
Abstract
Biological trace elements are essential for human health. Imbalance in trace element metabolism and homeostasis may play an important role in a variety of diseases and disorders. While the majority of previous researches focused on experimental verification of genes involved in trace element metabolism and those encoding trace element-dependent proteins, bioinformatics study on trace elements is relatively rare and still at the starting stage. This chapter offers an overview of recent progress in bioinformatics analyses of trace element utilization, metabolism, and function, especially comparative genomics of several important metals. The relationship between individual elements and several diseases based on recent large-scale systematic studies such as genome-wide association studies and case-control studies is discussed. Lastly, developments of ionomics and its recent application in human health are also introduced.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Introduction
Biological trace elements refer to those dietary elements which are required in very small amounts (less than 100 mg/day) for the proper growth, development, and physiology of an organism [1]. These micronutrients include iron (Fe), zinc (Zn), copper (Cu), molybdenum (Mo), cobalt (Co), nickel (Ni), manganese (Mn), chromium (Cr), vanadium (V), selenium (Se), iodine (I), and probably other elements. The majority of trace elements are metals. They provide proteins with unique coordination, catalytic, and electron transfer properties and are involved in critical enzymatic activities, immunological reactions, and physiological mechanisms [2, 3]. Due to the important roles these trace elements play in cells, efficient and specific mechanisms are needed to maintain and regulate their concentration, utilization, and storage, especially for those elements whose soluble forms are present in trace amounts in natural environments.
Trace element deficiencies are life-threatening health problems in some regions and populations of the world, which are responsible for a variety of clinical disorders, such as Fe deficiency in anemia patients [4]. Some groups of individuals, such as children, pregnant women, and the elderly, are more likely to develop trace element deficiency. On the other hand, accumulation of inappropriate amounts of certain metals (such as Cu) may result in overload disorders because of high toxicity of these metals [4]. Some trace elements may interact and could interfere with the essential functions of each other. For example, large doses of Zn supplementation can disrupt Cu uptake and lead to neurological problems [5]. In addition, trace element status can be altered in some clinical conditions and may interfere with the efficacy of the treatment [6]. Therefore, homeostasis of trace elements within the body should be carefully maintained to offer their adequate but not toxic levels for biological processes.
Research during the past 20 years has provided lots of evidence of how trace elements are utilized for humans. Marginal or severe trace element imbalances could be considered as risk factors for a variety of diseases, but mechanism of such cause and effect relationships needs a more complete understanding of basic metabolism, regulation, and function of these micronutrients. Previous studies of trace elements and genes involved in trace element metabolism have revealed the complexity of trace element utilization and function in nature. In the recent decade, with the rapid increase in the amount of biological data available (such as genomes, transcriptomes, proteomes, etc.) and a corresponding increase in computational approaches, omics-based and/or bioinformatics analysis of the relationship between trace elements and health or disease has become more and more important. Attempts have been made at a genome-wide level based on high-throughput sequencing techniques, which could improve our understanding of the utilization of trace elements in normal physiological conditions and their variations or dyshomeostasis in disease [7,8,9,10,11,12]. Very recently, the term ionome has been introduced, which is defined as all mineral nutrients and trace elements found in an organism. Several ionome-based studies have identified new features of elemental network for complex diseases such as diabetes and neurodegenerative diseases [13,14,15]. These contributions may not only provide important mechanistic insights into the metabolism and homeostasis of trace elements but facilitate development of new drugs and therapeutic strategies against some of the imbalanced elements.
This chapter focuses on the metabolism and function of several important trace elements in human health as well as their association with the onset and development of diseases mainly from the perspective of bioinformatics and systems biology, such as comparative genomics, genome-wide association study (GWAS), and population and/or cohort studies. Such information may achieve a more integrated and system-level picture of the critical roles these elements play in both physiological and pathological conditions. Recent developments in the study of ionome in diseases (disease ionomics) are also discussed.
4.2 Computational Resource for Trace Elements
4.2.1 Databases
Integration of genes/proteins that bind one or more trace elements for their biological function (say, metalloproteins) and those involved in trace element metabolism from multiple resources (such as large nucleotide/protein databases and literatures) is the basis for understanding their utilization and function in different organisms. In recent years, several trace element-specific web databases have been successfully built up, including MDB, MeRNA, MINAS, dbTEU, MetalPDB, and some other databases.
MDB (the Metalloprotein Database and Browser) is the first web-accessible resource for metalloprotein research, which offers quantitative information on metal-binding sites in protein structures available from the Protein Data Bank (PDB) [16]. MDB also provides tools for analysis of patterns in the metal-binding sites and for prediction of potential metal-binding sites from new protein structures.
MeRNA (metals in RNA) is a database of metal-binding sites identified in RNA structures. It focuses on eight known binding motifs and is used to aid in the study of the roles of metals in RNA biology such as RNA folding and catalysis [17]. Recently, another database of metal ions in nucleic acids (MINAS) has been developed to list all nucleic acid-bound metal ions contained in the PDB, which will be useful to identify new possible metal-binding motifs in nucleic acids [18].
Metal-MACiE is a web-based database that aims to collect the known information on the properties and the roles of metals in catalytic mechanisms of metalloenzymes [19]. This database can be used to advance our understanding of the chemistry underlying metal-dependent catalysis.
dbTEU (DataBase of Trace Element Utilization) is a large protein database of trace element utilization [20]. This manually curated database contains ~16,500 known transporters and user proteins for five trace elements (Cu, Mo, Co, Ni, and Se) in more than 700 organisms from the three domains of life. It also offers interactive tools for search and browse of trace elements, proteins, organisms, and sequences.
Mespeus is a newly developed database of metal interactions with proteins [21]. It lists metal and protein interactions whose geometry has been experimentally determined and could be further visualized.
MetalPDB is a novel resource of metal sites in biological macromolecular structures [22]. This database is achieved through the systematic and automated representation of metal-binding sites in proteins and nucleic acids by way of minimal functional sites (MFSs). The web interface allows access to a comprehensive overview of metal-containing structures, providing a basis to investigate the basic principles governing the properties of these systems.
SelenoDB provides full annotations of Se-containing protein (or selenoprotein) genes in at least 58 animal genomes, which is a valuable resource for addressing medical and evolutionary questions in Se biology [23].
4.2.2 Computational Tools for Trace Element Utilization
Identification of trace element-dependent proteins is not only useful for the inference of protein function but also important for understanding the roles of trace elements. To date, several bioinformatics algorithms and tools have been developed for identification of genes encoding metalloproteins (particularly for Zn and Fe) or selenoproteins in different organisms including humans. Unfortunately, considering that metal-binding properties still remain difficult to predict at the whole-proteome level, it is currently not possible to identify complete sets of metalloproteins in most organisms. Further efforts are needed to identify additional and reliable common features.
An early study reported a software named Zincfinder for the prediction of the Zn-binding proteins based on support vector machine (SVM) learning method [24]. This predictor identified some unprecedented Zn-binding sites and proteins which were further validated through structural modeling. Another SVM and homology-based algorithm was reported to provide higher precision at different levels compared to Zincfinder [25].
TEMSP (3D TEmplate-based Metal Site Prediction) is a structure-based method to predict Zn-binding sites in proteins [26]. This tool improves previously reported methods in predicting Zn-binding proteins with minimum overpredictions. In addition, TEMSP can also predict the Zn-bound local structures, which is helpful for functional analysis.
Zincidentifier software integrates multiple sequence and structural properties and graph-theoretic network features, followed by an efficient feature selection using random forest to improve prediction of Zn-binding sites and proteins [27]. This method can not only be applied to large-scale prediction of Zn-binding sites using structural information but also give valuable insights into new features for characterizing the Zn-binding sites.
ZincExplorer is a new hybrid method for the prediction of Zn-binding sites from protein sequences, which combines the outputs of different types of predictors [28]. It could also identify the interdependent relationships of the predicted Zn-binding sites bound to the same Zn ion.
SIREs (search for iron-responsive elements) is a user-friendly web-based tool for the prediction of iron-responsive elements (IREs) in query genome [29]. This web server provides structure analysis, predicted RNA folds, and an overall quality flag based on properties of well-characterized IREs.
HemeBIND is the first algorithm for heme (an Fe-porphyrin complex)-binding residue prediction in proteins by integrating structural and sequence information such as evolutionary conservation, solvent accessibility, depth, and protrusion [30]. A better performance has been reported when compared with individual classifier alone.
SCMHBP is a novel tool for the prediction and analysis of heme-binding proteins using propensity scores of dipeptides [31]. This approach is based on a scoring card method for predicting and analyzing heme-binding proteins from sequences. SCMHBP performs well relative to comparison with such methods as SVM, decision tree, and Bayes classifiers and improves our understanding of heme-binding proteins rather than merely improves the prediction accuracy in predicting them.
FINDSITE-metal is a threading-based method which is specifically used to detect metal-binding sites in protein structures [32]. It integrates evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. An accuracy of 70–90% could be achieved for Fe, Cu, Zn, and some other metal ions. This algorithm was applied to quantify the metal-binding proteins of the human proteome.
Compared to metalloprotein prediction, computational identification of selenoproteins and the complete set of selenoproteins (selenoproteome) in different organisms, including humans, have been reported. Several programs have been widely used for selenoprotein prediction in different kingdoms, such as SECISearch and bSECISearch tools for prediction of selenoprotein genes in eukaryotes and bacteria, respectively [33, 34]. In addition, a method named Seblastian was also developed to predict new selenoprotein genes in eukaryotes [35].
4.3 Metabolism and Homeostasis of Trace Elements and Their Association with Disease
Trace elements play important roles in all types of cells; as a consequence, the ability of the cell to tightly manage their homeostasis is very important. In eukaryotes, the major processes related to the metabolism of trace elements (especially transition metals) are similar, which include uptake, compartmentalization, storage, and export [36]. High-affinity transport systems have been identified for several metals [37]. Some metal ions could also be transported via unspecific cation influx systems [38]. Excessive uptake of certain elements can be toxic to cell growth. Thus, storage of these elements in inactive sites or forms and export systems are needed to prevent their overload in the cell. It is clear that homeostasis of trace elements should be carefully maintained to provide sufficient levels while preventing accumulation to toxic levels.
The majority of trace elements are directly incorporated into target proteins, whereas some have to form trace element-containing cofactors or complexes (e.g., molybdopterin for Mo, vitamin B12 for Co, and selenocysteine for Se) prior to their insertion into user proteins. A general scheme of metal utilization in eukaryotes is shown in Fig. 4.1. The following sections will focus on several essential trace elements and discuss recent progress on bioinformatics research of their metabolism, physiological roles, and correlation with diseases.

A general scheme of metal metabolism in eukaryotes. The major components involved in metal metabolism and homeostasis include transporters (importers and/or exporters), metal-binding chaperones, and user proteins (metalloproteins). Some metals (such as Mo and Co) have to form metal-containing cofactors prior to their insertion into user proteins
4.3.1 Iron
4.3.1.1 Iron Metabolism and Iron-Binding Proteins
Fe is the second most abundant metal (after aluminum) in the Earth’s crust and is an absolute requirement for all living organisms. This metal is needed for the function of a wide range of enzymes and pathways related to its rich coordination chemistry and redox properties [39]. Besides Fe ions, proteins can bind different forms of Fe-containing cofactors, such as heme or Fe-S clusters. In mammals, Fe is essential for cellular respiration, oxygen transport, energy production, xenobiotic detoxification, and DNA synthesis. On the other hand, redox properties of Fe contribute to its toxicity, which produces reactive oxygen species (ROS) that are harmful to biological molecules. To maintain Fe homeostasis at both the systemic and the cellular levels, mammals have developed a complex machinery to control its intake, utilization, and recycling. In the past several decades, several key findings have shaped our current understanding of Fe metabolism, including the identification of the transferrin (Tf) receptor (TfR), the iron-responsive element/iron-regulatory protein (IRE/IRP), the Fe-regulatory hormone hepcidin, and its target ferroportin (Fpn) [40,41,42,43]. Nevertheless, our current knowledge of Fe biology remains incomplete.
In general, inorganic Fe is initially reduced to Fe2+ by ferrireductase and transported through the cellular membrane by the divalent metal transporter 1 (DMT1) [44]. Organic heme Fe is transported into the cytosol and released by heme oxygenase 1. Excess intracellular Fe is then stored in the storage protein ferritin [45]. Cytosolic Fe is exported into the plasma by the basolateral Fe exporter Fpn [43]. Export of Fe from enterocytes into the blood requires the ferroxidase hephaestin, a multicopper oxidase that oxidizes Fe2+ to Fe3+ [46]. In the plasma, Fe3+ is bound to Tf which delivers Fe to different cells. Most cells acquire Fe via TfR1 (a high-affinity ubiquitously expressed receptor) and TfR2 (restricted to certain cell types with much lower affinity than TfR1). Fe is imported into mitochondria (the major site of Fe utilization) for heme biosynthesis by the transporter protein mitoferrin [47]. However, the mechanism by which heme passes through the mitochondria is poorly understood. Fpn is believed to be the only ferrous Fe exporter. In addition, hepcidin, the key circulating hormone mainly produced by the liver, can systematically modulate Fe homeostasis, which regulates cellular Fe efflux from different cells by binding to Fpn and inducing its internalization and degradation [42, 48].
Recent advances in the study of Fe metabolism have revealed multiple intricate pathways that are essential for maintaining Fe homeostasis. Thus, bioinformatics and systems biology approaches may represent a new strategy for understanding Fe metabolism and its function in proteins. Several computational and dynamic models have been developed to describe the Fe metabolic network and its homeostasis based on microarrays, high-throughput sequencing, and proteomics data, which may shed light on the mechanistic foundations of Fe regulation [49,50,51]. However, key parts of the system remain poorly understood.
To understand the function of Fe in various processes, a more important issue is to identify all Fe-binding proteins. So far, it is very difficult to identify the complete Fe-dependent metalloproteomes by computational approaches. Several bioinformatics studies have been conducted for understanding ferroproteomes, at least partially, in some organisms including humans. One comparative study investigated the occurrence of nonheme Fe-binding proteins based on Fe-binding pattern recognition in a selected number of organisms and found that 90% of Fe-binding proteins have homologs in all three domains of life [52]. The majority of Fe-binding proteins were involved in electron transfer or enzymes performing oxidoreductase functions, suggesting that Fe is mostly used in redox catalysis. Fe-S clusters were the cofactors in about 40% of nonheme Fe proteins retrieved. Another structural analysis of the protein environment around Fe-binding sites revealed that similar sites could be found in unrelated proteins [53]. Very recently, a new algorithm named MetalPredator has been developed for the prediction of the Fe-S proteome [54].
Heme constitutes 95% of functional Fe in the human body. It is a prosthetic group, comprising a ferrous Fe and protoporphyrin IX, and is an essential cofactor in various biological processes [55]. Heme-binding proteins (or hemoproteins) carry out a variety of important functions, such as oxygen transport, electron transfer, and enzyme catalysis. The utilization of heme requires a complex machinery for its biosynthesis, insertion into hemoproteins, and uptake from external sources [55, 56]. Several bioinformatics tools, such as SCMHBP and HemeBIND, have been developed to predict hemoproteins [30, 31]. A genome-wide study investigated the processes of heme biosynthesis and uptake in several hundreds of prokaryotic organisms, which allowed to identify organisms capable of performing none, one, or both processes [57]. Many Gram-positive pathogens support heme uptake from the host, implying that this process can be a potential target for a wide range of antibiotics. Another bioinformatics analysis of all known genes in the heme biosynthesis pathway in animals revealed that these genes are under strong purifying selection from cnidarians to mammals and that multiple-level controls on the activity of this pathway depend on the linear depth of these genes [58]. Further studies on hemoproteins in higher eukaryotes such as mammals are needed.
4.3.1.2 Iron Homeostasis and Diseases
Our knowledge of diseases associated with Fe mainly depends on our understanding of Fe homeostasis. Levels of Fe can be affected by many factors such as genetic variations, diet that contains insufficient or excessive Fe, reduced intake or absorption of Fe, and hemolysis. Therefore, a lot of epidemiological and omics-based studies have focused on examining the association of dietary Fe intake and gene mutations with the risks of common diseases. Many Fe-associated diseases or disorders are attributable to genetic malfunctions that influence the hepcidin/Fpn trait.
Fe overload may lead to Fe deposition in the liver, heart, brain, and some other organs, which promotes the formation of hydroxyl radicals and causes damage to DNA and protein or even cell death. Long-term Fe overload increases the risk of cancer, diabetes mellitus, liver cirrhosis, arthritis, cardiac arrhythmia, heart failure, retinal degeneration, neurodegenerative diseases, and premature death [59, 60]. The main treatments for Fe overload include phlebotomy and Fe chelation therapy [61].
Hemochromatosis is the most common genetic Fe overload disorder and results from mutations in several genes including hemochromatosis protein HFE (involved in transcriptional regulation of hepcidin), TfR2, hemojuvelin, Fpn, and hepcidin, all of which affect the hepcidin/Fpn regulatory axis [62,63,64,65]. The main characteristic of hemochromatosis is the Fe accumulation in vital organs where it may cause cell injury and organ dysfunction.
Aceruloplasminemia is an autosomal recessive disease caused by mutations in the gene encoding ceruloplasmin, a ferroxidase involved in the oxidation of Fe2+ into Fe3+, therefore assisting in Fe transport across the cell membrane [66]. This disease is characterized by a total absence of ceruloplasmin in the blood and accumulation of Fe in hepatocytes, neurons in the brain, and pancreatic islet cells, which in turn leads to diabetes mellitus, neurologic diseases, dementia, and some other diseases.
Fe deficiency is the major cause of anemia. Considering that the majority of total body Fe is used in hemoglobin synthesis, Fe deficiency may affect the production of healthy red blood cells. In addition, deficiency in this metal can result in premature birth, poor growth development, and weak cognitive skills and also affects the nervous system. Changes in diet and Fe supplements can treat minor Fe deficiency, while severe patients may require transfusion of red blood cells or intravenous Fe. It has been reported that a rare mutation in the gene TMPRSS6 encoding matriptase-2 may lead to Fe-refractory Fe-deficiency anemia [67]. As a result, Fe absorption from the intestine and Fe release from macrophages are inhibited, causing severe Fe deficiency [63].
Fe dyshomeostasis in cancer is well known for a long time. The relationship between elevated Fe levels and increased cancer incidence has been debated for years [68]. However, dietary Fe deprivation and Fe chelators have been suggested in cancer therapy, which implies a strong link between Fe-rich environment and cancer [69, 70]. Moreover, levels of TfR1 were observed to be elevated in cancer, which could be used as an anticancer drug target [71]. It was also found that levels of hepcidin were increased and levels of Fpn were decreased in both breast cancer cell lines and patients, implying a direct relationship between intracellular Fe homeostasis and tumor growth [72, 73]. A recent computational study defined the Fe regulatory gene signature which includes TfR1, HFE, and some other genes for outcome prediction in breast cancer patients [74]. Although cancer is definitely more than an Fe disorder, these findings indicate a clear relationship between Fe metabolism and cancer development.
4.3.2 Zinc
Zn plays a pivotal role as a structural, catalytic, and signaling component that can be found in numerous proteins, including enzymes, structural proteins, transcription factors, cytokines, and ribosomal proteins. A global search within the human genome with a bioinformatics approach showed that about 2800 proteins (10% of the human proteome) consist of potential Zn-binding sites [75]. In addition, Zn was suggested to be a fundamental element in the origin of life, and its bioavailability may have been a limiting factor in eukaryotic evolution [76]. Thus, it is expected that Zn metabolism and homeostasis in an organism are tightly controlled. Imbalance in Zn homeostasis has been found to be associated with a variety of human diseases.
4.3.2.1 Zinc Metabolism and Zinc-Dependent Proteome
The biological functions of Zn-binding proteins are maintained through cellular Zn levels, which are mainly regulated by Zn transporters, Zn-sensing molecules (such as metallothioneins), and metal-response element-binding transcription factor-1 (MTF-1) [77,78,79].
In mammals, two groups of Zn transporters have been identified on the basis of their structural and functional features: solute carrier family 39A (SLC39A) that includes mammalian ZRT/IRT-related proteins (ZIPs) [80] and solute carrier family 30A (SLC30A) that comprises mammalian ZnTs [81]. Fourteen ZIP homologs (Zip1 to Zip14) have been identified in the human genome, all of which mediate Zn uptake from the extracellular environment or intracellular vesicles into the cytoplasm. Ten members of the ZnT family (ZnT1 to ZnT10) are responsible for Zn efflux from the cytoplasm toward intracellular vesicles or the extracellular space. The expression of specific ZIPs and ZnTs is tissue dependent and related to specific cellular functions. A recent bioinformatics analysis of the distribution and evolutionary trends of all ZIP family members in eukaryotes suggested that Zip11 is the most ancient Zn transporter that might have originated in early eukaryotic ancestors [82].
Metallothioneins (MTs) are a class of small cytosolic metal-binding proteins that contain one-third cysteine (Cys) residues [83]. These proteins can bind Zn and some other metal ions with high affinity. MTs are thought to be involved in the intracellular regulation of Zn concentration and detoxification of nonessential heavy metals [84]. Four isoforms have been identified so far, the most widely expressed isoforms in mammals being MT1 and MT2. Synthesis of MT is strongly induced by metals, mediated by MTF-1, an important transcription factor for liver development and cell stress response [85]. Under pathological conditions, MTF-1 seems to be involved in tumor angiogenesis and drug resistance. It thus seems generally advisable to monitor MTF-1 activity in stress-related processes including aging and carcinogenesis.
Identification of Zn-binding proteins is important for understanding how Zn is used by different organisms. Thousands of genes encoding Zn-binding proteins have been reported, especially after the completion of genome projects, implying that a great number of biological processes are Zn dependent. In recent years, several comparative genomic studies have been carried out for prediction of Zn-dependent metalloproteomes in the human genome and in genomes of other organisms. Based on known Zn-binding domains and patterns, an early study investigated Zn proteomes in several organisms from the three domains of life [86]. The number of Zn-binding proteins correlated with the proteome size in an organism. In eukaryotes, the majority of Zn proteins are involved in performing enzymatic catalysis and in regulating DNA transcription, especially Zn-finger-containing transcription factors that are almost exclusively a privilege of eukaryotes.
The Zn-binding motifs could also be affected by different functions of Zn-dependent proteins. Four-ligand motifs are often observed in structural sites where Zn contributes to the stability of protein structure, whereas three-ligand motifs (the fourth is often water) are associated with catalytic sites where Zn participates in enzymatic reactions [87]. Moreover, conserved residues in these motifs are quite different. For example, among all predicted Zn-binding proteins in human, 97% have a structural Zn site with at least one Cys ligand and 40% have four Cys ligands [75]. On the other hand, one-third of human Zn proteins containing a three-ligand motif have three histidine (His) residues. The conservation of Zn-finger-binding sites could be associated with their more recent origin, whereas the differentiation of the catalytic Zn-binding sites could be the result of evolutionary processes that led to the development of different enzymatic reactions targeting different physiological substrates [88].
4.3.2.2 Zinc Homeostasis and Diseases
The importance of Zn in human metabolism is illustrated by increasing evidence that points to Zn metabolism as a critical player in the onset or progression of a growing number of multifactorial diseases such as diabetes, Alzheimer’s disease (AD), and asthma. While Zn deficiency is commonly caused by dietary factors, several genetic causes of Zn deficiency have been reported. Therefore, in order to evaluate the influence of Zn on disease risk, it is important to adopt population-based approaches that take into consideration the Zn status and/or genetic variations in the genes encoding proteins that regulate Zn metabolism.
Diabetes is a common metabolic disease characterized by impaired glucose homeostasis and long-term damage, dysfunction, and failure of various organs. It comprises several forms, all of which are associated with varying degrees of hyperglycemia. Type 1 diabetes (T1D) is caused by an autoimmune destruction of islet beta cells leading to little or no insulin production, whereas type 2 diabetes (T2D) is characterized by hyperinsulinemia caused by a failure in the insulin signaling pathway triggered by the activation of the insulin receptor [89]. Zn ions are essential for the normal processing and storage of insulin. Several epidemiological studies have demonstrated that whole-body Zn status is associated with diabetes, including significantly decreased serum Zn levels and increased urinary Zn excretion [90,91,92,93]. Zn supplementation could improve T2D symptoms, both in mice and diabetic patients [94]. However, there is no clear evidence if the use of Zn supplementation would have an effect for the prevention of T2D [95]. On the other hand, Zn imbalance can result not only from insufficient dietary intake but also from impaired function of proteins that regulate Zn metabolism. The first comprehensive GWAS study for T2D demonstrated a link between T2D development and a single nucleotide polymorphism (SNP) rs1326663 in the SLC30A8 gene encoding ZnT8 transporter in diabetic patients [96]. In recent years, several other SNPs in this gene were reported [97, 98]. It is unclear if some of these SNPs can affect glucose homeostasis. Other genetic mutations were also identified, such as a SNP in the promoter region of the MT2A gene which is closely associated with hyperglycemia in old patients with T2D [99]. These human genetic studies highlight the relationship between Zn and glucose homeostasis and diabetes, and further investigation in this direction is very important.
AD is an age-related neurodegenerative disease, characterized by progressive impairment of memory and cognitive abilities. It is commonly thought that AD is caused by the abnormal accumulation and deposition of extracellular senile plaques composed of Cu-Zn aggregates of the amyloid β-peptide (Aβ) [100]. Excess Zn was found to be associated with amyloid plaques, and several studies have indicated that Aβ deposition could be inhibited by Zn chelation [101, 102]. Expression levels of several Zn transporters such as ZnT1, ZnT4, and ZnT6 were increased in early- and late-stage AD subjects [103, 104]. To date, studies on Zn homeostasis and related genes in neurodegenerative diseases are still few and preliminary. Some of these proteins in maintaining brain Zn homeostasis are thought to be important in the onset and progression of AD. In the future, identification of genetic variations in genes controlling Zn homeostasis is essential to establish possible functional links between Zn metabolism and AD.
Asthma is a common chronic inflammatory disease of the airways caused by a combination of genetic and environmental factors. Zn deficiency may play a role in the pathogenesis, control, and severity of asthma [105, 106]. In addition, hair Zn levels were found to be significantly lower in wheezy infants than in healthy controls, implying that Zn deficiency may influence the risk of wheezing in early childhood [107].
Mutations in genes responsible for Zn metabolism have been reported to be associated with additional inherited disorders of Zn deficiency. For example, variations in two Zn transporters Zip4 and ZnT4 are linked to the Zn deficiency diseases acrodermatitis enteropathica syndrome in humans and lethal milk syndrome in mice, respectively [108, 109]. A point mutation in ZnT2 is associated with transient neonatal Zn deficiency [108]. Another population-based study on the genetics of Zn metabolism suggested that a SNP in the MTF-1 gene was associated with lymphoma susceptibility [110]. Mutations in some other human Zn transporters were also observed to be related to a range of diseases, including heart disease and mental illnesses [111]. To date, the functional consequences of these mutations and their interactions with dietary Zn are not known. It remains unclear whether some variations only increase the risk for diseases if dietary Zn levels are inadequate or exceeded.
4.3.3 Copper
Cu is essential for approximately a dozen of proteins and enzymes that carry out fundamental biological functions required for growth and development, such as mitochondrial oxidative phosphorylation, free-radical detoxification, pigmentation, neurotransmitter synthesis, and Fe metabolism [112]. As free cuprous ions react readily with hydrogen peroxide to yield the deleterious hydroxyl radical, it is important for Cu-dependent organisms to have a complex Cu regulatory network to prevent its deficiency and to limit its toxicity. Disrupted Cu homeostasis may lead to excess or toxicity of Cu, which is associated with the pathogenesis of hepatic disorder, neurodegenerative changes, and other disease conditions [113].
4.3.3.1 Copper Metabolism and Cuproproteins
In eukaryotes, cellular Cu trafficking and homeostasis are tightly regulated via a complex system which contains Cu transporters, chaperones, and other components. Cu is acquired by the high-affinity Cu transporter (Ctr) family [114]. Different organisms may possess multiple Ctr proteins located in different biological membranes. In humans, two Ctr proteins (hCtr1 and hCtr2) are identified. hCtr1 is the main Cu importer, which is located predominantly at the plasma membrane, but may also be present in intracellular vesicular compartments [115]. hCtr2 is localized in late endosomes and lysosomes and may be involved in the delivery of Cu ions to the cytosol [116]. Cu export is mediated by an important category of ATP-dependent transporters, the ATP7 family [117]. Mammals have two isoforms: ATP7A and ATP7B [118]. ATP7A is expressed in most tissues (such as the intestinal epithelium, heart, and brain) except the liver, which is required for the transport of Cu into the trans-Golgi network for biosynthesis of several secreted cuproproteins and for basolateral efflux of Cu in the intestine and other cells [119]. ATP7B is predominantly detected in the liver and is required for Cu metalation of ceruloplasmin and biliary Cu excretion [119].
Within the cell, Cu is delivered to specific compartments or cuproproteins by different metallochaperones, including CCS, COX17, and Atox1 [120]. CCS is the Cu chaperone for Cu-Zn superoxide dismutase (Cu-Zn SOD), which delivers Cu in the cytoplasm and intermitochondrial space. COX17 delivers Cu to mitochondria to cytochrome c oxidase (COX) via additional chaperones COX11, SCO1, and SCO2. Atox1 (antioxidant protein 1) is responsible for shuttling Cu from the cytosol to exporters ATP7A and ATP7B. Other proteins involved in Cu homeostasis may exist and might include COMMD1 (Cu metabolism MURR1 domain), metallothionein, and amyloid precursor protein [119, 120]. Plasma protein transport of Cu from the intestine to liver and in systemic circulation probably includes albumin and alpha-2-macroglobulin. Changes in the expression of some of these proteins may help to monitor Cu status of humans.
To date, a number of cuproproteins have been characterized in various organisms. The Cu sites in these proteins could be divided into three types based on spectroscopic and structural properties, and some cuproproteins (such as multicopper oxidases, MCOs) may contain multiple types of Cu centers [121, 122]. Type 1 Cu proteins play important roles in electron transfer in the respiratory and photosynthetic chains of bacteria and plants, including plastocyanin, plantacyanin, and several other proteins [121]. Type 1 Cu center is also found in some larger cuproproteins, such as COX I and COX II, nitrite reductase, and a variety of MCOs (ascorbate oxidase, hephaestin, ceruloplasmin, etc.). Type 2 cuproproteins include Cu-Zn SOD, Cu amine oxidase (CuAO), peptidylglycine R-hydroxylating monooxygenase (PHM), and dopamine β-monooxygenase (DBM) [122]. Other cuproproteins include tyrosinase, hemocyanin, galactose oxidase, and Cnx1G. A list of known cuproprotein families in eukaryotes is shown in Table 4.1.
In recent years, several bioinformatics studies have been carried out to characterize important features of Cu utilization and cuproproteomes (the whole set of cuproproteins) in a variety of organisms [123,124,125,126]. Based on a set of Cu-binding motifs derived from known cuproprotein sequences and domain recognition methods, a computational strategy was developed for examining the occurrence of cuproproteins in several sequenced prokaryotes and eukaryotes [124, 125]. The size of the cuproproteome is generally less than 1% of the total proteome. Recently, several comparative genomic studies examined the Cu utilization trait and cuproproteomes in hundreds of sequenced organisms, which revealed a more clear view of Cu utilization, especially in eukaryotes [123, 127]. Almost all sequenced eukaryotes could utilize Cu. Among all examined cuproprotein families, MCOs, COX I, COX II, and Cu-Zn SOD were the most abundant cuproproteins. The largest cuproproteomes in eukaryotes were found in land plants (e.g., 62 and 78 cuproproteins in Arabidopsis thaliana and Oryza sativa, respectively). Mammals may have approximately 20 known cuproproteins [127].
4.3.3.2 Copper Status and Human Diseases
There are few reports of Cu excess or deficiency in the general population except for formerly obese patients after Roux-en-Y gastric bypass surgery, in whom Cu deficiency was reported with an incidence of 18.8% [128]. On the other hand, several studies have indicated the relationship between dietary Cu and health issues.
High Cu level in the serum has been considered as a potential risk factor for cardiovascular disease in several case-control and population-based studies [129,130,131]. However, dietary Cu intake was not predictive of cardiovascular mortality in a cohort study of older British people [132]. In a cross-sectional study, a negative relationship between dietary or serum Cu and total and LDL (low-density lipoprotein) cholesterol was observed, implying that a high Cu intake and status are associated with a better lipoprotein profile [133]. A second cross-sectional study showed that serum Cu was positively associated with HDL (high-density lipoprotein) cholesterol [134]. Limited evidence also suggests that low Cu diet may lead to premature ventricular discharge and cardiac arrhythmia [135].
The hypothesis that Cu intake might be linked to cognitive decline (such as AD) is based on the well-recognized age-dependent accumulation of some metals (including Cu, Zn, and Fe) in key sites of the brain [136]. An inverse linear association between serum Cu concentrations and cognitive performance was observed in a large cohort of elderly healthy women [137]. A recent study showed a significant inverse correlation of the serum levels of free Cu with both Mini-Mental State Examination (MMSE) and attention-related neuropsychological test scores, suggesting that free Cu appears to be a player in cognitive decline [138, 139]. However, a controversy point was also reported that free serum Cu may increase even when total body Cu decreases, which questions the relevance of free Cu as a marker of Cu exposure [140]. In addition, Cu may promote Aβ aggregation in the brain, and unusually high concentration of Cu has been observed in AD senile plaques [141]. Cu-induced oxidative stress is another mechanism that may lead to profound neurodegenerative processes in AD [142].
The relationship between Cu and cancer has been investigated by some groups. For example, two cohort studies examined the link between Cu intake and lung cancer and lymphoma. In one large cohort study (482,875 subjects), there was no significant association between total Cu intake and lung cancer risk [143]. In another cohort study, no link could be identified between total or dietary Cu and the risk of non-Hodgkin’s lymphoma, diffuse large B-cell lymphoma, or follicular lymphoma [144]. In a study of diet and early breast cancer, the ceruloplasmin/total blood Cu ratio was found to be significantly related to the disease [145]. For other types of cancer, no cohort studies have assessed Cu intake. Thus, no conclusion can be drawn regarding Cu intake and cancers so far.
One cohort study reported that there was no relationship between total (diet and supplements) or dietary Cu intake and risk of rheumatoid arthritis [146]. In spite that the use of Cu supplements showed a weak but significant inverse association with rheumatoid arthritis, such association did not persist after further adjustment for confounders. Thus, no conclusion can be made regarding Cu intake and rheumatoid arthritis.
It has been suggested that Cu may influence the immune system. Animals with severe Cu deficiency have reduced populations of neutrophils and T cells, impaired proliferation of T lymphocytes in response to mitogens, and decreased activity of B lymphocytes, phagocytes, and natural killer cells. However, in humans, the impact of Cu supplementation on immune function is less well documented. The effect of low-Cu diets on immune function has been examined in healthy men, which could significantly inhibit the proliferation of peripheral blood mononuclear cells and increase the fraction of circulating B cells [147]. It seems that the impact of Cu on the immune system can only be observed in specific situations where Cu malabsorption may be combined with low Cu intakes, such as post-bariatric gastric bypass surgery patients [128].
Besides dietary Cu amounts, mutations in genes involved in Cu homeostasis and cuproprotein genes are also associated with severe pathology. It is well known that genetic variations in ATP7A and ATP7B underlie Menkes disease and Wilson’s disease, respectively [148]. Menkes disease is an X-linked inherited disorder, and it is caused by a mutation in the ATP7A gene. Mutations in this gene lead to hypothermia, neuronal degeneration, mental retardation, abnormalities in hair, bone fractures, and aortic aneurysms. Wilson’s disease is an autosomal recessive genetic disorder whose clinical manifestations are liver disease and neurological damage and is caused by disabling mutations in both copies of the ATP7B gene. In addition, mutations of Cu-Zn SOD have been connected with amyotrophic lateral sclerosis, where a gain of function is responsible for the underlying neurological symptomatology [149]. As mentioned above, mutations in the ceruloplasmin allele may lead to aceruloplasminemia [66].
4.3.4 Molybdenum
Mo is an essential transition metal for many living organisms as it is a key component of the active site of molybdoenzymes catalyzing key redox reactions in the metabolism of carbon-, nitrogen-, and sulfur-containing compounds [150,151,152]. With the exception of bacterial nitrogenase, all known molybdoenzymes use the pterin-based Mo cofactor (Moco) [150].
4.3.4.1 Molybdenum Uptake, Molybdenum Cofactor Biosynthesis, and Molybdoproteins
Studies on Mo uptake in eukaryotes are quite limited. Only two types of eukaryotic Mo transporters have been characterized, MOT1 and MOT2 [153, 154]. Mammals only have MOT2 protein. The function of MOT2 in Mo transport or homeostasis is not clear and needs to be examined in the future.
Moco is synthesized by a conserved multistep pathway which includes (i) conversion of GTP into cyclic pyranopterin monophosphate (cPMP), (ii) transformation of cPMP into molybdopterin, and (iii) adenylylation of molybdopterin and subsequent Mo insertion. At least seven proteins (MOCS1A, MOCS1B, MOCS2A, MOCS2B, MOCS3, GEPH-G, and GEPH-E as named in humans) are involved in Moco biosynthesis [155]. Details of these processes have been described in many review articles [150,151,152, 155]. In addition, a Moco sulfurase, catalyzing the generation of the sulfurylated form of Moco that is essential for activation of the xanthine oxidase family of proteins such as xanthine dehydrogenase (XDH) and aldehyde oxidase (AO), has been identified in plants and humans [156].
To date, more than 50 different molybdoenzymes have been found in bacteria. In contrast, only a limited number of molybdoenzymes are present in eukaryotes and can be divided into three classes: the xanthine oxidase (XO) family which is represented by XDH and AO, the sulfite oxidase (SO) family which includes SO and nitrate reductase (NR), and the mitochondrial amidoxime-reducing component mARC family (Table 4.1) [155, 157]. There are five different molybdoenzymes known in humans: XDH, AO, SO, and two isoforms of mARC (mARC1 and mARC2). SO and XDH catalyze catabolic reactions in Cys and purine metabolism, and their structures and reaction mechanisms have been studied intensively. In contrast, functions of AO and mARC enzymes remain unclear, both of which have been suggested to function in drug metabolism [157, 158].
In the recent decade, several bioinformatics studies have focused on the identification of genes involved in Mo uptake and Moco biosynthesis and genes encoding molybdoenzymes in a wide range of sequenced organisms [127, 159, 160]. In eukaryotes, Mo utilization pathway was mainly observed in animals, plants, algae, and certain fungi, whereas parasites and yeasts lack the Mo utilization trait. Essentially all Mo-utilizing organisms have members of the SO and XO families. Plants appeared to have the largest number of molybdoproteins among eukaryotes (10–11 molybdoproteins) [127].
4.3.4.2 Molybdenum Cofactor and Molybdoenzyme Deficiencies
Moco deficiency (MoCD) is a rare inborn error of metabolism causing the loss of all molybdoenzyme activities. The clinical manifestations of MoCD involve intractable neonatal seizures, severe developmental delay, progressive microcephaly with brain atrophy, and even early childhood death [161]. Most of the symptoms of MoCD are very similar to isolated SO deficiency, which is caused by mutations in the SUOX gene (the gene encoding SO) leading to the accumulation of sulfite. Therefore, SO is considered as the most important Moco-dependent enzyme, and sulfite accumulation presents the primary cause of neurological impairment in both disorders [162].
XDH deficiency results in the excessive excretion of xanthine in urine leading to a disease called xanthinuria, which includes type 1 and type 2 [163]. Type 1 xanthinuria is caused by the loss of activity of XDH resulting in an accumulation of xanthine. In contrast, type 2 xanthinuria is caused by the simultaneous loss of activities of XDH and AO due to mutations in the MCSU gene, whose protein product is essential for the sulfuration of Moco in enzymes of the XO family [164]. A very low level of plasma uric acid and high levels of xanthine are hallmarks of both types of xanthinuria. Patients of both groups have similar clinical presentation, mostly due to increased xanthine deposition; however, the mechanism involved in the disease is less clear.
MoCD is mainly caused by mutations in any steps of Mo biosynthetic pathway. Previous studies have identified two types of MoCD: type A and type B. It has been found that MoCD type A patients carry mutations in the MOCS1 gene, while type B patients are defective in MOCS2 [165]. Mutations in the gephyrin gene cause very severe forms of MoCD due to impaired synaptic inhibition.
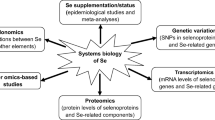
4.3.5 Selenium
Se is an important metalloid in many organisms from bacteria to humans. This micronutrient is known primarily for its functions in redox homeostasis and is recognized as one of the promising cancer chemopreventive agents [166]. It also has a role in antivirus activity, in anti-inflammatory activity, in preventing heart disease and other cardiovascular and muscle disorders, and in delaying the progression of AIDS [167,168,169]. In addition, Se is required for mammalian development, male reproduction, and immune function.
4.3.5.1 Selenocysteine Biosynthesis and Selenoproteins
Se exerts its functions in the form of selenocysteine (Sec), which is co-translationally incorporated into selenoproteins [170]. The biosynthesis of Sec and its incorporation into selenoproteins, which have been reviewed in many other articles, require a complex molecular machinery that recodes UGA codons from stop signals to Sec function [170,171,172]. In eukaryotes, this process needs a cis-acting Sec insertion sequence (SECIS) element which is located in the 3′-untranslated region (3′-UTR) of selenoprotein mRNAs, tRNA[Ser]Sec, and several trans-acting factors dedicated to Sec incorporation. In mammals, proteins and enzymes that are involved in Sec biosynthesis include selenophosphate synthetase 2 (SPS2), Sec synthase (SecS), O-phosphoseryl-tRNA[Ser]Sec kinase, eukaryotic Sec-specific elongation factor (eEFSec), Secp43, SECIS-binding protein 2, and ribosomal protein L30 [173]. Moreover, Sec is usually present in the active site of selenoproteins, being essential for their catalytic activity.
In the past several years, remarkable progress in genome sequencing projects provided an opportunity and resources for selenoprotein identification. Several bioinformatics algorithms have been developed to predict selenoprotein genes in a variety of prokaryotic and eukaryotic genomes [33,34,35]. The general strategy of these approaches is to find candidate SECIS elements and then analyze upstream regions to identify selenoprotein genes. Besides, additional SECIS-independent approaches were developed, which employ Cys-containing proteins and comprehensive protein databases to search nucleotide sequence databases for selenoprotein genes [174]. Based on these tools, a number of novel selenoproteins have been discovered in various organisms [23, 175,176,177]. A complete list of known eukaryotic selenoproteins is shown in Table 4.1. In mammals, a total of 25 and 24 selenoproteins were identified in human and mouse, respectively [175]. The main selenoprotein families include glutathione peroxidases (GPxs) that have oxidoreductase functions and also regulate immune response, thioredoxin reductases (TRs) which modulate transcription and signal transduction functions, iodothyronine deiodinases (Dios) that participate in thyroid hormone metabolism, selenoprotein P (SelP), 15-kDa selenoprotein (Sep15), SPS2, and methionine-R-sulfoxide reductase 1. However, the functions of many eukaryotic selenoproteins are unknown.
Recent comparative analyses of eukaryotic selenoproteomes revealed that significant differences in the composition of selenoproteomes could be seen even among related organisms [176, 178]. The number of selenoproteins varied from 0 (plants, fungi, and some protists) to 56 (Aureococcus anophagefferens) [179]. Among all selenoproteins, selenoprotein K (SelK) and selenoprotein W (SelW) were the two most widespread selenoproteins which are present in most eukaryotes that utilize Sec. The origin of many selenoproteins in mammals can be traced back to the ancestral, unicellular eukaryotes [176]. Many of these selenoproteins were preserved during evolution and remain in mammals and green algae, whereas many other organisms, including land plants, fungi, nematodes, insects, and some protists, manifested massive, independent selenoprotein gene losses. It seems that large selenoproteomes mainly occur in aquatic organisms, whereas the organisms that lack or have few selenoproteins are mostly terrestrial (with the exception of mammals) [176].
4.3.5.2 Selenium Metabolism and Human Disease
As an essential micronutrient, the range of Se intake for human health is narrow, such that low Se intake is associated with developmental defects and disease states and high Se results in toxicity. Recent Se supplementation trials have found that moderately higher Se intake may influence redox status through selenoprotein synthesis to cause T2D [180, 181]. Thus, Se homeostasis needs to be tightly regulated in humans.
Several diseases have been reported to be associated with severe Se deficiency, such as Keshan disease, Kashin-Beck disease, and myxedematous endemic cretinism. Keshan disease was first described as endemic cardiomyopathy with multiple foci of necrosis in the early 1930s in northeastern China, with higher incidence in women and children [182]. It was suggested that Se deficiency in combination with coxsackie virus infection might be required for the development of Keshan disease [182]. Kashin-Beck disease is a chronic, endemic osteochondropathy accompanied by joint necrosis, which affects individuals in Se-deficient areas of China, Siberia, and North Korea [183]. A polymorphism in the GPx1 gene was reported as a potential genetic risk factor in the development of this disease [184]. Myxedematous endemic cretinism is induced by thyroid atrophy and results in mental retardation, which has been observed in those areas of the world with both severe I and Se deficiencies [185].
Se toxicity (selenosis, blood Se level > 100 μg/dL) can be acute or chronic. The symptoms include vomiting, abdominal pain, diarrhea, hair loss, fatigue, irritability, and neurological impairment [186]. Selenosis in humans is a rare event except in very high-Se areas. The famous Se and Vitamin E Cancer Prevention Trial (SELECT) that involved more than 35,000 men revealed the potential risk of T2D, alopecia, and dermatitis due to Se supplements [180].
Se supplementation is prioritized for brain development and function as almost all selenoproteins are expressed in neurons [187]. Recently, mutations of the SecS gene were reported to cause autosomal-recessive progressive cerebellocerebral atrophy (PCCA) in Jews of Iraqi and Moroccan ancestry, which disrupt the biosynthesis of Sec and thus the production of selenoproteins [188]. This disease represents the first clinical syndrome related to Sec biosynthesis in humans.
As mentioned above, previous Se supplementation trials for cancer prevention revealed an over two-fold increase in T2D incidence in the Se-supplemented compared to the placebo group [181, 189, 190]. The SELECT project revealed a similar trend [180]. A recent study reported that SelP is associated with the development of T2D, which may induce insulin resistance in the liver and muscle, resulting in hyperglycemia [191]. Overproduction of GPx1 in mice also resulted in a T2D-like phenotype [192]. In addition, some selenoproteins that are related to ER stress, such as selenoprotein S (SelS) and Dio2, have been found to be involved in the development of T2D. Increased expression of SelS mRNA was observed in human subcutaneous adipocytes from T2D patients [193]. A SNP of human Dio2 (A/G at codon 92) has been identified, which is associated with greater insulin resistance in T2D patients [194].
The association between Se and other diseases, such as cancer, cardiovascular disease, neurodegenerative disease, thyroid disease, and reproductive system disease, has been reported in numerous studies [166, 195, 196]. Among different types of evidence, identification of genetic variants in selenoprotein genes or Se-related genes has shed light on the relationship between Se and disease risk, especially for cancer. Although the mechanistic links between Se levels, selenoproteins, and carcinogenesis are not clear, a significant number of GWAS studies have shown that a small number of SNPs in several selenoprotein genes may influence risk of several cancers, including colorectal, prostate, lung, breast, or bladder cancers [197]. For example, mutations in the coding regions or UTRs of GPx1, GPx4, and SelP genes have functional consequences and could be associated with breast cancer [197, 198]. More SNPs in the promoter, coding region, and UTRs of GPx1, GPx4, SelP, Sep15, SelK, SelS, TR1, and TR2 genes were considered as prostate cancer and/or colorectal cancer risks [197, 198]. Furthermore, mutations in SPS2 and SecS genes were significantly associated with Crohn’s disease [199]. Thus, continued research to study the effects of these mutations on selenoprotein synthesis and Se homeostasis could help to understand the relationship between Se metabolism and different diseases.
4.3.6 Other Trace Elements
Co is mainly used in the form of cobalamin (vitamin B12), a water-soluble cofactor involved in methyl group transfer and rearrangement reactions [200]. A recent comparative genomic analysis revealed that most B12-utilizing eukaryotic organisms are animals (except insects) [201]. Mammals have a unique absorption, delivery, and activation system for vitamin B12. In humans, only two enzymes bind vitamin B12: methionine synthase (MetH) and methylmalonyl coenzyme A mutase (MCM), both of which are important for health. MetH is essential in folate-mediated one-carbon metabolism, including DNA synthesis and chromatin methylation, whereas MCM catabolizes branched-chain and odd-chain fatty acids. Vitamin B12 is required for erythropoiesis, and the classic presentation of vitamin B12 deficiency is hematologic: megaloblastic anemia [202]. Vitamin B12 deficiency also leads to neurologic manifestations which may be irreversible. Cobalamin C disease (CblC) with methylmalonic aciduria and homocystinuria is the most frequent genetic disorder associated with vitamin B12 metabolism, which is caused by an inability of the cell to convert vitamin B12 to its active forms. The typical symptoms may include intrauterine growth retardation, microcephaly, failure to thrive, hypotonia, hydrocephalus, neurological deterioration, hematological abnormalities, and hemolytic uremic syndrome. The MMACHC gene is responsible for the CblC, which may act both as an intracellular vitamin B12 trafficking chaperone and as a decyanase catalyzing the reductive decyanation of cyanocobalamin [203]. To date, more than 75 mutations have been reported in this gene [204].
I is a chemical element required for thyroid hormone production. Early deficiency of this element in life impairs cognition and growth, but its status is also a primary determinant of benign thyroid disorders in adults, such as goiter, nodules, and hyper- and hypothyroidism [205]. In contrast, the role of I intake in thyroid cancer remains unclear, despite decades of studies and debates. To date, studies of thyroid cancer epidemiology in different populations are very challenging because it is still a relatively rare and, in most cases, indolent cancer. The available evidences from several case-control studies imply that I deficiency is a risk factor for thyroid cancer and that it particularly increases risk for follicular thyroid cancer and, possibly, anaplastic thyroid cancer [206].
With regard to other trace elements, a great number of experimental studies have been conducted for understanding their metabolism and function. However, bioinformatics analysis of their utilization in human health and disease is almost completely blank. Therefore, more research efforts are needed in this area.
4.4 Ionomics and Human Health
4.4.1 An Overview of Ionome and Ionomics
In the recent decade, a new term, ionome, has been introduced, which is defined as the mineral nutrients and trace elements of an organism [207]. Ionomics, the study of the ionome, involves quantitative analyses of elemental composition in living systems using high-throughput elemental analysis technologies and their integration with bioinformatics tools [208]. Such an approach has been widely applied in plants in response to physiological stimuli, developmental state, and genetic modifications. It has been shown that ionomics has the ability to help identify genes and gene networks that directly control the ionome. In addition, it may provide a powerful tool to investigate more complex gene networks that control developmental and physiological processes and influence the ionome indirectly [209].
The majority of experimental techniques for elemental analysis include inductively coupled plasma mass spectrometry (ICP-MS), inductively coupled plasma optical emission spectroscopy (ICP-OES), and X-ray fluorescence. Among them, ICP-MS is the most frequently used approach, which is capable of detecting metals and several nonmetals at very low concentration (such as part per trillion). Compared to ICP-OES, ICP-MS allows for a smaller sample size owing to its greater sensitivity and has the ability to detect different isotopes of the same element. Currently, ICP-MS has been successfully used for large-scale ionomic studies in yeast, plants, and mammals, which illustrate the power of ionomics to identify new aspects of trace element metabolism and homeostasis and how such information can be used to develop hypotheses regarding the functions of previously uncharacterized genes [210,211,212,213].
As large-scale ionomic studies may produce large amount of data due to the analysis of hundreds or thousands of samples over a period of time, it is important to develop appropriate information management systems and tools for genome-scale data acquisition, validation, storage, and analysis. The Purdue Ionomics Information Management System (PiiMS) is an example of such a workflow control system, which provides an open-access platform for data processing, mining, and discovery [214]. This system (http://www.ionomicshub.org/home/PiiMS) provides integrated workflow control, data storage, and analysis to facilitate high-throughput data acquisition, along with integrated tools for data search, retrieval, and visualization for hypothesis development. To promote rapid knowledge generation about the ionome and related genes/networks, it is also important that such information should be correctly annotated for further discovery. However, systems to allow researcher-driven annotation of genes involved in trace element metabolism and homeostasis are very limited. With the increase in the number of novel trace element-related genes and their functions, new approaches allowing for such systematized annotation are needed.
Very recently, mechanisms that regulate different trace elements in human HeLa cells were characterized by a genome-wide high-throughput siRNA/ionomics screen [213]. A computational strategy was developed for data processing and advanced analysis. Based on the primary screen data and gene network analysis, a secondary screen was performed, which revealed additional candidate genes involved in the homeostasis of Cu, Se, Fe, and some other trace elements. This ionomic dataset should be useful for further studies on trace element metabolism and homeostasis in humans.
4.4.2 Recent Application of Disease Ionomics
Before the birth of ionomics, ICP-MS has been applied to quantify the levels of multiple trace elements in samples of different diseases for years. For example, an early ICP-MS-based study examined 20 elements in brain tissue, cerebrospinal fluid, serum, and aqueous humor from AD patients and matched control subjects [215]. Another study which determined concentrations of 14 trace elements in blood samples of patients with coronary heart disease (CHD) showed that patients had elevated Co plasma as well as diminished Cu blood concentrations [216]. In recent years, ICP-MS-based elemental distribution analysis has been reported for some other diseases, such as T2D, Parkinson’s disease, viral infection, autism, atherosclerosis, and cancer [217,218,219,220,221]. In spite that related elements were reported for each of these diseases, the associations and interactions among these elements are unknown due to methodological limitation.
With the rapid development of systems biology and statistical approaches, advanced computational strategies have recently been used for systematic analysis of the ionome in several diseases such as T2D, which improves our understanding of the complex interactions among different elements.
Sun et al. measured the fasting plasma elemental concentrations to investigate associations of ion modules/networks with overweight/obesity, metabolic syndrome, and T2D in 976 middle-aged Chinese men and women [13]. Based on mutual information analysis, they constructed disease-related ion networks and found that Cu and phosphorus always ranked the first two among three specific ion networks associated with the above situations. In addition, three ionome patterns were also observed, which provide new clues for studying the relationship between plasma ionome and metabolic disorders. Very recently, another population-based study which analyzed urine ionome of 2115 Chinese aged 55–76 years revealed that increased urinary Ni concentration is associated with elevated prevalence of T2D [14].
Considering that disturbance in metal homeostasis is among many of the factors that lead to the development of malignancy of cancer, one study investigated the relationship between cancer risk and element status in order to support diagnosis of cancer [222]. They analyzed both essential elements (such as Ca, Mg, Zn, Cu, Mn, and Fe) and toxic metals (such as cadmium and lead) in the samples of hair and nails obtained from patients with larynx cancer and healthy subjects. Levels of the majority of examined essential elements were significantly decreased in patients, while the opposite trend was observed for the heavy metals. In addition, a variety of statistical data mining approaches have been used for the prediction of cancer probability, and the best results were obtained using logistic regression, artificial neural networks, and canonical discriminant analysis. These constructed classifiers can be useful for estimating cancer risk and early screening of the disease.
The utilization of ionomic techniques was also reported for some other diseases. For example, very recently one study examined the concentration of metals in saliva and blood for periodontal disease [223]. They used clustering approaches in the classification of samples of saliva based on the concentration of selected metals. The results of cluster analysis suggested that the metal profiles of saliva in those with periodontal disease are different from the controls, which may become a basis for the future development of diagnostic and prognostic biomarkers for periodontal disease. In another study, researchers quantified the concentrations of multiple trace elements in plasma from 238 patients of Parkinson’s disease and 302 controls, which is so far the largest cohort for measuring plasma levels of these elements [15]. It was found that lower plasma Se and Fe levels might reduce the risk for this disease, whereas lower plasma Zn was probably a disease risk factor. Finally, a SVM model was built to predict patients based on the plasma concentrations of several trace elements as well as other features such as sex and age, which achieved a good performance. In the future, new computational strategies and algorithms should be developed to improve ionomic studies.
4.5 Conclusions
Bioinformatics and system-level approaches have given powerful support for studying the metabolism, homeostasis, and function of trace elements as well as their relationship with a variety of diseases. This chapter describes recent studies that used bioinformatics and related methods to better understand the general principles of utilization of several essential trace elements. In addition, recent case-control- or population-based studies of individual elements in different diseases and disease ionomics have provided significant advances in discovering new relationship between trace element homeostasis and disease onset and progression. Nevertheless, it should be admitted that the usage of bioinformatics in the field of trace element research is still limited. In the future, with the increased availability of genome/transcriptome/proteome data and improved techniques for ionomics, bioinformatics and computational systems biology will play a significant role in studies on the roles that trace elements play in human health and disease.
References
Mertz W. The essential trace elements. Science. 1981;213:1332–8.
Xiu YM. Trace elements in health and diseases. Biomed Environ Sci. 1996;9:130–6.
Mertz W. Review of the scientific basis for establishing the essentiality of trace elements. Biol Trace Elem Res. 1998;66:185–91.
Van Gossum A, Neve J. Trace element deficiency and toxicity. Curr Opin Clin Nutr Metab Care. 1998;1:499–507.
Bremner I, Beattie JH. Copper and zinc metabolism in health and disease: speciation and interactions. Proc Nutr Soc. 1995;54:489–99.
West C, Hautvast J. Nutrition. From ‘whither’ to ‘wither’ micronutrient malnutrition. Lancet. 1997;350:111–5.
Pande MB, Nagabhushan P, Hegde ML, Rao TS, Rao KS. An algorithmic approach to understand trace elemental homeostasis in serum samples of Parkinson disease. Comput Biol Med. 2005;35:475–93.
Burkhead JL, Gray LW, Lutsenko S. Systems biology approach to Wilson’s disease. Biometals. 2011;24:455–66.
Lancaster WA, Praissman JL, Poole FL 2nd, Cvetkovic A, Menon AL, Scott JW, Jenney FE Jr, Thorgersen MP, Kalisiak E, Apon JV, et al. A computational framework for proteome-wide pursuit and prediction of metalloproteins using ICP-MS and MS/MS data. BMC Bioinformatics. 2011;12:64.
Mitchell S, Mendes P. A computational model of liver iron metabolism. PLoS Comput Biol. 2013;9:e1003299.
Andreini C, Bertini I. A bioinformatics view of zinc enzymes. J Inorg Biochem. 2012;111:150–6.
Zhu L, Chen X, Kong X, Cai YD. Investigation of the roles of trace elements during hepatitis C virus infection using protein-protein interactions and a shortest path algorithm. Biochem Biophys Acta. 2016.; pii: S0304–4165(16)30160-X
Sun L, Yu Y, Huang T, An P, Yu D, Yu Z, Li H, Sheng H, Cai L, Xue J, et al. Associations between ionomic profile and metabolic abnormalities in human population. PLoS One. 2012;7:e38845.
Liu G, Sun L, Pan A, Zhu M, Li Z, Wang Z, Liu X, Ye X, Li H, Zheng H, et al. Nickel exposure is associated with the prevalence of type 2 diabetes in Chinese adults. Int J Epidemiol. 2015;44:240–8.
Zhao HW, Lin J, Wang XB, Cheng X, Wang JY, Hu BL, Zhang Y, Zhang X, Zhu JH. Assessing plasma levels of selenium, copper, iron and zinc in patients of Parkinson’s disease. PLoS One. 2013;8:e83060.
Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: the Metalloprotein database and browser at the Scripps research institute. Nucleic Acids Res. 2002;30:379–82.
Stefan LR, Zhang R, Levitan AG, Hendrix DK, Brenner SE, Holbrook SR. MeRNA: a database of metal ion binding sites in RNA structures. Nucleic Acids Res. 2006;34:D131–4.
Schnabl J, Suter P, Sigel RK. MINAS—a database of metal ions in nucleic AcidS. Nucleic Acids Res. 2012;40:D434–8.
Andreini C, Bertini I, Cavallaro G, Holliday GL, Thornton JM. Metal-MACiE: a database of metals involved in biological catalysis. Bioinformatics. 2009;25:2088–9.
Zhang Y, Gladyshev VN. dbTEU: a protein database of trace element utilization. Bioinformatics. 2010;26:700–2.
Harding MM, Hsin KY. Mespeus—a database of metal interactions with proteins. Methods Mol Biol. 2014;1091:333–42.
Andreini C, Cavallaro G, Lorenzini S, Rosato A. MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2013;41:D312–9.
Romagné F, Santesmasses D, White L, Sarangi GK, Mariotti M, Hübler R, Weihmann A, Parra G, Gladyshev VN, Guigó R, et al. SelenoDB 2.0: annotation of selenoprotein genes in animals and their genetic diversity in humans. Nucleic Acids Res. 2014;42:D437–43.
Passerini A, Andreini C, Menchetti S, Rosato A, Frasconi P. Predicting zinc binding at the proteome level. BMC Bioinformatics. 2007;8:39.
Shu N, Zhou T, Hovmöller S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics. 2008;24:775–82.
Zhao W, Xu M, Liang Z, Ding B, Niu L, Liu H, Teng M. Structure-based de novo prediction of zinc-binding sites in proteins of unknown function. Bioinformatics. 2011;27:1262–8.
Zheng C, Wang M, Takemoto K, Akutsu T, Zhang Z, Song J. An integrative computational framework based on a two-step random forest algorithm improves prediction of zinc-binding sites in proteins. PLoS One. 2012;7:e49716.
Chen Z, Wang Y, Zhai YF, Song J, Zhang Z. ZincExplorer: an accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Mol BioSyst. 2013;9:2213–22.
Campillos M, Cases I, Hentze MW, Sanchez M. SIREs: searching for iron-responsive elements. Nucleic Acids Res. 2010;38:W360–7.
Liu R, Hu J. HemeBIND: a novel method for heme binding residue prediction by combining structural and sequence information. BMC Bioinformatics. 2011;12:207.
Liou YF, Charoenkwan P, Srinivasulu Y, Vasylenko T, Lai SC, Lee HC, Chen YH, Huang HL, Ho SY. SCMHBP: prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinformatics. 2014;15:S4.
Brylinski M, Skolnick J. FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins. 2011;79:735–51.
Kryukov GV, Kryukov VM, Gladyshev VN. New mammalian selenocysteine-containing proteins identified with an algorithm that searches for selenocysteine insertion sequence elements. J Biol Chem. 1999;274:33888–97.
Zhang Y, Gladyshev VN. An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics. 2005;21:2580–9.
Mariotti M, Lobanov AV, Guigo R, Gladyshev VN. SECISearch3 and Seblastian: new tools for prediction of SECIS elements and selenoproteins. Nucleic Acids Res. 2013;41:e149.
Ba LA, Doering M, Burkholz T, Jacob C. Metal trafficking: from maintaining the metal homeostasis to future drug design. Metallomics. 2009;1:292–311.
Bird AJ. Cellular sensing and transport of metal ions: implications in micronutrient homeostasis. J Nutr Biochem. 2015;26:1103–15.
Ackland ML, McArdle HJ. Cation-dependent uptake of zinc in human fibroblasts. Biometals. 1996;9:29–37.
Hider RC, Kong X. Iron: effect of overload and deficiency. Met Ions Life Sci. 2013;13:229–94.
Seligman PA. Structure and function of the transferrin receptor. Prog Hematol. 1983;13:131–47.
Henderson BR, Kühn LC. Interaction between iron-regulatory proteins and their RNA target sequences, iron-responsive elements. Prog Mol Subcell Biol. 1997;18:117–39.
Fleming RE, Sly WS. Hepcidin: a putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc Natl Acad Sci U S A. 2001;98:8160–2.
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–81.
Andrews NC. The iron transporter DMT1. Int J Biochem Cell Biol. 1999;31:991–4.
Chifman J, Laubenbacher R, Torti SV. A systems biology approach to iron metabolism. Adv Exp Med Biol. 2014;844:201–25.
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–9.
Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3.
Lao BJ, Kamei DT. A compartmental model of iron regulation in the mouse. J Theor Biol. 2006;243:542–54.
Lopes TJ, Luganskaja T, Vujić Spasić M, Hentze MW, Muckenthaler MU, Schümann K, Reich JG. Systems analysis of iron metabolism: the network of iron pools and fluxes. BMC Syst Biol. 2010;4:112.
Chifman J, Kniss A, Neupane P, Williams I, Leung B, Deng Z, Mendes P, Hower V, Torti FM, Akman SA, et al. The core control system of intracellular iron homeostasis: a mathematical model. J Theor Biol. 2012;300:91–9.
Andreini C, Banci L, Bertini I, Elmi S, Rosato A. Non-heme iron through the three domains of life. Proteins. 2007;67:317–24.
Andreini C, Bertini I, Cavallaro G, Najmanovich RJ, Thornton JM. Structural analysis of metal sites in proteins: non-heme iron sites as a case study. J Mol Biol. 2009;388:356–80.
Valasatava Y, Rosato A, Banci L, Andreini C. MetalPredator: a web server to predict iron-sulfur cluster binding proteomes. Bioinformatics. 2016;32:2850–2. pii:btw238
Barupala DP, Dzul SP, Riggs-Gelasco PJ, Stemmler TL. Synthesis, delivery and regulation of eukaryotic heme and Fe-S cluster cofactors. Arch Biochem Biophys. 2016;592:60–75.
Hooda J, Shah A, Zhang L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Forum Nutr. 2014;6:1080–102.
Cavallaro G, Decaria L, Rosato A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J Proteome Res. 2008;7:4946–54.
Tzou WS, Chu Y, Lin TY, Hu CH, Pai TW, Liu HF, Lin HJ, Cases I, Rojas A, Sanchez M, You ZY, Hsu MW. Molecular evolution of multiple-level control of heme biosynthesis pathway in animal kingdom. PLoS One. 2014;9:e86718.
Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341:1986–95.
Andrews NC. Iron metabolism: iron deficiency and iron overload. Annu Rev Genomics Hum Genet. 2000;1:75–98.
Gumienna-Kontecka E, Pyrkosz-Bulska M, Szebesczyk A, Ostrowska M. Iron chelating strategies in systemic metal overload, neurodegeneration and cancer. Curr Med Chem. 2014;21:3741–67.
Barton JC. Hemochromatosis and iron overload: from bench to clinic. Am J Med Sci. 2013;346:403–12.
Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117:4425–33.
Del-Castillo-Rueda A, Moreno-Carralero MI, Cuadrado-Grande N, Alvarez-Sala-Walther LA, Enríquez-de-Salamanca R, Méndez M, Morán-Jiménez MJ. Mutations in the HFE, TFR2, and SLC40A1 genes in patients with hemochromatosis. Gene. 2012;508:15–20.
Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180–6.
Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci U S A. 1995;92:2539–43.
Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, Mayo MM, Samuel SM, Strouse JJ, Markianos K, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet. 2008;40:569–71.
Torti SV, Torti FM. Ironing out cancer. Cancer Res. 2011;71:1511–4.
Hann HW, Stahlhut MW, Blumberg BS. Iron nutrition and tumor growth: decreased tumor growth in iron-deficient mice. Cancer Res. 1988;48:4168–70.
Corcé V, Gouin SG, Renaud S, Gaboriau F, Deniaud D. Recent advances in cancer treatment by iron chelators. Bioorg Med Chem Lett. 2016;26:251–6.
Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet. 1980;2:390–2.
Pinnix ZK, Miller LD, Wang W, D’Agostino R Jr, Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43ra56.
Pan X, Lu Y, Cheng X, Wang J. Hepcidin and ferroportin expression in breast cancer tissue and serum and their relationship with anemia. Curr Oncol. 2016;23:e24–6.
Miller LD, Coffman LG, Chou JW, Black MA, Bergh J, D’Agostino R Jr, Torti SV, Torti FM. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011;71:6728–37.
Andreini C, Banci L, Bertini I, Rosato A. Counting the zinc-proteins encoded in the human genome. J Proteome Res. 2006;5:196–201.
Dupont CL, Butcher A, Valas RE, Bourne PE, Caetano-Anollés G. History of biological metal utilization inferred through phylogenomic analysis of protein structures. Proc Natl Acad Sci U S A. 2010;107:10567–72.
Gaither LA, Eide DJ. Eukaryotic zinc transporters and their regulation. Biometals. 2001;14(3–4):251–70.
Maret W. The function of zinc metallothionein: a link between cellular zinc and redox state. J Nutr. 2000;130:1455S–8S.
Andrews GK. Cellular zinc sensors: MTF-1 regulation of gene expression. Biometals. 2001;14:223–37.
Eide DJ. The SLC39 family of metal ion transporters. Pflugers Arch. 2004;447:796–800.
Palmiter RD, Huang L. Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch. 2004;447:744–51.
Yu Y, Wu A, Zhang Z, Yan G, Zhang F, Zhang L, Shen X, Hu R, Zhang Y, Zhang K, et al. Characterization of the GufA subfamily member SLC39A11/Zip11 as a zinc transporter. J Nutr Biochem. 2013;24:1697–708.
Kimura T, Kambe T. The functions of Metallothionein and ZIP and ZnT transporters: an overview and perspective. Int J Mol Sci. 2016;17:336.
Vasák M, Hasler DW. Metallothioneins: new functional and structural insights. Curr Opin Chem Biol. 2000;4:177–83.
Lichtlen P, Schaffner W. The “metal transcription factor” MTF-1: biological facts and medical implications. Swiss Med Wkly. 2001;131:647–52.
Andreini C, Banci L, Bertini I, Rosato A. Zinc through the three domains of life. J Proteome Res. 2006;5:3173–8.
Vallee BL, Auld DS. Cocatalytic zinc motifs in enzyme catalysis. Proc Natl Acad Sci U S A. 1993;90:2715–8.
Andreini C, Bertini I, Rosato A. Metalloproteomes: a bioinformatic approach. Acc Chem Res. 2009;42:1471–9.
Chausmer AB. Zinc, insulin and diabetes. J Am Coll Nutr. 1998;17:109–15.
El-Yazigi A, Hannan N, Raines DA. Effect of diabetic state and related disorders on the urinary excretion of magnesium and zinc in patients. Diabetes Res. 1993;22:67–75.
Garg VK, Gupta R, Goyal RK. Hypozincemia in diabetes mellitus. J Assoc Physicians India. 1994;42:720–1.
Basaki M, Saeb M, Nazifi S, Shamsaei HA. Zinc, copper, iron, and chromium concentrations in young patients with type 2 diabetes mellitus. Biol Trace Elem Res. 2012;148:161–4.
Jansen J, Rosenkranz E, Overbeck S, Warmuth S, Mocchegiani E, Giacconi R, Weiskirchen R, Karges W, Rink L. Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J Nutr Biochem. 2012;23:1458–66.
Vardatsikos G, Pandey NR, Srivastava AK. Insulino-mimetic and anti-diabetic effects of zinc. J Inorg Biochem. 2013;120:8–17.
El Dib R, Gameiro OL, Ogata MS, Módolo NS, Braz LG, Jorge EC. Do Nascimento P Jr, Beletate V. Zinc supplementation for the prevention of type 2 diabetes mellitus in adults with insulin resistance. Cochrane Database Syst Rev. 2015;5:CD005525.
Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–5.
Davidson HW, Wenzlau JM, O’Brien RM. Zinc transporter 8 (ZnT8) and β cell function. Trends Endocrinol Metab. 2014;25:415–24.
Huang Q, Yin JY, Dai XP, Wu J, Chen X, Deng CS, Yu M, Gong ZC, Zhou HH, Liu ZQ. Association analysis of SLC30A8 rs13266634 and rs16889462 polymorphisms with type 2 diabetes mellitus and repaglinide response in Chinese patients. Eur J Clin Pharmacol. 2010;66:1207–15.
Giacconi R, Cipriano C, Muti E, Costarelli L, Maurizio C, Saba V, Gasparini N, Malavolta M, Mocchegiani E. Novel -209A/G MT2A polymorphism in old patients with type 2 diabetes and atherosclerosis: relationship with inflammation (IL-6) and zinc. Biogerontology. 2005;6:407–13.
Tõugu V, Tiiman A, Palumaa P. Interactions of Zn(II) and cu(II) ions with Alzheimer’s amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics. 2011;3:250–61.
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–91.
Cuajungco MP, Fagét KY, Huang X, Tanzi RE, Bush AI. Metal chelation as a potential therapy for Alzheimer’s disease. Ann N Y Acad Sci. 2000;920:292–304.
Lovell MA, Smith JL, Xiong S, Markesbery WR. Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer’s disease. Neurotox Res. 2005;7:265–71.
Smith JL, Xiong S, Markesbery WR, Lovell MA. Altered expression of zinc transporters-4 and -6 in mild cognitive impairment, early and late Alzheimer’s disease brain. Neuroscience. 2006;140:879–88.
Arik Yilmaz E, Ozmen S, Bostanci I, Misirlioglu ED, Ertan U. Erythrocyte zinc levels in children with bronchial asthma. Pediatr Pulmonol. 2011;46:1189–93.
Murgia C, Lang CJ, Truong-Tran AQ, Grosser D, Jayaram L, Ruffin RE, Perozzi G, Zalewski PD. Zinc and its specific transporters as potential targets in airway disease. Curr Drug Targets. 2006;7:607–27.
Tahan F, Karakukcu C. Zinc status in infantile wheezing. Pediatr Pulmonol. 2006;41:630–4.
Kasana S, Din J, Maret W. Genetic causes and gene–nutrient interactions in mammalian zinc deficiencies: acrodermatitis enteropathica and transient neonatal zinc deficiency as examples. J Trace Elem Med Biol. 2015;29:47–62.
Ackland ML, Michalczyk A. Zinc deficiency and its inherited disorders -a review. Genes Nutr. 2006;1:41–9.
Tamura Y, Maruyama M, Mishima Y, Fujisawa H, Obata M, Kodama Y, Yoshikai Y, Aoyagi Y, Niwa O, Schaffner W, et al. Predisposition to mouse thymic lymphomas in response to ionizing radiation depends on variant alleles encoding metal-responsive transcription factor-1 (Mtf-1). Oncogene. 2005;24:399–406.
Devirgiliis C, Zalewski PD, Perozzi G, Murgia C. Zinc fluxes and zinc transporter genes in chronic diseases. Mutat Res. 2007;622:84–93.
Peña MM, Lee J, Thiele DJ. A delicate balance: homeostatic control of copper uptake and distribution. J Nutr. 1999;129:1251–60.
Gaetke LM, Chow-Johnson HS, Chow CK. Copper: toxicological relevance and mechanisms. Arch Toxicol. 2014;88:1929–38.
Petris MJ. The SLC31 (Ctr) copper transporter family. Pflugers Arch. 2004;447:752–5.
Klomp AE, Tops BB, Van Denberg IE, Berger R, Klomp LW. Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1). Biochem J. 2002;364:497–505.
van den Berghe PV, Folmer DE, Malingré HE, van Beurden E, Klomp AE, van de Sluis B, Merkx M, Berger R, Klomp LW. Human copper transporter 2 is localized in late endosomes and lysosomes and facilitates cellular copper uptake. Biochem J. 2007;407:49–59.
Southon A, Palstra N, Veldhuis N, Gaeth A, Robin C, Burke R, Camakaris J. Conservation of copper-transporting P(IB)-type ATPase function. Biometals. 2010;23:681–94.
Linz R, Lutsenko S. Copper-transporting ATPases ATP7A and ATP7B: cousins, not twins. J Bioenerg Biomembr. 2007;39:403–7.
Prohaska JR. Role of copper transporters in copper homeostasis. Am J Clin Nutr. 2008;88:826S–9S.
Prohaska JR, Gybina AA. Intracellular copper transport in mammals. J Nutr. 2004;134:1003–6.
Sakurai T, Kataoka K. Structure and function of type I copper in multicopper oxidases. Cell Mol Life Sci. 2007;64:2642–56.
MacPherson IS, Murphy ME. Type-2 copper-containing enzymes. Cell Mol Life Sci. 2007;64:2887–99.
Ridge PG, Zhang Y, Gladyshev VN. Comparative genomic analyses of copper transporters and cuproproteomes reveal evolutionary dynamics of copper utilization and its link to oxygen. PLoS One. 2008;3:e1378.
Andreini C, Bertini I, Rosato A. A hint to search for metalloproteins in gene banks. Bioinformatics. 2004;20:1373–80.
Andreini C, Banci L, Bertini I, Rosato A. Occurrence of copper proteins through the three domains of life: a bioinformatic approach. J Proteome Res. 2008;7:209–16.
Decaria L, Bertini I, Williams RJ. Copper proteomes, phylogenetics and evolution. Metallomics. 2011;3:56–60.
Zhang Y, Gladyshev VN. General trends in trace element utilization revealed by comparative genomic analyses of co, cu, Mo, Ni, and se. J Biol Chem. 2010;285:3393–405.
Gletsu-Miller N, Broderius M, Frediani JK, Zhao VM, Griffith DP, Davis SS Jr, Sweeney JF, Lin E, Prohaska JR, Ziegler TR. Incidence and prevalence of copper deficiency following roux-en-y gastric bypass surgery. Int J Obes. 2012;36:328–35.
Singh MM, Singh R, Khare A, Gupta MC, Patney NL, Jain VK, Goyal SP, Prakash V, Pandey DN. Serum copper in myocardial infarction--diagnostic and prognostic significance. Angiology. 1985;36:504–10.
Kok FJ, Van Duijn CM, Hofman A, Van der Voet GB, De Wolff FA, Paays CH, Valkenburg HA. Serum copper and zinc and the risk of death from cancer and cardiovascular disease. Am J Epidemiol. 1988;128:352–9.
Salonen JT, Salonen R, Korpela H, Suntioinen S, Tuomilehto J. Serum copper and the risk of acute myocardial infarction: a prospective population study in men in eastern Finland. Am J Epidemiol. 1991;134:268–76.
Bates CJ, Hamer M, Mishra GD. Redox-modulatory vitamins and minerals that prospectively predict mortality in older British people: the National Diet and nutrition survey of people aged 65 years and over. Br J Nutr. 2011;105:123–32.
Bo S, Durazzo M, Gambino R, Berutti C, Milanesio N, Caropreso A, Gentile L, Cassader M, Cavallo-Perin P, Pagano G. Associations of dietary and serum copper with inflammation, oxidative stress, and metabolic variables in adults. J Nutr. 2008;138:305–10.
Ghayour-Mobarhan M, Taylor A, New SA, Lamb DJ, Ferns GA. Determinants of serum copper, zinc and selenium in healthy subjects. Ann Clin Biochem. 2005;42:364–75.
Milne DB, Nielsen FH. Effects of a diet low in copper on copper-status indicators in postmenopausal women. Am J Clin Nutr. 1996;63:358–64.
Zatta P, Drago D, Zambenedetti P, Bolognin S, Nogara E, Peruffo A, Cozzi B. Accumulation of copper and other metal ions, and metallothionein I/II expression in the bovine brain as a function of aging. J Chem Neuroanat. 2008;36:1–5.
Lam PK, Kritz-Silverstein D, Barrett Connor E, Milne D, Nielsen F, Gamst A, Morton D, Wingard D. Plasma trace elements and cognitive function in older men and women: the rancho Bernardo study. J Nutr Health Aging. 2008;12:22–7.
Salustri C, Barbati G, Ghidoni R, Quintiliani L, Ciappina S, Binetti G, Squitti R. Is cognitive function linked to serum free copper levels? A cohort study in a normal population. Clin Neurophysiol. 2010;121:502–7.
Brewer GJ. Copper toxicity in Alzheimer’s disease: cognitive loss from ingestion of inorganic copper. J Trace Elem Med Biol. 2012;26:89–92.
Klevay LM. Copper and cognition. Clin Neurophysiol. 2010;121:2177.
Noda Y, Asada M, Kubota M, Maesako M, Watanabe K, Uemura M, Kihara T, Shimohama S, Takahashi R, Kinoshita A, et al. Copper enhances APP dimerization and promotes Aβ production. Neurosci Lett. 2013;547:10–5.
Eskici G, Axelsen PH. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry. 2012;51:6289–311.
Mahabir S, Forman MR, Dong YQ, Park Y, Hollenbeck A, Schatzkin A. Mineral intake and lung cancer risk in the NIH-American Association of retired persons diet and health study. Cancer Epidemiol Biomark Prev. 2010;19:1976–83.
Thompson CA, Habermann TM, Wang AH, Vierkant RA, Folsom AR, Ross JA, Cerhan JR. Antioxidant intake from fruits, vegetables and other sources and risk of non-Hodgkin’s lymphoma: the Iowa Women’s health study. Int J Cancer. 2010;126:992–1003.
Dabek JT, Hyvönen-Dabek M, Kupila-Rantala T, Hãrkönen M, Adlercreutz H. Early breast cancer, diet, and plasma copper fractions. Ann Clin Lab Sci. 1996;26:215–26.
Cerhan JR, Saag KG, Merlino LA, Mikuls TR, Criswell LA. Antioxidant micronutrients and risk of rheumatoid arthritis in a cohort of older women. Am J Epidemiol. 2003;157:345–54.
Kelley DS, Daudu PA, Taylor PC, Mackey BE, Turnlund JR. Effects of low-copper diets on human immune response. Am J Clin Nutr. 1995;62:412–6.
de Bie P, Muller P, Wijmenga C, Klomp LW. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673–88.
Kaur SJ, McKeown SR, Rashid S. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene. 2016;577:109–18.
Rajagopalan KV, Johnson JL. The pterin molybdenum cofactors. J Biol Chem. 1992;267:10199–202.
Hille R, Hall J, Basu P. The mononuclear molybdenum enzymes. Chem Rev. 2014;114:3963–4038.
Schwarz G, Mendel RR. Molybdenum cofactor biosynthesis and molybdenum enzymes. Annu Rev Plant Biol. 2006;57:623–47.
Tejada-Jiménez M, Llamas A, Sanz-Luque E, Galván A, Fernández E. A high-affinity molybdate transporter in eukaryotes. Proc Natl Acad Sci U S A. 2007;104:20126–30.
Tejada-Jiménez M, Galván A, Fernández E. Algae and humans share a molybdate transporter. Proc Natl Acad Sci U S A. 2011;108:6420–5.
Mendel RR, Bittner F. Cell biology of molybdenum. Biochim Biophys Acta. 1763;2006:621–35.
Bittner F, Oreb M, Mendel RR. ABA3 is a molybdenum cofactor sulfurase required for activation of aldehyde oxidase and xanthine dehydrogenase in Arabidopsis thaliana. J Biol Chem. 2001;276:40381–4.
Ott G, Havemeyer A, Clement B. The mammalian molybdenum enzymes of mARC. J Biol Inorg Chem. 2015;20:265–75.
Garattini E, Terao M. Aldehyde oxidase and its importance in novel drug discovery: present and future challenges. Expert Opin Drug Discov. 2013;8:641–54.
Zhang Y, Gladyshev VN. Molybdoproteomes and evolution of molybdenum utilization. J Mol Biol. 2008;379:881–99.
Zhang Y, Rump S, Gladyshev VN. Comparative genomics and evolution of molybdenum utilization. Coord Chem Rev. 2011;255:1206–17.
Atwal PS, Scaglia F. Molybdenum cofactor deficiency. Mol Genet Metab. 2016;117:1–4.
Rupar CA, Gillett J, Gordon BA, Ramsay DA, Johnson JL, Garrett RM, Rajagopalan KV, Jung JH, Bacheyie GS, Sellers AR. Isolated sulfite oxidase deficiency. Neuropediatrics. 1996;27:299–304.
Ichida K, Amaya Y, Okamoto K, Nishino T. Mutations associated with functional disorder of xanthine oxidoreductase and hereditary xanthinuria in humans. Int J Mol Sci. 2012;13:15475–95.
Zhou Y, Zhang X, Ding R, Li Z, Hong Q, Wang Y, Zheng W, Geng X, Fan M, Cai G, et al. Using next-generation sequencing to identify a mutation in human MCSU that is responsible for type II Xanthinuria. Cell Physiol Biochem. 2015;35:2412–21.
Reiss J, Hahnewald R. Molybdenum cofactor deficiency: mutations in GPHN, MOCS1, and MOCS2. Hum Mutat. 2011;32:10–8.
Duntas LH, Benvenga S. Selenium: an element for life. Endocrine. 2015;48:756–75.
Beck MA, Matthews CC. Micronutrients and host resistance to viral infection. Proc Nutr Soc. 2000;59:581–5.
Rees K, Hartley L, Day C, Flowers N, Clarke A, Stranges S. Selenium supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;1:CD009671.
Sanmartin C, Plano D, Font M, Palop JA. Selenium and clinical trials: new therapeutic evidence for multiple diseases. Curr Med Chem. 2011;18:4635–50.
Stadtman TC. Selenocysteine. Annu Rev Biochem. 1996;65:83–100.
Hatfield DL, Gladyshev VN. How selenium has altered our understanding of the genetic code. Mol Cell Biol. 2002;22:3565–76.
Allmang C, Wurth L, Krol A. The selenium to selenoprotein pathway in eukaryotes: more molecular partners than anticipated. Biochim Biophys Acta. 1790;2009:1415–23.
Schmidt RL, Simonović M. Synthesis and decoding of selenocysteine and human health. Croat Med J. 2012;53:535–50.
Kryukov GV, Gladyshev VN. The prokaryotic selenoproteome. EMBO Rep. 2004;5:538–43.
Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–43.
Lobanov AV, Fomenko DE, Zhang Y, Sengupta A, Hatfield DL, Gladyshev VN. Evolutionary dynamics of eukaryotic selenoproteomes: large selenoproteomes may associate with aquatic life and small with terrestrial life. Genome Biol. 2007;8:R198.
Zhang Y, Gladyshev VN. Trends in selenium utilization in marine microbial world revealed through the analysis of the global ocean sampling (GOS) project. PLoS Genet. 2008;4:e1000095.
Mariotti M, Ridge PG, Zhang Y, Lobanov AV, Pringle TH, Guigo R, Hatfield DL, Gladyshev VN. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS One. 2012;7:e33066.
Gobler CJ, Berry DL, Dyhrman ST, Wilhelm SW, Salamov A, Lobanov AV, Zhang Y, Collier JL, Wurch LL, Kustka AB, et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc Natl Acad Sci U S A. 2011;108:4352–7.
Dunn BK, Richmond ES, Minasian LM, Ryan AM, Ford LG. A nutrient approach to prostate cancer prevention: the selenium and vitamin E cancer prevention trial (SELECT). Nutr Cancer. 2010;62:896–918.
Rayman MP, Stranges S. Epidemiology of selenium and type 2 diabetes: can we make sense of it? Free Radic Biol Med. 2013;65:1557–64.
Loscalzo J. Keshan disease, selenium deficiency, and the selenoproteome. N Engl J Med. 2014;370:1756–60.
Guo X, Ma WJ, Zhang F, Ren FL, Qu CJ, Lammi MJ. Recent advances in the research of an endemic osteochondropathy in China: Kashin-Beck disease. Osteoarthr Cartil. 2014;22:1774–83.
Xiong YM, Mo XY, Zou XZ, Song RX, Sun WY, Lu W, Chen Q, Yu YX, Zang WJ. Association study between polymorphisms in selenoprotein genes and susceptibility to Kashin-Beck disease. Osteoarthr Cartil. 2010;18:817–24.
Dumont JE, Corvilain B, Contempre B. The biochemistry of endemic cretinism: roles of iodine and selenium deficiency and goitrogens. Mol Cell Endocrinol. 1994;100:163–6.
Raisbeck MF. Selenosis. Vet Clin North Am Food Anim Pract. 2000;16:465–80.
Zhang Y, Zhou Y, Schweizer U, Savaskan NE, Hua D, Kipnis J, Hatfield DL, Gladyshev VN. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J Biol Chem. 2008;283:2427–38.
Agamy O, Ben Zeev B, Lev D, Marcus B, Fine D, Su D, Narkis G, Ofir R, Hoffmann C, Leshinsky-Silver E, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet. 2010;87:538–44.
Clark LC, Combs GF Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. JAMA. 1996;27:1957–63.
Stranges S, Marshall JR, Natarajan R, Donahue RP, Trevisan M, Combs GF, Cappuccio FP, Ceriello A, Reid ME. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: a randomized trial. Ann Intern Med. 2007;147:217–23.
Misu H, Takamura T, Takayama H, Hayashi H, Matsuzawa-Nagata N, Kurita S, Ishikura K, Ando H, Takeshita Y, Ota T, et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010;12:483–95.
Wang XD, Vatamaniuk MZ, Wang SK, Roneker CA, Simmons RA, Lei XG. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia. 2008;51:1515–24.
Olsson M, Olsson B, Jacobson P, Thelle DS, Björkegren J, Walley A, Froguel P, Carlsson LM, Sjöholm K. Expression of the selenoprotein S (SELS) gene in subcutaneous adipose tissue and SELS genotype are associated with metabolic risk factors. Metabolism. 2011;60:114–20.
Canani LH, Capp C, Dora JM, Meyer EL, Wagner MS, Harney JW, Larsen PR, Gross JL, Bianco AC, Maia AL. The type 2 deiodinase a/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2005;90:3472–8.
Kurokawa S, Berry MJ. Selenium. Role of the essential metalloid in health. Met Ions Life Sci. 2013;13:499–534.
Speckmann B, Grune T. Epigenetic effects of selenium and their implications for health. Epigenetics. 2015;10:179–90.
Méplan C, Hesketh J. Selenium and cancer: a story that should not be forgotten-insights from genomics. Cancer Treat Res. 2014;159:145–66.
Méplan C. Selenium and chronic diseases: a nutritional genomics perspective. Forum Nutr. 2015;7:3621–51.
Gentschew L, Bishop KS, Han DY, Morgan AR, Fraser AG, Lam WJ, Karunasinghe N, Campbell B, Ferguson LR. Selenium, selenoprotein genes and Crohn’s disease in a case-control population from Auckland, New Zealand. Nutrients. 2012;4:1247–59.
Yamada K. Cobalt: its role in health and disease. Met Ions Life Sci. 2013;13:295–320.
Zhang Y, Rodionov DA, Gelfand MS, Gladyshev VN. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics. 2009;10:78.
Finkelstein JL, Layden AJ, Stover PJ. Vitamin B-12 and perinatal health. Adv Nutr. 2015;6:552–63.
Kim J, Gherasim C, Banerjee R. Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci U S A. 2008;105:14551–4.
Lerner-Ellis JP, Tirone JC, Pawelek PD, Doré C, Atkinson JL, Watkins D, Morel CF, Fujiwara TM, Moras E, Hosack AR, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet. 2006;38:93–100.
Zimmermann MB, Boelaert K. Iodine deficiency and thyroid disorders. Lancet Diabetes Endocrinol. 2015;3:286–95.
Zimmermann MB, Galetti V. Iodine intake as a risk factor for thyroid cancer: a comprehensive review of animal and human studies. Thyroid Res. 2015;8:8.
Lahner B, Gong J, Mahmoudian M, Smith EL, Abid KB, Rogers EE, Guerinot ML, Harper JF, Ward JM, McIntyre L, Schroeder JI, Salt DE. Genomic scale profiling of nutrient and trace elements in Arabidopsis thaliana. Nat Biotechnol. 2003;21:1215–21.
Baxter I. Ionomics: the functional genomics of elements. Brief Funct Genomics. 2010;9:149–56.
Salt DE, Baxter I, Lahner B. Ionomics and the study of the plant ionome. Annu Rev Plant Biol. 2008;59:709–33.
Yu D, Danku JM, Baxter I, Kim S, Vatamaniuk OK, Vitek O, Ouzzani M, Salt DE. High-resolution genome-wide scan of genes, gene-networks and cellular systems impacting the yeast ionome. BMC Genomics. 2012;13:623.
Huang XY, Salt DE. Plant Ionomics: from elemental profiling to environmental adaptation. Mol Plant. 2016;9:787–97.
Ma S, Lee SG, Kim EB, Park TJ, Seluanov A, Gorbunova V, Buffenstein R, Seravalli J, Gladyshev VN. Organization of the Mammalian Ionome According to organ origin, lineage specialization, and longevity. Cell Rep. 2015;13:1319–26.
Malinouski M, Hasan NM, Zhang Y, Seravalli J, Lin J, Avanesov A, Lutsenko S, Gladyshev VN. Genome-wide RNAi ionomics screen reveals new genes and regulation of human trace element metabolism. Nat Commun. 2014;5:3301.
Baxter I, Ouzzani M, Orcun S, Kennedy B, Jandhyala SS, Salt DE. Purdue ionomics information management system. An integrated functional genomics platform. Plant Physiol. 2007;143:600–11.
Emmett SE. ICP-MS: a new look at trace elements in Alzheimer’s disease. Prog Clin Biol Res. 1989;317:1077–86.
Krachler M, Lindschinger M, Eber B, Watzinger N, Wallner S. Trace elements in coronary heart disease: impact of intensified lifestyle modification. Biol Trace Elem Res. 1997;60:175–85.
Alimonti A, Bocca B, Pino A, Ruggieri F, Forte G, Sancesario G. Elemental profile of cerebrospinal fluid in patients with Parkinson’s disease. J Trace Elem Med Biol. 2007;21:234–41.
Ilbäck NG, Frisk P, Tallkvist J, Gadhasson IL, Blomberg J, Friman G. Gastrointestinal uptake of trace elements are changed during the course of a common human viral (Coxsackievirus B3) infection in mice. J Trace Elem Med Biol. 2008;22:120–30.
Hanć A, Komorowicz I, Iskra M, Majewski W, Barałkiewicz D. Application of spectroscopic techniques: ICP-OES, LA-ICP-MS and chemometric methods for studying the relationships between trace elements in clinical samples from patients with atherosclerosis obliterans. Anal Bioanal Chem. 2011;399:3221–31.
Sarafanov AG, Todorov TI, Centeno JA, Macias V, Gao W, Liang WM, Beam C, Gray MA, Kajdacsy-Balla AA. Prostate cancer outcome and tissue levels of metal ions. Prostate. 2011;71:1231–8.
Flores CR, Puga MP, Wrobel K, Garay Sevilla ME, Wrobel K. Trace elements status in diabetes mellitus type 2: possible role of the interaction between molybdenum and copper in the progress of typical complications. Diabetes Res Clin Pract. 2011;91:333–41.
Golasik M, Jawień W, Przybyłowicz A, Szyfter W, Herman M, Golusiński W, Florek E, Piekoszewski W. Classification models based on the level of metals in hair and nails of laryngeal cancer patients: diagnosis support or rather speculation? Metallomics. 2015;7:455–65.
Herman M, Golasik M, Piekoszewski W, Walas S, Napierala M, Wyganowska-Swiatkowska M, Kurhanska-Flisykowska A, Wozniak A, Florek E. Essential and toxic metals in oral fluid-a potential role in the diagnosis of periodontal diseases. Biol Trace Elem Res. 2016;173:275–82.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31171233) and the Natural Science Foundation of Guangdong Province (No. 2015A030313555).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Zhang, Y. (2017). Trace Elements and Healthcare: A Bioinformatics Perspective. In: Shen, B. (eds) Translational Informatics in Smart Healthcare. Advances in Experimental Medicine and Biology, vol 1005. Springer, Singapore. https://doi.org/10.1007/978-981-10-5717-5_4
Download citation
DOI: https://doi.org/10.1007/978-981-10-5717-5_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-5716-8
Online ISBN: 978-981-10-5717-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)