
Abstract
Major depression (MD) is one of the most common psychiatric disorders worldwide. Currently, the first-line treatment for MD targets the serotonin system but these drugs, notably the selective serotonin reuptake inhibitors, usually need 4 to 6 weeks before the benefit is felt and a significant proportion of patients shows an unsatisfactory response. Numerous treatments have been developed to circumvent these issues as venlafaxine, a mixed serotonin-norepinephrine reuptake inhibitor that binds and blocks both the SERT and NET transporters. Despite this pharmacological profile, it is difficult to have a valuable insight into its ability to produce more robust efficacy than single-acting agents. In this review, we provide an in-depth characterization of the pharmacological properties of venlafaxine from in vitro data to preclinical and clinical efficacy in depressed patients and animal models of depression to propose an indirect comparison with the most common antidepressants. Preclinical studies show that the antidepressant effect of venlafaxine is often associated with an enhancement of serotonergic neurotransmission at low doses. High doses of venlafaxine, which elicit a concomitant increase in 5-HT and NE tone, is associated with changes in different forms of plasticity in discrete brain areas. In particular, the hippocampus appears to play a crucial role in venlafaxine-mediated antidepressant effects notably by regulating processes such as adult hippocampal neurogenesis or the excitatory/inhibitory balance. Overall, depending on the dose used, venlafaxine shows a high efficacy on depressive-like symptoms in relevant animal models but to the same extent as common antidepressants. However, these data are counterbalanced by a lower tolerance. In conclusion, venlafaxine appears to be one of the most effective treatments for treatment of major depression. Still, direct comparative studies are warranted to provide definitive conclusions about its superiority.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a mental disorder characterized by numerous and persistent symptoms such as anhedonia, anxiety, despair, social withdrawal, cognitive deficits, sleep disturbance, and feelings of guilt. According to the World Health Organization (WHO), more than 250 million patients worldwide will suffer from MDD in the coming years. The pathophysiological mechanisms underlying MDD are not yet fully understood, but the monoamine-deficiency hypothesis is now well accepted. This hypothesis stipulates that the occurrence of MDD is associated with deficiencies of the three main monoamine neurotransmitters, namely 5-hydroxytryptamine (5-HT), norepinephrine (NE), and dopamine (DA). On the contrary, stimulating monoaminergic transmission using monoamine reuptake inhibitors or monoamine oxidase inhibitors have proven to be effective in improving mood (Blier 2014). By inhibiting the serotonin transporter (SERT), the selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine, paroxetine, or (es)-citalopram block 5-HT reabsorption into neurons, thereby increasing extracellular serotonin concentrations in the synaptic cleft. Although the monoamine-deficiency hypothesis does not fully capture the complex dimension of MDD, efforts are still underway to improve the effectiveness of SSRIs (Liu et al. 2017). It is well documented that this class of antidepressants effectively relieves the symptoms of depression in 60%–65% of the cases. However, a significant proportion of depressed patients does not respond appropriately to these treatments (Rush et al. 2006). Moreover, the remission rate of ~30% is low, and even if antidepressants rapidly increase the extracellular levels of monoamines in the central nervous system (CNS), the first therapeutic signs often take several weeks (i.e., 4–6) to appear.
Owing to the etiological heterogeneity of MDD and the obvious therapeutic limits of SSRIs, it is difficult to conceive that modulating serotonergic neurotransmission alone can generate beneficial and enduring effects in all patients (Hasler 2010). In this context, research in neuropsychopharmacology aims to improve the therapeutic activity of pharmacological drugs that enhance serotonergic tone. It has been postulated that the concomitant inactivation of the SERT and NE transporters (NET) could produce more potent effects than single-acting compounds (Guiard et al. 2009). However, it is still unclear whether adding the noradrenergic component yields more robust antidepressant effects.
This review provides a synthetic overview of current knowledge of the pharmacological properties of venlafaxine from preclinical and clinical studies. Preclinical studies have focused on the mechanisms of action of venlafaxine and its behavioral effects. In contrast, clinical trials or meta-analyses were designed to compare this drug's efficacy with other conventional antidepressant treatment options and highlight its potential superiority.
Pharmacological profile of venlafaxine
The venlafaxine binding profile was initially conducted in vitro and its affinity for different pharmacological targets was evaluated. In rats and humans, a preferential interaction of venlafaxine with SERT compared to NET was reported, whereas its affinity for DAT is very low and insignificant in both species (Bymaster et al. 2001; Millan et al. 2001). Venlafaxine displays a preferential (30-fold higher) affinity for the SERT compared to NET (i.e., KiSERT: 82 nM vs. KiNET: 2480 nM) in the rat brain (Bymaster et al. 2001). The affinity of venlafaxine towards other pharmacological targets involved in the modulation of cerebral monoaminergic neurotransmission (serotonergic, adrenergic, dopaminergic, muscarinic, and histaminergic receptors) was also evaluated. It appears that venlafaxine displays a negligible off-target affinity supporting the idea that it could lack of side effects, notably those observed with the tricyclics (TCAs).
Regarding its functional activity, venlafaxine blocks the SERT at the dose of 8 mg/kg in mice, while a twice higher dose is necessary to inhibit the NET (Bacq et al. 2012). In line with the latter observations, it inhibits 5-HT reuptake in a potent and robust manner at the dose of 75 mg in depressed patients, whereas higher doses (i.e., 150–225 mg) are required to induce NE reuptake inhibition (Debonnel et al. 2007). These data demonstrate that depending on the dose, venlafaxine can specifically target serotonergic neurotransmission alone or both serotonergic and noradrenergic tones without acting on off-targets.
In vivo preclinical studies of venlafaxine properties
Electrophysiological properties of venlafaxine
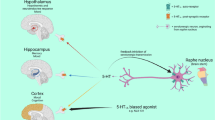
The electrophysiological properties of venlafaxine on monoaminergic neurons have been extensively investigated. Due to its ability to block the SERT and the NET, this compound produces a strong impact on both 5-HT and NE systems, associated with different acute and chronic effects. To understand the in vivo electrophysiological properties of venlafaxine, it is essential to bear in mind that monoamine transporters are expressed in presynaptic cell bodies regions (either on collaterals or directly on soma) and nerve terminals. Importantly, the acute inhibition of monoamines transporters reduces neuronal firing rate due to the accumulation of endogenous monoamines around presynaptic inhibitory somatodendritic autoreceptors (Fig. 1, Table 1).

Mechanism of action of Venlafaxine.5-HT and NE are stored into presynaptic vesicles. Incoming action potentials trigger their release into the synaptic cleft after melding with the cell membrane. 5-HT/NE can then bind to postsynaptic serotonergic and adrenergic G-coupled receptors. In parallel of the activation of these postsynaptic elements, 5-HT/NE can return into the presynaptic neurons through serotonin/norepinephrine transporters (SERT/NET) through a high affinity reuptake process. 5-HT/NE can also be removed from the synaptic cleft by neighboring glial cells such as astrocytes (not shown here). Into the presynaptic neuron, 5-HT and NE can penetrate into exocytosis vesicles through a vesicular monoamine transporter or degraded by the monoamine oxidase (MAO). Serotonergic and noradrenergic neurons have multiple ways to up- and downregulate monoamines response thereby maintaining a normal excitatory/inhibitory balance and protecting the brain from a sub-or over-stimulation. Receptors are not only found on the postsynaptic neuronal membrane, but also presynaptically. In particular, autoreceptors are found on axon terminals (i.e. 5HT1B and A2R) or on the soma (i.e. 5HT1A and A2R). If too many neurotransmitters accumulate in the synaptic cleft, these Gi-coupled autoreceptors are activated to mediate an inhibitory signal on the firing or release activities of the presynaptic serotonergic and adrenergic neurons. By inhibiting the SERT and NET transporters, venlafaxine can simultaneously increase extracellular 5-HT/NE levels in various brain regions involved and therefore to activate postsynaptic receptors involved in the regulation of emotional states. However, this action is limited by the recruitment of the autoreceptors after an acute administration of venlafaxine. While the treatment is prolonged, such inhibitory feedbacks desensitize thereby producing a higher rate of monoamines in the synaptic cleft necessary to promote beneficial behavioral effects.
Acute in vivo studies on monoaminergic neurons
Initial electrophysiological studies demonstrate that the acute administration of venlafaxine dose-dependently inhibits the spontaneous firing rate of dorsal raphe nucleus (DRN) 5-HT neurons with an ED50 value of 233 μg/kg i.v. in anesthetized rats (Béïque et al. 1999). Evidence shows that the 5-HT1A receptor antagonist WAY 100635 abolishes this inhibitory response (Millan et al. 2001; Artaiz et al. 2005; Béïque et al. 2000a; Gartside et al. 1997). This demonstrates that venlafaxine leads to an accumulation of 5-HT around 5-HT cell bodies, which in turn, stimulates inhibitory somatodendritic 5-HT1A autoreceptors. Regarding noradrenergic neurons, studies show that they are also dose-dependently inhibited by venlafaxine, but to a lesser degree. Indeed, the spontaneous firing of locus coeruleus (LC) NE neurons is inhibited by venlafaxine with an ED50 of 727 μg/kg i.v (Béïque et al. 1999). This response is abolished in the presence of alpha2-adrenergic receptor antagonist, idazoxan (Berrocoso and Mico 2007), raising the possibility that the over-activation of inhibitory somatodendritic alpha2-adrenergic receptors mediates such response.
Sub-chronic and chronic in vivo studies on monoaminergic neurons
Single-unit recordings in the rat DRN show that two days of subcutaneous administration of venlafaxine (10 mg/kg) decreases the activity of 5-HT neurons and to a lesser extent, NE neurons (Béïque et al. 2000b), as observed after acute administration (Béïque et al. 1999). In marked contrast, this effect is no longer observed after 21 days of treatment, suggesting a progressive attenuation of the inhibitory feedback exerted by somatodendritic 5-HT1A autoreceptors. This is evidenced by the fact that venlafaxine desensitizes these autoreceptors after 21 days of treatment at doses of 10 to 40 mg/kg (Béïque et al. 2000b). In contrast, single-unit recordings in the LC show that the inhibitory action of venlafaxine persists after 14 and 21 days of treatment (10–40 mg/kg). Although the reasons for such differences between the 5-HT and NE systems remain debatable, it was proposed that the somatodendrtic alpha-2-adrenergic autoreceptors do not have the peculiarity to desensitize (Berrocoso and Mico 2007; Béïque et al. 2000c).
The latter consideration is important since the time required for serotonergic antidepressants to desensitize somatodendritic 5-HT1A autoreceptors and thereby to significantly increase 5-HT neuronal firing, coincides with their onset of therapeutic activity. Consequently, given that venlafaxine displays the same electrophysiological properties than SSRIs in term of firing recovery, it is not surprising that the delay in venlafaxine response is similar to that of SSRIs. The lack of alpha2 adrenoceptor desensitization may be counterproductive for the beneficial effect of venlafaxine in term of efficacy or delay of action. In agreement with this hypothesis, electrophysiological studies showed that the pharmacological inactivation of alpah2 adrenoceptor with mirtazapine produces a rapid increase in noradrenergic but also serotonergic neurotransmission (Haddjeri et al. 1995). The latter results led to the hypothesis that the combination of venlafaxine with mirtazapine could produce more rapid improvement of depressive symptoms compared to SSRI alone. The interest for such combination derives from studies of mirtazapine augmentation of a variety of antidepressant agents (principally SSRIs) rather than specifically of venlafaxine (Carpenter et al. 1999, 2002).
In vivo studies on postsynaptic monoaminergic neurons
At the nerve terminals and particularly in the hippocampus, postsynaptic pyramidal CA3 neurons are sensitive to 5-HT and NE. These post-synaptic neurons express 5-HT1A and alpha-2 heteroreceptors which mediate inhibitory responses. Accordingly, acute intravenous administration of venlafaxine (5 mg/kg) in rats results in a massive (73%) suppression of CA3 pyramidal neurons firing, similar to the effects of the SSRI paroxetine (Béïque et al. 2000b). Interestingly, this effect is reversed by the 5-HT1A antagonist WAY100635 (Béïque et al. 2000b). Hence, any increase in extracellular 5-HT levels, but also of NE, results in the progressive attenuation of postsynaptic neuronal activity. This property has been successfully used to assess the in vivo potency of pharmacological agents known to inhibit monoaminergic transporters. Indeed, the time required for the firing activity to recover 50% of the initial firing rate after application of 5-HT or NE (recovery time: RT50 value) is an indication of reuptake inhibitors' ability to maintain 5-HT and/or NE in the synaptic cleft. The higher the RT50 value, the higher the ability to block the transporters. Of note, the subcutaneous administration of venlafaxine at 16 mg/kg significantly increased the RT50 after the microiontophoretic application of 5-HT or NE (Bacq et al. 2012). This highlights the ability of venlafaxine to prolong inhibitory actions of 5-HT and NE onto CA3 pyramidal cells by blocking their respective transporters. Interestingly, the genetic depletion of the organic cation transporter 2 (OCT2), a transporter exhibiting low affinity for monoamines, potentiates venlafaxine-induced increase in RT50 (Bacq et al. 2012). This suggests that residual 5-HT and NE could be removed from the synaptic cleft by OCT2.
Chronic administration of venlafaxine (21 days by osmotic minipumps at either 10 or 40 mg/kg/day) increases RT50 for 5-HT (Béïque et al. 2000b). The authors show an enhancement of serotonergic neurotransmission after 21-days of treatment with venlafaxine, and this effect is only achieved under conditions where the desensitization of the terminal 5-HT1B autoreceptors is appended to the somatodendritic 5-HT1A receptors (Béïque et al. 2000b). Remarkably, RT50 for NE is increased only at the dose of 40 mg/kg/day, corroborating a preferential effect on SERT inhibition over NET inhibition (Béïque et al. 2000b). Taken together, these data confirm the above-mentioned in vitro data that venlafaxine blocks the reuptake of 5-HT and to a lesser degree NE.
Taken together, these data suggest that venlafaxine blocks 5-HT and NE reuptake processes but its potency to target the 5-HT system is greater compared to the NE system. At low doses, venlafaxine act as an SSRI whereas higher doses are required to impact both systems. Chronic administration of venlafaxine leads to an overall increased 5-HT neurotransmission in response to a progressive desensitization of somatodendritic 5-HT1A autoreceptors as classical antidepressant drugs do (Quentin et al. 2018). Interestingly, although a 21-day treatment of venlafaxine desensitizes somatodendritic and terminal 5-HT1A/1B autoreceptors, it fails to do so on alpha2-adrenergic autoreceptors. This agrees with previous studies showing that different classes of antidepressants such as the NRI reboxetine but also the IMAO and the TCA do not modify the functional sensitivity of alpha2-adrenoceptors even after sustained administration (Blier and de Montigny 1985; Lacroix et al. 1991; Szabo and Blier 2001).
Neurochemical properties of venlafaxine
The neurochemical effects of venlafaxine on extracellular monoamines have been assessed in different brain structures using in vivo microdialysis in rodents.
In the rat frontal cortex, Millan and collaborators examined the acute effects of increasing doses of subcutaneous venlafaxine on the extracellular concentrations of the three monoamines ([5-HT]ext, [NE]ext, and [DA]ext). In agreement with venlafaxine ability to block the SERT and the NET, the authors observed a significant and dose-dependent increase of [5HT]ext starting from 0.63 mg/kg up to 40 mg/kg of venlafaxine with a maximal increase of 400% from baseline. Likewise, venlafaxine increases [NE]ext approximately by 500% which remains elevated for up to 3 hours after subcutaneous administration of the antidepressant at the dose of 40 mg/kg. Although venlafaxine has no affinity for the DAT, increases of cortical [DA]ext are also detected in response to venlafaxine from 2.5 mg/kg up to 40 mg/kg (Millan et al. 2001). It was proposed that the pharmacological inactivation of the NET might prevent DA reuptake by a heterologous reuptake process (Guiard et al. 2008; Morón et al. 2002). Regarding NE, a significant increase of [NE]ext was detected in the rat brain following administration of 3 to 30 mg/kg of venlafaxine (Koch et al. 2003; Beyer et al. 2002). Similar results were obtained from mice in which intraperitoneal administration of venlafaxine at the dose of 30 mg/kg also increased the cortical extracellular concentrations of the three monoamines (Higashino et al. 2014). In mice, the effects of venlafaxine on [5HT]ext (but not on [NE]ext) were even observed at a lower dose (i.e. 8 mg/kg) (David et al. 2003). It should be noted that all these data originate from non-depressed mice. In the rat hippocampus, another brain region critically involved in MDD, the acute subcutaneous administration of venlafaxine (5, 10, and 20 mg/kg) fosters [5HT]ext and [NE]ext (Millan et al. 2001; Sánchez et al. 2007). In the rat striatum and nucleus accumbens, venlafaxine (10 mg/kg, s.c) induces an increase in [5HT]ext whereas [DA]ext remains unchanged (Millan et al. 2001).
Only a few studies assessed the effect of chronic venlafaxine on monoaminergic neurotransmission. Surprisingly, a 14 or 21 day-period of subcutaneous or intraperitoneal administration of venlafaxine at the doses of 10 and 30 mg/kg does not affect cortical [5HT]ext, [NE]ext, and [DA]ext basal levels (Millan et al. 2001; Higashino et al. 2014). Gur and colleagues also evaluated the effect of the intraperitoneal administration of venlafaxine at the dose of 5 mg/kg for 28 days and again, they did not observe significant effects on [5HT]ext (Gur et al. 1999). However, another study reports that the chronic subcutaneous administration of venlafaxine for 14 days at the dose of 10 mg/kg increases cortical [5HT]ext and [NE]ext and that such effects could be potentiated by an OCT2 pharmacological blocker (Rahman et al. 2008). This finding further confirms that OCT2 contributes to the clearance of the remaining 5-HT and NE in the synaptic cleft.
To conclude, it appears that acute venlafaxine has a positive impact on monoaminergic systems due to its ability to block the SERT and the NET. These transporters are present in brain regions involved in the regulation of mood and their pharmacological blockade enhances extracellular 5HT and NE levels. Evidence also suggests that venlafaxine may act on dopaminergic transmission through the inactivation of a heterologous reuptake process involving the NET, notably in the frontal cortex. An increase in monoamine levels is also observed in the cell body areas (i.e., in the DRN and LC) and this favors the importunate binding and activation of inhibitory 5HT1A/alpha2 autoreceptors. Surprisingly and contrary to what is observed in electrophysiological studies, microdialysis data do not show a preferential effect of venlafaxine on [5HT]ext compared to [NE]ext (even at low doses) excepted in one study (David et al. 2003). In these microdialysis experiments, literature reports a great variability from one study to another that could depend on different parameters. Although neurochemical data come from rat studies, we noticed differences between species in response to venlafaxine treatment. One might ask whether the levels of expression of the SERT and/or NET is different in rats and mice for example. Doses and routes of administration could be other factors determining the impact venlafaxine on the 5-HT and NE systems. Finally, the precise site of implantation of the microdialysis probes could also be a critical point because brain structures display different monoaminergic inputs and different levels of monoaminergic transporters.
Regarding chronic exposure, electrophysiological studies report a functional desensitization of 5-HT1A autoreceptors after chronic venlafaxine treatment. However, this effect is not accompanied by an enhancement of serotonergic neurotransmission. A large number of studies have failed to show increases in basal 5-HT level in terminal areas after chronic administration of venlafaxine but also with other 5-HT reuptake inhibitors, unless the sample collections were performed less than 24 h after the last drug administration (Bel and Artigas 1993; Kihara and Ikeda 1995; Bosker et al. 1995; Invernizzi et al. 1995, 1996; Hjorth and Auerbach 1999; Jaskiw et al. 2006). This lack of effect could reflect adaptive changes. Indeed, according to the homeostasis rules, it is possible the release process is attenuated. 5-HT synthesis and degradation can also be modified which could explain this lack of significant action of venlafaxine on 5-HT tone. Finally, it is important to note that there are no studies assessing the chronic effects of venlafaxine on animal models of depression. One would expect that chronic venlafaxine could exert its beneficial effects specifically in pathological conditions characterized by low extracellular 5-HT levels. Another explanation can be a technical issue with intracerebral microdialysis. It is well known that the gold standard technique to probe monoaminergic tone after chronic exposure is the “no net flux” (Guiard et al. 2005) but this approach was not applied in the currently available publications.
Behavioral effects of venlafaxine
Preclinical studies have explored its antidepressant properties in naïve non-stressed animals and in various animal models of depression. These include animals submitted to unpredictable chronic mild stress (UCMS), chronic social defeat (CSD), maternal separation (MS), olfactory bulbectomy (OB), ovariectomy (OVX), or prolonged administration of corticosterone (CORT). This part of the review focuses on the behavioral effects of venlafaxine on mice.
In naïve mice
The efficacy of venlafaxine on mice depressive-like behavior was mainly evaluated using the FST and the TST tests and a wide range of doses was screened (from 1 up to 80 mg/kg). Michel Bourin’s group was the first to evaluate the behavioral effects of venlafaxine in mice and reported that its acute venlafaxine (8 to 64 mg/kg) elicits antidepressant-like effects in the FST and TST as evidenced by a reduction of animal’s immobility time. This study also showed that 8 mg/kg of venlafaxine blocks 5-HT reuptake inhibition, while doses above 16 mg/kg seem to inhibit the SERT and the NET (Redrobe et al. 1998). Such beneficial effects are unlikely to result from a psychostimulant effect since high doses of venlafaxine do not influence locomotor activity (Berrocoso and Mico 2007). Of note, most of the subsequent studies did not demonstrate the antidepressant-like effects of venlafaxine when doses below 8 mg/kg were used (Bortolatto et al. 2010; Castagné et al. 2009; Bourin et al. 2009; David et al. 2001) (Table 2). However, some studies report beneficial effects of venlafaxine intraperitoneal administration at doses of 4 (Kulkarni and Dhir 2007) (Table 2). To explain these mixed results, the importance of mice strain and gender was explored (variables not shown in Table 2). It appears that most studies used males and that C57BL6 and albino (CD1/SWISS/LACA) mice were more prone to respond to low doses (<8 mg/kg) of venlafaxine. The mode of administration could be another important parameter of venlafaxine efficacy since in acute experiments performed in mice, its antidepressant-like effects appear at 80 mg/kg but not at 20 mg/kg when oral administration was performed (Yamada et al. 2013).
Collectively, these data suggest that in non-pathological conditions, the acute administration of venlafaxine produces antidepressant-like effects at doses ranging from 8 to 80 mg/kg and that the expression of its beneficial effects does not necessarily require the noradrenergic system.
With respect to sub-chronic (< 7 days) administration, two studies tested whether prolonged treatment with venlafaxine could produce more robust effects than those observed after acute administration (Ren et al. 2018; Ide et al. 2010). It is noteworthy that the antidepressant-like effects of venlafaxine (10 mg/kg; i.p.) were observed in both FST and TST, following daily drug administration for three consecutive days (Ren et al. 2018). In another work, venlafaxine was administered for 5 days at either 10 or 30 mg/kg, and despair was evaluated each day in the FST (Ide et al. 2010). Remarkably, at 10 mg/kg, venlafaxine produced antidepressant-like effects observable from the first day of treatment, and this response persisted to the fourth day. In contrast, at 30 mg/kg, venlafaxine triggered antidepressant-like effects only after 3 and 4 days of treatment. Considering these data, one would anticipate that chronic administration of venlafaxine also promotes positive effects in the FST and TST. Indeed, such beneficial effects were observed from the seventh day of treatment in the TST and after 2 weeks of exposure in the FST and TST (Liu et al. 2012). It is plausible that such delay coincides with the time required for the cellular and structural adaptations that underlie the therapeutic effect of antidepressants and contribute to the adaptive plasticity induced in the brain by these drugs. In another study, 2 weeks of venlafaxine at the dose of 16 mg/kg had no antidepressant-like effect (Liang et al. 2016). However, after 3 weeks of treatment, venlafaxine 4 (Thomas et al. 2016a) and 5 (Abdel-Wahab and Salama 2011) mg/kg elicited antidepressant-like effects in the FST and TST. Higher doses were also tested (10–20 mg/kg), revealing a dose-response relationship (Abdel-Wahab and Salama 2011). After 4 weeks of treatment with venlafaxine (10 mg/kg, p.o.) antidepressant-like effects were also observed in the TST (Carlini et al. 2012). Although interesting, these data focused on despair and further experiments are needed to determine whether venlafaxine also acts on other symptoms of depression.
The acute anxiolytic effect of venlafaxine was assessed using a large spectrum of concentrations (i.e. 10 – 60 mg/kg) and found from 30mg/kg (Li et al. 2006). Despite the beneficial effect on anxiety of a single administration of venlafaxine, several studies failed to demonstrate anxiolytic-like effects of this drug when administered chronically. Indeed, 2 or 4 weeks of venlafaxine at the dose of 10 or 16 mg/kg had no effect on the time spent in the open arms or in the center of the arena in the EPM, OF, or EZM test whereas some studies reported beneficial effects after the administration of the SSRIs fluoxetine and paroxetine or the SNRI duloxetine (Carlini et al. 2012; Hu et al. 2018; Mirza et al. 2007).
Altogether, studies using acute, sub-chronic, and chronic administration of venlafaxine in naive mice suggest that over 8 mg/kg, venlafaxine elicits antidepressant-like effects on despair (Table 2). Furthermore, additional anxiolytic effects of venlafaxine are observed after acute administration when administered at high dose, whereas prolonged treatment does not. Moreover, when many doses were evaluated, several studies observed a dose-response relationship with greater effects in response to high doses and this could be correlated with electrophysiological or neurochemical measures. This dual effect of venlafaxine on serotonergic (low/high doses) and noradrenergic (only high dose) can explain such a dose-response relationship.
In mouse models of depression
A review of the literature reveals a dozen of publications evaluating the effects of venlafaxine in mouse models of depression. Among them, 3 used the chronic forced swimming test (cFST) model which involves subjecting mice to repeated swimming stress. In one study, mice are submitted to this protocol for 21 consecutive days while receiving low doses of venlafaxine (from 2 to 16 mg/kg; i.p.)(Thomas et al. 2016a). Antidepressant-like effects in the FST and tail suspension test (TST) were observed only after 3 weeks of treatment at the dose of 4 mg/kg. At higher doses (8 and 16 mg/kg), the antidepressant-like effects occur after 2 weeks of treatment (Thomas et al. 2016a, 2017). In another study, the cFST is applied for one week while mice receive venlafaxine at doses of 5 and 10 mg/kg. An immediate beneficial effect persisting for seven days was revealed (Kumar et al. 2010). At the end of this experimental procedure, anxiolytic effects were also found in the elevated plus maze (EPM) and the mirror chamber test (approach-avoidance test when animals are confronted by a mirror), whatever the dose administered (5 and 10 mg/kg; i.p.) (Kumar et al. 2010).
The behavioral effects of venlafaxine were also tested in the chronic social defeat (CSD) model of depression which consists in repeatedly exposing naïve mice to aggressor mice. Venzala and colleagues treated mice with venlafaxine for 4 weeks at a dose of 20 mg/kg (i.p.) before submitting them to a battery of behavioral tests. They observed antidepressant-like effects in the FST and sucrose preference test (SPT), anxiolytic responses in the EPM and novelty-suppressed feeding (NSF). However, venlafaxine did not abolish the deficit in the social interaction test (Venzala et al. 2012). In a different study, mice were treated with venlafaxine for six weeks at the dose of 16 mg/kg (i.p.) before evaluating their behavior (Bai et al. 2017). While no antidepressant nor anxiolytic effects were seen in the FST and TST or EPM, an improved capacity for social interaction was observed in these animals (Bai et al. 2017). Moreover, in postoperative cognitive dysfunction (POCD), venlafaxine (16 mg/kg; i.p.) was shown to increase working memory in the Y-maze (Li and Zhang 2021). Furthermore, mice submitted to mechanical and cold hyperalgesia induced by repeated injection of oxaliplatin, developed traits characteristic of an anxiety-depressive phenotype, whereas the acute venlafaxine treatment (16 mg/kg, s.c) alleviated neuropathic pain and promoted antidepressant-like effects in these animals (Hache et al. 2015). In summary, the 16 mg/kg i.p. chronically administered is the optimal dose to treat depression and anxiety but also to improve cognitive performances in several animal models of depression.
In two other studies, mice submitted to the Unpredictable chronic mild stress (UCMS) were used to address venlafaxine’s antidepressant properties. After 7 days of venlafaxine (10 mg/kg, i.p.) treatment, antidepressant-like effects were observed in the FST and TST, while only 3 days of treatment were not sufficient to elicit such effects (Ren et al. 2018). As expected, in UCMS mice, prolonged venlafaxine treatment (i.e., 2 weeks, i.p.) using the same dose also produced antidepressant-like effects, notably by improving social interaction (Wang et al. 2020).
Chronic administration of corticosterone (CORT) in the drinking water of mice is a widely used model of depression, which allows screening the effects of antidepressant drugs. For instance, an 8-week period of CORT exposure induces depressive-like behaviors which are reversed by monoaminergic antidepressant drugs (David et al. 2009). Indeed, 3 weeks (16mg/kg) chronic administration of venlafaxine exerted antidepressant- and anxiolytic-like effects in the splash test (ST) and the zero maze respectively (Bacq et al. 2012). Interestingly, studies showed that ovariectomy (OVX)-induced depressive-like symptoms in females are reversed by long-term intragastric administration of venlafaxine. In a first study, depressive-like behaviors occurred 8 weeks only after ovariectomy, and venlafaxine (70 mg/kg, intragastric) therapeutic action in the TST is observed as early as after 1 and 2 weeks of treatment in OVX mice compared to vehicle mice while depressive-like behavior occurred only 8 weeks after ovariectomy (Ye et al. 2016). More recently, these authors reported that intragastric administration of venlafaxine (9.75 mg/kg) in OVX females had no effect in the EPM (Xu et al. 2019). Lastly, Poretti et al. (2016) used bulbectomy-induced depressive-like behavior in mice. In this model, the oral administration of venlafaxine (5 to 20 mg/kg) elicited antidepressant effects in the TST after 3weeks of chronic treatment at the highest dose (20 mg/kg) while antidepressant effects were observed after 4weeks of treatment at 10 to 20 mg/kg (Poretti et al. 2016). The lowest dose (5 mg/kg) did not induce a change in immobility time in this test.
The literature review indicates that venlafaxine displays antidepressant-like effects in various mouse models of depression, when administered acutely, sub-chronically as well as chronically (Table 3). The effective dose, i.e., the dose that promotes beneficial behavioral effects on anxiety, was found to start at 8 mg/kg, although a few studies have documented antidepressant-like effects at lower doses. This indicates that targeting the 5HT system is sufficient to elicit beneficial effects and there is no evidence showing that a high dose of venlafaxine promotes greater antidepressant-like responses. The possibility that low and high doses promote distinct effects warrant further investigations. Furthermore, the different routes of administration (i.p. vs. p.o.) elicit similar behavioral effects in the mouse models of depression. Finally, treatment duration is another parameter that should be considered. Most studies show that venlafaxine becomes effective after a minimum of 3 weeks of treatment, as is the case with conventional antidepressants (i.e., SSRI).
Effects of venlafaxine on sleep
Links between sleep, depression, and antidepressant treatments are strong. About three-quarters of depressed patients have insomnia symptoms, and hypersomnia is present in about 40% of young depressed adults. As well as the subjective experience of sleep symptoms, there are well-documented changes in sleep architecture in depression (Nutt et al. 2008). Evidence shows that sleep architecture is modified by an increase in rapid eye movement (REM) sleep propensity in the early night resulting in an increased REM sleep quantity and sleep fragmentation. This leads to poor sleep quality (Grønli et al. 2004). Interestingly, venlafaxine is able to modify sleep architecture. For instance, in naïve rats, it was shown that venlafaxine produces a dose-related suppression of REM sleep as TCA or SSRIs do (Wichniak et al. 2017). Venlafaxine also increases wake time (Salin-Pascual and Moro-Lopez 1997). Although these effects were found in non-depressed rodents, the reduction of total sleep duration and sleep efficiency (hypersomnia) observed in the mouse CORT model of depression (Le Dantec et al. 2014) strongly suggests that venlafaxine could have a beneficial action on this parameter, notably due to its neurochemical properties. Indeed, at the neurochemical levels, it is well known that the serotonergic and noradrenergic systems are involved in the regulation of sleep and wakefulness, their activity being at maximum during the awake state and minimum during sleep (Adrien 2002). Because the production of REM sleep depends on the decrease of serotonergic and noradrenergic tones in brain stem structures, it is possible that the ability of this antidepressant to favor inhibition of REM relies on its ability to increase monoaminergic tone. Evidence demonstrated that both SSRIs and SNRIs have the same effects on the suppression of REM (Wichniak et al. 2017) suggesting that the elevation of 5-HT neurotransmission is sufficient to impact this parameter and therefore to counterbalance sleep disturbances observed in depression. Whether or not the effects of venlafaxine on sleep differ according to the dose and its ability to increase 5-HT and/or NE extracellular levels has yet to be solved.
Mechanism of action venlafaxine: emphasis on brain plasticity
Impact of venlafaxine on adult hippocampal neurogenesis
Ex vivo studies evaluated the influence of animal models of depression on Brain-Derived Neurotrophic Factor (BDNF). They showed that corticosterone, mimicking an increased HPA reactivity and LPS induce a significant reduction of BDNF gene and protein expression in the frontal cortex and hippocampus (Lin et al. 2019; Huang et al. 2011). Going one step further, several studies evaluated the effects of venlafaxine on BDNF expression and on hippocampal adult neurogenesis. After 7 days of administration, venlafaxine increases BDNF expression in the hippocampus (Larsen et al. 2008) while fluoxetine fails to do so. In another study, 7 days of venlafaxine (10 mg/kg, p.o.) fails to modify hippocampal or cortical BDNF protein level (Cooke et al. 2009). Nevertheless, the authors reported enhanced cortical BDNF protein levels in response to the chronic administration of venlafaxine (21 days, 10 mg/kg, p.o.), while citalopram had no effect. Cortical and hippocampal BDNF levels were also increased after a 5-week period of venlafaxine treatment in rat (20 mg/kg, p.o) (Czubak et al. 2009). Interestingly, a 28-day period of venlafaxine treatment (10 mg/kg, p.o.) followed by 28 days of chronic restrained stress (CRS) without treatment in rat, enhanced BDNF expression (Lapmanee et al. 2017). In the same model of depression, systemic administrations of venlafaxine (5 mg/kg) for 14 and 21 days rescued hippocampal BDNF expression (Xu et al. 2004). Following UCMS, a marked increase of BDNF was observed in the rat hippocampus after 28 of venlafaxine(Wang et al. 2020; Huang et al. 2014).
Regarding the influence of venlafaxine on hippocampal neurogenesis, it was shown that the administration of a high (but not low) dose of venlafaxine (40 mg/kg) for 14 days increased cell proliferation in the dentate gyrus of rats (Mostany et al. 2008). In the Chronic Restraint Stress (CRS) rat model, a 21-day treatment with venlafaxine (5 mg/kg, i.p.) rescues the impairment of hippocampal progenitors’ proliferation (Xu et al. 2004). Accordingly, Zhang and colleagues showed that the deleterious effects of UCMS on hippocampal cell proliferation were abolished by 21 days of venlafaxine (15 mg/kg, i.p.) (Zhang et al. 2015). Similar effects were found in the MS and UCMS models of depression after 14 days of venlafaxine treatment (respectively 20 & 5 mg.kg; p.o.) (Belovicova et al. 2017; Martisova et al. 2015).
Another important step of adult neurogenesis is the survival of adult-born cells. While the expression of pro-apoptotic or anti-apoptotic molecules such as Bax or Bcl-xl are respectively up- and downregulated in the hippocampus of UCMS submitted animals, a 21-day treatment with venlafaxine (15 mg/kg, p.o.) counteracts these effects (Wang et al. 2011). Two studies confirm the anti-apoptotic role of venlafaxine. Indeed, in the UCMS, 28 days of venlafaxine administration (10 mg/kg, i.p.) prevents the increase of apoptotic neurons in the hippocampus (Huang et al. 2014). Moreover, Saad et al. (2019) report that after 28-day of venlafaxine treatment, ovariectomized rats exhibit a marked reduction of hippocampal Bax/Bcl2 ratio, caspase-3 activity, and tumor necrosis factor alpha levels (Saad et al. 2019).
The ability of venlafaxine to influence cell fate choice during adult neurogenesis process was also studied. For this, primary astrocyte cultures were treated with different antidepressants including venlafaxine, imipramine, and fluoxetine (Cabras et al. 2010). In the presence of each of these drugs, cultured astrocytes rapidly acquired a neuronal morphology and expressed neuronal markers. This reveals a process that might contribute to the antidepressant-like effect of venlafaxine. Several studies have shown that the response of chronic antidepressants is mediated by the stimulation of adult hippocampal neurogenesis (Hanson et al. 2011) whereas the ablation of this process attenuates the antidepressant-like effect of some antidepressants as described with SSRI and TCA (Santarelli et al. 2003; David et al. 2009). However, this proof of concept is still lacking using venlafaxine.
Impact of venlafaxine on fast-spiking interneurons extracellular matrix remodeling
Emerging evidence suggests that MDD impacts the remodeling of extracellular matrix in various brain regions. The extracellular matrix is the major constituent of perineuronal nets (PNNs) enwrapping parvalbumin (PV) expressing cells (Sorg et al. 2016), a subpopulation of GABAergic interneurons. Although the role of PNNs is not fully understood, it has been proposed that they contribute to reinforce the activation of these GABAergic interneurons, through a better sensitivity to glutamatergic inputs (Tewari et al. 2018; Frischknecht et al. 2009). As PV interneurons contact excitatory pyramidal cells in the hippocampus, PNNs significantly influence the excitatory/inhibitory (E/I) balance. Most notably, they alter this balance (Yizhar et al. 2011), an endpoint relevant to stress-induced depressive-like state (Albrecht et al. 2016; Wang et al. 2019). In support of this assumption, an increase of PNNs has been reported in several animal models of depression such as the CSD or CORT models (Riga et al. 2017).
On the contrary, the digestion of PNNs with chondroitinase (ChABC) reduces the excitability of GABAergic interneurons (Tewari et al. 2018; Favuzzi et al. 2017) thereby increasing hippocampal activity. Interestingly, a chronic treatment with venlafaxine has been shown to reverse CORT-induced increase in PNNs as the result of an upregulation of metalloproteases (MMPs) such as MMP-9 (Bijata et al. 2017; Alaiyed et al. 2019; Alaiyed et al. 2020). MMPs are enzymes that cleave PNNs and subsequently reduce the activation of inhibitory PV interneurons (Bozzelli et al. 2020). As depicted in Fig. 2, a new hypothesis in the field of neuropsychopharmacology posits that venlafaxine would attenuate the PNNs which in turn would restore a normal E/I balance in CORT-exposed mice (Fig. 2). Interestingly, elevated MMP-9 levels were found in autopsy-derived prefrontal cortex samples of MDD patients treated with antidepressants compared to controls (Alaiyed et al. 2020). The involvement of MMP-9 in the regulation of emotional state is further supported by pharmacological or genetic studies showing that the inactivation of MMP-9 increases basal anxiety (Ringland et al. 2021) but also despair and sociability in stressed animals (Vafadari et al. 2019). Moreover, single nucleotide polymorphisms of MMP-9 have been unveiled in human and they have been associated with MDD (Bobińska et al. 2016a; Bobińska et al. 2016b) but also to treatment response (Rybakowski et al. 2011).

Impact of venlafaxine on the excitatory/inhibitory (E/I) balance in the hippocampus. Stress increases perineuronal nets (PNNs) deposition around parvalbumin positive (PV+) GABAergic interneurons in the hippocampus due to the inactivation of metalloproteases (MMP) such as MMP9. This reinforces the excitatory synapses upon PV+ GABAergic interneurons thereby leading to their hyperactivity. Such a process would be associated to an increased release of GABA in the hippocampus. As PV+ GABAergic interneurons contact excitatory pyramidal cells, their hyperactivity induces a local inhibition of pyramidal cells. Overall altered E/I balance is observed in animal models of depression (left panel). Venlafaxine, through the inhibition of serotonin (5-HT) and norepinephrine (NE) reuptake, promotes the accumulation of these monoamines in the synaptic cleft. Evidence demonstrates that both 5-HT and NE favor the activity of MMP9, which in turn, leads to PNNs degradation. In that condition, the activation of PV+ GABAergic interneurons is attenuated. A decreased release of GABA is then expected causing the disinhibition of pyramidal glutamatergic neurons. As a functional consequence, the E/I balance is rescued (right panel), a process necessary for antidepressant response.
Efficacy of venlafaxine in the treatment of depressed patients
This is, now, well-established in clinics that venlafaxine is an efficient antidepressant treatment. Compared with placebo, a recent meta-analysis highlighted an odds ratio (OR) of 1.78 [95% confidence intervals (95% CI)1.61–1.96] (Cipriani et al. 2018). In the early 1990s, a number of studies compared venlafaxine with SSRIs. A first study showed a greatest improvement of depressive symptoms (Montgomery Asberg Rating Scale (MADRS) and Hamilton Depressive Rating Scale (HDRS)) in the venlafaxine group (200mg/day) versus fluoxetine (40mg/day) after 4 and 6 weeks treatment, in a population with melancholic features (Clerc et al. 1994). However, this first study focused only in 34 patients in each group. A second study compared venlafaxine 75mg (n=153) to fluoxetine 25mg (n=161). A clinical improvement was reported in the two groups, without difference between groups. There was a subgroup analysis focusing on 88 patients for which there was an increase of venlafaxine from 75mg to 150mg and so targeting both monoaminergic systems. Significant differences were described in favor of venlafaxine in the HDRS total from weeks 3 to 8 (Dierick et al. 1996). A first meta-analysis focusing on the efficacy of venlafaxine compared with SSRIs and other antidepressants highlighted that venlafaxine seemed more efficient than SSRIs. However, these results were less clear, focusing on other antidepressants such as TCAs. Moreover, this meta-analysis showed that venlafaxine carried an advantage of about 1.2 HDRS points compared with all other antidepressants (total size effect: 0.14) and even a little more, when focusing specifically on SSRIs (size effect: 0.17). This decrease seemed too small to be clinically pertinent. In addition, the NNT (number needed to treat) was of 19 (95%CI 11–63) for response and of 14 (95%CI 9–29) for remission (Smith et al. 2002). Another meta-analysis, focusing on 17 randomized controlled studies, showed a trend towards superiority of venlafaxine over SSRIs in remission rates (risk ratio [RR]= 1.07, 95%CI=0.99 to 1.15), and a slight superiority in response rates (RR=1.06, 95%CI=1.01 to 1.12) over SSRIs. However, the NNT were 34 and 27 for remission and response, respectively. There was also a modest advantage for venlafaxine in change scores (using MADRS or HDRS) (effect size=−0.09, 95% CI=−0.16 to −0.02, p=0.013) (Weinmann et al. 2008). In addition, a meta-analysis of data from 39 published and unpublished clinical trials randomized 8659 patients (n = 4644 to venlafaxine; n = 4015 to an SSRI). This meta-analysis found that venlafaxine had a higher response rate than SSRIs (RR=1.084 95% CI=1.019–1.101) (Papakostas et al. 2007). Another meta-analysis of 34 randomized, double-blind studies focusing only on remission rates, included 8744 patients (4191 patients treated with venlafaxine and 3621 treated with SSRIs). It showed that the difference in remission rates was 5.9% in favor of venlafaxine over SSRIs, as a class (95% CI= 0.038–0.081: p= 0.001). The NNT was 17 (95% CI: 12–26). Focusing on specific SSRIs, the difference from fluoxetine was significant (6.6% [95% CI: 0.030–0.095]). The difference between paroxetine, sertraline, and citalopram was not significant (Nemeroff et al. 2008). More recently, two other meta-analyses have addressed the efficacy of venlafaxine versus SSRIs. One focused on published studies. De Silva et al. noted that venlafaxine was superior to SSRIs by focusing on remission [odds ratio (OR= 1.13, 95% CI = 1.0–1.28, p = 0.05)] and response (OR = 1.17, 95% CI = 1.03–1.34, p = 0.02). In addition, venlafaxine seemed to have a significantly better response rate than fluoxetine (OR = 1.28, 95% CI = 1.05–1.55, p = 0.01). There were no significant differences in response or remission between venlafaxine and other individual SSRIs (de Silva and Hanwella 2012). The second meta-analysis included unpublished data. It showed that response rates were significantly higher for venlafaxine than for SSRIs (OR = 1.20, 95% CI 1.07–1.35). But, the remission rate was not higher for venlafaxine than for SSRIs (OR = 1.12, 95% CI 0.98–1.28) (Schueler et al. 2011). Recently, Cipriani et al. showed, in a network meta-analysis focused on comparing the efficacy of 21 antidepressant drugs for the acute treatment of adults with MDD, that fluoxetine is less effective than venlafaxine (OR= 0.84, 95% CI = 0.73–0.97) (Cipriani et al. 2018). Interestingly, a very recent meta-analysis of venlafaxine and duloxetine showed that non-RCTs (random clinical trials) are generally better suited to describe a drug efficacy in clinical practice than RCTs. However, it appears that non-RCTs are associated with a smaller size-effect than RCT (Schneider et al. 2021). In conclusion, it seems that venlafaxine is more effective than SSRIs (mainly fluoxetine which is one of the most prescribed) in terms of response rate, despite a relatively weak size effect measured on depression scales. Interestingly, in treatment-resistant patients, it is possible to switch from SSRI to SNRI treatment like venlafaxine to observe a therapeutic response. In this way, a retrospective study showed that patients who remain severely depressed following SSRI treatment may gain benefit from a high dose of venlafaxine (i.e., 225 mg/daily) which likely improve both 5-HT and NE tone, rather than switching to another SSRI (Barak et al. 2011).
Finally, it is noteworthy that there is no evidence for the occurrence of serotonin syndrome with venlafaxine alone even in high doses. To the best of our knowledge, only one study reported that a 29-year-old depressed Taiwanese woman developed serotonin syndrome at the dose of 37.5 mg/d (Pan and Shen 2003). Several cases, however, were described when venlafaxine is combined with other pharmacological compounds including mirtazapine (Decoutere et al. 2012), tranylcypromine (Brubacher et al. 1996), fluoxetine (Bhatara et al. 1998), trazodone (McCue and Joseph 2001), or tramadol (Albiñana Pérez et al. 2012). Although the underlying mechanisms remain unknown, it is likely that the functional and reciprocal interactions between the monoaminergic systems or between 5-T/NE and the opioid system may precipitate such a syndrome.
Concluding remarks
In vitro binding studies report that venlafaxine displays a strong affinity for the human/rat SERT although a higher affinity is observed with an SSRI and a TCA (i.e., citalopram and clomipramine). Similarly, human NET/rat binding assays indicate that venlafaxine displays a good affinity for this target but to a lower extent than the NRI reboxetine (Millan et al. 2001). Compared with other SNRIs (duloxetine and levomilnacipram), venlafaxine has also less affinity for the SERT and NET transporters (Auclair et al. 2013; Béïque et al. 1998) indicating that it represents a reliable SERT and NET blocker but not the best. In vivo, electrophysiological approaches show that venlafaxine and the SSRI paroxetine share the same potency at inhibiting the SERT even though there is no direct comparison (Béïque et al. 1998). This property worth mentioning because paroxetine is recognized as the most potent SSRI currently available. However, this remarkable pharmacological property is limited to the 5HT system because studies demonstrate that several antidepressants are more potent than venlafaxine at blocking the NET. Indeed, venlafaxine is less potent at blocking the NET than reboxetine or desipramine (Auclair et al. 2013; Béïque et al. 1998). This in vivo electrophysiological profile can be compared with neurochemical studies using microdialysis. Comparing the minimal effective doses (MED) required to increase cortical extracellular 5-HT concentrations, Millan and collaborators showed that venlafaxine has a similar profile than citalopram but is four-fold lower than that of clomipramine (Millan et al. 2001), levomilnacipran, and duloxetine (Auclair et al. 2013). As regard cortical extracellular NE concentrations, similar MED are found between venlafaxine and other SNRIs levomilnacipran and duloxetine (Auclair et al. 2013) which is quite unexpected considering the in vitro binding studies and in vivo electrophysiological data. This likely emphasizes the fact that mechanisms occurring at the nerve terminals might change our predictions regarding the net effects of venlafaxine noradrenergic neurotransmissions and behavioral properties. Indeed, in terms of behavior, only few differences were detected between antidepressants when comparing venlafaxine with the SSRIs fluoxetine, sertraline, escitalopram, and fluvoxamine; the SNRI duloxetine; the TCAs desipramine/imipramine; or other atypical antidepressants such as bupropion. Nevertheless, behavioral studies showed that venlafaxine has the lowest MED of the above-mentioned antidepressants in the TST and/or FST (Castagné et al. 2009; Socała et al. 2012). Such a difference could negatively reverberate on the onset of action of venlafaxine. In support of this hypothesis, it was shown in an animal model of depression, that the antidepressant-like effects of venlafaxine (20 mg/kg) occurred as soon as 21 days of treatment whereas a 14-day period is required with fluoxetine (20 mg/kg) to promote beneficial behavioral effects (Poretti et al. 2016). On the contrary, regarding anxiolytic response, venlafaxine is less effective than the SSRI fluoxetine or citalopram and the SNRI duloxetine when assessed in marble burying test, nestlet shredding, or zero maze (Li et al. 2006; Mirza et al. 2007)
Collectively, it appears that venlafaxine is a potent SERT inhibitor and at higher doses, is also able to block NET as NRIs do. Behavioral studies did not yield strong evidence regarding the greater effects of venlafaxine compared to the other antidepressants. With respect to clinical practice, it appears that at higher doses, thus acting as a mixed SERT/NET inhibitor, venlafaxine shows superior effects than SSRIs on response rate, but these results have yet to be confirmed. However, these potential beneficial effects are accompanied by a weaker tolerance than SSRIs, especially during the first week of treatment, thus reducing compliance and possibly attenuating its response rate (Dierick et al. 1996; Weinmann et al. 2008).
Abbreviations
- 5-HT:
-
5-hydrotryptamine or serotonin
- 5-HT1A:
-
Serotonin 1A receptor
- 5-HT1B:
-
Serotonin 1B receptor
- [5-HT]ext :
-
Extracellular serotonin
- BDNF:
-
Brain-derived neurotrophic factor
- CA3:
-
Ammon’s horn 3
- cFST:
-
Chronic forced swim test
- CI:
-
Confidence interval
- CNS:
-
Central nervous system
- CORT:
-
Corticosterone
- CSD:
-
Chronic social defeat
- DA:
-
Dopamine
- DAT:
-
Dopamine transporter
- [DA]ext :
-
Extracellular dopamine
- DRN:
-
Dorsal raphe nucleus
- ED50:
-
Median effective dose
- EPM:
-
Elevated plus maze
- EZM:
-
Elevated zero maze
- FST:
-
Forced swim test
- HDRS:
-
Hamilton depressive rating scale
- HPA:
-
Hypothalamic–pituitary–adrenal axis
- LC:
-
Locus coeruleus
- LPS:
-
Lipopolysaccharide
- MADRS:
-
Montgomery-Asberg depression rating scale
- MD:
-
Mean difference
- MDD:
-
Major depressive disorder
- MED:
-
Minimal effective dose
- MMP:
-
Matrix metalloproteinase
- MS:
-
Maternal separation
- NE:
-
Norepinephrine or noradrenaline
- NET:
-
Norepinephrine transporter
- [NE]ext :
-
Extracellular norepinephrine
- NNT:
-
Number needed to treat
- NRI:
-
Norepinephrine reuptake inhibitor
- NSF:
-
Novelty-suppressed feeding
- OB:
-
Olfactory bulbectomy
- OCT2:
-
Organic cation transporter 2
- OF:
-
Open field
- OR:
-
Odds ratio
- OVX:
-
Ovariectomy
- PNN:
-
Perineuronal net
- POCD:
-
Post-operative cognitive dysfunction
- PV:
-
Parvalbumine
- RCT:
-
Randomized clinical trial
- RR:
-
Risk ratio
- RT50:
-
Recovery time 50
- SERT:
-
Serotonin transporter
- SMD:
-
Standard mean difference
- SNRI:
-
Serotonin-noradrenaline reuptake inhibitor
- SPT:
-
Sucrose preference test
- SSRI:
-
Selective serotonin reuptake inhibitor
- ST:
-
Splash test
- TCA:
-
Tricyclic antidepressant
- TST:
-
Tail suspension test
- UCMS:
-
Unpredictable chronic mild stress
- VTA:
-
Ventral tegmental area
- WHO:
-
World Health Organization
References
Abdel-Wahab BA, Salama RH (2011) Venlafaxine protects against stress-induced oxidative DNA damage in hippocampus during antidepressant testing in mice. Pharmacol Biochem Behav 100:59–65
Adrien J (2002) Neurobiological bases for the relation between sleep and depression. Sleep Med Rev 6:341–351
Alaiyed S, Bozzelli PL, Caccavano A, Wu JY, Conant K (2019) Venlafaxine stimulates PNN proteolysis and MMP-9-dependent enhancement of gamma power; relevance to antidepressant efficacy. J Neurochem 148:810–821
Alaiyed S, McCann M, Mahajan G, Rajkowska G, Stockmeier CA, Kellar KJ et al (2020) Venlafaxine stimulates an MMP-9-dependent increase in excitatory/inhibitory balance in a stress model of depression. J Neurosci 40:4418–4431
Albiñana Pérez MS, Cea Pereira L, Bilbao Salcedo J, Rodríguez PI (2012) Possible serotonin syndrome associated with administration of venlafaxine and tramadol. Farm Hosp 36:548
Albrecht A, Ivens S, Papageorgiou IE, Çalışkan G, Saiepour N, Brück W et al (2016) Shifts in excitatory/inhibitory balance by juvenile stress: a role for neuron-astrocyte interaction in the dentate gyrus. Glia. 64:911–922
Artaiz I, Zazpe A, Innerárity A, Del Olmo E, Díaz A, Ruiz-Ortega JA et al (2005) Preclinical pharmacology of F-98214-TA, a novel potent serotonin and norepinephrine uptake inhibitor with antidepressant and anxiolytic properties. Psychopharmacology 182:400–413
Auclair AL, Martel JC, Assié MB, Bardin L, Heusler P, Cussac D et al (2013) Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 70:338–347
Bacq A, Balasse L, Biala G, Guiard B, Gardier AM, Schinkel A et al (2012) Organic cation transporter 2 controls brain norepinephrine and serotonin clearance and antidepressant response. Mol Psychiatry 17:926–939
Bai S, Zhang X, Chen Z, Wang W, Hu Q, Liang Z et al (2017) Insight into the metabolic mechanism of Diterpene Ginkgolides on antidepressant effects for attenuating behavioural deficits compared with venlafaxine. Sci Rep 7:9591
Barak Y, Swartz M, Baruch Y (2011) Venlafaxine or a second SSRI: switching after treatment failure with an SSRI among depressed inpatients: a retrospective analysis. Prog Neuro-Psychopharmacol Biol Psychiatry 35:1744–1747
Béïque JC, de Montigny C, Blier P, Debonnel G (1998) Blockade of 5-hydroxytryptamine and noradrenaline uptake by venlafaxine: a comparative study with paroxetine and desipramine. Br J Pharmacol 125:526–532
Béïque JC, de Montigny C, Blier P, Debonnel G (1999) Venlafaxine: discrepancy between in vivo 5-HT and NE reuptake blockade and affinity for reuptake sites. Synapse. 32:198–211
Béïque JC, Blier P, de Montigny C, Debonnel G (2000a) Potentiation by (-)Pindolol of the activation of postsynaptic 5-HT(1A) receptors induced by venlafaxine. Neuropsychopharmacology. 23:294–306
Béïque J, de Montigny C, Blier P, Debonnel G (2000b) Effects of sustained administration of the serotonin and norepinephrine reuptake inhibitor venlafaxine: I. in vivo electrophysiological studies in the rat. Neuropharmacology. 39:1800–1812
Béïque J, de Montigny C, Blier P, Debonnel G (2000c) Effects of sustained administration of the serotonin and norepinephrine reuptake inhibitor venlafaxine: II. In vitro studies in the rat. Neuropharmacology. 39:1813–1822
Bel N, Artigas F (1993) Chronic treatment with fluvoxamine increases extracellular serotonin in frontal cortex but not in raphe nuclei. Synapse. 15:243–245
Belovicova K, Bogi E, Koprdova R, Ujhazy E, Mach M, Dubovicky M (2017) Effects of venlafaxine and chronic unpredictable stress on behavior and hippocampal neurogenesis of rat dams. Neuro Endocrinol Lett 38:19–26
Berrocoso E, Mico JA (2007) In vivo effect of venlafaxine on locus coeruleus neurons: role of opioid, alpha(2)-adrenergic, and 5-hydroxytryptamine(1A) receptors. J Pharmacol Exp Ther 322:101–107
Beyer CE, Boikess S, Luo B, Dawson LA (2002) Comparison of the effects of antidepressants on norepinephrine and serotonin concentrations in the rat frontal cortex: an in-vivo microdialysis study. J Psychopharmacol 16:297–304
Bhatara VS, Magnus RD, Paul KL, Preskorn SH (1998) Serotonin syndrome induced by venlafaxine and fluoxetine: a case study in polypharmacy and potential pharmacodynamic and pharmacokinetic mechanisms. Ann Pharmacother 32:432–436
Bijata M, Labus J, Guseva D, Stawarski M, Butzlaff M, Dzwonek J et al (2017) Synaptic remodeling depends on signaling between serotonin receptors and the extracellular matrix. Cell Rep 19:1767–1782
Blier P (2014) Rational site-directed pharmacotherapy for major depressive disorder. Int J Neuropsychopharmacol 17:997–1008
Blier P, de Montigny C (1985) Serotoninergic but not noradrenergic neurons in rat central nervous system adapt to long-term treatment with monoamine oxidase inhibitors. Neuroscience. 16:949–955
Bobińska K, Szemraj J, Czarny P, Gałecki P (2016a) Role of MMP-2, MMP-7, MMP-9 and TIMP-2 in the development of recurrent depressive disorder. J Affect Disord 205:119–129
Bobińska K, Szemraj J, Czarny P, Gałecki P (2016b) Expression and activity of metalloproteinases in depression. Med Sci Monit 22:1334–1341
Bortolatto CF, Jesse CR, Wilhelm EA, Nogueira CW (2010) Involvement of potassium channels in the antidepressant-like effect of venlafaxine in mice. Life Sci 86:372–376
Bosker FJ, van Esseveldt KE, Klompmakers AA, Westenberg HG (1995) Chronic treatment with fluvoxamine by osmotic minipumps fails to induce persistent functional changes in central 5-HT1A and 5-HT1B receptors, as measured by in vivo microdialysis in dorsal hippocampus of conscious rats. Psychopharmacology 117:358–363
Bourin M, Chenu F, Prica C, Hascoët M (2009) Augmentation effect of combination therapy of aripiprazole and antidepressants on forced swimming test in mice. Psychopharmacology 206:97–107
Bozzelli PL, Caccavano A, Avdoshina V, Mocchetti I, Wu J-Y, Conant K (2020) Increased matrix metalloproteinase levels and perineuronal net proteolysis in the HIV-infected brain; relevance to altered neuronal population dynamics. Exp Neurol 323:113077
Brubacher JR, Hoffman RS, Lurin MJ (1996) Serotonin syndrome from venlafaxine-tranylcypromine interaction. Vet Hum Toxicol 38:358–361
Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG, Shaw JL, Thompson L, Nelson DL et al (2001) Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 25:871–880
Cabras S, Saba F, Reali C, Scorciapino ML, Sirigu A, Talani G et al (2010) Antidepressant imipramine induces human astrocytes to differentiate into cells with neuronal phenotype. Int J Neuropsychopharmacol 13:603–615
Carlini VP, Poretti MB, Rask-Andersen M, Chavan RA, Ponzio MF, Sawant RS et al (2012) Differential effects of fluoxetine and venlafaxine on memory recognition: possible mechanisms of action. Prog Neuro-Psychopharmacol Biol Psychiatry 38:159–167
Carpenter LL, Jocic Z, Hall JM, Rasmussen SA, Price LH (1999) Mirtazapine augmentation in the treatment of refractory depression. J Clin Psychiatry 60:45–49
Carpenter LL, Yasmin S, Price LH (2002) A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry 51:183–188
Castagné V, Porsolt RD, Moser P (2009) Use of latency to immobility improves detection of antidepressant-like activity in the behavioral despair test in the mouse. Eur J Pharmacol 616:128–133
Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y et al (2018) Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 391:1357–1366
Clerc GE, Ruimy P, Verdeau-Pallès J (1994) A double-blind comparison of venlafaxine and fluoxetine in patients hospitalized for major depression and melancholia. The Venlafaxine French Inpatient Study Group. Int Clin Psychopharmacol 9:139–143
Cooke JD, Grover LM, Spangler PR (2009) Venlafaxine treatment stimulates expression of brain-derived neurotrophic factor protein in frontal cortex and inhibits long-term potentiation in hippocampus. Neuroscience. 162:1411–1419
Czubak A, Nowakowska E, Kus K, Burda K, Metelska J, Baer-Dubowska W et al (2009) Influences of chronic venlafaxine, olanzapine and nicotine on the hippocampal and cortical concentrations of brain-derived neurotrophic factor (BDNF). Pharmacol Rep 61:1017–1023
David DJ, Bourin M, Hascoët M, Colombel MC, Baker GB, Jolliet P (2001) Comparison of antidepressant activity in 4- and 40-week-old male mice in the forced swimming test: involvement of 5-HT1A and 5-HT1B receptors in old mice. Psychopharmacology 153:443–449
David DJP, Bourin M, Jego G, Przybylski C, Jolliet P, Gardier AM (2003) Effects of acute treatment with paroxetine, citalopram and venlafaxine in vivo on noradrenaline and serotonin outflow: a microdialysis study in Swiss mice. Br J Pharmacol 140:1128–1136
David DJ, Samuels BA, Rainer Q, Wang J-W, Marsteller D, Mendez I et al (2009) Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 62:479–493
de Silva VA, Hanwella R (2012) Efficacy and tolerability of venlafaxine versus specific serotonin reuptake inhibitors in treatment of major depressive disorder: a meta-analysis of published studies. Int Clin Psychopharmacol 27:8–16
Debonnel G, Saint-André E, Hébert C, de Montigny C, Lavoie N, Blier P (2007) Differential physiological effects of a low dose and high doses of venlafaxine in major depression. Int J Neuropsychopharmacol 10:51–61
Decoutere L, De Winter S, Vander Weyden L, Spriet I, Schrooten M, Tournoy J et al (2012) A venlafaxine and mirtazapine-induced serotonin syndrome confirmed by de- and re-challenge. Int J Clin Pharm 34:686–688
Dierick M, Ravizza L, Realini R, Martin A (1996) A double-blind comparison of venlafaxine and fluoxetine for treatment of major depression in outpatients. Prog Neuro-Psychopharmacol Biol Psychiatry 20:57–71
Favuzzi E, Marques-Smith A, Deogracias R, Winterflood CM, Sánchez-Aguilera A, Mantoan L et al (2017) Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron. 95:639–655.e10
Frischknecht R, Heine M, Perrais D, Seidenbecher CI, Choquet D, Gundelfinger ED (2009) Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci 12:897–904
Gartside SE, Umbers V, Sharp T (1997) Inhibition of 5-HT cell firing in the DRN by non-selective 5-HT reuptake inhibitors: studies on the role of 5-HT1A autoreceptors and noradrenergic mechanisms. Psychopharmacology 130:261–268
Grønli J, Murison R, Bjorvatn B, Sørensen E, Portas CM, Ursin R (2004) Chronic mild stress affects sucrose intake and sleep in rats. Behav Brain Res 150:139–147
Guiard BP, Froger N, Hamon M, Gardier AM, Lanfumey L (2005) Sustained pharmacological blockade of NK1 substance P receptors causes functional desensitization of dorsal raphe 5-HT 1A autoreceptors in mice. J Neurochem 95:1713–1723
Guiard BP, El Mansari M, Blier P (2008) Cross-talk between dopaminergic and noradrenergic systems in the rat ventral tegmental area, locus ceruleus, and dorsal hippocampus. Mol Pharmacol 74:1463–1475
Guiard B, Mansari M, Blier P (2009) Prospect of a dopamine contribution in the next generation of antidepressant drugs: the triple reuptake inhibitors. CDT. 10:1069–1084
Gur E, Dremencov E, Lerer B, Newman ME (1999) Venlafaxine: acute and chronic effects on 5-hydroxytryptamine levels in rat brain in vivo. Eur J Pharmacol 372:17–24
Hache G, Guiard BP, Nguyen TH, Quesseveur G, Gardier AM, Peters D et al (2015) Antinociceptive activity of the new triple reuptake inhibitor NS18283 in a mouse model of chemotherapy-induced neuropathic pain. Eur J Pain 19:322–333
Haddjeri N, Blier P, de Montigny C (1995) Noradrenergic modulation of central serotonergic neurotransmission: acute and long-term actions of mirtazapine. Int Clin Psychopharmacol 10(Suppl 4):11–17
Hanson ND, Owens MJ, Nemeroff CB (2011) Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology. 36:2589–2602
Hasler G (2010) Pathophysiology of depression: do we have any solid evidence of interest to clinicians? World Psychiatry 9:155–161
Higashino K, Ago Y, Umehara M, Kita Y, Fujita K, Takuma K et al (2014) Effects of acute and chronic administration of venlafaxine and desipramine on extracellular monoamine levels in the mouse prefrontal cortex and striatum. Eur J Pharmacol 729:86–93
Hjorth S, Auerbach SB (1999) Autoreceptors remain functional after prolonged treatment with a serotonin reuptake inhibitor. Brain Res 835:224–228
Hu Q, Shen P, Bai S, Dong M, Liang Z, Chen Z et al (2018) Metabolite-related antidepressant action of diterpene ginkgolides in the prefrontal cortex. Neuropsychiatr Dis Treat 14:999–1011
Huang Z, Zhong X-M, Li Z-Y, Feng C-R, Pan A-J, Mao Q-Q (2011) Curcumin reverses corticosterone-induced depressive-like behavior and decrease in brain BDNF levels in rats. Neurosci Lett 493:145–148
Huang X, Mao Y-S, Li C, Wang H, Ji J-L (2014) Venlafaxine inhibits apoptosis of hippocampal neurons by up-regulating brain-derived neurotrophic factor in a rat depression model. Int J Clin Exp Pathol 7:4577–4586
Ide S, Fujiwara S, Fujiwara M, Sora I, Ikeda K, Minami M et al (2010) Antidepressant-like effect of venlafaxine is abolished in μ-opioid receptor-knockout mice. J Pharmacol Sci 114:107–110
Invernizzi R, Bramante M, Samanin R (1995) Extracellular concentrations of serotonin in the dorsal hippocampus after acute and chronic treatment with citalopram. Brain Res 696:62–66
Invernizzi R, Bramante M, Samanin R (1996) Role of 5-HT1A receptors in the effects of acute chronic fluoxetine on extracellular serotonin in the frontal cortex. Pharmacol Biochem Behav 54:143–147
Jaskiw GE, Kirkbride B, Bongiovanni R (2006) In rats chronically treated with clozapine, tyrosine depletion attenuates the clozapine-induced in vivo increase in prefrontal cortex dopamine and norepinephrine levels. Psychopharmacology 185:416–422
Kihara T, Ikeda M (1995) Effects of duloxetine, a new serotonin and norepinephrine uptake inhibitor, on extracellular monoamine levels in rat frontal cortex. J Pharmacol Exp Ther 272:177–183
Koch S, Hemrick-Luecke SK, Thompson LK, Evans DC, Threlkeld PG, Nelson DL et al (2003) Comparison of effects of dual transporter inhibitors on monoamine transporters and extracellular levels in rats. Neuropharmacology. 45:935–944
Kulkarni SK, Dhir A (2007) Effect of various classes of antidepressants in behavioral paradigms of despair. Prog Neuro-Psychopharmacol Biol Psychiatry 31:1248–1254
Kumar A, Garg R, Gaur V, Kumar P (2010) Venlafaxine involves nitric oxide modulatory mechanism in experimental model of chronic behavior despair in mice. Brain Res 1311:73–80
Lacroix D, Blier P, Curet O, de Montigny C (1991) Effects of long-term desipramine administration on noradrenergic neurotransmission: electrophysiological studies in the rat brain. J Pharmacol Exp Ther 257:1081–1090
Lapmanee S, Charoenphandhu J, Teerapornpuntakit J, Krishnamra N, Charoenphandhu N (2017) Agomelatine, venlafaxine, and running exercise effectively prevent anxiety- and depression-like behaviors and memory impairment in restraint stressed rats. PLoS One 12:e0187671
Larsen MH, Hay-Schmidt A, Rønn LCB, Mikkelsen JD (2008) Temporal expression of brain-derived neurotrophic factor (BDNF) mRNA in the rat hippocampus after treatment with selective and mixed monoaminergic antidepressants. Eur J Pharmacol 578:114–122
Le Dantec Y, Hache G, Guilloux JP, Guiard BP, David DJ, Adrien J et al (2014) NREM sleep hypersomnia and reduced sleep/wake continuity in a neuroendocrine mouse model of anxiety/depression based on chronic corticosterone administration. Neuroscience. 274:357–368
Li L, Zhang C (2021) Venlafaxine attenuated the cognitive and memory deficit in mice exposed to isoflurane alone. Front Neurol 12:591223
Li X, Morrow D, Witkin JM (2006) Decreases in nestlet shredding of mice by serotonin uptake inhibitors: Comparison with marble burying. Life Sci 78:1933–1939
Liang Z, Bai S, Shen P, Hu Q, Wang X, Dong M et al (2016) GC-MS-based metabolomic study on the antidepressant-like effects of diterpene ginkgolides in mouse hippocampus. Behav Brain Res 314:116–124
Lin LY, Luo SY, Al-Hawwas M, Herselman MF, Zhou XF, Bobrovskaya L (2019) The long-term effects of ethanol and corticosterone on the mood-related behaviours and the balance between mature BDNF and proBDNF in mice. J Mol Neurosci 69:60–68
Liu J, Qiao W, Yang Y, Ren L, Sun Y, Wang S (2012) Antidepressant-like effect of the ethanolic extract from Suanzaorenhehuan Formula in mice models of depression. J Ethnopharmacol 141:257–264
Liu B, Liu J, Wang M, Zhang Y, Li L (2017) From serotonin to neuroplasticity: evolvement of theories for major depressive disorder. Front Cell Neurosci 11:305
Martisova E, Aisa B, Tordera RM, Puerta E, Solas M, Ramirez MJ (2015) Venlafaxine reverses decreased proliferation in the subventricular zone in a rat model of early life stress. Behav Brain Res 292:79–82
McCue RE, Joseph M (2001) Venlafaxine- and trazodone-induced serotonin syndrome. Am J Psychiatry 158:2088–2089
Millan MJ, Gobert A, Lejeune F, Newman-Tancredi A, Rivet JM, Auclair A et al (2001) S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J Pharmacol Exp Ther 298:565–580
Mirza NR, Nielsen EØ, Troelsen KB (2007) Serotonin transporter density and anxiolytic-like effects of antidepressants in mice. Prog Neuro-Psychopharmacol Biol Psychiatry 31:858–866
Morón JA, Brockington A, Wise RA, Rocha BA, Hope BT (2002) Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci 22:389–395
Mostany R, Valdizán EM, Pazos A (2008) A role for nuclear beta-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology. 55:18–26
Nemeroff CB, Entsuah R, Benattia I, Demitrack M, Sloan DM, Thase ME (2008) Comprehensive analysis of remission (COMPARE) with venlafaxine versus SSRIs. Biol Psychiatry 63:424–434
Nutt D, Wilson S, Paterson L (2008) Sleep disorders as core symptoms of depression. Dialogues Clin Neurosci 10:329–336
Pan J-J, Shen WW (2003) Serotonin syndrome induced by low-dose venlafaxine. Ann Pharmacother 37:209–211
Papakostas GI, Thase ME, Fava M, Nelson JC, Shelton RC (2007) Are antidepressant drugs that combine serotonergic and noradrenergic mechanisms of action more effective than the selective serotonin reuptake inhibitors in treating major depressive disorder? A meta-analysis of studies of newer agents. Biol Psychiatry 62:1217–1227
Poretti MB, Sawant RS, Rask-Andersen M, de Cuneo MF, Schiöth HB, Perez MF et al (2016) Reduced vasopressin receptors activation mediates the anti-depressant effects of fluoxetine and venlafaxine in bulbectomy model of depression. Psychopharmacology 233:1077–1086
Quentin E, Belmer A, Maroteaux L (2018) Somato-dendritic regulation of raphe serotonin neurons; a key to antidepressant action. Front Neurosci 12:982
Rahman Z, Ring RH, Young K, Platt B, Lin Q, Schechter LE et al (2008) Inhibition of uptake 2 (or extraneuronal monoamine transporter) by normetanephrine potentiates the neurochemical effects of venlafaxine. Brain Res 1203:68–78
Redrobe JP, Bourin M, Colombel MC, Baker GB (1998) Dose-dependent noradrenergic and serotonergic properties of venlafaxine in animal models indicative of antidepressant activity. Psychopharmacology 138:1–8
Ren Z, Yan P, Zhu L, Yang H, Zhao Y, Kirby BP et al (2018) Dihydromyricetin exerts a rapid antidepressant-like effect in association with enhancement of BDNF expression and inhibition of neuroinflammation. Psychopharmacology 235:233–244
Riga D, Kramvis I, Koskinen MK, van Bokhoven P, van der Harst JE, Heistek TS et al (2017) Hippocampal extracellular matrix alterations contribute to cognitive impairment associated with a chronic depressive-like state in rats. Sci Transl Med 9:eaai8753
Ringland C, Schweig JE, Eisenbaum M, Paris D, Ait-Ghezala G, Mullan M et al (2021) MMP9 modulation improves specific neurobehavioral deficits in a mouse model of Alzheimer’s disease. BMC Neurosci 22:39
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D et al (2006) Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163:1905–1917
Rybakowski JK, Skibinska M, Suwalska A, Leszczynska-Rodziewicz A, Kaczmarek L, Hauser J (2011) Functional polymorphism of matrix metalloproteinase-9 (MMP-9) gene and response to lithium prophylaxis in bipolar patients. Hum Psychopharmacol 26:168–171
Saad MA, El-Sahar AE, Sayed RH, Elbaz EM, Helmy HS, Senousy MA (2019) Venlafaxine mitigates depressive-like behavior in ovariectomized rats by activating the EPO/EPOR/JAK2 signaling pathway and increasing the serum estradiol level. Neurotherapeutics. 16:404–415
Salin-Pascual RJ, Moro-Lopez ML (1997) Effects of venlafaxine in the sleep architecture of rats. Psychopharmacology 129:295–296
Sánchez C, Brennum LT, í Stórustovu S, Kreilgård M, Mørk A (2007) Depression and poor sleep: The effect of monoaminergic antidepressants in a pre-clinical model in rats. Pharmacol Biochem Behav 86:468–476
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 301:805–809
Schneider AC, Breilmann J, Reuter B, Becker T, Kösters M (2021) Systematic evaluation of the “efficacy-effectiveness gap” in the treatment of depression with venlafaxine and duloxetine. Acta Psychiatr Scand 144(2):113–124
Schueler Y-B, Koesters M, Wieseler B, Grouven U, Kromp M, Kerekes MF et al (2011) A systematic review of duloxetine and venlafaxine in major depression, including unpublished data. Acta Psychiatr Scand 123:247–265
Smith D, Dempster C, Glanville J, Freemantle N, Anderson I (2002) Efficacy and tolerability of venlafaxine compared with selective serotonin reuptake inhibitors and other antidepressants: a meta-analysis. Br J Psychiatry 180:396–404
Socała K, Nieoczym D, Wyska E, Poleszak E, Wlaź P (2012) Influence of sildenafil on the antidepressant activity of bupropion and venlafaxine in the forced swim test in mice. Pharmacol Biochem Behav 103:273–278
Sorg BA, Berretta S, Blacktop JM, Fawcett JW, Kitagawa H, Kwok JCF et al (2016) Casting a wide net: role of perineuronal nets in neural plasticity. J Neurosci 36:11459–11468
Szabo ST, Blier P (2001) Effects of the selective norepinephrine reuptake inhibitor reboxetine on norepinephrine and serotonin transmission in the rat hippocampus. Neuropsychopharmacology. 25:845–857
Tewari BP, Chaunsali L, Campbell SL, Patel DC, Goode AE, Sontheimer H (2018) Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat Commun 9:4724
Thomas J, Khanam R, Vohora D (2016a) Augmentation of effect of venlafaxine by folic acid in behavioral paradigms of depression in mice: evidence of serotonergic and pro-inflammatory cytokine pathways. Pharmacol Rep 68:396–403
Thomas J, Khanam R, Vohora D (2016b) Augmentation of antidepressant effects of venlafaxine by agomelatine in mice are independent of kynurenine pathway. Neurochem Int 99:103–109
Thomas J, Khanam R, Vohora D (2017) Activation of indoleamine 2, 3- dioxygenase pathway by olanzapine augments antidepressant effects of venlafaxine in mice. Psychiatry Res 258:444–448
Vafadari B, Mitra S, Stefaniuk M, Kaczmarek L (2019) Psychosocial stress induces schizophrenia-like behavior in mice with reduced MMP-9 activity. Front Behav Neurosci 13:195
Venzala E, García-García AL, Elizalde N, Delagrange P, Tordera RM (2012) Chronic social defeat stress model: behavioral features, antidepressant action, and interaction with biological risk factors. Psychopharmacology 224:313–325
Wang Y, Xiao Z, Liu X, Berk M (2011) Venlafaxine modulates depression-induced behaviour and the expression of Bax mRNA and Bcl-xl mRNA in both hippocampus and myocardium. Hum Psychopharmacol 26:95–101
Wang H-L, Sun Y-X, Liu X, Wang H, Ma Y-N, Su Y-A et al (2019) Adolescent stress increases depression-like behaviors and alters the excitatory-inhibitory balance in aged mice. Chin Med J 132:1689–1699
Wang J-L, Wang Y, Gao T-T, Liu L, Wang Y-J, Guan W et al (2020) Venlafaxine protects against chronic stress-related behaviors in mice by activating the mTORC1 signaling cascade. J Affect Disord 276:525–536
Weinmann S, Becker T, Koesters M (2008) Re-evaluation of the efficacy and tolerability of venlafaxine vs SSRI: meta-analysis. Psychopharmacology 196:511–520 discussion 521-522
Wichniak A, Wierzbicka A, Walęcka M, Jernajczyk W (2017) Effects of antidepressants on sleep. Curr Psychiatry Rep 19:63
Xu H, Luo C, Richardson JS, Li XM (2004) Recovery of hippocampal cell proliferation and BDNF levels, both of which are reduced by repeated restraint stress, is accelerated by chronic venlafaxine. Pharm J 4:322–331
Xu H, Li W, Zhang B, Huang S, Liu X (2019) Long-term estrogen deprivation changes the response to antianxiety drugs in mice in the elevated plus maze test. Gynecol Endocrinol 35:1054–1058
Yamada K, Kobayashi M, Mori A, Jenner P, Kanda T (2013) Antidepressant-like activity of the adenosine A(2A) receptor antagonist, istradefylline (KW-6002), in the forced swim test and the tail suspension test in rodents. Pharmacol Biochem Behav 114–115:23–30
Ye Y, Liu C, Liu X, Huang S (2016) Ovariectomy changes the response to antidepressant drugs in tail suspension test in mice. Gynecol Endocrinol 32:986–990
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ et al (2011) Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 477:171–178
Zhang X, Li X, Li M, Ren J, Yun K, An Y et al (2015) Venlafaxine increases cell proliferation and regulates DISC1, PDE4B and NMDA receptor 2B expression in the hippocampus in chronic mild stress mice. Eur J Pharmacol 755:58–65
Acknowledgements
Illustrations were created using Biorender image database.
Author information
Authors and Affiliations
Contributions
B.P. Guiard and C. Rampon designed the study. B. Coutens and A. Yrondi managed the literature searches and wrote the first draft of the manuscript. All authors contributed to and have approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Coutens, B., Yrondi, A., Rampon, C. et al. Psychopharmacological properties and therapeutic profile of the antidepressant venlafaxine. Psychopharmacology 239, 2735–2752 (2022). https://doi.org/10.1007/s00213-022-06203-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-022-06203-8