
Abstract
Rationale
Major depressive disorder (MDD) is a highly prevalent illness that affects large populations across the world, and increasing evidence suggests that neuroinflammation and levels of brain-derived neurotrophic factor (BDNF) are closely related to depression. Dihydromyricetin (DHM) is a kind of flavonoid natural product that has been reported to display multiple pharmacological effects, including anti-inflammatory and anti-oxidative properties, and these may contribute to ameliorate MDD.
Objective
This study investigated the effect of DHM on depression-related phenotypes in various experimental animal models.
Methods
The antidepressant-like effect of DHM was validated via depression-related behavioral tests in naïve male C57BL/6 mice, as well as in the acute lipopolysaccharide-induced mouse model of depression. The chronic unpredicted mild stress (CUMS) mouse model of depression was also used to assess the rapid antidepressant-like effect of DHM by tail suspension test (TST), forced swimming test (FST), locomotor activity, and sucrose preference test (SPT). The expression of BDNF and inflammatory factors were determined through Western blotting and enzyme-linked immunosorbent assay, respectively.
Results
DHM reduced immobility time in the TST and FST both in mice and the acute LPS-induced mouse model of depression. Seven days of DHM treatment ameliorated depression-related behaviors induced by CUMS, whereas similar treatment with the typical antidepressant venlafaxine did not. DHM activated the ERK1/2-CREB pathway and increased glycogen synthase kinase-3 beta (GSK-3β) phosphorylation at ser-9, with upregulation of BDNF expression, in both hippocampal tissues and cultured hippocampal cells.
Conclusion
The present data indicate that DHM exerts a more rapid antidepressant-like effect than does a typical antidepressant, in association with enhancement of BDNF expression and inhibition of neuroinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a highly prevalent illness that affects large populations across the world (Fang et al. 2013; Pesarico et al. 2016; Tao et al. 2016). Although the pathobiological basis of MDD is not understood, increasing evidence suggests that neuroinflammation and levels of brain-derived neurotrophic factor (BDNF) in the hippocampus are closely related to depression-like behavior (Chan et al. 2016; Wohleb 2016). Structural and functional changes in the hippocampus may play a critical role in the development of MDD (Dale et al. 2016; Harrisberger et al. 2015); decreased BDNF expression is evident in MDD patients (Nuernberg et al. 2016), and antidepressant drugs can restore expression of BDNF in either patients with MDD or experimental models of depression (Cheng and Salton 2013; Dwivedi 2013; Gimenez-Cassina et al. 2012; Manosso et al. 2015). The prefrontal cortex (Zhang et al. 2014) and nucleus accumbens (Yang et al. 2016) may also play a role in such experimental models. In addition, numerous studies have demonstrated that neuroinflammation plays an important role in the development of MDD (Eyre et al. 2016; Kaufmann et al. 2017; Vogelzangs et al. 2014). Some inflammatory mediators may also constitute biomarkers and indicate therapeutic response to antidepressant treatment (Hughes et al. 2016; O’Connor et al. 2009; Rapaport et al. 2015). Therefore, regulation of neuroinflammation has also been considered as a potential therapeutic target for MDD.
Dihydromyricetin (DHM) is a flavonoid extracted from Ampelopsis grossedentata in southern China (Chen et al. 2015; Tong et al. 2015) that has been reported to display multiple pharmacological effects, including anti-tumor, anti-inflammatory, and anti-oxidative properties (Jiang et al. 2014; Kao et al. 2017; Liu et al. 2016; Tang et al. 2016; Xie et al. 2015). We have previously shown that DHM protects dopaminergic neurons against MPTP-induced neurodegeneration in a mouse model of Parkinson’s disease by suppressing the activity of glycogen synthase kinase-3 beta (GSK-3β) (Ren et al. 2016) and protects neurons from ischemia-reperfusion-induced apoptosis by activating ERK1/2 and the cAMP response element-binding (CREB) pathway (Zhao et al. 2017) in vivo and in vitro. In addition to direct protection of neurons, we have recently shown that DHM inhibited microglia-mediated inflammation in affording protection from ischemic insult (Zhao et al. 2017). In agreement with our observation, recent studies also showed that DHM suppresses inflammatory responses in RWA 264.7 cells through inhibiting activation of NF-κB and MAPK signaling pathways (Hou et al. 2015) and ameliorates behavioral deficits in animal models of Alzheimer’s disease (Liang et al. 2014). In addition, activities against alcohol intoxication and to maintain glucose homeostasis have also been reported (Chen et al. 2015; Le et al. 2016; Shen et al. 2012).
Given the evidence for important neuroprotective, anti-inflammatory, and anti-oxidative effects in MDD treatment, we investigated the potential therapeutic effects of DHM, a substance that readily crosses the blood-brain barrier (Shen et al. 2012; Youdim et al. 2003), on depression-related phenotypes in experimental mouse models. Our results indicate that DHM treatment ameliorates depression-related behavior in both the acute lipopolysaccharide (LPS)-induced and the chronic, unpredicted mild stress (CUMS)-induced models of depression in mice. We further demonstrate that DHM activates the MAPK/CREB signaling pathway and inhibits the activity of GSK-3β, which resulted in upregulation of BDNF both in vivo and in cultured hippocampal neurons.
Materials and methods
Animals
Male C57BL/6J mice (age 6–8 weeks; 22 ± 3 g) were purchased from Shanghai SLAC Laboratory Animals Co. Ltd. (Shanghai, China) and housed in groups of 4–6 per cage in a humidity- and temperature-controlled vivarium on a 12:12 h light/dark cycle (lights on at 8:00 a.m.). Food and water were available ad libitum except during weeks when animals were trained to perform the sucrose preference test (Wang et al. 2016). All animal protocols were proved by Animal Care and Use Committee of Soochow University and were in compliance with the Guidelines for the Care and Use of Laboratory Animals (Chinese-National-Research-Council, 2006) and the “ARRIVE” (Animals in Research: Reporting In Vivo Experiments) guidelines. Every effort was made to minimize animal suffering and to reduce the number of animals used in the experiments (Ren et al. 2016).
Drugs and reagents
DHM ((2R,3R)-3,5,7-trihydroxy-2-(3,4,5-trihydroxyphenyl)chroman-4-one; catalog number ST03840120MG; purity by HPLC ≥ 98%)was obtained from Shanghai Standard Biotech Co., Ltd., Shanghai, China; it was initially dissolved in dimethylsulfoxide (DMSO) for stock solutions and diluted with saline for intraperitoneal (i.p.) injection (final trace concentration of DMSO ≤ 0.2%) (Ren et al. 2016) at an injection volume of 1 ml/kg body weight. DHM powder was stored in brown bottle covered by silver paper at 4 °C, away from light and kept dry. The DHM solution was stored at − 40 °C away from light. Both DHM powder and solution were prepared immediately prior to use. For cell culture, DHM was dissolved in DMSO to 100 mM as stock solution and diluted with cell culture medium to concentrations from 10 to 50 μM. Lipopolysaccharide (LPS), venlafaxine hydrochloride, and U0126 were purchased from Sigma-Aldrich, Co. (Darmstadt, Germany) and LY294002 (10 mg/ml) was purchased from Beyotime (Shanghai, China). LPS and venlafaxine hydrochloride were dissolved in PBS or saline, and U0126 was dissolved in DMSO for stock solutions.
LPS-induced model of depression
Mice were randomly divided into 4 groups of 12 animals each: saline, LPS (i.p., 0.83 mg/kg), LPS + DHM 10.0 mg/kg, and LPS + DHM 20.0 mg/kg. In DHM treatment groups, 10.0 or 20.0 mg/kg DHM was administered i.p. once daily for 3 days before LPS treatment. On the last day, 0.83 mg/kg LPS was injected 30 min after DHM administration; 24 h later, the tail suspension test (TST) and forced swim test (FST) were performed and locomotor activity was measured (Jangra et al. 2016; Wang et al. 2010; Zhu et al. 2016). Also, the body weight of mice was measured before and 24 h post-injection of LPS.
Chronic unpredicted mild stress model of depression
The CUMS procedure was carried out as described previously (Wang et al. 2016). Mice were randomly divided into five groups of at least 12 mice each: saline, CUMS, CUMS + DHM 10.0 mg/kg, CUMS + DHM 20.0 mg/kg, and CUMS + venlafaxine 10.0 mg/kg. In brief, mice were subjected to a variety of stresses for 8 weeks, with the stressors and stressor sequences applied at random each week to ensure unpredictability. Additionally, mice were subjected daily to two of the following stressors: light/dark cycle inversion (for 48 h in a different room), damp sawdust (over-night), sawdust-free cage (over-night), white noise (2 h), movement restraint (in a small tube), 45° cage tilting (over-night), cold or hot water swimming (5 min), water deprivation (24 h), tail pinch (2 min), overcrowding cage (over-night), and electric stimulus (0.5 mA, 30 times in 15 s). In the CUMS model, stressors were applied over 8 weeks and then mice in treatment groups were administered DHM 10.0 or 20.0 mg/kg i.p. or venlafaxine10.0 mg/kg i.p. once daily for 7 days; the TST and FST were performed on the third and seventh days, respectively (Abe-Higuchi et al. 2016; Mao et al. 2017).
Behavioral tests
Tail suspension test
Mice were subjected to the TST as described previously (Pesarico et al. 2016; Tao et al. 2016; Wang et al. 2016). Briefly, animals were suspended at least 80 cm above the floor by adhesive tape placed approximately 1 cm from the tip of the tail, under both acoustic and visual isolation. The total TST procedure was 6 min, and immobility time was recorded during the last 4 min; mice were considered immobile only when hanging passively and motionless.
Forced swim test
Mice were subjected to the FST as described previously (Pesarico et al. 2016; Tao et al. 2016; Wang et al. 2016), subject to minor modifications. Briefly, each mouse was placed individually into a 4000 ml Pyrex glass beaker containing 3000 ml of warm water (temperature 25 ± 1 °C). The total FST procedure was 6 min, and immobility time was recorded during the last 4 min; mice were considered immobile only when motionless or floating with only small movements necessary to keep the head above water.
Locomotor activity
An infrared photobeam activity cage system (Jiliang Ltd., Shanghai, China) was used to record and analyze mouse horizontal locomotor activity. Mice were kept in the test room for 30 min before the experiment to habituate to the new environment. Mice were then placed individually in a 25 × 25 × 30 cm plexiglass/polyvinyl arena. Total distance traveled (mm) and time (s) spent in the center area were counted for 20 and 5 min, respectively, using videotracking software (ANY-maze; Stoelting, IL, USA) (Pesarico et al. 2016; Ren et al. 2016; Tao et al. 2016; Wang et al. 2016).
Sucrose preference test
This test was performed as described previously (Wang et al. 2016), subject to minor modifications. Briefly, mice were placed individually into a two-bottle, free-choice cage. One of the bottles contained water and the other 1% (w/v) sucrose solution. Prior to initiation of the test, all mice were acclimatized to 1% sucrose solution and drinking water for 48 h. Before the sucrose preference test, mice were deprived of water and food for 24 h, after which they were allowed free access to the two bottles over a 2-h period. The sucrose preference test was performed weekly during the CUMS procedure, with sucrose preference (%) = sucrose consumption/(sucrose consumption + water consumption).
Primary neuron culture
Primary mouse neurons were derived from the hippocampus of C57BL/6 mice at embryonic day 17 or 18, as described previously (Wang et al. 2016). Dissected hippocampus tissue was digested with 2% trypsin and DNAse-I for 30 min in 37 °C and resuspended in Neurobasal medium (Gibco, NY, USA) containing 2% B-27. After filtration with a 70-μm sieve, collected cells were seeded in poly-d-lysine-coated plates and cultured at 37 °C in a 5% CO2 incubator for 14 days. Culture medium was replaced every other day.
Western blotting
Hippocampus tissue and cultured hippocampal neuron were lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl,1% NP-40, 1% Na deoxycholate, 1% Triton X-100, 1 mM PMSF, EDTA, and protease inhibitor) for 30 min on ice and then incubated at 95 °C for 5 min (Ren et al. 2016). Western blotting was performed using standard protocols, and blots were probed with anti-pCREB (S133) (1:800; 9198S, Santa Cruz), anti-CREB (1:1000; 9197S, Santa Cruz), anti-pERK1/2 (1:1000; 4370S, Cell Signaling Technology), anti-ERK1/2 (1:1000; 4695S, Cell Signaling Technology), anti-BDNF (1:200; SC-546, Santa Cruz), anti-pGSK3β (S9) (1:1000; 9323S, Cell Signaling Technology), anti-GSK3β (1:1000; 9832S, Cell Signaling Technology), and anti-α-tubulin (1:10,000; T6074, Sigma-Aldrich) antibodies. Data were analyzed and quantified by ChemiScope 3300 Mini (CLINX, Shanghai, China) and densitometry with ImageJ software. Phosphorylated ERK, GSK3β, and CREB were related to tubulin. Each experiment was replicated at least three times and signals normalized to control in each independent experiment.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from cultured hippocampal neurons using RNAiso plus reagent (TaKaRa Biotechnology, Dalian, China), and 1 μg RNA was reverse-transcribed into cDNA using moloney murine leukemia virus reverse transcriptase, deoxynucleotide triphosphate, recombinant RNAse inhibitor, and oligo (dT) according to the manufacturer’s instructions. cDNAs were amplified using the following specific primers: GAPDH, forward primer: 5′-TGTGTCCGTCGT GGATCTGA-3′, reverse primer: 5′-TTGCTGTTGAAGTCGCAGGAG-3′; TNF-α, forward primer: 5′-CAGGAGGGAGAACAGAAACTCCA-3′, reverse primer: 5′-CCTGGTTGGCTGCTTGCTT-3′; and IL-6, forward primer:5′-TTCCATCCAGTTGCCTTCTT-3′, reverse primer:5′-CAGAATTGCCATTGCACAAC-3′. Quantitative tests of mRNA expression were performed by SYBR Premix II (TaKaRa, Dalian, China) according to the manufacturer’s instructions. Expression of target genes was normalized to GAPDH and quantified relative to expression in the respective control. Results were analyzed and expressed as previously described (Ni et al. 2015; Wu et al. 2015).
Enzyme-linked immunosorbent assay
To measure TNF-α and IL-6, hippocampal tissue was collected from mice following the LPS-induced model of depression and controls. Samples were homogenized ultrasonically in RIPA buffer with protease inhibitor (Roche, Mannheim, Germany). Supernatants were collected after centrifugation (12,000 rpm, 20 min, 4 °C) and protein concentration measured by BCA protein kit (Tiangen, Beijing, China). TNF-a and IL-6 commercial ELISA kits were purchased from Boster Biosciences Co., Wuhan, China, and protein expression measured according to the manufacturer’s instructions. OD values were assessed by Microplate Reader (Infinite M200 PRO, Tecan, Switzerland) (Ni et al. 2015).
Statistical analysis
Data were analyzed by GraphPad Prism® (version 6.0) software and presented as means ± SEM. One-way ANOVAs were followed by Dunnett’s test for comparisons with vehicle control groups or Newman-Keul’s test for multiple-group comparisons. For data on weight gain during the CUMS procedure, repeated measures ANOVA was employed. A probability value of p < 0.05 was considered statistically significant.
Results
DHM reduces immobility time in FST and TST
We first examined the antidepressant-like effect of DHM using the FST and TST in naïve mice. As shown in Fig. 1a, b, a 2-h pretreatment with DHM (10.0 and 20.0 mg/kg) did not alter immobility time in either FST or TST, while the typical antidepressant venlafaxine (10.0 mg/kg) significantly decreased immobility time. However, pretreatment with DHM (10.0 and 20.0 mg/kg) once per day for 3 days resulted in a marked reduction in immobility time in dose-dependent manner (Fig. 1c, d). In contrast, pretreatment with DHM for 1 or 2 days did not elicit similar antidepressant effects. These results indicated that the antidepressant effect of DHM requires repeated administration over 3 days.

Effects of treatment with DHM or venlafaxine on immobility time in the tail suspension test (TST) and forced swimming test (FST) in naïve mice. Male C57BL/6 mice were divided into five groups: saline; DHM 5.0, 10.0, and 20.0 mg/kg; and venlafaxine10.0 mg/kg. Mice received intraperitoneal (i.p.) injections of DHM or venlafaxine 2 h before behavioral tests: a TST and b FST immobility times after a single administration of DHM or venlafaxine and c TST and d FST immobility times after repeated administration of DHM or venlafaxine once daily for three consecutive days. At least 12 mice were used in each group. Values are presented as means ± SEM. Statistical analyses were performed by one-way ANOVA followed by Dunnett’s test: *p < 0.05, **p < 0.01, ***p < 0.001 vs saline
DHM attenuates LPS-induced depression-related behaviors and inflammatory markers
We next employed an inflammation-induced, depression-related model in mice, induced by acute injection of LPS. As expected, acute LPS injection (0.83 mg/kg) induced depression-like behavior in mice in terms of increases in immobility times in FST and TST and reduced locomotion (Fig. 2a, b) (Guan et al. 2015; Kurosawa et al. 2016; Wohleb 2016). DHM (10.0 and 20.0 mg/kg) reversed the LPS-induced increase in immobility time in both TST and FST (Fig. 2a, b) and markedly attenuated LPS-induced inhibition of locomotion in terms of total distance traveled (Fig. 2c). DHM (10.0 and 20.0 mg/kg) attenuated LPS-induced weight loss 24 h after LPS injection (Fig. 2d). DHM (10.0 and 20.0 mg/kg) also attenuated the LPS-induced increase in hippocampal TNF-α and IL-6 mRNA (Fig. 2e, f) and protein levels (Fig. 2g, h).

Effects of DHM on LPS-induced changes in the tail suspension test (TST) and forced swimming test (FST) and on locomotor activity, body weight, TNF-α, and IL-6. Male C57BL/6 mice were divided into four groups: saline, LPS 0.83 mg/kg, LPS 0.83 mg/kg + DHM 10.0 mg/kg, and LPS 0.83 mg/kg + DHM 20.0 mg/kg. Mice received intraperitoneal (i.p.) injections of DHM once daily for three consecutive days. On the last day, LPS was injected i.p. 30 min after DHM treatment. Immobility times in a TST and b FST, and c locomotor activity, as total distance traveled, were evaluated 24 h after LPS injection. d Body weight (g) was recorded before and 24 h after LPS administration and the difference calculated. At least 12 mice were used in each group. Following the behavioral studies, mice were sacrificed and hippocampal tissue collected for assay of e TNF-α mRNA, f IL-6 mRNA, g TNF-α protein, and h IL-6 protein after 3 days of treatment with DHM treatment, 10.0 and 20.0 mg/kg. At least 12 mice were used in each group. Values are presented as means ± SEM. Statistical analyses for a–h were performed by one-way ANOVA followed by the Newman-Keuls test. *p < 0.05, **p < 0.01, ***p < 0.001
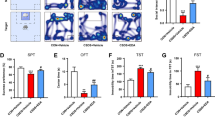
DHM attenuates CUMS-induced depression-related and anxiety-related behaviors and disruption to the ERK1/2-CREB-BDNF pathway
The experimental procedure for the CUMS model is shown in Fig. 3a. There were no differences in sucrose preference between the five groups at the beginning of the experiment. At 3 and 7 days following CUMS for 8 weeks, mice showed the expected marked reduction in sucrose preference (Fig. 3b, e) and increase in immobility time in both FST and TST (Fig. 3c, d, f, g), together with a decrease in time exploring the center area of the activity monitor but without alteration in distance traveled (Fig. 3i, j). Three days of DHM treatment (10.0 and 20.0 mg/kg) did not alter CUMS-induced decrease in sucrose preference (Fig. 3b) or increase in FST and TST time (Fig. 3c, d). However, 7 days of DHM treatment reversed CUMS-induced decrease in sucrose preference (Fig. 3e) and increase in FST and TST time (Fig. 3f, g), together with reversal of CUMS-induced decrease in time exploring the center area of the activity monitor but without alteration in distance traveled (Fig. 3i, j); 7 days of DHM treatment also reversed CUMS-induced weight loss (Fig. 3h). In contrast, 7 days of treatment with venlafaxine (10.0 mg/kg) did not influence any of these CUMS-induced effects (Fig. 3b–j).

Effects of treatment with DHM or venlafaxine in the chronic unpredictable mild stress (CUMS) model. a Experimental design. Mice were subjected to CUMS over 8 weeks and then divided randomly into four treatment groups: CUMS, CUMS + DHM 10.0 mg/kg i.p., CUMS + DHM20.0 mg/kg i.p., or venlafaxine 10.0 mg/kg i.p. once daily for 7 days; naïve mice received saline as controls. Sucrose preference test (SPT), tail suspension test (TST), and forced swimming test (FST) were carried out after three (b–d) and seven (e–g) days of drug treatment. h Body weight was recorded weekly. Total distance traveled (i) and time spend in the center area (j) were recorded after 7 days of drug treatment. At least 12 mice were used in each group. Values are presented as means ± SEM. Statistical analyses for b–d were performed by one-way ANOVA followed by Dunnett’s test; statistical analysis for e–j were performed by one-way ANOVA (repeated measures ANOVA for h) followed by the Newman-Keuls test; *p < 0.05, **p < 0.01, ***p < 0.001; # p < 0.05, ## p < 0.01,### p < 0.001 vs saline
Phosphorylation of ERK1/2, CREB, and GSK-3β (ser-9) were decreased, and the expression of BDNF was downregulated in the hippocampus of mice exposed to CUMS (Fig. 4a, b). DHM treatment (10.0 and 20.0 mg/kg daily for 7 days) reversed these CUMS-induced alterations in ERK1/2, CREB, GSK-3β, and BDNF (Figs. 4a, b). To further confirm these observations, we studied the effects of DHM in naïve mice and primary cultured hippocampal neurons. DHM (5.0, 10.0, and 20.0 mg/kg in hippocampus tissue; 5, 25, and 50 μM in primary cultured hippocampal neurons) increased phosphorylation of ERK1/2, CREB, and GSK-3β at ser-9 (Fig. 4c, d).

Effects of DHM on ERK1/2, CREB, and GSK-3β pathways and stimulated BDNF expression in vivo and in vitro. Following the behavioral studies of Fig. 4, mice were sacrificed and hippocampal tissue dissected and processed for assay. a, b Western blots with respective antibodies. c Naïve mice were treated i.p. with DHM 5.0, 10.0, and 20.0 mg/kg, sacrificed after 2 h and hippocampal tissue dissected for immunoblot assays. d Primary cultured hippocampal neurons were treated with DHM 10.0, 25.0, and 50.0 μM for 1 h and cells collected for immunoblot assays. For each of a, b, c, and d, representative images for immunoblots are shown in the left panel and quantitative data shown in the right panels. Values are presented as means ± SEM from at least three independent experiments. Statistical analyses for a and b were performed by one-way ANOVA followed by the Newman-Keuls test. Statistical analyses for c and d were performed by one-way ANOVA followed by Dunnett’s test. *p < 0.05, **p < 0.01, ***p < 0.001 vs control
Since a 7-day treatment with DHM elevated hippocampal BDNF expression in mice exposed to CUMS (Fig. 4a), we further investigated DHM-stimulated expression of BDNF in vivo and in primary cultured hippocampal neurons. Administration of DHM (20.0 mg/kg) to naïve mice increased expression of hippocampal BDNF in a time-dependent manner, with this effect evident by day 3 and continuing to day 7; in contrast, venlafaxine (10.0 mg/kg) was without effect (Fig. 5a, b). Primary cultured hippocampal neurons treated with DHM (10–50 μM) for 48 h also exhibited increased expression of BDNF (Fig. 5c). In agreement with in vivo observations, expression of BDNF in primary cultured neurons was enhanced in a time-dependent manner following treatment with 25 μM DHM, with this effect evident by day 2 and continuing to day 3 (Fig. 5d). To confirm the signaling pathways for DHM-stimulated BDNF expression, we employed U0126, a ERK1/2 inhibitor, and LY294002, a selective inhibitor of Akt, which activates GSK-3β indirectly. In primary cultured hippocampal neurons, stimulation of BDNF expression by 25 μM DHM was inhibited by each of 10 μM LY294002 and 300 nM U0126 (Fig. 6a), indicating that stimulation of BDNF expression by DHM is dependent on ERK1/2 activation and GSK-3β inhibition.

Effects of a DHM and b venlafaxine on BDNF expression in hippocampus. Mice were treated i.p. with 20.0 mg/kg DHM or 10.0 mg/kg venlafaxine once daily for 1, 3, and 7 days and sacrificed at these different times. BDNF expression in hippocampal tissue was measured by immunoblot. Primary cultured hippocampal neurons were c treated with 10.0, 25.0, and 50.0 μM DHM for 48 h, or d treated with 25.0 μM DHM for 1, 2, or 3 days, before collection for immunoblot assays to determine BDNF expression. For each of a, b, c, and d, representative images for immunoblots are shown in the upper panels and quantitative data shown in the lower panels. Values are presented as means ± SEM from at least three independent experiments. Statistical analyses were performed by one-way ANOVA followed by Dunnett’s test: *p < 0.05, **p < 0.01, ***p < 0.001 vs control

Effects of the selective Akt inhibitor LY294002 and the selective MEK1/2 inhibitor U0126 on DHM-stimulated BDNF expression. a Primary cultured hippocampal neurons were pretreated in the presence or absence of 10 μM LY294002 or 300 nM U0126 prior to and during DHM treatment for an additional period of 48 h. For a, representative images for immunoblots are shown in the left panel and quantitative data shown in the right panel. Values are presented as means ± SEM. Statistical analyses were performed by one-way ANOVA followed by the Newman-Keuls test: # p < 0.05, ## p < 0.01, ### p < 0.001. b Potential signaling mechanisms for DHM-mediated antidepressant-like activity: DHM increases phosphorylation of ERK1/2 to activate CREB and enhance expression of BDNF; DHM also inhibits GSK-3β, via phosphorylating GSK-3β at ser-9, to enhance BDNF expression. However, how DHM regulates ERK1/2 and GSK-3β remains unclear
Discussion
Currently available antidepressant drugs require administration for a number of weeks to attain full clinical efficacy. In addition, these drugs can also cause a number of serious adverse effects. Thus, there is an urgent need to develop novel antidepressant drugs. The present study employed depression-related phenotypes in experimental mouse models and demonstrated that DHM, a natural occurring flavonoid purified from Ampelopsis grossedentata, elicited antidepressant-like effects. In the inflammation (LPS)-induced mouse model of depression, DHM reduced immobility time in both the TST and FST and inhibited inflammatory cytokines. We also observed that DHM attenuated depression-related behaviors in the CUMS mouse model in a time-dependent manner. This antidepressant-like effect was evident following 7 days of DHM treatment. Relative to the serotonin and norepinephrine reuptake inhibitor venlafaxine, our results indicate that 7 days of treatment with DHM elicited an antidepressant-like effect on CUMS-induced depression-related behaviors without central stimulatory effects (Supplementary figure S1). We further demonstrated that DHM promoted hippocampal BDNF expression both in vivo and in primary cultured hippocampal neurons in a time-dependent manner. Additionally, we demonstrated that activation of ERK1/2 and CREB and inhibition of GSK-3β contribute to DHM-stimulated BDNF expression. The present data indicate that DHM may be a promising antidepressant candidate.
Neuroinflammation is increasingly recognized as an important factor in the development of depression and/or as a potential index of therapeutic response in depressive illness. Acute LPS is a reliable model for inducing inflammation-related depression-like phenotypes; it also elicits the expression and secretion of inflammatory cytokines such as TNF-α and IL-6, which appear critical in the pathological processes of inflammation-related depression (Kurosawa et al. 2016; O’Connor et al. 2009; Park et al. 2011; Sulakhiya et al. 2016; Wang et al. 2010). We demonstrated that LPS-induced increases in immobility time and weight loss were prevented by a 3-day pretreatment with DHM. In addition, DHM inhibited LPS-stimulated expression and secretion of TNF-αand IL-6 in mice. These results indicated that 3 days of pretreatment with DHM prevented LPS-induced depression-related behavior and attenuated associated LPS-induced increases in hippocampal inflammatory processes. Thus, inhibition of inflammation may also contribute to the antidepressant-like action of DHM.
It is interesting to note that antidepressant-like activity on LPS-induced depression-related behavior in mice was observed after 3 days of DHM treatment; acute administration of DHM for 1 or 2 days did not elicit antidepressant-like activity. This may be associated with BDNF expression (BoJiang et al. 2016), as increased expression of BDNF in the hippocampus was evident after 3 days of DHM treatment and elevated BDNF expression in primary cultured hippocampal neurons was evident only after 2–3 days of DHM treatment. DHM activates ERK1/2 and GSK-3β, and both ERK1/2 and GSK-3β are known to be involved in the regulation of BDNF expression (Beurel and Jope 2010; Bui et al. 2012; Gimenez-Cassina et al. 2012; Omata et al. 2011; Screaton et al. 2004; Tsai et al. 2008; Xue et al. 2016; Zunszain et al. 2013). Such DHM-induced activation of the ERK1/2-CREB-BDNF pathway may contribute to its antidepressant-like activity, and there is growing evidence that BDNF plays an important role in many neuropsychiatric diseases, including MDD (Duman and Voleti 2012; Mai et al. 2002; Sun et al. 2016; Xu et al. 2016; Xue et al. 2016).
Potential signaling mechanisms for DHM-mediated antidepressant-like activity are indicated in Fig. 6b, but the question of how DHM upregulates the phosphorylation level of ERK1/2 andGSK-3β needs further research. Furthermore, the mechanism(s) underlying the time-dependent induction of BDNF expression by DHM remain to be determined. While our studies focus on BDNF expression in the hippocampus, future studies should also include investigation of pro-BDNF and any role(s) for the prefrontal cortex and nucleus accumbens in these processes.
In summary, the present work demonstrates that (a) DHM elicits a more rapid antidepressant-like effect than does the typical antidepressant venlafaxine and that (b) this effect may involve stimulation of BDNF expression, possibly via activation of the ERK1/2-CREB pathway and inhibition of GSK-3β, and inhibition of neuroinflammation. These findings provide the first evidence that DHM may be a promising candidate for the treatment of MDD.
References
Abe-Higuchi N, Uchida S, Yamagata H, Higuchi F, Hobara T, Hara K, Kobayashi A, Watanabe Y (2016) Hippocampal Sirtuin 1 signaling mediates depression-like behavior. Biol Psychiatry 80:815–826
Beurel E, Jope RS (2010) Glycogen synthase kinase-3 regulates inflammatory tolerance in astrocytes. Neuroscience 169:1063–1070
BoJiang, LuSong, Cheng-NiuWang, WeiZhang, ChaoHuang, Li-JuanTong (2016) Antidepressant-like effects of GM1 ganglioside involving the BDNF signaling cascade in mice. International Journal of Neuropsychopharmacology 19: pyw046
Bui C, Barter MJ, Scott JL, Xu Y, Galler M, Reynard LN, Rowan AD, Young DA (2012) cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. Faseb Journal Official Publication of the Federation of American Societies for. Exp Biol 26:3000–3011
Chan SY, Matthews E, Burnet PW (2016) ON or OFF?: modulating the N-methyl-D-aspartate receptor in major depression. Front Mol Neurosci 9:169
Chen S, Zhao X, Wan J, Ran L, Qin Y, Wang X, Gao Y, Shu F, Zhang Y, Liu P, Zhang Q, Zhu J, Mi M (2015) Dihydromyricetin improves glucose and lipid metabolism and exerts anti-inflammatory effects in nonalcoholic fatty liver disease: a randomized controlled trial. Pharmacol Res 99:74–81
Cheng J, Salton SR (2013) The role of neurotrophins in major depressive disorder. Transl Neurosci 4:46–58
Dale E, Pehrson AL, Jeyarajah T, Li Y, Leiser SC, Smagin G, Olsen CK, Sanchez C (2016) Effects of serotonin in the hippocampus: how SSRIs and multimodal antidepressants might regulate pyramidal cell function. CNS Spectrums 21:143–161
Duman RS, Voleti B (2012) Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci 35:47–56
Dwivedi Y (2013) Involvement of brain-derived neurotrophic factor in late-life depression. Am J Geriatr Psychiatr : Off J Am Assoc Geriatr Psychiatr 21:433–449
Eyre HA, Lavretsky H, Kartika J, Qassim A, Baune BT (2016) Modulatory effects of antidepressant classes on the innate and adaptive immune system in depression. Pharmacopsychiatry 49:85–96
Fang X, Guo L, Jia J, Jin GZ, Zhao B, Zheng YY, Li JQ, Zhang A, Zhen XC (2013) SKF83959 is a novel triple reuptake inhibitor that elicits anti-depressant activity. Acta Pharmacol Sin 34:1149–1155
Gimenez-Cassina A, Lim F, Diaz-Nido J (2012) Chronic inhibition of glycogen synthase kinase-3 protects against rotenone-induced cell death in human neuron-like cells by increasing BDNF secretion. Neurosci Lett 531:182–187
Guan XT, Lin WJ, Tang MM (2015) Comparison of stress-induced and LPS-induced depressive-like behaviors and the alterations of central proinflammatory cytokines mRNA in rats. PsyCh J 4:113–122
Harrisberger F, Smieskova R, Schmidt A, Lenz C, Walter A, Wittfeld K, Grabe HJ, Lang UE, Fusar-Poli P, Borgwardt S (2015) BDNF Val66Met polymorphism and hippocampal volume in neuropsychiatric disorders: a systematic review and meta-analysis. Neurosci Biobehav Rev 55:107–118
Hou XL, Tong Q, Wang WQ, Shi CY, Xiong W, Chen J, Liu X, Fang JG (2015) Suppression of inflammatory responses by dihydromyricetin, a flavonoid from Ampelopsis grossedentata, via inhibiting the activation of NF-kappaB and MAPK signaling pathways. J Nat Prod 78:1689–1696
Hughes MM, Connor TJ, Harkin A (2016) Stress-related immune markers in depression: implications for treatment. Int J Neuropsychopharmacol
Jangra A, Sriram CS, Lahkar M (2016) Lipopolysaccharide-induced behavioral alterations are alleviated by sodium phenylbutyrate via attenuation of oxidative stress and neuroinflammatory cascade. Inflammation 39:1441–1452
Jiang B, Le L, Pan H, Hu K, Xu L, Xiao P (2014) Dihydromyricetin ameliorates the oxidative stress response induced by methylglyoxal via the AMPK/GLUT4 signaling pathway in PC12 cells. Brain Res Bull 109:117–126
Kao SJ, Lee WJ, Chang JH, Chow JM, Chung CL, Hung WY, Chien MH (2017) Suppression of reactive oxygen species-mediated ERK and JNK activation sensitizes dihydromyricetin-induced mitochondrial apoptosis in human non-small cell lung cancer. Environ Toxicol 32:1426–1438
Kaufmann FN, Costa AP, Ghisleni G, Diaz AP, Rodrigues AL, Peluffo H, Kaster MP (2017) NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain Behav Immun
Kurosawa N, Shimizu K, Seki K (2016) The development of depression-like behavior is consolidated by IL-6-induced activation of locus coeruleus neurons and IL-1beta-induced elevated leptin levels in mice. Psychopharmacology 233:1725–1737
Le L, Jiang B, Wan W, Zhai W, Xu L, Hu K, Xiao P (2016) Metabolomics reveals the protective of dihydromyricetin on glucose homeostasis by enhancing insulin sensitivity. Sci Rep 6:36184
Liang J, López-Valdés HE, Martínez-Coria H, Lindemeyer AK, Shen Y, Shao XM, Olsen RW (2014) Dihydromyricetin ameliorates behavioral deficits and reverses neuropathology of transgenic mouse models of Alzheimer’s disease. Neurochem Res 39:1171–1181
Liu S, Ai Q, Feng K, Li Y, Liu X (2016) The cardioprotective effect of dihydromyricetin prevents ischemia-reperfusion-induced apoptosis in vivo and in vitro via the PI3K/Akt and HIF-1alpha signaling pathways. Apoptosis : An Int J Programmed Cell Death 21:1366–1385
Mai L, Jope RS, Li X (2002) BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem 82:75–83
Manosso LM, Moretti M, Ribeiro CM, Goncalves FM, Leal RB, Rodrigues AL (2015) Antidepressant-like effect of zinc is dependent on signaling pathways implicated in BDNF modulation. Prog Neuro-Psychopharmacol Biol Psychiatry 59:59–67
Mao Q, Gong X, Zhou C, Tu Z, Zhao L, Wang L, Wang X, Sun L, Xia J, Lian B, Chen J, Mu J, Yang D, Xie P (2017) Up-regulation of SIRT6 in the hippocampus induced rats with depression-like behavior via the block Akt/GSK3beta signaling pathway. Behav Brain Res 323:38–46
Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X, Cheng J, Jia J, Zhen X (2015) MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun 49:75–85
Nuernberg GL, Aguiar B, Bristot G, Fleck MP, Rocha NS (2016) Brain-derived neurotrophic factor increase during treatment in severe mental illness inpatients. Transl Psychiatry 6:e985
O'Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R (2009) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14:511–522
Omata N, Chiu CT, Moya PR, Leng Y, Wang Z, Hunsberger JG, Leeds P, Chuang DM (2011) Lentivirally mediated GSK-3beta silencing in the hippocampal dentate gyrus induces antidepressant-like effects in stressed mice. Int J Neuropsychopharmacol 14:711–717
Park SE, Lawson M, Dantzer R, Kelley KW, McCusker RH (2011) Insulin-like growth factor-I peptides act centrally to decrease depression-like behavior of mice treated intraperitoneally with lipopolysaccharide. J Neuroinflammation 8:179
Pesarico AP, Sartori G, Bruning CA, Mantovani AC, Duarte T, Zeni G, Nogueira CW (2016) A novel isoquinoline compound abolishes chronic unpredictable mild stress-induced depressive-like behavior in mice. Behav Brain Res 307:73–83
Rapaport MH, Nierenberg AA, Schettler PJ, Kinkead B, Cardoos A, Walker R, Mischoulon D (2015) Inflammation as a predictive biomarker for response to omega-3 fatty acids in major depressive disorder: a proof-of-concept study. Mol Psychiatry 21:71
Ren ZX, Zhao YF, Cao T, Zhen XC (2016) Dihydromyricetin protects neurons in an MPTP-induced model of Parkinson’s disease by suppressing glycogen synthase kinase-3 beta activity. Acta Pharmacol Sin 37:1315–1324
Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Rd YJ, Takemori H (2004) The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119:61
Shen Y, Lindemeyer AK, Gonzalez C, Shao XM, Spigelman I, Olsen RW, Liang J (2012) Dihydromyricetin as a novel anti-alcohol intoxication medication. J Neurosci: Off J Soc Neurosci 32:390–401
Sulakhiya K, Keshavlal GP, Bezbaruah BB, Dwivedi S, Gurjar SS, Munde N, Jangra A, Lahkar M, Gogoi R (2016) Lipopolysaccharide induced anxiety- and depressive-like behaviour in mice are prevented by chronic pre-treatment of esculetin. Neurosci Lett 611:106–111
Sun J, Wang F, Hong G, Pang M, Xu H, Li H, Tian F, Fang R, Yao Y, Liu J (2016) Antidepressant-like effects of sodium butyrate and its possible mechanisms of action in mice exposed to chronic unpredictable mild stress. Neurosci Lett 618:159–166
Tang N, Ma J, Wang KS, Mi C, Lv Y, Piao LX, GH X, Li X, Lee JJ, Jin X (2016) Dihydromyricetin suppresses TNF-alpha-induced NF-kappaB activation and target gene expression. Mol Cell Biochem 422:11–20
Tao W, Dong Y, Su Q, Wang H, Chen Y, Xue W, Chen C, Xia B, Duan J, Chen G (2016) Liquiritigenin reverses depression-like behavior in unpredictable chronic mild stress-induced mice by regulating PI3K/Akt/mTOR mediated BDNF/TrkB pathway. Behav Brain Res 308:177–186
Tong Q, Hou X, Fang J, Wang W, Xiong W, Liu X, Xie X, Shi C (2015) Determination of dihydromyricetin in rat plasma by LC-MS/MS and its application to a pharmacokinetic study. J Pharm Biomed Anal 114:455–461
Tsai SJ, Liou YJ, Hong CJ, YW Y, Chen TJ (2008) Glycogen synthase kinase-3beta gene is associated with antidepressant treatment response in Chinese major depressive disorder. The Pharmacogenomics J 8:384–390
Vogelzangs N, Beekman AT, van Reedt Dortland AK, Schoevers RA, Giltay EJ, de Jonge P, Penninx BW (2014) Inflammatory and metabolic dysregulation and the 2-year course of depressive disorders in antidepressant users. Neuropsychopharmacol : Off Publ Am Coll Neuropsychopharmacol 39:1624–1634
Wang Y, Guo L, Jiang HF, Zheng LT, Zhang A, Zhen XC (2016) Allosteric modulation of sigma-1 receptors elicits rapid antidepressant activity. CNS Neurosci Ther 22:368–377
Wang Y, Lawson MA, Dantzer R, Kelley KW (2010) LPS-induced indoleamine 2,3-dioxygenase is regulated in an interferon-gamma-independent manner by a JNK signaling pathway in primary murine microglia. Brain Behav Immun 24:201–209
Wohleb ES (2016) Neuron-microglia interactions in mental health disorders: “for better, and for worse”. Front Immunol 7:544
Wu Z, Li L, Zheng LT, Xu Z, Guo L, Zhen X (2015) Allosteric modulation of sigma-1 receptors by SKF83959 inhibits microglia-mediated inflammation. J Neurochem 134:904–914
Xie J, Liu J, Chen TM, Lan Q, Zhang QY, Liu B, Dai D, Zhang WD, LP H, Zhu RZ (2015) Dihydromyricetin alleviates carbon tetrachloride-induced acute liver injury via JNK-dependent mechanism in mice. World J Gastroenterol 21:5473–5481
Xu LZ, Xu DF, Han Y, Liu LJ, Sun CY, Deng JH, Zhang RX, Yuan M, Zhang SZ, Li ZM, Xu Y, Li JS, Xie SH, Li SX, Zhang HY, Lu L (2016) BDNF-GSK-3beta-beta-catenin pathway in the mPFC is involved in antidepressant-like effects of Morinda officinalis oligosaccharides in rats. Int J Neuropsychopharmacol
Xue W, Wang W, Gong T, Zhang H, Tao W, Xue L, Sun Y, Wang F, Chen G (2016) PKA-CREB-BDNF signaling regulated long lasting antidepressant activities of Yueju but not ketamine. Sci Rep 6:26331
Yang B, Zhang JC, Han M, Yao W, Yang C, Ren Q, Ma M, Chen QX, Hashimoto K (2016) Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology 233:3647–3657
Youdim KA, Dobbie MS, Kuhnle G, Proteggente AR, Abbott NJ, Rice-Evans C (2003) Interaction between flavonoids and the blood-brain barrier: in vitro studies. J Neurochem 85:180
Zhang JC, Wu J, Fujita Y, Yao W, Ren Q, Yang C, Li SX, Shirayama Y, Hashimoto K (2014) Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int J Neuropsychopharmacol 18
Zhao Y, Wang P, Chen S, Han C, Yan Q, Zheng L, Jia J, Ren Z, Zhen X (2017) Dihydromyricetin protects against cerebral ischemia/reperfusion injury via suppressing microglia-mediated neuroinflammation and activation of ERK1/2-CREB signaling pathway. J Funct Foods 33:76–84
Zhu L, Nang C, Luo F, Pan H, Zhang K, Liu J, Zhou R, Gao J, Chang X, He H, Qiu Y, Wang J, Long H, Liu Y, Yan T (2016) Esculetin attenuates lipopolysaccharide (LPS)-induced neuroinflammatory processes and depressive-like behavior in mice. Physiol Behav 163:184–192
Zunszain PA, Horowitz MA, Cattaneo A, Lupi MM, Pariante CM (2013) Ketamine: synaptogenesis, immunomodulation and glycogen synthase kinase-3 as underlying mechanisms of its antidepressant properties. Mol Psychiatry 18:1236–1241
Acknowledgements
This work was supported by funds from the National Science Foundation of China (81130023, 30825042). Support provided from the Priority Academic Program Development of Jiangsu Higher Education Institutes (PAPD) and the Jiangsu key laboratory grant (BM2013003) is also appreciated.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Fig. S1
(DOC 43 kb)
Rights and permissions
About this article

Cite this article
Ren, Z., Yan, P., Zhu, L. et al. Dihydromyricetin exerts a rapid antidepressant-like effect in association with enhancement of BDNF expression and inhibition of neuroinflammation. Psychopharmacology 235, 233–244 (2018). https://doi.org/10.1007/s00213-017-4761-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4761-z