
Abstract
Aims/hypothesis
We previously observed hyperglycaemia, hyperinsulinaemia, insulin resistance and obesity in Gpx1-overexpressing mice (OE). Here we determined whether these phenotypes were eliminated by diet restriction, subsequently testing whether hyperinsulinaemia was a primary effect of Gpx1 overexpression and caused by dysregulation of pancreatic duodenal homeobox 1 (PDX1) and uncoupling protein-2 (UCP2) in islets.
Methods
First, 24 male OE and wild-type (WT) mice (2 months old) were given 3 g (diet-restricted) or 5 g (full-fed) feed per day for 4 months to compare their glucose metabolism. Thereafter, several mechanistic experiments were conducted with pancreas and islets of the two genotypes (2 or 6 months old) to assay for beta cell mass, reactive oxygen species (ROS) levels, mitochondrial membrane potential (Δψm) and expression profiles of regulatory proteins. A functional assay of islets was also performed.
Results
Diet restriction eliminated obesity but not hyperinsulinaemia in OE mice. These mice had greater pancreatic beta cell mass (more than twofold) and pancreatic insulin content (40%) than the WT, along with an enhanced Δψm and glucose-stimulated insulin secretion in islets. With diminished ROS production, the OE islets displayed hyperacetylation of H3 and H4 histone in the Pdx1 promoter, elevated PDX1 and decreased UCP2.
Conclusions/interpretation
Overproduction of the major antioxidant enzyme, glutathione peroxidase 1, caused seemingly beneficial changes in pancreatic PDX1 and UCP2, but eventually led to chronic hyperinsulinaemia by dysregulating islet insulin production and secretion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic beta cells are considered to be low in antioxidant capacity and susceptible to oxidative stress [1]. Consequently, oxidative injury of beta cells has become a new aetiological focus of diabetes and insulin resistance [2]. Indeed, insulin synthesis or secretion in insulin-producing or -secreting cells is impaired by elevated intracellular reactive oxygen species (ROS), this impairment being partially alleviated by overproduction of antioxidant enzymes [3, 4]. However, overproduction of antioxidant proteins in animals, either specifically in pancreatic beta cells [5, 6] or ubiquitously in various tissues [7, 8], has generated highly variable or even negative phenotypes [9]. Most strikingly, we have observed spontaneous development of hyperglycaemia, hyperinsulinaemia, insulin resistance and obesity in mice overproducing Se-dependent cellular glutathione peroxidase-1 (GPX1), a major intracellular antioxidant enzyme [10].
While the physiological relevance of our paradoxical finding was shown by a strong correlation between increased erythrocyte GPX1 activity and insulin resistance in gestational diabetic women [11], our initial study [10] did not sort out the metabolic sequence of the phenotype in the Gpx1-overexpressing (OE) mice. It remained unclear whether hyperinsulinaemia and insulin resistance in OE mice were caused by or largely confounded by obesity. Because obesity may be eliminated or controlled by diet restriction [12], it is logical to examine whether obesity in OE mice can be prevented by restricted feeding, and whether their altered insulin status is independent of obesity.
Hyperinsulinaemia may be induced by elevated beta cell mass and/or excessive insulin synthesis and secretion [13, 14]. The transcriptional factor pancreatic duodenal homeobox 1 (PDX1) plays a pivotal role in pancreatic beta cell differentiation as well as insulin gene expression and synthesis [14–17]. Uncoupling protein 2 (UCP2) serves as a negative regulator of mitochondrial membrane potential (Δψm) [18], which positively correlates with glucose-stimulated insulin secretion (GSIS) [19]. Levels and function of PDX1 and UCP2 are affected by intracellular ROS status [20, 21], glucotoxicity [22] and antioxidants [6, 23, 24]. However, it is not known whether in vivo global overexpression of Gpx1 could overly diminish intracellular production of hydroperoxides and subsequently dysregulate production of PDX1 and UCP2 and their role in insulin synthesis and secretion.
Although post-transcriptional regulation of PDX1 by ROS or antioxidants has been postulated [20, 23–25], new mechanisms such as epigenetic modification [26] of Pdx1 have not been studied. The proximal region of Pdx1 promoter consists of an islet-specific expression consensus E-box motif, which predominantly binds the upstream transcription factor [27]. Site-specific acetylation or deacetylation of nucleosomal histones H3 and H4 is central to the switch between permissive and repressive chromatin structure and thus activation or repression of transcription [28]. With a high affinity for upstream transcription factor binding [29], H3 and H4 are the core histones with high levels of acetylation at the active transcriptional loci [30]. It would be of great interest to determine whether the presumed diminished intracellular ROS production in OE islets enhances Pdx1 transcription via hyper-acetylating H3 and H4 in its proximal promoter region, causing hypertrophy of beta cells and overproduction of insulin. In addition, Pdx1 transcription and PDX1 stability are affected by three key insulin signal proteins: c-jun terminal kinase (JNK), protein kinase B (AKT) and protein tyrosine phosphatase 1B (PTP1B) [20, 31, 32]. Because these proteins are highly sensitive to ROS [9, 33, 34], elevated GPX1 activity may impact on PDX1 via alteration of their production or phosphorylation.
In the present study, we first demonstrated that hyperinsulinaemia was a primary effect of Gpx1 overexpression and was not prevented by diet restriction. Thereafter, we conducted a series of experiments to test whether: (1) hyperinsulinaemia in OE mice was attributable to increased beta cell mass, insulin synthesis, mitochondrial potential and GSIS in islets; (2) OE islets phenotype was due to altered PDX1 and UCP2 levels; and (3) the dysregulation of PDX1 and UCP2 was mediated by overly scavenged ROS level, hyperacetylation of H3 and H4 histone of Pdx1 promoter and alterations of JNK, AKT and PTP1B. Our data indicate that hyperinsulinaemia in OE mice was associated with upregulated PDX1 and a downregulated UCP2 protein in islets, and that hyperacetylation of H3 and H4 histone of Pdx1 promoter in response to Gpx1 overexpression was a novel regulatory mechanism for this key transcriptional factor in vivo.
Methods
Transgenic mice and animal care
All mouse experiments were approved by the Institutional Animal Care and Use Committee at Cornell University. The OE mice were derived from a B6C3 (C57B1×C3H) hybrid line (Taconic, Germantown, NY, USA) [35] and carried three additional copies of the Gpx1 gene. The wild-type (WT) was derived from the non-transgenic littermates. Elevated GPX1 activity in the OE mice was confirmed in a number of tissues and showed no significant effect on other selenoproteins and antioxidant enzymes [35]. All mice were weaned at 3 weeks of age, given free access to a Torula yeast and sucrose-based diet (0.4 mg Se/kg) [35] and distilled water. Mice were individually reared in plastic cages in an animal room with constant temperature (22°C) and a 12 h light–dark cycle. All assays were conducted with male mice at 6 months of age unless otherwise indicated.
Diet restriction experiment
A total of 12 WT and 12 OE male mice (2 months of age) were fed 5 g (full-fed) or 3 g (diet-restricted) of the diet per day [36]. During the 4 month trial, body weights of mice were recorded monthly. Fasting (8 h, overnight) blood glucose concentrations of mice were measured monthly from tail blood using a glucometer (Bayer, Elkhart, IN, USA). At the end of the trial period, all mice were killed to collect blood (plasma), liver, pancreas and gastrocnemius muscle to assay for activities of GPX1, GPX3, thioredoxin reductase and total superoxide dismutase [10]. At 1 week prior to the end of the study, fasted mice were tested for insulin tolerance (0.5 units per kg body weight; Humulin R; Eli Lilly, Indianapolis, IN, USA), glucose tolerance (1 g/kg, d-glucose) and GSIS (1 g/kg) (tests conducted at 2 day intervals). Plasma insulin concentration was determined using a rat/mouse insulin ELISA kit (Crystal Chem, Downers Grove, IL, USA) [10].
In vitro experiments and assays
Detailed experimental protocols and complete information on reagents, materials and instruments are described in Electronic supplementary material (ESM). Briefly, total pancreatic insulin concentration was determined (n = 4 mice per genotype) using the above-described kit after acid-ethanol extraction [37]. Pancreatic beta cell mass was determined by immunostaining of paraffin-embedded pancreatic sections (n = 3 mice × three slides per genotype) [38] with a guinea pig polyclonal antibody against mouse insulin (Zymed, San Francisco, CA, USA). Immunostaining of pancreatic GPX1 protein (n = 3 mice × three slides per genotype) was conducted using a rabbit polyclonal antibody against bovine GPX1 (Lab Frontier, Seoul, Korea). Islets were isolated from mice using a standard procedure [39] with minor modifications. Total GPX1 activity was assayed in homogenised islets isolated from the OE and WT mice (n = 3 per genotype) [10]. For insulin secretion assays, islets (30 per sample, n = 3 per genotype by treatment) were incubated for 1 h at 37°C in 1 ml Hanks’ balanced salt solution containing 2.8 or 16.7 mmol/l glucose with or without 50 μmol/l H2O2. The released insulin (after 60 min incubation) in the medium supernatant fraction was quantified as described above. Intracellular ROS levels were detected using dichlorodihydrofluorescein diacetate (Molecular Probes, Eugene, OR, USA) [6]. Islet Δψm was determined using a mitochondrial membrane sensor kit (ApoAlert; Clontech Laboratories, Mountain View, CA, USA).
The mRNA levels of four genes in islets were determined by Q-PCR (7900HT; Applied Biosystems, Foster City, CA, USA). Total RNA was prepared from freshly isolated islets (200 per sample, n = 6 mice per genotype) using Trizol (Invitrogen, Carlsbad, CA, USA). Protein concentrations of eight insulin-related signal molecules in homogenates of pancreas (n = 4 per genotype) and islets (400 per sample, n = 4 mice per genotype) were determined as previously described [10].
Two experiments were conducted to determine how elevated GPX1 activity in the OE islets affected acetylation of the H3 and H4 core histones in the Pdx1 proximal promoter region [40] and also to determine their response to the H2O2-induced hypoacetylation. In both experiments, islets (n = 500 per sample) were isolated from 16 mice (n = 4 per genotype by age) and cultured for 48 h (RPMI 1640 medium) before the assay. In the second experiment, islets were further incubated with or without 50 μmol/l of H2O2 for 24 h. The histone cross-linking and chromatin immunoprecipitation (ChIP) were carried out by using a ChIP assay kit (Upstate/ Millipore, Temecula, CA, USA) following the manufacturer’s instructions.
Ebselen, a seleno-organic compound that mimics the enzymatic function of GPX1 [41], was used to determine whether the decreased UCP2 protein levels in the OE islets were related to the enhanced GPX1 activity. Islets isolated from three WT mice were pooled and divided into three groups (200 islets per sample, n = 3 per treatment): medium only (RPMI 1640, 10 mmol/l glucose and 10% [vol./vol.] fetal bovine serum), solvent vehicle control (0.25% [vol./vol.] DMSO) and 50 μmol/l ebselen. After 12 h incubation, islets were collected for western blot analysis of UCP2 as described above.
Statistical analyses
Quantitative data were analysed using SAS (release 6.11; SAS Institute, Cary, NC, USA). Genotype or treatment effects were tested by one or two-way ANOVA and Student’s t test. Data are presented as mean ± SE and significance was set at p < 0.05.
Results
Dietary restriction eliminated obesity, but not hyperinsulinaemia in OE mice
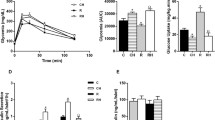
Compared with the WT, the full-fed OE mice became heavier (p < 0.05) (Fig. 1a) and exhibited hyperglycaemia (p < 0.05) (Fig. 1c), insulin resistance (Fig. 1e) and hyperinsulinaemia with elevated GSIS (Fig. 1i) at various time-points. Diet restriction in the OE mice prevented all of these phenotypes (Fig. 1b,d,f) except for hyperinsulinaemia. The diet-restricted OE mice still had higher (p < 0.05) plasma insulin concentrations at 0 (baseline, 66%) and 15 min (222%) after the glucose challenge (Fig. 1j) than did the WT mice. Despite slightly higher (p < 0.05) blood glucose concentrations than WT at several time-points during the glucose tolerance test (Fig. 1g,h), the OE mice actually exhibited greater (p < 0.05) glucose tolerance relative to their initial concentration (data not shown). Compared with the WT, the elevation of GPX1 activity in the OE mice (Table 1) was relatively greater in pancreas (23-fold) than in liver (24%) or muscle (4.2-fold), while there was no difference in activities of other enzymes.

Effects of full feeding (5 g/day) on body weight (a), fasting (overnight, 8 h) blood glucose concentrations (c), insulin tolerance (0.5 units/kg) (e), glucose tolerance (1 g/kg) (g) and GSIS (1 g glucose/kg) (i) of OE and WT mice. b, d, f, h, j Effects of diet restriction (3 g/day) on variables as above. Data are means ± SE (n = 6). *p < 0.05 for difference between two genotypes at given time points. White circles, WT; white triangles, OE (both full feeding); black circles, WT; black triangles, OE (both diet restriction)
The OE mice had elevated beta cell mass, insulin synthesis and GSIS
Pancreatic insulin content was 40% higher (p < 0.05) in the OE than the WT mice (Fig. 2a). After incubation with 2.8 and 16.7 mmol/l glucose, OE islets released 67 and 85% more (p < 0.05) insulin into the media, respectively than those of WT (Fig. 2b). The H2O2 treatment decreased (p < 0.05, 37%) GSIS in the WT islets treated with 16.7 mmol/l glucose, but not in the OE islets. Elevated GSIS was also seen in OE islets at 15 min incubation and different ages of mice (2 to 10 months old; data not shown). Pancreatic insulin staining showed a stronger colour and a larger area in the OE than in the WT mice (Fig. 2c). The beta cell mass represented only 0.48% of the total pancreas in the WT, but was elevated (p < 0.05) to 1.27% in the OE mice.

Effects of Gpx1 overexpression on pancreatic total insulin concentration (a), islet insulin secretion (b) and pancreatic beta cell mass (c) with representative image of insulin staining of pancreas sections and quantification of beta cell mass as indicated (WT, 0.48 ± 0.08%; OE, 1.27 ± 0.36% of total pancreas). Data are mean ± SE. *p < 0.05 for difference between genotype (within treatment); †p<0.05 for H2O2 treatment effect in WT islets at the high glucose incubation. White bars, WT; black bars, OE
Overproduction of GPX1 in the OE islets was accompanied by an attenuated ROS production and an elevated mitochondrial membrane potential
The immunostaining of GPX1 protein in pancreatic sections (Fig. 3a) was stronger in the OE than in the WT mice. Islet GPX1 activity was 22-fold greater (p < 0.05) in the OE than in the WT (Fig. 3b). As shown by the intensity of green fluorescence (Fig. 3c,), intracellular ROS production in the OE islets treated with 5 mmol/l glucose was lower than that of the WT islets. The green fluorescence in the WT islets was increased by exposure to 25 mmol/l glucose for 12 h and further intensified by treatment with 50 μmol/l H2O2 for 5 h. In contrast, these treatments in the OE islets induced no apparent increase in green fluorescence over baseline. At 5 mmol/l glucose, the OE islet image showed yellow instead of green fluorescence as in the WT, implying a greater Δψm in the OE than in the WT (Fig. 3d). At 25 mmol/l glucose, islet mitochondria from both genotypes were highly energised (red fluorescence).

Effects of Gpx1 overexpression on pancreatic GPX1 level (a), islet GPX1 activity (b), islet intracellular ROS production (c) and islet mitochondrial membrane potential (d). a Representative images of immunostaining of GPX1 protein in pancreatic sections of WT and OE mice as indicated. The dark brown staining in OE mice indicates a high level of GPX1 protein. Islet GPX1 activity (b) was enhanced 20-fold (*p < 0.05) in the OE (black column) versus WT (white column) mice. c Decreased intracellular ROS production in islets of the OE compared with those of WT mice under various treatments as indicated. d OE islets had higher mitochondrial membrane potential than the WT islets under different treatments. In all panels, experiments were conducted on islets isolated from 6-month-old mice (n = 3 to 5 per genotype). Scale bar, 10 µm. Magnification, 40×
The OE mice showed opposite changes in islet
PDX1 and UCP2 The relative mRNA level of Pdx1 in OE islets was approximately threefold (p < 0.05) that in WT islets (Fig. 4). The mRNA levels of preproinsulin 1 and preproinsulin 2 in the OE islets were also double (p < 0.05 to 0.07) those of the WT. In contrast, the mRNA level of Ucp2 in the OE islets was 23% lower (p < 0.05) than that in the WT islets. Western blot analyses of the islet homogenates indicated a 67% (p < 0.05) increase in PDX1 and 31 to 57% decreases (p < 0.05) in UCP2, phosphorylated JNK, phosphorylated AKT on Thr-308, and PTP1B in the OE than the WT mice (Fig. 5a). Similar genotype differences in PDX1 and UCP2 proteins were detected in islets of 2-month-old mice (ESM Fig. 1). These assayed proteins were chosen to verify their genotype differences initially detected in the pancreas homogenates (ESM Fig. 2). In addition, the OE showed a 24% (p < 0.05) decrease in total phosphorylated PDX1 protein in pancreas compared with WT, but the two genotypes had similar levels of pancreatic total JNK, total AKT and phosphorylated AKT on Ser-473. Compared with islets treated with medium only or the solvent vehicle-controls, the WT islets treated with 50 μmol/l ebselen had their UCP2 protein reduced to a minimal level (Fig. 5b). This implied that the decreased UCP2 protein in the OE islets was associated with elevated GPX1 activity.

Effects of Gpx1 overexpression on islet mRNA levels of Pdx1, preproinsulin 1 (preproins 1), preproinsulin 2 (preproins 2) and Ucp2 in 6-month-old mice. For each gene, the values were normalised to that of 18S rRNA in the same reaction. The gene expression level in the OE islets was presented as relative to the mean (set at 1) of WT. Data are means ± SE (n = 6). *p < 0.05; †p ≤ 0.06; ‡p ≤ 0.07 for difference between genotypes

Effects of Gpx1 overexpression on islet protein levels of insulin-related signal molecules (a) and responses of islet UCP2 protein to GPX1 mimic ebselen (b). a Representative (n = 4) immunoblots showing the protein levels (mean ± SE, n = 4) in OE islets relative to the WT islets (as 100%): PDX1 (167 ± 17), UCP2 (43 ± 16), phosphorylated JNK (JNK-P, 69 ± 22), phosphorylated AKT on Thr-308 (AKT-P308, 49 ± 27) and PTP1B (59 ± 28) (all p < 0.05 for difference between genotype). The relative protein level was normalised to that of ß-actin on the same membrane. b Representative (n = 3) immunoblot showing responses of islet UCP2 protein to ebselen. The relative UCP2 protein level (mean ± SE, n = 3) was 100 ± 5.4 for the blank control, 111.9 ± 7.6 for the vehicle control, and 5.3 ± 2.8 for the ebselen treatment, respectively (p < 0.05 for effect of ebselen compared with vehicle and blank controls)
The OE islets exhibited hyperacetylation of H3 and H4 histone at the proximal promoter of Pdx1
Both H3 and H4 acetylation were increased (p < 0.05) in OE islets compared with WT islets (Fig. 6a,b). The genotype difference was numerically greater at 6 than at 2 months of age. Treating the islets with H2O2 decreased H3 and H4 acetylation by approximately 50% (p < 0.05) in the WT, but not in the OE group (Fig. 6c,d). Apparently, hyperacetylation of H3 and H4 histone in the OE islets was associated with their enhanced capacity of H2O2 degradation resulting from Gpx1 overexpression.

Effects of Gpx1 overexpression on islet H3 (a) and H4 (b) histone acetylation (Ac) in the proximal region of Pdx1 promoter in mice at 2 and 6 months of age (n = 4 per genotype by age) and on the H2O2-induced hypoacetylation of islet H3 (c) and H4 (d) histone in 6-month-old mice (n = 4 per genotype by treatment). Values are mean ± SE (n = 4) and expressed as per cent of WT. *p < 0.05 for difference between genotypes within a given age or treatment; †p < 0.05 for H2O2 treatment effect within the WT. White bars, WT; black bars, OE mice
Discussion
Our most important finding is that hyperinsulinaemia in OE mice was not directly linked to obesity. Diet restriction eliminated obesity, hyperglycaemia and insulin resistance in the OE mice, but could not prevent fasting hyperinsulinaemia or elevated GSIS. In fact, similar outcomes were also produced by diet restriction of 6-month-old OE mice for 4 months (data not shown). Because the OE and WT mice shared similar activities in tissues for all the assayed antioxidant enzymes other than GPX1, hyperinsulinaemia in the OE mice was mainly, if not solely, related to the elevated GPX1 activity. Without insulin resistance, hyperinsulinaemia in the diet-restricted OE mice was probably due to a non-compensatory overproduction and/or hypersecretion of insulin.
Indeed, the OE mice had a higher pancreatic beta cell mass and insulin content, and thus a greater capacity or potential for insulin production than the WT. The elevated islet mRNA levels of preproinsulin 1 and preproinsulin 2 were consistent with the increased pancreatic insulin content in the OE mice. Comparatively, neither baseline insulin concentration nor insulin gene expression in islets was altered by overexpressing catalase by up to 50-fold [9], two forms of metallothionein by up to 30-fold [9, 42] or three forms of superoxide dismutase enzymes by up to 10-fold [5, 7]. Furthermore, overproduction of metallothionein and catalase in beta cells actually accelerated cytokine-induced beta cell death [9]. Thus, the hypertrophic effect of GPX1 on beta cell mass and insulin synthesis is rather unique. Infection of rat islets with adenovirus encoding human GPX1 gene protected against the ribose-induced loss of insulin mRNA, content and secretion. However, a sixfold increase in GPX1 activity and 72 h infection seemed to be insufficient to alter baseline levels of these three parameters [6, 24].
An enhanced functional PDX1 protein in the OE islets helps explain the hypertrophic effect of Gpx1 overexpression on beta cell mass and insulin synthesis. Because in previous research only post-transcriptional regulation of Pdx1 expression and function by ROS or antioxidant has been suggested [6, 20, 23, 25], our finding on the hyperacetylation of H3 and H4 histone at the proximal promoter of Pdx1 in the OE mice reveals not only a novel, in vivo epigenetic, but also a potential transcriptional regulation for this key factor by antioxidant enzymes. Preceding transcriptional activation [43], hyperacetylation of H3 and H4 helps remodel the chromatin at Pdx1 promoter to form a more accessible structure for transcription [44]. This modification helps explain the increased Pdx1 mRNA levels in the OE islet and is strongly associated with the GPX1 function of H2O2 degradation. The latter statement is based on the negative correlation between histone acetylation and islet intracellular ROS production at 2 and 6 months of age, and on the fact that H2O2-induced histone hypoacetylation was prevented in the OE islets. Along with a modest decrease (24%) in pancreatic PDX1 phosphorylation (degradation) [25], the upregulation of Pdx1 gene expression enabled the OE mice to maintain a higher level of functional PDX1 protein than the WT mice to promote beta cell differentiation and insulin synthesis. While similar positive effects on PDX1 protein were produced by three dietary antioxidant supplements in C57BL/KsJ-db/db mice [23], overproduction of catalase in beta cells accelerated cytokine-induced PDX1 protein disappearance [9]. It is intriguing to see such different impacts on PDX1 protein by GPX1 and catalase, given the so similar catalytic functions.
Decreased protein levels of phosphorylated JNK, phosphorylated AKT on Thr-308 and PTP1B in the OE islets were consistent with the attenuated ROS production, but presented mixed impacts on PDX1 abundance and stability. Suppressed phosphorylation of JNK was likely to promote PDX1 function by inhibiting ROS-induced nucleocytoplasmic translocation of PDX1 [20, 45] and to enhance Pdx1 transcription by protecting ROS-induced nuclear localisation of forkhead box O1 (an inhibitor of PDX1 transcription) [46]. The decreased levels of phosphorylated AKT on Thr-308 might attenuate H2O2-mediated phosphorylation (degradation) of PDX1 [25], but could also inhibit Pdx1 transcription via reduced phosphorylation of forkhead box O1 [9, 47]. While the decrease of PTP1B protein with concomitant increases of PDX1 protein, beta cell mass and insulin synthesis in the OE islets resembled the phenotype of PTP1B-null mice [32], it does not support a possible increase of ROS inhibition of PTP1B as the ultimate mechanism for suppressing the AKT/PDX1 pathway by catalase overexpression [9].
The decreased expression of Ucp2 mRNA and UCP2 protein in the OE islets may explain the enhanced Δψm [18] and the elevated GSIS [48]. A strong link between decreased levels of UCP2 and Gpx1 overexpression is supported by the response of the WT islets to GPX1 mimic, ebselen, and the resistance of OE islets to H2O2-suppressed GSIS. However, the UCP2 decrease may not be simply attributable to the diminished intracellular ROS because of a possible two-way feedback mechanism for the interaction of UCP2 and H2O2 [49]. While expression and function of UCP2 are activated by ROS [21], mitochondrial H2O2 generation can be decreased by UCP2 [49]. Therefore, the concurrent decreases in both intracellular ROS and UCP2 in OE mice may reflect a disruption of the feedback mechanism. Probably, the overly scavenging intracellular H2O2 in islets of the OE mice allowed co-existence of highly coupled or energised mitochondria with lower levels of UCP2, nudging up the well-controlled GSIS and contributing to hyperinsulinaemia [18]. Ucp2, coincidentally, is located in regions of human chromosome 11 and mouse chromosome 7, which have been linked to hyperinsulinaemia and obesity [18].
In summary, chronic hyperinsulinaemia in OE mice was associated with a dysregulated functional production of PDX1 and UCP2. It is striking that seemingly beneficial effects of GPX1 on these two key factors, beta cell mass, and insulin synthesis and secretion eventually led to chronic hyperinsulinaemia. Apparently, our in vivo results are in stark contrast to the transient benefits of upregulating antioxidant capacity as shown in vitro [6, 7] or during short-term situations [23]. Therefore, this study reveals a long-term metabolic risk of disturbing the balance between intracellular antioxidant defence and ROS formation [9] and cautions against antioxidant strategies to prevent or treat diabetes [9, 50]. We have illustrated a decreased UCP2 protein concomitant with an increased mitochondrial potential in the OE islets, suggesting a new function of GPX1 in mitochondria. In contrast to the postulated post-transcriptional regulation of PDX1 [6, 20, 23, 25], we have revealed that histone hyperacetylation occurs at the Pdx1 promoter and is a novel epigenetic regulation mechanism of the gene in vivo. This will offer a new view for interpreting outcomes associated with long-term human interventions of antioxidant supplementation to reduce insulin resistance and diabetes. Separating hyperinsulinaemia from obesity and insulin resistance in diet-restricted OE mice may help develop a specific model for the study of hyperinsulinaemia.
Abbreviations
- AKT:
-
protein kinase B
- ChIP:
-
chromatin immunoprecipitation
- GPX1:
-
glutathione peroxidase 1
- GSIS:
-
glucose-stimulated insulin secretion
- JNK:
-
c-jun terminal kinase
- OE:
-
Gpx1-overexpressing mice
- Δψm :
-
mitochondrial membrane potential
- PDX1:
-
pancreatic and duodenal homeobox 1
- PTP1B:
-
protein tyrosine phosphatase 1B
- ROS:
-
reactive oxygen species
- UCP2:
-
uncoupling protein 2
- WT:
-
wild-type
References
Grankvist K, Marklund SL, Täljedal IB (1981) Cu, Zn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J 199:393–398
Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2003) Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta cell dysfunction? Diabetes 52:1–8
Lortz S, Tiedge M, Nachtwey T, Karlsen AE, Nerup J, Lenzen S (2000) Protection of insulin-producing RINm5F cells against cytokine-mediated toxicity through overexpression of antioxidant enzymes. Diabetes 49:1123–1130
Moriscot C, Richard M, Favrot MC, Benhamou PY (2003) Protection of insulin-secreting INS-1 cells against oxidative stress through adenoviral-mediated glutathione peroxidase overexpression. Diabetes Metab 29:145–151
Chen H, Li X, Epstein PN (2005) MnSOD and catalase transgenes demonstrate that protection of islets from oxidative stress does not alter cytokine toxicity. Diabetes 54:1437–1446
Tanaka Y, Tran PO, Harmon J, Robertson RP (2002) A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. PNAS 99:12363–12368
Mysore TB, Shinkel TA, Collins J et al (2005) Overexpression of glutathione peroxidase with two isoforms of superoxide dismutase protects mouse islets from oxidative injury and improves islet graft function. Diabetes 54:2109–2116
Sandstrom J, Jonsson LM, Edlund H, Holmberg D, Marklund SL (2002) Overexpression of extracellular-SOD in islets of nonobese diabetic mice and development of diabetes. Free Radic Biol Medi 33:71–75
Li X, Chen H, Epstein PN (2006) Metallothionein and catalase sensitize to diabetes in nonobese diabetic mice: reactive oxygen species may have a protective role in pancreatic beta cells. Diabetes 55:1592–1604
McClung JP, Roneker CA, Mu W et al (2004) Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. PNAS 101:8852–8857
Chen X, Scholl TO, Leskiw MJ, Donaldson MR, Stein TP (2003) Association of glutathione peroxidase activity with insulin resistance and dietary fat intake during normal pregnancy. J Clin Endocrinol Metab 88:5963–5968
Drenick EJ, Brickman AS, Gold EM (1972) Dissociation of the obesity–hyperinsulinism relationship following dietary restriction and hyperalimentation. Am J Clin Nutr 25:746–755
Jetton TL, Lausier J, LaRock K et al (2005) Mechanisms of compensatory beta cell growth in insulin resistant rats: roles of Akt kinase. Diabetes 54:2294–2304
Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA (2001) Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 108:1631–1638
Offield MF, Jetton TL, Labosky PA et al (1996) PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122:983–995
Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H (1998) Beta cell specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta cell phenotype and maturity onset diabetes. Genes Dev 12:1763–1768
German M, Ashcroft S, Docherty K et al (1995) The insulin gene promoter. A simplified nomenclature. Diabetes 44:1002–1004
Fleury C, Neverova M, Collins S et al (1997) Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet 15:269–272
Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE (2006) Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am J Physiol Endocrinol Metab 290:E143–E148
Kawamori D, Kajimoto Y, Kaneto H et al (2003) Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH2-terminal kinase. Diabetes 52:2896–2904
Krauss S, Zhang C-Y, Scorrano L et al (2003) Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest 112:1831–1842
Olson LK, Sharma A, Peshavaria M et al (1995) Reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to a supraphysiologic glucose concentration is associated with loss of STF-1 transcription factor expression. PNAS 92:9127–9131
Kaneto H, Kajimoto Y, Miyagawa J et al (1999) Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta cells against glucose toxicity. Diabetes 48:2398–2406
Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP (1999) Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. PNAS 96:10857–10862
Boucher M-J, Selander L, Carlsson L, Edlund H (2006) Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem 281:6395–6403
Rahman I, Gilmour PS, Jimenez LA, MacNee W (2002) Oxidative stress and TNF-α induce histone acetylation and NF-κB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem 234(235):239–248
Sharma S, Leonard J, Lee S, Chapman HD, Leiter EH, Montminy MR (1996) Pancreatic islet expression of the homeobox factor STF-1 relies on an E-box motif that binds USF. J Biol Chem 271:2294–2299
Eberharter A, Becker BP (2002) Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 3:224–229
Vettese-Dadey M Grant PA, Hebbes TR, Crane-Robinson C, Allis CD, Workman JL (1996) Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J 15:2508–2518
Tazi J, Bird A (1990) Alternative chromatin structure at CpG islands. Cell 60:909–920
Kitamura T, Nakae J, Kitamura Y et al (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 110:1839–1847
Kushner JA, Haj FG, Klaman LD et al (2004) Islet-sparing effects of protein tyrosine phosphatase-1b deficiency delays onset of diabetes in IRS2 knockout mice. Diabetes 53:61–66
Meng T-C, Buckley DA, Galic S, Tiganis T, Tonks NK (2004) Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem 279:37716–37725
Goldstein BJ, Mahadev K, Wu X (2005) Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 54:311–321
Cheng W-H, Ho Y-S, Ross DA, Han Y, Combs GF Jr, Lei XG (1997) Overexpression of cellular glutathione peroxidase does not affect expression of plasma glutathione peroxidase or phospholipid hydroperoxide glutathione peroxidase in mice offered diets adequate or deficient in selenium. J Nutr 127:675–680
Pugh TD, Klopp RG, Weindruch R (1999) Controlling caloric consumption: protocols for rodents and rhesus monkeys. Neurobiol Aging 20:157–165
Rhodes CJ, Halban PA (1987) Newly synthesized proinsulin/insulin and stored insulin are released from pancreatic beta cells predominantly via a regulated, rather than a constitutive, pathway. J Cell Biol 105:145–153
Sund NJ, Vatamaniuk MZ, Casey M et al (2001) Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev 15:1706–1715
Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monaco AP (1985) An improved method for isolation of mouse pancreatic islets. Transplantation 40:437–438
Gerrish K, Van Velkinburgh JC, Stein R (2004) Conserved transcriptional regulatory domains of the pdx-1 gene. Mol Endocrinol 18:533–548
Dawson DA, Masayasu H, Graham DI, Macrae IM (1995) The neuroprotective efficacy of ebselen (a glutathione peroxidase mimic) on brain damage induced by transient focal cerebral ischemia in the rat. Neurosci Lett 185:5–69
Chen H, Carlson EC, Pellet L, Moritz JT, Epstein PN (2001) Overexpression of metallothionein in pancreatic beta cells reduces streptozotocin-induced DNA damage and diabetes. Diabetes 50:2040–2046
Lee DY, Hayes JJ, Pruss D, Wolffe AP (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72:73–84
Norton VG, Marvin KW, Yau P, Bradbury EM (1990) Nucleosome linking number change controlled by acetylation of histones H3 and H4. J Biol Chem 265:19848–19852
Kajimoto Y, Kaneto H (2004) Role of oxidative stress in pancreatic beta cell dysfunction. Ann NY Acad Sci 1011:168–176
Kawamori D, Kaneto H, Nakatani Y et al (2006) The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem 281:1091–1098
Bernal-Mizrachi E, Fatrai S, Johnson JD et al (2004) Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest 114:928–936
Zhang C-Y, Baffy G, Perret P et al (2001) Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105:745–755
Negre-Salvayre A, Hirtz C, Carrera G et al (1997) A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11:809–815
Pi J, Bai Y, Zhang Q et al (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56:1783–1791
Acknowledgements
The breeding stocks of OE and WT mice were provided by Y. S. Ho (Wayne State University, Detroit, MI, USA). We thank H. S. Niu, O. Vatamaniuk and Q. Long for technical assistance, and C. V. Wright and H. Edlund for kindly providing the PDX1 and pPDX1 antibodies, respectively. This work was supported by National Institutes of Health Grants DK53018 (to X. G. Lei) and DK55704 (R. A. Simmons).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 94.6 KB)
ESM Fig. 1
Western blot analysis of PDX1 and UCP2 in 2-month-old islets. Representative immunoblots of PDX1 and UCP2 proteins in islets of 2-month-old males (n = 4), with quantitative analysis of the relative levels normalised to β-actin (mean ± SE, n = 4, *p ≤ 0.05). The islet homogenates were centrifuged at 14,000×g for 30 min at 4°C. The supernatant fractions (20 μg protein) were subjected to Western blot analyses. Relative density of protein bands was quantified using the Alpha-Imager 2200 System (PDF 26.5 KB)
ESM Fig. 2
Effects of GPX1 overabundance on pancreatic proteins involved in insulin homeostasis. Western blot analysis of proteins PDX1/AKT insulin signalling pathway in pancreas of 6-month-old OE mice. Representative (n = 4) immunoblots of PDX1, phosphorylated PDX1 (PDX1-P), UCP2, JNK, phosphorylated JNK (JNK-P), PTP1B, AKT, and phosphorylated AKT on Thr-308 (AKT-P308) or on Ser-473 (AKT-P473) proteins in pancreas of 6-month-old male mice, with corresponding densitometric quantitative analysis of the relative levels normalised to beta actin (mean ± SE, n = 4, *p ≤ 0.05). The islet homogenates were centrifuged at 14,000×g for 30 min at 4°C. Protein concentrations were estimated using the Bradford method. The supernatant fractions (20 μg protein) were subjected to western blot analyses. Relative density of protein bands was quantified by using the Alpha-Imager 2200 System (PDF 143 KB)
Rights and permissions
About this article
Cite this article
Wang, X.D., Vatamaniuk, M.Z., Wang, S.K. et al. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia 51, 1515–1524 (2008). https://doi.org/10.1007/s00125-008-1055-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1055-3