes for the screening and characterization of cold-adapted lipases can bring new opportunities in the enzyme industry. In this study, a cold-active lipase was characterized from a psychrotrophic Pseudomonas sp. CRBC14 from a north-western Himalayan glacial soil. The 16S rRNA gene was used for the identification of the isolated bacterium, which is most widely used universal gene marker for psychrophilic and psychrotrophic bacterial identification (Farooq et al. 2021; Rafiq et al. 2017).
Tributyrin has previously been used to determine the lipase activity in psychrophilic and psychrotrophic Pseudomonas sp. by analyzing the clear zone around the bacterial colonies grown on tributyrin agar (Maharana and Ray 2015; Salwoom et al. 2019). Bacterial lipases are mostly active in alkaline conditions (Gupta et al. 2004). We also observed the maximum lipase activity (zone of hydrolysis) at pH 8.0 on tributyrin agar. Similar observations were made in cold-active lipases from Pseudomonas sp. LSK25 (Salwoom et al. 2019), Pseudomonas sp. AKM-LS (Maharana and Ray 2015), Pseudomonas sp. KB700A (Rashid et al. 2020), Pseudomonas antartica and Pseudomonas meridian (Reddy et al. 2004). Furthermore, Pseudomonas sp. CRBC14 displayed its optimum lipase activity at 20 °C. This temperature was lower than reported in Pseudomonas sp. 7323 (Zhang et al. 2008), Pseudoalteromonas sp. 643A (Cieśliński et al. 2007), Pseudoalteromonas sp. NJ 70 (Wang et al. 2012), Pseudoalteromonas Haloplanktis TAC125, CR9 (De Pascale et al. 2008) where optimal activity was observed at 30, 35 and 40 °C, respectively. While as, cold-active lipases previously isolated from psychrophilic/psychrotrophic Pseudomonas fragi X14033 (Alquati et al. 2002), Pseudomonas sp. KB700A (Rashid et al. 2020) and Pseudomonas sp. 7323 (Zhang et al. 2013) showed optimal activity at 20 °C. Besides, some lipases were reported to show better catalytic activity at low temperatures. For instance, Guo et al (2021) reported a cold-active lipase from Pseudomonas marinensis with optimal activity at 4 °C and pH 8.0. In another study, a novel cold-adapted lipase (LipI.3_KE38) from Pseudomonas fluorescence KE38 manifested optimal activity at 25 °C and pH 8.5 (Karakaş and Arslanoğlu, 2020).
In silico research has become an important means for discovering and detecting novel enzymes for industrial application (Kwoun Kim et al. 2004). Many analytical tools have been developed to classify conserved genes based on their identical relationships in order to extract the most information from the genome sequences that are currently available (De Pascale et al. 2008). In earlier studies, such approachs have been used to amplify the gene coding cold-active protease and lipase from a psychrophilic Psychrobacter sp. 94-6 PB (Perfumo et al. 2020) and Psychrobacter sp. G (Xuezheng et al. 2010). The structure of some cold-active lipases from psychrotrophic bacteria has previously been predicted using template-guided modeling methods (Abd. Jalil et al. 2018; Kumar et al. 2020). Moreover, simulation approaches have been used to check the stability of substrates like Triton X-100/toluene with a cold-active lipase from Pseudomonas sp. AMS8 (Abd. Jalil et al. 2018). A similar approach was thus used to amplify the gene coding for cold-active lipase from a psychrotrophic bacterium CRBC14 and predict its binding affinity towards two different substrates (tributyrin and oleic acid).
The molecular mass of current cold-active lipase was around 63.71 kDa; Rashid et al (2020) previously reported a similar lipase (CALip) from Pseudomonas sp. KB700A with a molecular mass of 49.92 kDa. In addition, psychrotolerant lipases from Pseudomonas fluorescens KE38 and Pseudomonas sp. AKM-L5 had a molecular mass of 43 kDa and 57 kDa, respectively (Gökbulut and Arslanoğlu 2013; Maharana and Ray 2015). The primary structure of present lipase indicated that it was a member of bacterial lipases (Arpigny and Jaeger 1999). Besides, the amino acid sequence of CRBC14 lipase showed 89% similarity with lipases belonging to group III (subfamily I.3).
The stereochemical quality of model proved to be acceptable as over 92% of the residues were spaced in most favored regions (Mushtaq et al. 2021). While testing the compatibility of primary and 3D structure of model above 98% residues (Fig. 4) scored over or equal to 0.2 further stamping model correctness (Farooq et al. 2021). Apart from this, the z-score of model aligned well with the z-scores of similar length proteins (Fig. 5), having experimentally determined structures suggesting the overall model quality to be highly reliable for docking studies (Wiederstein and Sippl 2007). Moreover, the RMSD was estimated by way of comparing model with a template (Fig. 3). A very low RMSD (0.055 Å) was obtained on comparing these structures, again corroborating model accuracy (Farooq et al. 2021; Pettersen et al. 2004).
On the whole, molecular docking studies coupled with binding free energy estimations revealed that the lipase has more binding inclination towards tributyrin than oleic acid. These results were consistent with our in-vitro studies, where the zone of hydrolysis was found to be more on tributyrin substrate than olive oil (oleic acid). As per the available literature, this was the first approach to use an in silico strategy to characterize the protein sequence of a target gene coding a cold-active lipase from psychrotrophic Pseudomonas sp. CRBC14. With a low optimal temperature profile, this lipase could be a potential candidate for industrial uses. Further, investigation of these unexplored Himalayan glaciers could lead to the discovery of new cold-active lipases with unique properties and applications.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary file.
Code availability
Not applicable.
References
Abd. Jalil F, Raja Abd. Rahman R, Salleh A, Mohamad Ali M (2018) Optimization and in silico analysis of a cold-adapted lipase from an Antarctic Pseudomonas sp. strain AMS8 reaction in Triton X-100 reverse micelles. Catalysts 8:289. https://doi.org/10.3390/catal8070289
Al-Ghanayem AA, Joseph B (2020) Current prospective in using cold-active enzymes as eco-friendly detergent additive. Appl Microbiol Biotechnol 104:2871–2882. https://doi.org/10.1007/s00253-020-10429-x
Alquati C, De Gioia L, Santarossa G, Alberghina L, Fantucci P, Lotti M (2002) The cold-active lipase of Pseudomonas fragi: heterologous expression, biochemical characterization and molecular modeling. Eur J Biochem 269:3321–3328
Angkawidjaja C, You DJ, Matsumura H, Kuwahara K, Koga Y, Takano K, Kanaya S (2007) Crystal structure of a family I.3 lipase from Pseudomonas sp. MIS38 in a closed conformation. FEBS Lett 581(26):5060–5064. https://doi.org/10.1016/j.febslet.2007.09.048
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Baweja M, Nain L, Kawarabayasi Y, Shukla P (2016) Current technological improvements in enzymes toward their biotechnological applications. Front Microbiol 7:965. https://doi.org/10.3389/FMICB.2016.00965
Bharathi D, Rajalakshmi G (2019) Microbial lipases: an overview of screening, production and purification. Biocatal Agric Biotechnol 22:1878–8181. https://doi.org/10.1016/j.bcab.2019.101368
Bhatia RK, Ullah S, Hoque MZ, Ahmad I, Yang Y-H, Bhatt AK, Bhatia SK (2020) Psychrophiles: a source of cold-adapted enzymes for energy efficient biotechnological industrial processes. J Environ Chem Eng 9(1):104607. https://doi.org/10.1016/j.jece.2020.104607
Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4(1):1–13. https://doi.org/10.1038/nprot.2008.197
Chandra P, Enespa SR, Arora PK (2020) Microbial lipases and their industrial applications: a comprehensive review. Microb Cell Factories 191:1–42. https://doi.org/10.1186/S12934-020-01428-8
Cieśliński H, Białkowska AM, Długołęcka A, Daroch M, Tkaczuk KL, Kalinowska H, Kur J, Turkiewicz M (2007) A cold-adapted esterase from psychrotrophic Pseudoalteromas sp. strain 643A. Arch Microbiol 188:27–36. https://doi.org/10.1007/s00203-007-0220-2
De Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34:362–365. https://doi.org/10.1093/nar/gkl124
De Pascale D, Cusano AM, Autore F, Parrilli E, Di Prisco G, Marino G, Tutino ML (2008) The cold-active Lip1 lipase from the Antarctic bacterium Pseudoalteromonas haloplanktis TAC125 is a member of a new bacterial lipolytic enzyme family. Extremophiles 12:311–323. https://doi.org/10.1007/s00792-008-0163-9
Farooq S, Nazir R, Ganai SA, Ganai BA (2021) Isolation and characterization of a new cold-active protease from psychrotrophic bacteria of Western Himalayan glacial soil. Sci Rep 11(1):12768. https://doi.org/10.1038/s41598-021-92197-w
Fatima S, Faryad A, Ataa A, Joyia FA, Parvaiz A (2020) Microbial lipase production: a deep insight into the recent advances of lipase production and purification techniques. Biotechnol Appl Bioc 68(3):445–458. https://doi.org/10.1002/bab.2019
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49(21):6177–6196. https://doi.org/10.1021/jm051256o
Ganai SA (2021) Characterizing binding intensity and energetic features of histone deacetylase inhibitor pracinostat towards class I HDAC isozymes through futuristic drug designing strategy. In Silico Pharmacol 9(1):18. https://doi.org/10.1007/s40203-021-00077-y
Gökbulut AA, Arslanoğlu A (2013) Purification and biochemical characterization of an extracellular lipase from psychrotolerant Pseudomonas fluorescens KE38. Turkish J Biol 37:538–546
Guo C, Zheng R, Cai R, Sun C, Wu S (2021) Characterization of two unique cold-active lipases derived from a novel deep-sea cold seep bacterium. Microorganisms 9(4):1–16. https://doi.org/10.3390/microorganisms9040802
Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol 64:763–781. https://doi.org/10.1007/S00253-004-1568-8
Joseph B, Ramteke PW, Thomas G (2008) Cold active microbial lipases: some hot issues and recent developments. Biotechnol Adv 26:457–470. https://doi.org/10.1016/j.biotechadv.2008.05.003
Joseph B, Shrivastava N, Ramteke PW (2012) Extracellular cold-active lipase of Microbacterium luteolum isolated from Gangotri glacier, western Himalaya: isolation, partial purification and characterization. J Genet Eng Biotechnol 10:137–144. https://doi.org/10.1016/j.jgeb.2012.02.001
Kamble A, Srinivasan S, Singh H (2018) In-Silico bioprospecting: finding better enzymes. Mol Biotechnol 61:53–59. https://doi.org/10.1007/S12033-018-0132-1
Karakaş F, Arslanoğlu A (2020) Gene cloning, heterologous expression, and partial characterization of a novel cold-adapted subfamily I.3 lipase from Pseudomonas fluorescence KE38. Sci Rep 10(1):1–13. https://doi.org/10.1038/s41598-020-79199-w
Kavitha M (2016) Cold active lipases – an update. Front Life Sci 9(3):226–38. https://doi.org/10.1080/21553769.2016.1209134
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE (2020) PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res 49:1388–1395. https://doi.org/10.1093/nar/gkaa971
Kumar S, Stecher G, Tamura K, Dudley J (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Kumar A, Mukhia S, Kumar N, Acharya V, Kumar S, Kumar R (2020) A broad temperature active lipase purified from a psychrotrophic bacterium of Sikkim Himalaya with potential application in detergent formulation. Front Bioeng Biotechnol 8:1–16. https://doi.org/10.3389/fbioe.2020.00642
Kwoun Kim H, Jung Y-J, Choi W-C, Ryu HS, Oh T-K, Lee J-K (2004) Sequence-based approach to finding functional lipases from microbial genome databases. FEMS Microbiol Lett 235:349–355. https://doi.org/10.1111/j.1574-6968.2004.tb09609.x
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. https://doi.org/10.1107/s0021889892009944
Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA (2011) The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins 79:2794–2812. https://doi.org/10.1002/prot.23106
Lüthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356(6364):83–85. https://doi.org/10.1038/356083a0
Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27(3):221–234. https://doi.org/10.1007/s10822-013-9644-8
Maharana A, Ray P (2015) A novel cold-active lipase from psychrotolerant Pseudomonas sp. AKM-L5 showed organic solvent resistant and suitable for detergent formulation. J Mol Catal B Enzym 120:173–178. https://doi.org/10.1016/J.MOLCATB.2015.07.005
Mhetras N, Mapare V, Gokhale D (2021) Cold active lipases: biocatalytic tools for greener technology. Appl Biochem Biotechnol 193:2245–2266. https://doi.org/10.1007/S12010-021-03516-W
Mir MA, Ganai SA, Mansoor S, Jan S, Mani P, Masoodi KZ, Amin H, Rehman UM, Ahmad P (2020) Isolation, purification and characterization of naturally derived crocetin beta-d-glucosyl ester from Crocus sativus L. against breast cancer and its binding chemistry with ER-alpha/HDAC2. Saudi J Biol Sci 27(3):975–984. https://doi.org/10.1016/j.sjbs.2020.01.018
Mushtaq H, Jehangir A, Ganai SA, Farooq S, Ganai BA, Nazir R (2021) Biochemical characterization and functional analysis of heat stable high potential protease of Bacillus amyloliquefaciens strain HM48 from soils of Dachigam National Park in Kashmir Himalaya. Biomolecules 11:117. https://doi.org/10.3390/biom11010117
Nardini M, Lang DA, Liebeton K, Jaeger KE, Dijkstra BW (2000) Crystal structure of Pseudomonas aeruginosa lipase in the open conformation. The prototype for family I.1 of bacterial lipases. J Biol Chem 275:31219–31225. https://doi.org/10.1074/jbc.M003903200
Perfumo A, Freiherr von Sass GJ, Nordmann E-L, Budisa N, Wagner D (2020) Discovery and characterization of a new cold-active protease from an extremophilic bacterium via comparative genome analysis and in vitro expression. Front Microbiol 11:881. https://doi.org/10.3389/fmicb.2020.00881
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Rafiq M, Hayat M, Anesio AM, Jamil SUU, Hassan N, Shah AA, Hasan F (2017) Recovery of metallo-tolerant and antibiotic resistant psychrophilic bacteria from Siachen glacier. Pakistan Plos One. https://doi.org/10.1371/journal.pone.0178180
Rashid N, Shimada Y, Ezaki S, Atomi H, Imanaka T (2020) Low-temperature lipase from psychrotrophic Pseudomonas sp. Strain KB700A. Appl Environ Microbiol 67:4064–4069. https://doi.org/10.1128/AEM.67.9.4064-4069.2001
Reddy GSN, Matsumoto GI, Schumann P, Stackebrandt E, Shivaji S (2004) Psychrophilic pseudomonads from Antarctica: Pseudomonas antarctica sp. nov., Pseudomonas meridiana sp. nov. and Pseudomonas proteolytica sp. nov. Int J Syst Evol Microbiol 54:713–719. https://doi.org/10.1099/IJS.0.02827-0
Ribeiro BD, De Castro AM, Coelho MAZ, Freire DMG (2011) Production and use of lipases in bioenergy: a review from the feedstocks to biodiesel production. Enzyme Res 2011:615803–615819. https://doi.org/10.4061/2011/615803
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. https://doi.org/10.1093/nar/gku316
Salwoom L, Rahman RNZRA, Salleh AB, Shariff FM, Convey P, Pearce D, Ali MSM (2019) Isolation, characterisation, and lipase production of a cold-adapted bacterial strain Pseudomonas sp. LSK25 isolated from Signy Island, Antarctica. Molecules 24:1–14. https://doi.org/10.3390/molecules24040715
Sathish Yadav KN, Adsul MG, Bastawde KB, Jadhav DD, Thulasiram HV, Gokhale DV (2011) Differential induction, purification and characterization of cold active lipase from Yarrowia lipolytica NCIM 3639. Bioresour Technol 102(22):10663–70. https://doi.org/10.1016/J.BIORTECH.2011.09.013
Schrödinger Release 2021-1: LigPrep, Schrödinger, LLC, New York, NY, 2021
Shankaran KS, Ganai SA, Arun KP, Brindha P, Mahadevan V (2016) In silico and in vitro evaluation of the anti-inflammatory potential of Centratherum punctatum Cass-A. J Biomol Struct Dyn 35:765–780. https://doi.org/10.1080/07391102.2016.1160840
Shivaji S, Reddy GS, Aduri RP, Kutty R, Ravenschlag K (2004) Bacterial diversity of a soil sample from Schirmacher Oasis, Antarctica. Cell Mol Biol (Noisy-le-grand) 50:525–536
Srinivas TNR, Singh SM, Pradhan S, Pratibha MS, Kishore KH, Singh AK, Begum Z, Prabagaran SR, Reddy GSN, Shivaji S (2011) Comparison of bacterial diversity in proglacial soil from Kafni Glacier, Himalayan Mountain ranges, India, with the bacterial diversity of other glaciers in the world. Extremophiles 15:673–690. https://doi.org/10.1007/s00792-011-0398-8
Tian W, Chen C, Lei X, Zhao J, Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46:363–367. https://doi.org/10.1093/nar/gky473
Wang Q, Hou Y, Ding Y, Yan P (2012) Purification and biochemical characterization of a cold-active lipase from Antarctic sea ice bacteria Pseudoalteromonas sp. NJ 70. Mol Biol Rep 39:9233–9238. https://doi.org/10.1007/s11033-012-1796-4
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407–W410. https://doi.org/10.1093/nar/gkm290
Xuezheng L, Shuoshuo C, Guoying X, Shuai W, Ning D, Jihong S (2010) Cloning and heterologous expression of two cold-active lipases from the Antarctic bacterium Psychrobacter sp. G. Polar Res 29:421–429. https://doi.org/10.3402/polar.v29i3.6087
Yadav AN, Sachan SG, Verma P, Kaushik R (2016) Cold active hydrolytic enzymes production by psychrotrophic Bacilli isolated from three sub-glacial lakes of NW Indian Himalayas. J Basic Microbiol 56:294–307. https://doi.org/10.1002/jobm.201500230
Zhang JW, Zeng RY (2008) Molecular cloning and expression of a cold-adapted lipase gene from an antarctic deep sea psychrotrophic bacterium Pseudomonas sp. 7323. Mar Biotechnol 10:612–621. https://doi.org/10.1007/s10126-008-9099-4
Zhang J, Lin S, Zeng R (2007) Cloning, expression and characterization of a cold-adapted lipase gene from an antarctic deep-sea psychorotrophic bacterium, Psychobacter sp. 7195. J Microbiol Biotechnol 17:604–610
Zhang D-C, Brouchkov A, Griva G, Schinner F, Margesin R (2013) Isolation and characterization of bacteria from ancient Siberian permafrost sediment. Biology (Basel) 2:85–106. https://doi.org/10.3390/biology2010085
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
SF: data curation, investigation, methodology, writing original draft. SAG: molecular docking, MMGBSA, validation, writing original draft. BAG: supervision, feedbacks, manuscript revision. SM: software license, resources. BU.: formal analysis, visualization. RN: methodology, writing original draft, project administration, resources, supervision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical approval
The authors in this study carried out no animal or human studies.
Consent to participate
Not applicable.
Consent for publication
The publication of this manuscript has been approved by all the authors.
Additional information
Communicated by Michael Polymenis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Farooq, S., Ganai, S.A., Ganai, B.A. et al. Molecular characterization of lipase from a psychrotrophic bacterium Pseudomonas sp. CRBC14. Curr Genet 68, 243–251 (2022). https://doi.org/10.1007/s00294-021-01224-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-021-01224-w