
Abstract
Filamentous fungi are frequently used as hosts for heterologous production of industrial enzymes and bioactive secondary metabolites. This role will likely increase in the post-genomic era as a multitude of fungal genome sequences are mined for new valuable products. The trend for constructing fungal cell factories is to use synthetic biology approaches. In simplicity, this includes puzzling available bio-blocks together using seamless DNA fusions to generate vectors, gene-expression cassettes, and protein fusions. Moreover, it also sets the stage to expand natural enzymatic functions and chemistry by combining functional domains into synthetic proteins with novel enzymatic properties or by combining enzymes from different organisms in a common host to establish new biosynthetic pathways. In this chapter, we describe the components of a modern synthetic biology toolbox and how they have been used to construct and optimize fungal cell factories for heterologous production of natural and synthetic enzymes and secondary metabolites.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
I. Introduction
Fungi are well-established producers of foods, food additives, industrial enzymes, and pharmaceutical drugs and contribute significantly to human health and economy. Production of industrial enzymes alone had an annual worth of €3.5 billion in 2015, which is a mere fraction of the general white biotechnology products estimated to reach €450 billion in 2020 (Meyer et al. 2016). The vast majority of fungal processes are performed with species that have not been genetically engineered. Hence, the fungi have either been domesticated through millennials of human use, or their performance and yields have been improved via classical genetic methods like mutagenesis. The importance of fungi has sparked several multi-species genome sequencing projects where one aim is to uncover novel fungal genes involved in enzyme secretion and secondary metabolite (SM) formation. The global fungal diversity currently encompasses 144,000 classified species and estimates reaching 3.8–6.0 million species (Taylor et al. 2014; Willis 2018), of which ~1500 species have been genome sequenced (de Vries et al. 2018; Grigoriev et al. 2014). As such, it is becoming increasingly clear that fungi represent a vast reservoir of potentially useful enzymes and secondary metabolites yet to be discovered. As part of the sequencing project of genus Aspergillus, Vesth et al. analyzed the genomes of 36 species of Aspergillus section Nigri, predicting 40,424 unique genes, including 17,903 carbohydrate-active enzymes and an average of 70 putative secondary metabolite gene clusters per species (near 2700 total), comprising 450 distinct compound classes (Vesth et al. 2018). With the increasing number of fungal genome sequences, many new genes and potentially interesting products will be uncovered in this post-genomic era.
However, many of these new products are likely made by natural fungal producers that are challenging to incorporate into industrial processes, either because they are problematic to propagate efficiently in bioreactors, difficult to engineer, or opportunistic human pathogens. Based on the logical assumption that fungi by evolution likely are the best producers of fungal products, we therefore envision that there will be an increasing demand of transferring relevant genes and pathways from novel, exotic fungi to well-characterized fungal cell factories.
Filamentous fungi have been widely used for heterologous production of industrial enzymes, taking advantage of their large protein secretory capacity. The most commonly used fungal species for this purpose are Trichoderma reesei and members of the genus Aspergillus, e.g., Aspergillus niger and Aspergillus oryzae , and thus, most studies have been performed with these species. More recently, filamentous fungi have also been employed for heterologous production of SMs, typically as a part of an SM pathway elucidation strategy. For these studies, classical model fungi like Aspergillus nidulans and Neurospora crassa have been added to the repertoire of cell factories. The usefulness of fungal industrial workhorses and general model systems is also reflected by the development of substantial genetic toolboxes and methodologies for heterologous gene expression for these species.
The current trend for heterologous gene expression is to implement a synthetic biology approach. Hence, a host strain is transformed with a gene-expression cassette, generated from libraries of bio-blocks (see Sect. III), which are individually functional molecular units that can be combined by simple and seamless DNA fusion strategies (Fig. 10.1). According to this concept, we will first describe strategies to set up synthetic biology-based expression systems followed by an overview of popular bio-blocks that can be used for construction of expression cassettes. Next, we will provide two sections with examples where these concepts have been partly or entirely used to produce either enzymes or secondary metabolites in a heterologous fungal host.

Assembly of a basic gene-expression cassette. Bio-blocks are PCR amplified using primers (arrows) with tails containing sequences that are complementary to the tails of the adjacent bio-block (indicated by color); see main text for details. This allows matching overhangs (small boxes) to be formed ensuring that the bio-blocks are fused in the correct order. Tails for inserting the gene-expression cassette into a vector are not shown
II. Expression Systems
Heterologous expression is achieved by transforming the new fungal host with a suitable gene-expression cassette , which is integrated into a chromosome or an extrachromosomal vector and thereby maintained during growth. Below, we review strategies for assembling gene-expression cassettes for heterologous enzyme or pathway expression, along with methods for introducing these into the fungal host and the benefits of these methods. The functional bio-blocks for fungal heterologous gene expression are presented in Sect. III.
A. Construction of Simple Gene-Expression Cassettes
In the simplest setups, heterologous production depends on the expression of a single gene of interest (GOI). In these cases, gene-expression cassettes (GEC) can be constructed by fusing the GOI bio-block to relevant components of a basic set of bio-blocks that includes promoters, terminators, and selectable markers (Fig. 10.1). If necessary, this set can be expanded with additional bio-blocks for specialized purposes such as sequences encoding secretion signals, epitope- and purification tags, fluorescent proteins etc., or bio-blocks that are designed to target the expression cassette for integration at a specific site in the genome by homologous recombination. Since many individual bio-blocks need to be combined in a single cloning step, it is important that they can be joined with high efficiency. Several reliable systems are available for this task, e.g., In-Fusion assembly (Zhu et al. 2007), Gibson assembly (Gibson et al. 2009), USER fusion (Bitinaite et al. 2007), and Golden Gate cloning (Engler and Marillonnet 2014). Often these systems are directly compatible with accompanying vectors dedicated to gene transfer into a desirable host and the construction work commonly facilitated by ligation and propagation in Escherichia coli . Alternatively, bio-blocks can be assembled by in vivo homologous recombination using, e.g., the yeast Saccharomyces cerevisiae as a host (Finnigan and Thorner 2015).
B. Introducing Gene-Expression Cassettes into Fungal Hosts
The gene-expression cassettes can be introduced into the fungal host according to different principles. Below, we briefly review how gene-expression cassettes can be maintained by being inserted into an extrachromosomal vector or by being integrated into a random or defined position on a chromosome (Fig. 10.2).

Strategies for introducing gene-expression cassettes into fungi. The GEC can be (a) inserted into a self-replicating AMA1 plasmid via ligation or by in vivo HR or (b) integrated into the host genome by random or (c) targeted integration through NHEJ or HR, respectively
1. Plasmid-Based Expression Systems
Plasmid-based expression systems utilize self-replicating plasmids carrying autonomous replication sequences (Fig. 10.2a). In many filamentous fungi, this can be achieved via the AMA1 (autonomous maintenance in Aspergillus ) element. AMA1 was originally discovered in a lab strain of A. nidulans (Gems et al. 1991) in a search for genetic elements enhancing transformation in A. nidulans (Johnstone et al. 1985). In subsequent studies, it has been shown that AMA1-based vectors increase fungal transformation efficiency in A. nidulans up to 2000 times in comparison to earlier developed ectopically integrating vectors (Aleksenko and Clutterbuck 1997). Moreover, AMA1 plasmids propagate in other Aspergillus species as well as in species belonging to other fungal genera, including Penicillium (Fierro et al. 1996), Talaromyces (Nielsen et al. 2017), Trichoderma (Kubodera et al. 2002), and Rosellinia (Shimizu et al. 2012).
Gene-expression cassettes can easily be incorporated into extrachromosomal AMA1-based shuttle vectors, using the cloning systems highlighted above. As an extreme case, fungal AMA1 plasmids have been combined with a bacterial artificial chromosome (Zhu et al. 1997) to form fungal artificial chromosomes (FACs). These vectors have been instrumental in the search for new secondary metabolite gene clusters as they allow up to 300 kbp of fungal DNA to be cloned into the FAC in E. coli and subsequently transferred into a new fungal host for product discovery (Bok et al. 2015; Clevenger et al. 2017). Importantly, AMA1-based plasmids are not very stable and their maintenance requires selection, thereby limiting their use to small-scale exploratory heterologous production experiments. However, we note that for some purposes, such as cas9 expression in CRISPR experiments, plasmid instability is desirable as plasmid loss allows for transient gene expression, diminishing potential Cas9 off-target effects (Zhang et al. 2015).
2. Chromosome-Based Expression Systems
Integration of the expression cassette into the host genome provides a more stable expression, as compared to AMA1-based expression, and attenuates the need to maintain selection pressure. Chromosomal integration of foreign DNA occurs via one of the two DNA repair mechanisms (Krappmann 2007), either non-homologous end joining (NHEJ), where the DNA integrates into a random locus (Fig. 10.2b), or homologous recombination (HR), where the DNA integrates into a defined locus (Fig. 10.2c). Although both pathways are active in all fungi, foreign DNA typically integrates more efficiently via the NHEJ pathway in most filamentous fungal species (Meyer et al. 2007; Nødvig et al. 2015).
a) Random Chromosomal Integration of Gene-Expression Cassettes
Since NHEJ is the dominant pathway for chromosomal integration of DNA in most filamentous fungi, random chromosomal integration of gene-expression cassettes via NHEJ is therefore straightforward and can be efficiently performed in essentially all transformable fungal species. A drawback of the method is that the chromatin structures of the insertion sites are unpredictable and expression levels may therefore vary substantially between different transformants (Lubertozzi and Keasling 2006). On the other hand, if several gene-expression cassettes enter a nucleus, they may undergo recombination prior to genomic integration. As a result, transformants may contain multiple copies of the GOI inserted into a single locus. In these cases, the transformants will often be better producers of the product due to the many gene copies. However, since the cassettes will be organized as direct and/or inverted repeats, expression levels of such strains may be unstable as gene copies may be lost due to direct repeat recombination or formation of hairpin structures (Leach 1994; Petes 1988). Lastly, it is important to note that when gene-expression cassettes integrate randomly into the genome, they may disrupt important genes, or alter the expression levels of neighboring genes, causing undesired phenotypes. In summary, with this integration method, it is advisable to screen for transformants that produce high yields over many generations and at the same time do not display undesirable fitness defects.
b) Defined Chromosomal Integration of Gene-Expression Cassettes
The use of defined and well-characterized loci as integration sites for GECs allows for gene expression that is much less prone to clonal differences. Hence, insertion of gene-expression cassettes into defined loci enables comparative screening of expression levels or enzyme activities based on different genetic elements employed in the cassette, such as promoter, secretion signal, and terminator sequences. Similarly, the effects achieved by changing the GOI sequence can be directly compared. In this way it is possible to address whether changes in the GOI codon composition increase yields or whether mutations infer changes in the heterologous protein that influences its activity, specificity, folding, and/or stability (Hansen et al. 2011b; Holm 2013).
Gene-expression cassettes can be inserted into integration sites in the genome by HR. To ensure high expression levels, it may be useful to position integration sites in intergenic regions located in transcriptionally highly active sections of a chromosome. With this method, the gene-expression cassettes must be flanked by up- and downstream targeting sequences matching the genomic expression site. Since fungi rarely prefer to integrate foreign DNA into its genome by the HR pathway, extensive screening for the desired transformant may be necessary. The screening workload can be reduced by using bipartite gene-targeting substrates that select for HR proficient protoplasts (Nielsen et al. 2006) or avoided by using NHEJ-deficient strains (Meyer et al. 2007). Alternatively, methods based on restriction enzymes or CRISPR technology can be employed (Ouedraogo et al. 2016; Zheng et al. 2017). Importantly with the latter technology, GOIs can be inserted in a marker-free manner and multiplexing is possible (Liu et al. 2015; Nødvig et al. 2018; Zhang et al. 2016a). In fact, CRISPR mediates very efficient gene targeting even in NHEJ-proficient strains (Nødvig et al. 2018), eliminating the risks of working with strains with a defective DNA repair pathway. We therefore envision that CRISPR-based methods will deliver the preferred tools for introducing gene-expression cassettes into defined chromosomal expression platforms.
C. Bio-Block-Based Multi-GOI Expression Strategies
In many cases, two or more genes are required for the synthesis of a desired product. For example, for heterologous production of SMs, several genes are commonly required to synthesize the product. Above, we described that it is possible to transfer large chromosomal fragments containing entire gene clusters from one fungus to another (Sect. II.B.1). However, in many cases the activation mechanism for the genes in the cluster is not known and may depend on unknown or even host-specific transcription factors (TF) (Keller 2019); see Chap. 11. Moreover, not all genes contributing to the biosynthetic pathway of interest may be situated in the cluster (Schäpe et al. 2019). It may therefore be desirable to reconstruct the genes involved in formation of the heterologous compound using a setup that allows necessary GOIs to be equipped with known and well-characterized promoter and terminator sequences. Here we present three strategies that allow for bio-block-based multi-GOI expression (Fig. 10.3). Although multi-GOI expression cassettes can be assembled on AMA1-based vectors, we recommend using defined chromosomal expression sites for GOI insertion to gain genetic stability and to reduce dependency on selectable markers.

Strategies for multi-GOI expression cassette assembly. Individual GECs composed of a promoter (Px), a gene of interest (GOIx), a terminator (Tx), and a selection marker (Mx). Individual GECs can be (a) integrated in different loci, or (b) assembled into a combined multi-GOI cassette for single locus integration, or (c) assembled as a single polycistronic GOI where coding sequences of individual polypeptides are separated by the sequence encoding the picornavirus 2A peptide (A2p)—see main text for details
In the first strategy (Fig. 10.3a), individual expression cassettes for different GOIs are inserted into a number of unique integration sites. This strategy is advisable if all of the individual gene-expression cassettes contain an identical bio-block, such as a specific promoter or terminator. In this way, copy loss due to direct repeat recombination between the identical bio-blocks is avoided. With conventional gene-targeting methods, insertion of gene-expression cassettes into individual sites requires a number of selectable markers that matches the number of gene-expression cassettes required for establishing the biosynthetic pathway. Alternatively, the genes can be inserted by iterative gene targeting using a recyclable marker (see Sect. III.B.5). Strain construction with these methods is cumbersome and restricts the method to biosynthetic pathways composed of a small number of genes. However, these limitations can be dramatically reduced by constructing the strains via CRISPR-based multiplexed marker-free gene insertions.
In the second strategy (Fig. 10.3b), all genes required to support a biosynthetic pathway are inserted into the same expression site. Arranging the GOIs as a synthetic gene cluster may be preferred if only few selectable markers are available. Note that if two markers are available, then an infinite number of consecutive gene-targeting events into the same locus can be performed. Hence, even very large synthetic gene clusters can be constructed at a defined expression site via multiple integration steps. In this case, the selectable marker used in a given integration step replaces the marker used in the previous integration step (see Sect. III.B.5). In addition, if the heterologous host has a sexual cycle, strains that contain a synthetic gene cluster in a single defined locus can be crossed to other strains without risking that the individual genes of the cluster segregates during meiosis. This feature can be used to combine the heterologous pathway with other beneficial traits harbored by other strains.
In the third strategy (Fig. 10.3c), the GOIs are arranged as a polycistronic unit where each open reading frame (ORF) is separated by a bio-block, encoding the 2A peptide from the Picornaviridae virus family; see Table 10.3 (Schuetze and Meyer 2017). This strategy is based on the facts that; firstly, the ribosome fails to link glycine and proline residues during translation of the 2A spacer, thereby resulting in a break in the polypeptide chain. Secondly, this error does not result in release of the ribosome from the mRNA, thereby allowing for continued translation. Hence, in a simple manner, several proteins can be encoded from a single transcript generated from a single expression cassette. One drawback of this method may be that the final size of the polycistronic gene-expression cassette makes construction work difficult. Additionally, “cleavage” at the 2A spacer results in a partial 2A peptide sequence remaining at the C-terminus of the protein, which may interfere with protein function and therefore needs to be removed, e.g., by combinatory usage with other proteolytic sites (Hoefgen et al. 2018). Moreover, as “cleavage” at the 2A spacer is not 100%, the relative efficiency of protein production often depends on the position of the proteins ORF in the transcript (Schuetze and Meyer 2017).
Lastly, we note that for all strategies, two genes can be inserted simultaneously as a single cassette if bidirectional promoters (Wiemann et al. 2018; Rendsvig et al. 2019) are used to control gene expression of the heterologous gene pairs. This feature can be used to speed up strain construction.
III. Bio-Blocks
A bio-block is a DNA sequence that encompasses a molecularly functional unit. This includes nucleotide sequences, e.g., promoters and terminators, or sequences encoding a protein, e.g., the GOI and selection marker, or shorter protein sequences such as secretion signals and purification tags. Generation of the basic gene-expression cassette for production of a heterologous protein requires several bio-blocks, including the GOI, promoter, and terminator. Selection markers may also be included in the construct, offering versatile applications in different scenarios. In recent years, the use of -omics data has driven the discovery of natural promoters, which are active during the desired cultivation conditions for production. While the majority of strain engineering has been based on natural genetic elements for controlling gene expression, significant headway has been made in the development of synthetic promoters and terminators. In extension, even full synthetic gene-expression systems have been established, in which several individual genetic elements (natural or modified) are combined, to facilitate controlled gene expression and enhance productivity. Moreover, to enable secretion of the protein, a transport signal must be included as an additional bio-block (Sect. IV.A.1). For specialized purposes like protein visualization or purification, protein tags fulfilling such tasks are also included in the construct, thereby increasing the pool of bio-blocks.
A. Gene of Interest
Since the goal of heterologous expression is to produce products derived from one or more genes of interest, this element can also be considered the most important bio-block. As the GOIs coding potential in most cases should not be changed, it therefore constitutes the least flexible bio-block; however, its design requires several considerations. The GOI sequence may be derived from different sources. In the simplest scheme, if the donor strain is related to the new host, genomic DNA can typically serve as a template to make a functional PCR-derived GOI bio-block. However, if the donor strain is more distantly related, intron splicing and codon bias may compromise gene expression and translation, and we will briefly review the two latter issues below.
1. Introns
The presence of introns in a heterologous gene may result in reduced mRNA production due to mis- and incomplete splicing (He and Cox 2016; Zhao et al. 2013). In principle, the problem can be solved by employing cDNA as a PCR template for making the GOI bio-block or alternatively fuse PCR-derived exons by another round of PCR or by a one-step cloning method that allows for seamless assembly of multiple fragments (An et al. 2007). Alternatively, an entirely synthetic gene may be acquired from a commercial source. However, transcription, splicing, polyadenylation, and mRNA export are a coordinated process (Bentley 2014) and as a result the presence of introns may affect gene expression.
Indeed, sequential elimination of three introns in a gene encoding a protease from the thermophilic fungus Malbranchea cinnamomea cumulatively reduced production levels in T. reesei (Paloheimo et al. 2016). Interestingly, the largest reduction was observed when the intron closest to the transcriptional start site was removed. Similar results were obtained when a gene encoding an antifungal protein from Aspergillus giganteus was expressed in Trichoderma viride (Xu and Gong 2003). Moreover, the inclusion of an artificial intron was required for the heterologous production of GFP and mRFP in the basidiomycete Armillaria mellea (Ford et al. 2016).
Since it is not straightforward to predict the impact of retaining introns in a foreign gene on the final yield of mRNA, it may be useful to try different gene variants containing no, a few selected, or all introns, to optimize expression levels from the GOI.
2. Codon Optimization
Codon usage may vary dramatically between species (Iriarte et al. 2012) and, consequently, influence how well a heterologous mRNA is translated into a protein, reviewed in (Hanson and Coller 2018; Tanaka et al. 2014). For example, codon composition may change the chromatin structure of the GOI affecting gene transcription efficiency (Zhou et al. 2016), whereas rare codons in the beginning of the transcript may determine the efficiency of translation initiation (Pop et al. 2014). Moreover, if the mRNA contains abnormally high levels of rare codons, this may result in premature transcription termination and production of reduced length mRNA for the ORF (Zhou et al. 2018). Conversely, rare codons may be used in the native host as translation pause sites required for proper and timely folding of subdomains of the protein structure. If such codons are replaced by frequently used codons, overall folding may be compromised (Yu et al. 2015; Zhou et al. 2015).
Different algorithms exist in order to address the different challenges concerning optimal codon choices for heterologous protein production (Gould et al. 2014). Construction of a codon-optimized gene usually requires de novo synthesis of the entire gene, which can be done by fusing a set of overlapping oligonucleotides (Sect. III.E) (Hoover and Lubkowski 2002) or simply be acquired from a commercial source. Codon optimization may benefit heterologous protein production significantly, especially if the source of the foreign gene and the new host are distantly related species. However, it is important to stress that predicting the optimal codon composition for heterologous gene expression is a discipline still in its infancy. It is therefore often an advantage to test more than one gene variant during cell factory construction. This can be exemplified by the extracellular yield from T. reesei expressing a codon-optimized mammalian monoclonal immunoglobulin (IgG) antibody, which showed a 40-fold difference, dependent on which of two companies had performed the codon optimization (Lin et al. 2006).
B. Selection Markers
Selection markers are mainly used to facilitate identification of transformants containing the gene-expression cassette (Dave et al. 2015). Since targeted integration of the gene-expression cassette can be conducted in a marker-free manner via CRISPR technology, the presence of this bio-block in the cassette is no longer essential. Nevertheless, selective markers will likely remain a common feature in a gene-expression cassette since strain identification is simplified and more efficient with a marker and since the marker may also serve useful post-transformation roles. For example, a selectable marker can be used to quickly identify strains, or to promote breeding via sexual and parasexual cycles, or to maintain AMA1 plasmid-based expression. Moreover, to achieve high expression levels, it may be desirable to obtain a fungus with many copies of the expression cassette. Such strains can be selected by employing cassettes containing a weak selection marker as the strains with a high copy number will gain a fitness advantage at heavy selection pressure (Wernars et al. 1985). In some cases, it is necessary to eliminate the marker after transformation, for example, if a marker-free strain is desirable or if a marker needs to be recycled during iterative genetic engineering.
The many uses of selectable markers in strain development are reflected in the fact that several different types of markers have been developed, which can be divided into four main categories: resistance, visual, auxotrophic/nutritional, and counter-selectable markers. Importantly, the choice of selectable marker depends on the strain that needs to be engineered and on the purpose of the experiment. Below, we will briefly review the different categories of selectable markers and discuss in which situations a given marker can be advantageous.
1. Resistance Markers
Resistance markers encode proteins that neutralize the toxic effect of antimicrobials and thereby convey resistance as a dominant trait. Since the functionality of these markers (like some visual markers, see below) typically do not require any modifications of the host genome, they can be used with wild-type model fungi or with fungi where no or few genetic tools are available. Several antimicrobial/resistance-marker systems have been introduced for fungal genetic engineering (Table 10.1).
The mechanism of these antimicrobials typically involves interfering with protein translation, inhibition of metabolism, or induction of lethal DNA double-stranded breaks (DSBs). The resistance to the antimicrobials is achieved through protein-based mechanisms including 1:1 drug binding, drug turnover by chemical modification, and drug-resistant target enzymes; see Table 10.1.
The presence of an array of different marker systems is highly useful because it potentially supports that several cycles of genetic engineering steps can be performed. Perhaps more importantly, it also expands the number of species that can be engineered due to the fact that many fungal species possess an inherent resistance to certain antimicrobials due to cell wall impermeability, native efflux pumps, or catalytic activities (Garneau-Tsodikova and Labby 2016). Sometimes, susceptibility depends on the media composition (Roller and Covill 1999) exemplified by Aspergillus species, which show resistance toward hygromycin B and bleomycin at pH-values below five and in complex or hypertonic media (Punt and van den Hondel 1992). Hence, before using resistance markers, it is necessary to conduct susceptibility assays of the fungus to determine appropriate selective antimicrobials and have consistent properties of selective media batches. It should be noted that the use of antimicrobials may be undesirable, especially for large-scale cultivations, due to the risk of developing drug resistant strains, the price of the antimicrobial, and that some of the compounds are toxic to humans.
2. Visual Markers
Visual markers provide a phenotypic trait that can be easily visualized by conferring, e.g., a color change due to disruption of a host gene, or by heterologous expression of a color, fluorescent, or bioluminescent marker. Like with antimicrobial markers, visual markers can be used for selection in wild-type strains, but without posing any risks in relation to compound toxicity or transference of resistance genes to other organisms.
Three types of heterologous visual markers are frequently used, all of which may be applied for spectrophotometric assays. Firstly, β-galactosidase LacZ (lacZ) (Lubertozzi and Keasling 2006) and β-glucuronidase GusA (uidA) of E. coli (Tada et al. 1991), respectively, convert the synthetic substrates X-gal and X-gluc to 5-bromo-4-chloro-3-hydroxyindole that undergoes dimerization forming a blue pigment. Secondly, luciferases convert substrates into bioluminescent products. Thirdly, fluorescent proteins derived from jellyfish Aequorea victoria GFP, which was adapted for fungal use by codon optimization and by eliminating a non-conventional/cryptic intron splice site (Lorang et al. 2001), or from RFP.
Visual markers do not offer any fitness advantage, and, if the goal is to detect transformants containing the expression cassette, selection requires visual screening. Selection by solely visual inspection can be achieved by inserting the cassette into conidial pigmentation genes (e.g., homologs of A. nidulans wA and yA genes), resulting in a distinct change in conidial pigmentation from the native black or green spores of Aspergillus species to white or yellow (Jørgensen et al. 2011; Nielsen et al. 2006). Similarly, the expression cassette can be inserted into adeA or adeB as this will produce easy detectable red colonies due to polymerization and oxidation of 4-amino-imidazole ribotide, the intermediate resulting from the disrupted purine biosynthetic pathway (Jin et al. 2004). Alternatively, the gene-expression cassette can be equipped with a heterologous marker that produces color, fluorescence, or bioluminescence to allow for visual selection. This is useful if the goal is to insert the cassette into an intergenic section of the genome by HR, thereby avoiding host gene disruption. In cases where correct transformants are rare, fluorescent markers set the stage for high-throughput detection schemes via FACS analysis, which can be employed if the fungus produces discrete entities like conidia (Bleichrodt and Read 2019; Vlaardingerbroek et al. 2015). Color, fluorescent, or bioluminescent markers can also be advantageously used if the goal is to insert the gene-expression cassette in multiple copies, e.g., via integration events catalyzed by the NHEJ pathway, as transformants with a high copy number can be selected for by the strength of the marker signal (Throndset et al. 2010).
When heterologous enzymes and fluorescent proteins are used as selective markers, it should be noted that some fungi produce enzymes that may catalyze the same reactions as those provided by the marker enzyme, or the fungi may display auto-fluorescence. It is therefore always necessary to test whether significant native background signals exist.
3. Nutritional Markers
Usage of nutritional markers relay on auxotrophies in the host organism, which thereby requires supplementation with specific metabolites to sustain growth. Introducing a functional copy of the given nutritional marker into the host complements the defective native gene function, allowing for growth without supplementation. In this way, nutritional markers offer selective pressure without the negative effects of antibiotics. On the other hand, prior to cell factory construction, the host needs to be mutagenized or genetically engineered to create the relevant auxotrophic mutations. If the starting point is a wild-type strain, this may require the use of mutagens, traditional genetic engineering using antimicrobial or visual markers, or CRISPR technology. Complementing homologous or heterologous genes can be used as markers, but often a heterologous marker is preferred as the sequence differences reduce the risk of generating false positives due to homologous recombination between the marker and the corresponding mutated locus.
Some commonly used nutritional markers include argB (Buxton et al. 1985), trpC (Goosen et al. 1989), adeA and adeB (Jin et al. 2004), pyroA (Osmani et al. 1999), and pyrG (Goosen et al. 1987), which are required for synthesis of arginine, tryptophan, adenine, pyridoxine, and uracil/uridine, respectively. Other frequently used markers are niaD required for nitrate assimilation (Unkles et al. 1989) and the amdS gene from A. nidulans allowing growth on acetamide as the sole nitrogen source (Kelly and Hynes 1985).
Like visual markers, the design of the nutritional marker can be used to select for strains containing multiple expression cassettes. For example, the complementary marker gene can be equipped with a poor promoter to ensure that only strains with many copies produce sufficient amounts of the missing enzyme.
Often several auxotrophies are introduced in the host to allow for several rounds of genetic engineering using a set of complementing nutritional markers. It is important to note that nutritional markers may change the metabolism of the host cell despite that the defects are compensated by addition of the missing metabolites to the growth medium. For physiological characterizations of the engineered strains, this may lead to undesired artefacts; and for cell factories, it may lead to suboptimal growth, or growth, which is restricted to specific media. It is therefore advisable to restore the functionality of those nutritional markers after genetic engineering. To facilitate this process, it has been recently shown that it is possible to functionally revert several mutated genes by multiplexing CRISPR technology (Nødvig et al. 2018).
4. Counter-Selectable Markers
Some nutritional markers can be counter-selected by using analogs of their natural substrates that are converted into antimetabolites. The two most frequently used counter-selectable wild-type markers are pyrG and amdS, which are lethal in the presence of 5-fluoroorotic acid (5-FOA) and 5′-fluoroacetamide (FAA), respectively. For both pyrG, and to lesser extent amdS, a gene copy is naturally present in wild-type fungi. Hence, the endogenous gene needs to be disrupted before the marker can be applied in selection/counter-selection experiments. In pyrG+ strains, 5-FOA is converted by the orotidine-5′-phosphate decarboxylase (PyrG) into 5-fluorouracil, which is further metabolized into toxic substances that interfere with RNA and DNA synthesis (Boeke et al. 1984; Longley et al. 2003). Similarly, in amdS+ strains, FAA is converted by the acetamidase (AmdS) into the toxic compound fluoroacetate (Hynes and Pateman 1970), which forms a stable complex with Coenzyme A (fluoroacetyl-CoA), hence hindering normal functionality of the tricarboxylic acid cycle. Other counter-selectable markers include genes in the tryptophan pathway, most often trpA (Foureau et al. 2012), and genes in the lysine pathway, most often lysB (Alberti et al. 2003), which can be counter-selected in the presence of 5-fluoroanthranilic acid, 5-FAA, and α-aminoadipate, respectively. Similarly, expression of genes encoding herpes simplex virus 1 thymidine kinase can be counter-selected as the viral thymidine kinase, unlike the thymidine kinases of the hosts, activates nucleoside analogs like 5-fluoro-2′-deoxyuridine for toxic incorporation into nucleic acids (Lupton et al. 1991). Counter-selectable markers are highly useful as they facilitate marker recycling and iterative genetic engineering.
5. Marker Recycling for Iterative Engineering
In most fungi, the number of available markers is a limiting factor for multi-step genetic engineering strategies. If, e.g., an entire biosynthetic pathway needs to be implemented into a new host, marker recycling is therefore often a necessity. Specifically, if two markers are available for transformation of a host strain, an endless number of genes can in principle be inserted into the same site in the host genome by iterative marker-swapping. In this method, the marker gene, which was used for gene integration in the previous transformation step, is replaced by a new gene targeting construct that contains new GOIs and another marker (Nielsen et al. 2013).
A marker gene can also be eliminated from the genome by spontaneous direct-repeat recombination if the marker is flanked by sufficiently long direct repeats, typically 500–1000 bp (Nielsen et al. 2006). This method may be preferred if it is desirable to leave a genetically neutral DNA sequence (i.e. the direct repeat) as the only scar after genetic engineering, or if it is desirable to integrate several genes or sets of genes into several different loci in the genome. As spontaneous direct recombination events are rare, this method requires that marker loss is selectable, e.g., by using a counter-selectable marker; see above (Sect. III.B.4). Alternatively, marker loss can be achieved by induced recombination involving either site-specific recombinases like Cre and a marker, which is flanked by its target sequences, or gene deletion catalyzed by site-specific nucleases like I-SceI or a CRISPR nuclease (Ouedraogo et al. 2016; Zhang et al. 2013).
C. Promoters
Among the different bio-blocks, promoters have drawn most attention since they control gene transcription initiation. Promoters are typically divided in two groups: those that are constitutively active, i.e., promoters that are active under all/most circumstances, and those that are inducible/repressible. Often constitutive promoters are preferred for large-scale heterologous production as they are active throughout the fermentation process in inexpensive media. In cases where the product is toxic or unstable, it may be necessary to restrict production to a specific growth phase to maximize yields; and in these cases, it is necessary to use an inducible/repressible promoter. However, as addition of inducing or repressing agents comes with an additional cost, use of this type of promoters may be restricted to small scale production or exploratory studies.
The vast majority of heterologous gene-expression studies are based on natural promoters of which we have listed sets of frequently used promoters derived from different fungal genera (Table 10.2). Interestingly, many promoters involved in basic metabolism do not seem to be species specific. Hence, promoter versatility allows the same genetic element to be used in many different species, even species belonging to different genera. However, to our knowledge, no comparative studies have systematically analyzed how active a given promoter is in different species. Based on the fair assumption that a promoter is most active in the species from which it originates, and less active when applied in other fungi, it is advisable to equip the GOI with a promoter derived from the intended production host. Importantly, the efficiency of promoters, even constitutive promoters, often depends on the growth environment and growth phase of the host. This has motivated establishment of synthetic promoters that offer orthogonal setups that work independently of the host metabolism. Below we present examples of natural and synthetic promoters.
1. Natural Promoters
A wide selection of natural fungal promoters for species belonging to Aspergillus, Penicillium, and Trichoderma genera as well as others (Table 10.2) have been experimentally characterized as constitutive or inducible, along with their mode of induction and repression, as reviewed in Fitz et al. (2018), Fleissner and Dersch (2010), and Kluge et al. (2018). Typical constitutive promoters for heterologous expression in fungal cell factories are derived from genes involved in major metabolic pathways or cell functions that require high steady protein levels. Examples are the gpdA promoter (PgpdA) and the stronger tef1 promoter (Ptef1), which control production of glyceraldehyde-3-phosphate dehydrogenase acting in glycolysis and translation elongation factor 1α assisting in protein synthesis, respectively. Promoters catalyzing basic reactions in the cell are often functional if transferred to other species. In line with this view, PgpdA and Ptef1 from A. nidulans have been shown to work in other species of Aspergilli (Nødvig et al. 2015), in Talaromyces atroroseus (Nielsen et al. 2017), and the PgpdA of A. nidulans was even applied in the fungus Metarhizium anisopliae (Nakazato et al. 2006). Inducible/repressible promoters are also recruited from basic metabolic genes. This includes the commonly used alcohol and threonine inducible alcA promoter, which controls expression of alcohol dehydrogenase I involved in alcohol catabolism, and the thiamine repressible thiA promoter, controlling expression of thiamine thiazole synthase involved in thiamine synthesis. This type of promoters can typically also be expected to function in other species, such as the PthiA of A. oryzae shown to also be functional in A. nidulans (Shoji et al. 2005) and the A. nidulans PalcA applied in A. fumigatus (Romero et al. 2003). Some very strong inducible promoters depend on degradation of the feedstock, e.g., the starch-inducible promoter of A. niger glaA, controlling expression of a secreted glucoamylase/1,4-alpha-glucosidase for starch hydrolysis. Similarly, a favorite promoter from T. reesei is the cel7a promoter controlling expression of secreted cellobiohydrolase I, which is induced on media containing cellulose or lactose. For promoters that normally drive expression of secreted biomass-degrading enzymes, the conditions inducing the gene expression typically involve co-induction of other endogenous genes of secreted biomass-degrading enzymes. Thereby, this may lead to production of these enzymes as undesired side-products. The activation mechanisms of such promoters are more specialized than for genes of the central metabolism and may not be directly transferrable from species to species. For example, activation of T. reesei Pcel7a involves recruitment of a general transcription factor of cellulase-encoding genes, Xyr1 (Castro et al. 2016), in complex with a non-coding RNA, HAX1 (Till et al. 2018; Table 10.2).
a) Approaches for Identification of Natural Promoters
Classical promoters may often be chosen due to their availability and history rather than due to considerations on whether their expression profile and strength fit the heterologous production process. Hence, in many cases the default choice is to employ the strongest promoter available as a basis for heterologous production. However, in some cases, maximum transcription levels are not desirable. For example, if the goal is to produce a complex secondary metabolite, the set of genes required to synthesize this compound may need to be expressed at different levels to achieve a balanced biosynthetic pathway. Expanding the library of fungal promoters to include members displaying a wide range of different expression strengths and other properties is therefore desirable. Fortunately, their discovery is accelerated by the availability of fully or partially sequenced fungal genomes. For example, by analyzing genome-wide transcription profiles obtained at different growth stages on different relevant growth media, it is possible to identify promoters with properties that are tailored to fit a given process. In the simplest scheme, one may apply small-scale targeted transcriptomic approaches, like RT-qPCR to determine the promoter strength under certain conditions (Li et al. 2012). Alternatively, large publically available datasets can be applied, such as the A. niger transcriptome microarrays covering several different growth conditions (Andersen et al. 2008; Breakspear and Momany 2007). Currently, transcriptomic microarray datasets covering 155 different cultivation conditions are available for A. niger (Schäpe et al. 2019). Using this approach to their advantage, Blumhoff and co-workers identified six novel constitutive A. niger promoters with varying expression strength (Blumhoff et al. 2013). The library contains promoters that are stronger and weaker than A. niger PgpdA displaying ten-fold differences in expression levels of native genes, and when applied to produce the β-glucuronidase (uidA) from E. coli , 1000-fold in specific activity yields were obtained. A similar set of T. reesei promoters displaying varying expression strengths has been derived from its glycolytic genes (Li et al. 2012), and in Penicillium chrysogenum both homologous and heterologous promoters have been evaluated for expression efficiency (Polli et al. 2016). It is important to note that although genome-wide transcription profiles provide mRNA levels for specific genes, they do not define the exact promoter sequences that control these genes. To this end, a global map of transcription initiation sites in A. nidulans was generated based on full-scale RNA-sequencing of gene transcripts obtained at six different growth conditions (Sibthorp et al. 2013), and this will facilitate extraction of new A. nidulans promoters as the map simplifies promoter sequence identification.
2. Synthetic Promoters and Gene-Expression Systems
Natural promoters are often influenced by the state of metabolism of the host, and this may be undesirable for heterologous production. Synthetic biology-based approaches are therefore adopted to generate artificial/synthetic gene-expression systems that ideally act independent of the host metabolism.
For example, Gressler et al. developed an expression system based on a fusion of the maltose-inducible amyB promoter from A. oryzae with the ORF encoding the transcriptional activator TerR from the Aspergillus terreus terrein gene cluster (Gressler et al. 2015). In addition, the system contains an expression cassette with a bidirectional promoter that binds TerR. Subsequently, the system was used to heterologously express two GOIs in A. niger by addition of maltose.
Another synthetic expression system was developed in T. reesei to facilitate cellulose degradation. Cre1 and Ace1; two general glucose stimulated repressors of genes involved in production of cellulolytic enzymes were fused to the activation domain of herpes simplex virus protein 16 (VP16 ), hence turning glucose repression into gene activation (Zhang et al. 2018).
More advanced orthogonal systems based on synthetic transcriptional regulators and matching synthetic promoter sequences have made gene expression much more controllable. For example, synthetic biology based methods have elegantly been applied to develop a constitutive expression system, which is almost equally functional in distantly related fungi including A. niger , T. reesei, and several yeasts (Rantasalo et al. 2018). Such synthetic tools bypass the organism specificity of conventional promoters and may become a valuable bio-block for multi-species studies. This system consists of a synthetic transcription factor (sTF) composed of the LexA repressor from E. coli fused to the activation domain from VP16. Expression of sTF is controlled by promoters with different arrays of LexA binding sites positioned upstream of a fungal core promoter, which was selected for functionality in several species. Low constitutive expression of the gene encoding the synthetic transcription factor is ensured via another universally active fungal core promoter. Importantly, promoter strength can be regulated by varying the number of LexA binding sites in the synthetic promoter. The system was recently combined with CRISPR-Cas9 multiplexing technology enabling triple targeted integration of the GOI cassette in three different loci in T. reesei (Rantasalo et al. 2019).
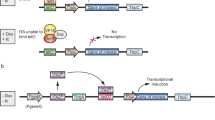
Orthogonal inducible expression platforms based on the Tet-on and Tet-off systems have been implemented in fungi (Fig. 10.4) (Meyer et al. 2011; Wanka et al. 2016). The Tet-off system is based on the synthetic transcription factor (tTA) a fusion of the E. coli repressor TetR with the activation domain of VP16 from herpes simplex virus, which binds to tetO operator elements in the absence of the tetracycline derivative doxycycline, Dox (Gossen and Bujard 1992). In contrast, the Tet-on system is based on rtTA-M2 (or other variants of rtTA) (Gossen et al. 1995; Urlinger et al. 2000), which are mutated versions of tTA that binds to tetO operator elements in the presence of Dox. A Dox responding synthetic promoter is made by positioning tetO elements upstream of a core promoter, which provides low or undetectable basal expression level in the absence of the synthetic transcription factors tTA and rtTA-M2 (Wanka et al. 2016). In the Tet-on system, which constitutively produces rtTA-M2, the promoter is activated by addition of Dox, whereas in the Tet-off system, which constitutively produces tTA, the promoter is activated in the absence of Dox. Note that the Tet-on system is often preferred over Tet-off due to its faster response time (Wanka et al. 2016).

Graphic representations of the Tet-on and Tet-off expression systems. (a) Tet-on system. (b) Tet-off system. In the Tet-on system, the Dox ligands acts as an activator of the synthetic TF rtTA2s-M2 allowing it to bind to the tetO7 sites of the promoter. In the Tet-off system, the Dox ligands repress binding of the synthetic TF tTA2s to the tetO7 sites of the promoter; see main text for details
D. Terminators
Terminators coordinate transcription termination and the extent of polyadenylation at the 3′-end of the new transcript, which in turn is important for its nuclear export and stability (Bentley 2014). In fungi, gene-expression cassettes have typically employed terminators derived from tef1 and trpC from Aspergilli, and it appears that terminators can be functionally transferred from one fungal species or genera to another. For example, A. nidulans tef1 and trpC terminators have successfully been used in various Aspergilli (Nødvig et al. 2015) and T. atroroseus (Nielsen et al. 2017), while the terminators of T. reesei cbhII and pdc were applied in A. niger (Blumhoff et al. 2013) and the basidiomycete Ganoderma lucidum (Qin et al. 2017), respectively. Since functionality is often conserved during heterologous application of terminators, an alternative approach for gene expression uses the GOI and its natural terminator directly as a functional unit for assembly of the gene-expression cassette (Gressler et al. 2015; Li et al. 2018).
We note that no thorough comparative analyses of the impact of terminators on the overall protein production have been performed for filamentous fungi despite the prominent roles of terminators in the RNA life-cycle. However, in other expression systems, e.g., in yeasts, the choice of terminator has been demonstrated to significantly influence production yields (Curran et al. 2013; Morse et al. 2017), and it may therefore be useful to assemble expression cassettes with terminator variants if production yields are suboptimal. To this end, we also note that small functional synthetic terminator bio-blocks have been developed for S. cerevisiae to facilitate gene-expression cassette assembly and control of production yields (Curran et al. 2015).
E. Protein Tags and Linkers
In many cases, it is desirable to expand the sequence of a protein with additional sequences encoding domains that provide new properties to the protein to facilitate its secretion, purification, or visibility (Table 10.3). These new domains often need to be attached to the main protein via linker sequences, which may act solely as spacers (Chen et al. 2013), or contain a proteolytic site that allows for removal of the attached domain (in vivo or in vitro) when its function is no longer required. This adds to the complexity of the gene-expression cassette structure as additional bio-blocks need to be designed and incorporated into the cassette at the appropriate positions.
The design of these bio-blocks depends on the size of the new functional unit and strategies for their assembly are presented in Fig. 10.5. Firstly, bio-blocks encoding large size addendums (>30 amino acid residues) like fluorescent proteins can be made as individual bio-blocks by PCR. Similar to the assembly of the basic gene-expression cassette (in Fig. 10.1), proper incorporation of the new bio-block is ensured by sequences in the tails of the primers used to generate the individual bio-blocks, which can be enzymatically cut to provide overhangs that direct the correct fusion order of all components. Secondly, for medium size bio-blocks encoding sequences of 10–30 amino acid residues (e.g. secretion signals), the information can be incorporated into each of two oligonucleotides that are annealed in vitro to form the complete bio-block. Note that small sequence extensions may be added to the oligonucleotides to produce bio-blocks, which are directly equipped with short ssDNA overhangs for bio-block assembly. Thirdly, for shorter sequences (<10 amino acid residues), e.g. purification tags or proteolytic sites, the information may be incorporated into the primer tails of neighboring bio-blocks. For example, if the C-terminus of a protein needs to be extended with a poly-histidine tag for purification, the primer tail used to link the GOI to the next bio-block could be elongated with the sequence encoding the tag. The combinations in which these basic strategies can be employed to construct GECs are numerous, and as the prices decrease, it is likely that even large size protein tags will be synthesized de novo rather than by PCR amplification.

Assembly strategies for employing protein tags. (a) Bio-blocks can be generated by three general strategies depending on their size: PCR amplification, annealing of two oligonucleotides, or incorporation into the tail of a primer used to PCR amplify another bio-block. (b) The resulting bio-blocks can be fused in a directed manner as illustrated in Fig. 10.1
IV. Heterologous Protein Production in Filamentous Fungi
Filamentous fungi serve as favorite hosts for production of industrial enzymes due to their superior protein secretory potential. This potential has been further improved by classical mutagenesis yielding strains with superior protein secretion properties. This includes A. niger CBS 513.88 and T. reesei RUT-C30, which can reach titers of 30 and 100 g/L of endogenous cellulolytic enzymes by optimized cultivations, respectively (Cairns et al. 2018; Cherry and Fidantsef 2003). Detailed mechanistic insights into the secretory pathway combined with fully sequenced genomes and a diverse range of omics data set the stage for rational strain engineering. In this section, we will present examples of genetically modified strains, which serve as platforms for heterologous protein production. This includes strains with improved production and secretion physiology obtained by introducing defined mutations influencing processes ranging from the delivery of amino acid building blocks for protein synthesis to processes involved in exocytosis.
Specifically, we will treat two major topics: firstly, engineering the secretory pathway focusing on transport signals, the associated processes of protein glycosylation and folding, and vesicular trafficking and secondly, expression in protease-deficient strains as a method for reducing degradation of the heterologous protein.
A. Engineering the Secretory Pathway
The protein secretion pathway is complex and provides several processes that can be engineered to enhance production yields. Generally, protein secretion processes can be grouped into three major themes. Firstly, secretion signals that mediate translocation into the endoplasmic reticulum (ER). Secondly, folding and glycosylation of secretory proteins. Thirdly, the complex machinery, which transports proteins through the secretory pathway and leads to secretion by exocytosis. Below, we provide examples showing how each of these steps can be optimized by rational genetic engineering.
1. Transport Signals
A significant obstacle in optimizing secretion of a heterologous protein is choosing the right pre- and pro-sequence, as protein secretion is inefficient when suboptimal signals are applied. Pre-sequence s encode secretion signals that mediate protein translocation into the ER, while pro-sequence s may facilitate the folding process and maintain proteins as inactive forms until its removal. One of the three general approaches is typically used to ensure secretion of a heterologous protein. In the first approach, secretion of the recombinant protein simply relies on the secretion signals of the native protein. In the second approach, the N-terminus of the recombinant protein is extended with either a pre- or pre-pro-sequence from a highly secreted protein of the production host (or from a closely related species) or thirdly N-terminal extension with a secreted carrier protein.
The first, and most simple, approach employs the native secretion signal of the heterologous protein. However, the fact that the sorting signal are from a different organism raises questions concerning their ability to support efficient secretion in the host. The observation that heterologous proteins originating from species closely related to the host often sort quite efficiently suggests that their secretion signals are functional.
Secretion signals may also be functional between more distantly related fungi, as observed with two basidiomycete laccases, Lcc1 of Pycnoporus coccineus and Lcc of Pycnoporus sanguineus , successfully secreted by the hosts A. oryzae and A. nidulans using the proteins native secretion signals, albeit with low yields (Hoshida et al. 2005; Li et al. 2018). More efficient secretion may be achieved by using host endogenous signals as they are expected to be more proficiently recognized and processed by the host. The second approach therefore employs the pre- or pre-pro-sequence of a highly secreted protein originating from the host, or a closely related species, to enhance secretion of the heterologous protein.
A successful example of this approach is production of basidiomycete laccase, Lac1, from Pycnoporus cinnabarinus in A. niger (Record et al. 2002). In this study, an 80-fold increase in extracellular activity was achieved by replacing the natural pre-sequence of Lac1 with the pre-pro-sequence of A. niger glaA.
In the final approach, a carrier protein is fused to the heterologous protein to improve production by alleviating post-translational bottlenecks and, in some cases, by increasing mRNA levels (Gouka et al. 1997). Comparative DNA microarrays of A. oryzae strains expressing heterologous bovine chymosin, with and without a carrier protein (AmyB), showed that inclusion of the carrier protein promoted induction of the UPR (see Sect. IV.A.2.b) and increased expression of genes encoding secretory chaperones and proteins involved in intracellular trafficking, thereby facilitating folding and secretion (Ohno et al. 2011). Examples of proteins that have been used as carriers are GlaA, AmyB, and CbhI from A. niger , A. oryzae, and T. reesei , respectively. To liberate the heterologous protein from the carrier protein in vivo, the two proteins are fused via a linker containing a Kex2 cleavage site (KR/RR) to enable proteolytic separation in the late-Golgi (Jin et al. 2007; Landowski et al. 2016).
Employing this approach, Gouka et al. used A. niger GlaA as carrier protein for heterologous production of human interleukin-6 in Aspergillus awamori , thereby improving the extracellular protein yield 100-fold (Gouka et al. 1997).
In an interesting variant of this approach, Jin et al. produced human lysozyme in A. oryzae using a gene cassette containing the amyB gene fused to tandem gene copies of HLY encoding lysosome, which were separated by sequences encoding Kex2 cleavage sites (Jin et al. 2007). However, releasing the heterologous protein from the carrier protein may be a bottleneck. For example, during production of human interferon (IFNα-2b) in T. reesei using Cbh1 as a carrier protein, 44% of the fusion protein was not cleaved by Kex2 (Landowski et al. 2016). More efficient Kex2 cleavage has been achieved by overexpressing kex2 (Landowski et al. 2016) or by optimizing Kex2 cleavage sites (Lin et al. 2006; Nakajima et al. 2006; Yang et al. 2013), e.g., by using the entire pro-sequence of GlaA, NVISKR, as a Kex2 cleavage site (Landowski et al. 2016).
It is important to stress that the choice of optimal signal sequences is not straightforward. For example, Rantasalo et al. optimized secretion of lipase B from Candida antarctica (CalB) in T. reesei (Rantasalo et al. 2019) by using natural secretion signals of the host proteins CbhI and CbhII and secretion signals from heterologous AmyA and GlaA from A. awamori and A. niger , respectively, and by using CbhI as a carrier protein. When the lipase yields obtained with the different strains were compared, the strain employing the secretion signal of AmyA performed best. On the other hand, studies on heterologous production of Cbh1 in A. awamori using different secretion signals showed no differences illustrating that gains may be dependent on the heterologous protein and/or the host (Adney et al. 2003; Chou et al. 2004).
2. Glycosylation and Folding
Glycosylation of the heterologous protein is important for the subsequent folding, and without validity of these properties, the protein activity may be impaired or abolished. In the heterologous production host, these processes may be executed in alternate fashion compared to the native producer, leading to reduced yields or non-functional proteins. Therefore, substantial efforts have been conducted to improve both processes by increasing or reducing expression of genes encoding proteins involved in the glycosylation and folding machinery of the production host to enhance protein production.
a) Glycosylation
Protein glycosylation has multifaceted impact on heterologous protein production. Firstly, it affects protein folding and secretion efficiencies; secondly, it influences the final properties of the enzyme including its stability, solubility, and catalytic parameters; and finally, it acts to protect the enzymes from proteases (Gupta and Shukla 2018; Li and d’Anjou 2009). In agreement with this view, mutation of the four N-glycosylation sites of A. terreus β-glucosidase resulted in reduced thermal stability and 15–35% decreased specific activity and catalytic rate as compared to the wild-type variant when heterologously produced by T. reesei (Wei et al. 2013). Hence, proper glycosylation is often required to achieve high production yields. For example, analysis of bovine chymosin produced by A. niger showed that an N-glycosylation site was poorly glycosylated by this host. Importantly, the extracellular activity yields could be increased by up to 100% in A. niger by optimizing the glycosylation efficiency of this site by site-directed mutagenesis (van den Brink et al. 2006). Similarly, production yields and quality may also be increased by engineering entirely new glycosylation sites into proteins. Using this approach, heterologous production activity yields of cellobiohydrolase Cel7A from Penicillium funiculosum in A. awamori were increased by 70% by engineering a novel N-glycosylation motif into the protein at position N194 (Adney et al. 2009). On the other hand, production yields and quality may be decreased if non-native glycosylation sites are unintentionally or aberrantly glycosylated by the glycosylation machinery of the heterologous host. Hence, a 70% increase of T. reesei Cel7A activity yields were achieved by removing a glycosylation site (N384), which contained a larger glycan structure when using A. awamori as production host, than in the native organism T. reesei (Adney et al. 2009). Fungi may also potentially be used as hosts for production of therapeutic proteins. The fact that different species add different sugar structures to proteins poses an additional production challenge, as therapeutic proteins containing aberrant sugar moieties may cause immune responses in patients. In yeasts, this challenge has been successfully addressed by humanizing the glycosylation pathway by genetic engineering (Gupta and Shukla 2018) indicating that production of therapeutic proteins may also be possible in filamentous fungi after dedicated strain engineering (Anyaogu and Mortensen 2015).
b) Chaperones and ER Stress
Overexpression of secretory proteins may result in accumulation of misfolded proteins in the ER, and this problem poses a severe bottleneck toward high yields of secreted proteins. Cells respond to misfolded proteins in the ER by triggering the unfolded protein response (UPR), which is sensed by the ER transmembrane protein Ire1. Normally, Ire1 forms an inactive complex with the chaperone BipA, but as BipA is increasingly recruited to assist in folding, Ire1 forms a nucleolytically active dimer that catalyzes splicing of the non-conventional intron in the hac1 mRNA (Krishnan and Askew 2014). The processed hac1 mRNA encodes mature Hac1 transcription factor, which in turn triggers transcription of UPR genes. This includes genes encoding ER-resident molecular chaperones like BipA, the heat-shock protein family of chaperones (Hsp104, Hsp70, Hsp90) and the protein disulfide-isomerase PdiA, thus satisfying the increased demand for folding capacity (Zubieta et al. 2018). Persistently misfolded proteins are deleterious to the cell and are degraded through the ERAD (ER-associated degradation) pathway that includes transport to the cytoplasm and ubiquitin-mediated degradation by the proteasome (Carvalho et al. 2011).
Several strategies have been pursued to optimize folding. These include defensive methods, like lowering the growth temperature or applying weaker promoters, but also potentially more awarding methods based on stimulating the folding potential via genetic engineering of the host organism folding machinery.
To promote heterologous protein production in A. awamori , the UPR pathway has been constitutively activated by overexpressing an intron-free variant of the hac1 gene. Using this strain, production of Trametes versicolor laccase (Lcc1) was increased sevenfold and bovine chymosin 2.8-fold (Valkonen et al. 2003). Similarly, heterologous production of A. niger glucose oxidase (Gox) in a T. reesei strain overexpressing intron-free hac1 increased production 1.8-fold (Wu et al. 2017). In comparison, overexpression of the bipA chaperone alone in T. reesei increased production of Gox by 1.5-fold.
Folding of proteins containing multiple disulfide bonds may be particularly challenging and may benefit from increased levels of the protein disulfide isomerase PdiA. In agreement with this, production of thaumatin, a sweet-tasting plant protein containing eight disulfide bonds, was increased twofold by using an A. awamori strain overexpressing pdiA (Moralejo et al. 2001). Since heterologous protein production appears to benefit from increased levels of host chaperones, it is tempting to speculate that more process-specific folding assistance could be achieved by co-expressing genes encoding one or more chaperones from the natural source of the protein of interest.
3. Vesicular Trafficking and Polarized Growth
Filamentous fungi propagate as hyphae and the general view is that most protein secretion occurs at the hyphal tip (Cairns et al. 2019). Secreted proteins are packed into vesicles, either by cargo-receptors or as the result of bulk flow, thereby mediating transport from the ER to the Golgi and from the Golgi to the plasma membrane (Barlowe and Miller 2013). Most vesicles accumulate at the Spitzenkörper at the apex of the hyphae before fusing to the membrane in a process that depends on the exocyst octamer (Ahmed et al. 2018; Riquelme and Sánchez-León 2014). Since proteins are synthesized throughout the hyphae, vesicles are transported by an elaborate transport system based on actin filaments and microtubules to ensure efficient transport to the hyphal apex (Steinberg et al. 2017). Many of the individual steps in the secretory pathway are understood in molecular detail, and a popular strategy aiming at enhancing secretory transport is to overexpress genes encoding proteins that are directly involved in transport. For example, loading of cargo proteins into vesicles and targeting vesicles to a destination membrane have been engineered to increase secretion as exemplified below.
To stimulate secretion of a heterologous protein, the vesicular transport was enhanced by overexpressing rabD, encoding a Rab GTPase involved in transport of exocytic post-Golgi vesicles (Pantazopoulou et al. 2014), the deletion of which reduces protein secretion (Punt et al. 2001). Specifically, a fusion of carrier glucoamylase to mRFP via a Kex2 cleavable linker (Kex2cl) increased production of mRFP in A. nidulans by 40% (Schalén et al. 2016) in the rabD overexpression background. Hoang and co-workers showed that Vip36, a putative lectin-type cargo receptor inferred to be involved in vesicular cargo loading of glycoproteins between ER and Golgi, facilitates secretion of the recombinant fusion proteins AmyB-Kex2cl-GFP and AmyB-Kex2cl-chymosin in A. oryzae , presumably by reducing protein retention in the ER (Hoang et al. 2015). Importantly, overexpression of vip36 in strains producing the recombinant GFP fusion protein led to sevenfold increase in the levels of extracellular GFP (Hoang et al. 2015). Finally, docking of vesicles to a target membrane was stimulated by overexpressing snc1 encoding a vSNARE, and this feature increased secretion of A. niger glucose oxidase in T. reesei by 2.2-fold (Wu et al. 2017).
Similarly, specific genes involved in the secretory pathway can be deleted to positively influence heterologous protein production. For example, the vacuolar protein sorting receptor Vps10 mediates transport of proteins from the Golgi to the vacuoles; and it has been observed that heterologous proteins designated for secretion partially accumulate in the vacuoles (Masai et al. 2003). This principle has been exploited in A. oryzae where a deletion of vps10 improved production of recombinant human lysosome and bovine chymosin 2.2- and 3-fold, respectively (Yoon et al. 2010). Even the micromorphology of the mycelium can be manipulated to enhance protein secretion. In an elegant study, Fiedler et al. produced a hyperbranching A. niger strain by deleting the gene encoding the Rho G-protein RacA, a key protein in maintaining polarized growth (Kwon et al. 2011); and this feature increased glucoamylase production four-fold (Fiedler et al. 2018).
C. Building Blocks for Protein Synthesis
Many filamentous fungi secrete large amounts of enzymes and production of these proteins consume building blocks that could be used for the desired heterologous protein. Hence, additional building blocks for heterologous protein production can be obtained by deleting genes encoding the most abundant secreted proteins. This strategy has the further advantages that it reduces pressure on the secretory pathway machinery and that subsequent product purification is simplified. In its simplest scheme, this approach can be utilized by deleting genes encoding major secreted products, such as α-amylase A and B of A. oryzae , thereby providing a strain with no detectable α-amylase activity (Kitamoto et al. 2015). Toward the same goal, genes encoding transcriptional activators of major cellulolytic enzymes have been deleted in both A. niger and T. reesei .
In A. niger , deletion of amyR reduced the total amount of secreted protein 16.4-fold (Zhang et al. 2016b). Likewise, deletion of xyr1 in T. reesei abolished expression of the genes cbhI and cbhII encoding the major cellobiohydrolases CBHI and CBHII, which may constitute up to 80% of the total amount of secreted protein (Bergquist et al. 2004; Stricker et al. 2006).
While these approaches diminished expression of endogenous enzymes, it may also be advantageous to modify the GOI expression cassette. To this end, for driving secretion of a heterologously produced protein, Rantasalo et al. exchanged usage of a carrier protein for a smaller secretion signal, thereby reducing the total secreted protein twofold and inferring a substantial increase in recombinant enzyme purity (Rantasalo et al. 2019).
Comparative transcriptomics approaches have been used to uncover engineering targets that enhance heterologous protein production. For example, processes related to biosynthesis of amino acids and tRNAs are upregulated in strains of A. nidulans overexpressing heterologous enzymes (Zubieta et al. 2018) and in A. niger CBS 513.88, a classic enzyme production strain (Andersen et al. 2011). Hence, building block availability may be a limiting factor that could be improved by genetic engineering, e.g., by increasing expression of genes encoding specific transporters or genes required for making aminoacryl-tRNAs.
D. Protease-Deficient Strains
The saprophytic lifestyle of filamentous fungi is key to their high secretion capacity. In addition to facilitating heterologous protein production, a consequence of this lifestyle is secretion of large amounts of proteases , which the fungi use for extracellular biomass degradation. For example, proteomic analysis of the T. reesei QM6a secretome under submerged cultivation showed pH-dependent secretion of 39 peptidases and proteinases, with more than 20 secreted simultaneously (Adav et al. 2011). Since these proteases need to digest a broad spectrum of substrates, chances are that the heterologous protein is also degraded leading to decreasing activity of the recombinant protein over time (Kamaruddin et al. 2018). In addition, intracellular proteases in the secretory pathway, such as Kex2, may also degrade recombinant proteins, which can be resolved by mutagenizing the cleavage site (Lin et al. 2006). The fact that addition of protease inhibitors, such as trypsin and aspartic protease inhibitor (Landowski et al. 2015), or use of strains with different protease profiles (Sun et al. 2016) can lead to significantly higher protein yields suggests that a strategy aiming at producing protease-deficient strains could be advantageous. However, whereas elimination of some proteases does not impact fitness, some may be involved in cell wall maintenance, and their absence often causes an altered undesired morphology. Hence, a gain in product yield may be lost in growth rate or overall productivity, and the relevant strain modifications vary from product to product even with the same host (Landowski et al. 2015).
In a simple case, Li et al. applied A. nidulans as host for heterologous production of a P. sanguineus laccase (Pslcc) and demonstrated that deletion of two genes encoding major proteolytic activities, proteases dipeptidyl-peptidase DppV and aspartic protease PepA, increased laccase production 13-fold (Li et al. 2018).
In a more elaborate study, Kitamoto et al. generated an A. oryzae strain with five protease gene deletions (alp1, npI, npII, pepA, pepE) thereby reducing extracellular protease activity to 1% (Kitamoto et al. 2015) and providing a production strain with minimal proteolysis.
In the post-genomic era, it is possible to make a rational gene deletion strategy based on proteomics where abundant proteases can be identified and linked to their genes. Using this strategy in T. reesei , nine target genes (pep1, tsp1, slp1, gap1, gap2, pep4, pep3, pep5, amp2) were identified and deleted to produce a strain where the majority of proteolytic product degradation is abolished (Landowski et al. 2015, 2016). The authors employed this strain for production of human interferon alpha-2b (IFNα-2b) and obtained 2.4 g/L of correctly processed IFNα-2b.
In contrast to deleting individual protease genes, similar effects may be achieved by RNAi silencing of individual genes (Kitamoto et al. 2015; Qin et al. 2012). In another approach, reduction of a set of proteases was achieved by deleting a general transcription factor gene, namely, prtT in A. niger , which reduced the levels of several secreted proteases, including PepA, PepB, and PepF. After producing Glomerella cingulata cutinase in this A. niger strain a 25-fold increase of residual activity was observed in culture filtrates after 2 weeks (Kamaruddin et al. 2018).
V. Heterologous Production of Secondary Metabolites in Filamentous Fungi
The genome sequencing projects have uncovered a vast repertoire of fungal SMs, which serves as an underexploited source of new food additives and drug candidates. This process has been facilitated by the fact that all genes required for production of a specific SM are typically organized in a biosynthetic gene cluster (BGC) (Keller 2019; Keller et al. 2005; Rokas et al. 2018). However, it is difficult to exploit the fungal SM potential for several reasons. Firstly, the vast majority of the fungal SM BGCs are silent; secondly, many producer species are difficult to propagate in bioreactors; thirdly, many compounds are produced in tiny amounts; fourthly, no genetic toolbox exists for new natural production hosts; and finally, the “generally recognized as safe” (GRAS) production status may be more difficult to achieve with a new species.
Heterologous expression of fungal BCGs using synthetic biology based approaches in well-characterized fungal cell factories provides avenues to speed up SM discovery, characterization, and production. On the other hand, heterologous SM production is challenged by several features including toxicity of some SMs, poor gene annotations, intron splicing differences, compartmentalization of pathways, and the requirement for simultaneous expression of many SM genes. Unlike heterologous production of industrial enzymes, heterologous production of SMs is still in its infancy, and most studies are aiming at product discovery and pathway elucidation rather than large-scale production. In this section, we will present examples on how heterologous SM-gene expression has contributed to expand our insights into SM biosynthesis, as well as challenges toward their production.
A. Challenges in Heterologous Secondary Metabolite Production
1. Product Toxicity
Many SMs are antimicrobials, and they may therefore impair the growth of, or even kill, the new heterologous producer strain. In case the task is to produce large amounts of a known SM, the first experiment should therefore be to test whether the host can tolerate the desired SM. If SM toxicity is a problem, it may be necessary to develop a resistant strain. If the resistance mechanism is known in the native producer species, it may be possible to transfer the resistance mechanism from the native host to the new producer strain.
For example, mycophenolic acid (MPA) from Penicillium brevicompactum kills A. nidulans by inhibiting its inosine-5′-monophosphate dehydrogenase (IMPDH). However, A. nidulans can be engineered to tolerate mycophenolic acid by inserting mpaF of the P. brevicompactum mpa BCG, which encodes an MPA-insensitive IMPDH, into its genome (Hansen et al. 2011a).
Toxicity can also be avoided or reduced by introducing a pump that exports the new compound. For example, the gliotoxin sensitivity of a sirodesmin transporter-deficient strain of Leptosphaeria maculans can be rescued by introduction of the transporter gene gliA from A. fumigatus (Gardiner et al. 2005). Similarly, S. cerevisiae expressing the mlcE efflux pump gene from the compactin BGC is protected against statins (Ley et al. 2015). A different mode of detoxification is based on glycosylation of the SM, a principle which is commonly used in plants (Sandermann Jr. 1992), but which has also been observed in fungi. For example, during yanuthone production by A. niger , a toxic intermediate is glycosylated (Holm et al. 2014), or during co-culturing, Trichoderma species uses glycosylation as a defense against deoxynivalenol, a toxin produced by Fusarium graminearum (Tian et al. 2016). For cell factory construction, this strategy has been applied for vanillin production in S. cerevisiae by expressing a gene encoding a glycosyltransferase from Arabidopsis thaliana (Brochado et al. 2010).
If the task of heterologous production is product discovery , rather than large-scale production, toxicity issues may be less important as only small amounts are sufficient for product identification. However, if weak growth of the new cell factory is observed, it may be advantageous to use inducible promoters to control expression of key genes in the pathway. For example, the inducible alcA promoter was fused to genes in the asperfuranone BGC to avoid toxicity issues during pathway elucidation (Chiang et al. 2013).
2. Genome Annotation and Intron Splicing
Heterologous production requires that the genes in the native producers are correctly annotated and for many sequenced genes, this may not be the case. In cases of doubt, it may be necessary to determine the correct 5′-end of the transcript by the rapid amplification of cDNA ends (RACE) technique (Frohman et al. 1988). Subsequently, introns and the correct stop codon can be identified by generating a complete cDNA of the gene by RT-PCR exploiting that the 3′-end of the transcript is polyadenylated. However, in cases where the GOI is silent, this strategy is not possible, and it may therefore be advisable to produce a set of gene constructs covering different combinations of start and stop codon possibilities.
Intron recognition and splicing differs between species, especially for phylogenetically distant organisms (Kupfer et al. 2004), and flawed splicing may reduce or even prevent formation of the desired protein as described in Sect. III.A.1. For example, introns are more abundant in basidiomycetes than in ascomycetes (Stajich et al. 2007). Moreover, introns from basidiomycetes may not be recognized properly in ascomycetes.
In agreement with this, a recent study using A. oryzae as a cell factory demonstrated that only half of the mRNA species produced by 30 terpene synthase genes from two basidiomycetes, Clitopilus pseudo-pinsitus and Stereum hirsutum , exhibited correct splicing patterns (Nagamine et al. 2019).
Even among species that are more closely related, e.g., within ascomycetes , transfer of genes between species may cause splicing errors. For example, heterologous expression of the 3-methylorcinaldehyde synthase gene from Acremonium strictum (Fisch et al. 2010), the avirulence gene ACE1 from Magnaporthe oryzae (Song et al. 2015), and the citrinin synthase gene from Monascus ruber (He and Cox 2016) in A. oryzae resulted in incorrect processing of introns.
Moreover, some mRNAs of filamentous fungi are regulated by alternative splicing (Kempken 2013; Zhao et al. 2013). Hence, populations of different splice variants were detected in the transcriptome (Wang et al. 2010) with consequences for protein levels (Chang et al. 2010). Indeed, new splice variants may be inactive or the correct variant may be produced only in small amounts (He and Cox 2016; Kempken and Windhofer 2004). Mapping mRNA splicing in the native producer, if possible, is therefore advisable; and if it is different in the new host, it may be necessary to produce different synthetic gene variants covering different splice variants to achieve heterologous production.
3. Compartmentalization of Secondary Metabolite Biosynthetic Pathways
Fungal secondary metabolite biosynthetic pathways are often compartmentalized into different subcellular locations, such as ER, vesicles, peroxisomes, cytoplasm, or vacuoles (Kistler and Broz 2015; Roze et al. 2011). The reason for this is either to channel the pathway into the compartment with relevant precursors to increase biosynthesis efficiency as in the case of penicillin in A. nidulans (Herr and Fischer 2014) or to sequester the toxic product or intermediates from the rest of the cell as in the case of aflatoxin synthesis in Aspergillus parasiticus (Chanda et al. 2009). Little is known about how SM biosynthetic pathways are compartmentalized in fungi, and it is unclear how often this occurs. However, in cases where compartmentalization is important for product formation or to avoid toxicity effects, it is likely essential that the pathway is organized in a similar manner in a new host. Otherwise, it may result in lack of production or even cell death. In the future, we expect that pathway compartmentalization will achieve significant attention in SM cell factory construction. As a standard control experiment, we advise to determine whether individual enzymes in the pathway localize to the same compartments in the natural producer and in the new host, e.g., by GFP tagging. In cases where the host sorts an enzyme incorrectly, it may be possible to ensure proper localization of the relevant enzyme by fusing it to transport signals or a carrier protein that is sorted to the correct compartment.
B. Secondary Metabolite Discovery via Different Gene-Expression Systems
Multiple gene-expression strategies and different fungal hosts have been used for SMs discovery and pathway elucidation by heterologous gene expression. A. oryzae , unlike most other common fungal cell factories, produces few SMs. A. oryzae is therefore often chosen as a host as the subsequent detection of the new SM in the metabolite profile is relatively simple. However, with A. oryzae genetic engineering is complicated by the fact that its asexual spores contain several nuclei (Maruyama et al. 2001), which makes purification of transformed strains more laborious. In contrast, A. nidulans produces asexual spores that contain only a single nucleus (Yuill 1950), and transformants are therefore relatively easy to purify. Hence, if pathway elucidation requires more elaborate genetic engineering, species like A. nidulans may be a better choice of host. Moreover, in some cases, the more complex host-chemistry can advantageously contribute to formation of novel synthetic and potentially useful compounds. Alternatively, it may facilitate construction of a new synthetic pathway by delivering a missing activity in the desired pathway. Below we will provide successful examples of heterologous SM production using different hosts and expression systems, which may serve as an inspiration for construction of new fungal SM cell factories.
1. Heterologous Expression of Secondary Metabolite Synthase Genes
To investigate whether a new SM can be produced in a given cell factory, it may often be advantageous to establish the first step of a multi-enzyme pathway, which produces the scaffold of the final compound. Indeed, the first demonstration of a heterologously produced SM was 6-methylsalicylic acid (6-MSA) (Fujii et al. 1996), which serves as a precursor of several SMs including patulin and yanuthones (Beck et al. 1990; Holm et al. 2014; Petersen et al. 2015; Read and Vining 1968). Hence, it is of interest to develop efficient systems for single gene transfer. In A. oryzae , a plasmid was constructed to allow a gene to be inserted ectopically into the genome in multiple copies (Fujii et al. 1995).
This plasmid has been widely applied for heterologous gene expression, and it has been used to deliver basic SM scaffolds including production of the naphthopyrone YWA1 (Watanabe et al. 1998, 1999), alternapyrone (Fujii et al. 2005), ferrirhodin (Munawar et al. 2013), and astellifadiene (Matsuda et al. 2016).
In A. nidulans , where genetic engineering is easier, genes have often been inserted into a defined locus to facilitate strain characterization and to obtain better gene-expression control. To ensure high expression levels, it may be useful to position integration sites in intergenic regions located in transcriptionally highly active sections of a chromosome.
Using this approach, it has been demonstrated that MpaC from P. brevicompactum produces 5-methylorsellinic acid (5-MOA) as the first intermediate toward production of the immunosuppressant drug mycophenolic acid by expressing mpaC in A. nidulans (Hansen et al. 2011b).
The same setup was used to make a cell factory yielding 1.8 g/l of 6-MSA by expressing yanA from A. niger in A. nidulans (Knudsen 2015), which is sixfold higher as compared to ectopic integration in A. oryzae reported by Fujii et al. (1996). Using the A. nidulans 6-MSA cell factory, it was possible to produce 13C-labelled 6-MSA, which was subsequently used to clarify the biosynthetic pathway for yanuthone D production in A. niger (Holm et al. 2014). The wA locus of A. nidulans has also been used as a gene-expression site. In this case, correct transformants can easily be identified as they produce white conidia. Using this approach, Chiang and co-workers discovered several new compounds by expressing A. terreus polyketide synthase genes in A. nidulans (Chiang et al. 2013).
Finally, we note that synthesis of some scaffolds requires the action of additional enzymes, such as a trans-enoyl reductase, a trans-acyltransferase, a trans-thioesterase, or another synthase. For example, tenellin synthetase from Beauveria bassiana requires trans-acting enoyl reductase for correct polyketide scaffold assembly (Halo et al. 2008); during lovastatin synthesis, both acyltransferase and thioesterase are necessary in order to release the polyketide products from the synthases LovF (Xie et al. 2009) and LovB (Xu et al. 2013), respectively, whereas for the synthesis of the first intermediate toward asperfuranone, two polyketide synthases are needed (Chiang et al. 2009). In these cases, a multi-gene insertion strategy is required; see Sect. II.C.
2. Reconstitution of Secondary Metabolite Pathways in a Heterologous Host
Establishing entire pathways is more challenging as several genes need to be functionally expressed. In the following section, we will describe various strategies that have been used to ensure expression of some or all genes from a BGC.
In an early study by Sakai et al., a 20 kb fragment of DNA containing the citrinin BGC from Monascus purpureus was inserted into an E. coli -Aspergillus shuttle cosmid and ectopically integrated into the genome of A. oryzae to produce 4 μg/L of citrinin (Sakai et al. 2008). This yield was increased approximately 400-fold to 1.48 mg/L when the cluster-specific transcriptional regulator encoding gene (ctnA) was constitutively expressed under control of the A. nidulans trpC promoter. Pathways may also be transferred using a vector set developed by Pahirulzaman et al., which allows up to 12 different genes to be ectopically integrated into the genome via three vectors containing different selectable markers (Pahirulzaman et al. 2012). Hence, pathways may be partly or entirely established in a fungus to produce key intermediates for pathway elucidation as well as to identify the final product. Using this system and A. oryzae as a host, He and Cox produced a detailed model of the citrinin pathway and generated a cell factory producing citrinin to final titers of 19.1 mg/L (He and Cox 2016).
Defined integration of entire BGCs into fungal chromosomal expression sites has also been used to establish functional SM pathways. One advantage of this method is that reverse genetics can be applied to dissect the pathway once the pathway has been functionally established in a new host. This principle has been exploited to successfully transfer all 12 biosynthetic genes required for geodin production from A. terreus to A. nidulans . Additionally, to activate the biosynthetic genes, the TF gene of the cluster (gedR) was equipped with a constitutive A. nidulans promoter prior to genomic integration. Subsequently, the efficient gene deletion toolbox of A. nidulans was used to identify gedL as the gene encoding the halogenase necessary for geodin production (Nielsen et al. 2013).
A recent screening-friendly method for linking BGC to metabolites uses FACs as vectors for gene transfer (see Sect. II.B.1) as they can accommodate DNA fragments containing even large BGCs. FAC-based BGC libraries from A. terreus , Aspergillus wentii, and Aspergillus aculeatus have been transferred and analyzed in A. nidulans resulting in the assignment of 17 compounds to BGCs (Bok et al. 2015; Clevenger et al. 2017).
3. Synthetic Pathway Setups for Secondary Metabolite Production
For some compounds of interest, it is not possible to establish a cell factory based on the natural pathway. This is the case if the biosynthetic pathway is unknown or it has only been partly elucidated or if enzymes are compartmentalized in the native producer in a manner that cannot be implemented in the new host. In these cases, it may be possible to make a cell factory based on a semi-synthetic or synthetic pathway with enzymes of known (or predicted) activities from various species. In this way, it may be possible to synthesize the desired compound, but in a manner, which is different from the pathway in the native host. For example, production of the pharmacologically relevant meroterpenoid daurichromenic acid (DCA) from the plant Rhododendron dauricum in A. oryzae represents a successful case of a functional semi-synthetic pathway (Okada et al. 2017). Using fungal expression vectors for ectopic genome integration, the production of DCA was achieved by expressing two fungal genes from Stachybotrys bisbyi encoding a polyketide synthase (StbA) and a prenyltransferase (StbC), as well as a plant gene from the native producer encoding the DCA synthase. Similarly, production of carminic acid , an important food colorant produced by the scale insect Dactylopius coccus , has been achieved in A. nidulans via a semi-synthetic pathway (Frandsen et al. 2018). In this case, the five-step pathway was based on a single gene from the natural producer encoding a C-glucosyltransferase , a plant gene from Aloe arborescens encoding a type III octaketide synthase, two bacterial genes from Streptomyces sp. R1128 encoding a cyclase (ZhuI) and a aromatase (ZhuJ), and finally an unknown fungal gene from A. nidulans , which putatively encodes a monooxygenase. Importantly, unlike the DCA cell factory, all non-fungal genes were inserted into fungal expression cassettes and integrated into defined expression sites. These examples highlight the potential of puzzling together pathways by combining genes from different organisms. Similar strategies may likely gain an increasing role in future development fungal cell factories for production of known and novel small molecules.
VI. Concluding Remarks and Perspectives
This review demonstrates that heterologous production in fungi is already well established and that synthetic biology-based methods are increasingly used to create new cell factories, typically via bio-block-based strategies. In the future, we envision that this trend will gather momentum and that libraries of bio-blocks containing mutated genes, scrambled homologous genes, or synthetic genes encoding new combinations of functional domains will serve as common resources for developing cell factories that produce, e.g., industrial enzymes with new or improved properties or SMs that may target new diseases. In addition, increasingly efficient high-throughput CRISPR-based methods will allow for the generation of genome-wide mutant-strain libraries. Hence, rather than using a single strain for construction of a heterologous cell factory, mutant libraries may serve as the preferred starting point toward developing new cell factories. In combination, these strategies will create an enormous number of potential cell factories; and screening for the successful candidates will very likely constitute a bottleneck. The expanding synthetic biology toolbox may also contribute to address this challenge. For example, it may offer biosensors that can be used to screen for high-yielding strains. In agreement with this view, membrane-bound RFP was recently used as a biosensor in allowing for high-throughput FACS-based screening of T. reesei mutants to identify strains with improved enzyme secretion potential (Gao et al. 2018). Interestingly, biosensors that monitor the levels of specific intracellular metabolites are already available for E. coli and S. cerevisiae (Adeniran et al. 2015; Morris 2010; Rogers et al. 2016; Zhang and Keasling 2011), and it is likely that this type biosensors will be used in the future to identify superior fungal cell factories for production of specific SMs.
Novel methods for controlling gene function during fermentation are highly desirable. To this end, we envision that new improved orthogonal TFs with programmable specificities, e.g., by using catalytically dead Cas9 variants as TFs (Qi et al. 2013), will be developed. Such TFs have already been shown in other organisms to facilitate the activation of silent genes (Cheng et al. 2013; Perez-Pinera et al. 2013), to regulate metabolism of a heterologous cell factories for increased production yields (Deaner and Alper 2017; Jensen et al. 2017; Vanegas et al. 2017), to study genetic interactions (Du et al. 2017; Peters et al. 2016), or even to induce directed evolution (Hess et al. 2016; Ma et al. 2016). We also envision that synthetic biology tools will change the fungal production platforms dramatically. Specifically, highly efficient genetic engineering tools will allow for the development of cell factories with minimal genomes, where all undesirable cell functions that do not significantly impact fitness during fermentation have been eliminated. For this purpose, multiplex gene editing has already been implemented in fungi (Foster et al. 2018; Liu et al. 2015; Nødvig et al. 2018; Pohl et al. 2016; Shi et al. 2019). Deletion of a number of BGCs in A. nidulans with the aim of increasing the pool of available SM precursors and to simplify metabolite analysis (Chiang et al. 2013, 2016) already serve as a small scale example of this strategy. More ambitiously, one may envision that entirely synthetic filamentous fungi, inspired by the synthetic yeast 2.0 project (Pretorius and Boeke 2018), may be developed. Altogether, we conclude that the rapidly expanding synthetic biology toolbox in combination with the large number of fully sequenced genomes will set the stage for a very exciting future of fungal heterologous product production.
References
Adav SS, Ravindran A, Chao LT, Tan L, Singh S, Sze SK (2011) Proteomic analysis of pH and strains dependent protein secretion of Trichoderma reesei. J Proteome Res 10:4579–4596
Adeniran A, Sherer M, Tyo KEJ (2015) Yeast-based biosensors: design and applications. FEMS Yeast Res 15:1–15
Adney WS, Chou YC, Decker SR, Ding SY, Baker JO, Kunkel G, Vinzant TB, Himmel ME (2003) Heterologous expression of Trichoderma reesei 1,4-β-D-glucan cellobiohydrolase (Cel 7A). In: Mansfield SD, Saddler JN (eds) Applications of enzymes to lignocellulosics, vol 855. American Chemical Society, Washington, DC, pp 403–437
Adney WS, Jeoh T, Beckham GT, Chou YC, Baker JO, Michener W, Brunecky R, Himmel ME (2009) Probing the role of N-linked glycans in the stability and activity of fungal cellobiohydrolases by mutational analysis. Cellulose 16:699–709
Ahmed SM, Nishida-Fukuda H, Li Y, McDonald WH, Gradinaru CC, Macara IG (2018) Exocyst dynamics during vesicle tethering and fusion. Nat Commun 9:1–17
Alberti A, Ferrero I, Lodi T (2003) LYS2 gene and its mutation in Kluyveromyces lactis. Yeast 20:1171–1175
Aleksenko A, Clutterbuck AJ (1997) Autonomous plasmid replication in Aspergillus nidulans: AMA1 and MATE elements. Fungal Genet Biol 21:373–387
An X, Lu J, Huang J, Zhang B, Liu D, Zhang X, Chen J, Zhou Y, Tong Y (2007) Rapid assembly of multiple-exon cDNA directly from genomic DNA. PLoS One 2:1–7
Andersen MR, Vongsangnak W, Panagiotou G, Salazar MP, Lehmann L, Nielsen J (2008) A trispecies Aspergillus microarray: comparative transcriptomics of three Aspergillus species. Proc Natl Acad Sci USA 105:4387–4392
Andersen MR, Salazar MP, Schaap PJ, Van De Vondervoort PJI, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, Van Dijck PWM, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, Van Ooyen AJJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A, Van Den Brink JM, Atkins A, Aerts A, Shapiro H, Pangilinan J, Salamov A, Lou Y, Lindquist E, Lucas S, Grimwood J, Grigoriev IV, Kubicek CP, Martinez D, van Peij NNME, Roubos JA, Nielsen J, Baker SE (2011) Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21:885–897
Anyaogu DC, Mortensen UH (2015) Manipulating the glycosylation pathway in bacterial and lower eukaryotes for production of therapeutic proteins. Curr Opin Biotechnol 36:122–128
Archer D, Jeenes D, MacKenzie D, Brightwell G, Lambert N, Lowe G, Radford S, Dobson C (1990) Hen egg white lysozyme expressed in, and secreted from, Aspergillus niger is correctly processed and folded. Biotechnology 8:741–745
Austin B, Hail RM, Tyler BM (1990) Optimized vectors and selection for transformation of Neurospora crassa and Aspergillus nidulans to bleomycin and phleomycin. Gene 93:157–162
Avalos J, Geever RF, Case ME (1989) Bialaphos resistance as a dominant selectable marker in Neurospora crassa. Curr Genet 16:369–372
Barlowe C, Miller EA (2013) Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193:383–410
Barredo JL, Cantoral JM, Alvarez E, Díez B, Martín JF (1989) Cloning, sequence analysis and transcriptional study of the isopenicillin N synthase of Penicillium chrysogenum AS-P-78. Mol Gen Genet 216:91–98
Beck J, Ripka S, Siegner A, Schiltz E, Schweizer E (1990) The multifunctional 6-methylsalicylic acid synthase gene of Penicillium patulum. Its gene structure relative to that of other polyketide synthases. Eur J Biochem 192:487–498
Bentley DL (2014) Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15:163–175
Bergquist PL, Te’o VSJ, Gibbs MD, Curach NC, Nevalainen KMH (2004) Recombinant enzymes from thermophilic micro-organisms expressed in fungal hosts. Biochem Soc Trans 32:293–297
Bitinaite J, Rubino M, Varma KH, Schildkraut I, Vaisvila R, Vaiskunaite R (2007) USER friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res 35:1992–2002
Bleichrodt RJ, Read ND (2019) Flow cytometry and FACS applied to filamentous fungi. Fungal Biol Rev 33:1–15
Blumhoff M, Steiger MG, Marx H, Mattanovich D, Sauer M (2013) Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl Microbiol Biotechnol 97:259–267
Boeke J, Lacroute F, Fink G (1984) A positive selection for mutants lacking orotidine-5′ phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197:346–346
Boel E, Hansen MT, Hjort I, Høegh I, Fiil NP (1984) Two different types of intervening sequences in the glucoamylase gene from Aspergillus niger. Eur Mol Biol Organ 3:1581–1585
Bok JW, Ye R, Clevenger KD, Mead D, Wagner M, Krerowicz A, Albright JC, Goering AW, Thomas PM, Kelleher NL, Keller NP, Wu CC (2015) Fungal artificial chromosomes for mining of the fungal secondary metabolome. BMC Genomics 16:1–10
Breakspear A, Momany M (2007) The first fifty microarray studies in filamentous fungi. Microbiology 153:7–15
Brochado AR, Matos C, Møller BL, Hansen J, Mortensen UH, Patil KR (2010) Improved vanillin production in baker’s yeast through in silico design. Microb Cell Factories 9:1–15
Bulakhov AG, Volkov PV, Rozhkova AM, Gusakov AV, Nemashkalov VA, Satrutdinov AD, Sinitsyn AP (2017) Using an inducible promoter of a gene encoding Penicillium verruculosum glucoamylase for production of enzyme preparations with enhanced cellulase performance. PLoS One 12:1–13
Buxton FP, Gwynne DI, Davies RW (1985) Transformation of Aspergillus niger using the argB gene of Aspergillus nidulans. Gene 37:207–214
Cairns TC, Nai C, Meyer V (2018) How a fungus shapes biotechnology: 100 years of Aspergillus niger research. Fungal Biol Biotechnol 5:1–14
Cairns TC, Zheng X, Zheng P, Sun J, Meyer V (2019) Moulding the mould: understanding and reprogramming filamentous fungal growth and morphogenesis for next generation cell factories. Biotechnol Biofuels 12:1–18
Carrington JC, Dougherty WG (1988) A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing. Biochemistry 85:3391–3395
Carvalho NDSP, Arentshorst M, Kooistra R, Stam H, Sagt CM, Van Den Hondel CAMJJ, Ram AFJ (2011) Effects of a defective ERAD pathway on growth and heterologous protein production in Aspergillus niger. Appl Microbiol Biotechnol 89:357–373
Chaga G, Bochkariov DE, Jokhadze GG, Hopp J, Nelson P (1999) Natural poly-histidine affinity tag for purification of recombinant proteins on cobalt(II)-carboxymethylaspartate crosslinked agarose. J Chromatogr A 864:247–256
Chanda A, Roze LV, Kang S, Artymovich KA, Hicks GR, Raikhel NV, Calvo AM, Linz JE (2009) A key role for vesicles in fungal secondary metabolism. Proc Natl Acad Sci USA 106:19533–19538
Chang KY, Georgianna RD, Heber S, Payne GA, Muddiman DC (2010) Detection of alternative splice variants at the proteome level in Aspergillus flavus. J Proteome Res 9:1209–1217
Chen X, Zaro J, Shen WC (2013) Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65:1357–1369
Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R (2013) Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23:1163–1171
Cherry JR, Fidantsef AL (2003) Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol 14:438–443
Chiang YM, Szewczyk E, Davidson AD, Keller N, Oakley BR, Wang CCC (2009) A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J Am Chem Soc 131:2965–2970
Chiang Y-M, Oakley CE, Ahuja M, Entwistle R, Schultz A, Chang S-L, Sung CT, Wang CCC, Oakley BR (2013) An efficient system for heterologous expression of secondary metabolite genes in Aspergillus nidulans. J Am Chem Soc 135:7720–7731
Chiang YM, Ahuja M, Oakley CE, Entwistle R, Asokan A, Zutz C, Wang CCC, Oakley BR (2016) Development of genetic dereplication strains in Aspergillus nidulans results in the discovery of aspercryptin. Angew Chem Int Ed 55:1662–1665
Chou YC, Adney WS, Decker SR, Baker JO, Kunkel G, Templeton DW, Himmel ME (2004) Cloning and heterologous expression of the gene encoding a family 7 glycosyl hydrolase from Penicillium funiculosum. In: Saha BC, Hayashi K (eds) Lignocellulose biodegradation, vol 889. American Chemical Society, Washington, DC, pp 170–193
Clevenger KD, Bok JW, Ye R, Miley GP, Verdan MH, Velk T, Chen C, Yang K, Robey MT, Gao P, Lamprecht M, Thomas PM, Islam MN, Palmer JM, Wu CC, Keller NP, Kelleher NL (2017) A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat Chem Biol 13:895–901
Collis CM, Hall RM (1985) Identification of a Tn5 determinant conferring resistance to phleomycins, bleomycins, and tallysomycins. Plasmid 14:143–151
Cullen D, Leong SA, Wilson LJ, Henner DJ (1987) Transformation of Aspergillus nidulans with the hygromycin-resistance gene, hph. Gene 57:21–26
Curran KA, Karim AS, Gupta A, Alper HS (2013) Use of high capacity terminators in Saccharomyces cerevisiae to increase mRNA half-life and improve gene expression control for metabolic engineering applications. Metab Eng 19:88–97
Curran KA, Morse NJ, Markham KA, Wagman AM, Gupta A, Alper HS (2015) Short synthetic terminators for improved heterologous gene expression in yeast. ACS Synth Biol 4:824–832
Czymmek KJ, Bourett TM, Howard RJ (2005) Fluorescent protein probes in fungi. In: Savidge T, Pothulakis C (eds) Microbial imaging, vol 34. Academic, San Diego, pp 27–62
Dave K, Prabha VL, Ahuja M, Dave K, Tejaswini S, Punekar NS (2015) Expanding the repertoire of selectable markers for Aspergillus transformation. In: van den Berg MA, Maruthachalam K (eds) Genetic transformation systems in fungi, vol 1. Springer International Publishing, Cham, pp 141–153
de Graaff L, Visser J (1988) Structure of the Aspergillus nidulans pyruvate kinase gene. Curr Genet 14:553–560
de Vries RP, Grigoriev IV, Tsang A (2018) Introduction: overview of fungal genomics. In: de Vries RP, Tsang A, Grigoriev IV (eds) Fungal genomics. Methods in molecular biology, vol 1775. Humana Press, New York, NY
Deaner M, Alper HS (2017) Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab Eng 40:14–22
Díez B, Mellado E, Rodríguez M, Bernasconi E, Barredo JL (1999) The NADP-dependent glutamate dehydrogenase gene from Penicillium chrysogenum and the construction of expression vectors for filamentous fungi. Appl Microbiol Biotechnol 52:196–207
dos Santos Castro S, de Paula RG, Antoniêto ACC, Persinoti GF, Silva-Rocha R, Silva RN (2016) Understanding the role of the master regulator XYR1 in Trichoderma reesei by global transcriptional analysis. Front Microbiol 7:1–16
Du D, Roguev A, Gordon DE, Chen M, Chen SH, Shales M, Shen JP, Ideker T, Mali P, Qi LS, Krogan NJ (2017) Genetic interaction mapping in mammalian cells using CRISPR interference. Nat Methods 14:577–583
Eberle J, Russo V (1994) Neurospora crassa blue light-inducible gene bli-3. Biochem Mol Biol Int 34:737–744
Engler C, Marillonnet S (2014) Golden gate cloning. In: Valla S, Lale R (eds) DNA cloning and assembly methods, vol 1116. Springer Science Business Media, New York, pp 119–131
Fiedler MRM, Barthel L, Kubisch C, Nai C, Meyer V (2018) Construction of an improved Aspergillus niger platform for enhanced glucoamylase secretion. Microb Cell Factories 17:1–12
Fierro F, Kosalková K, Gutiérrez S, Martín JF (1996) Autonomously replicating plasmids carrying the AMA1 region in Penicillium chrysogenum. Curr Genet 29:482–489
Finnigan GC, Thorner J (2015) Complex in vivo ligation using homologous recombination and high-efficiency plasmid rescue from Saccharomyces cerevisiae. Bio-protocol 5:1–13
Fisch KM, Skellam E, Ivison D, Cox RJ, Bailey AM, Lazarus CM, Simpson TJ (2010) Catalytic role of the C-terminal domains of a fungal non-reducing polyketide synthase. Chem Commun 46:5331–5333
Fitz E, Wanka F, Seiboth B (2018) The promoter toolbox for recombinant gene expression in Trichoderma reesei. Front Bioeng Biotechnol 6:1–15
Fleissner A, Dersch P (2010) Expression and export: recombinant protein production systems for Aspergillus. Appl Microbiol Biotechnol 87:1255–1270
Ford KL, Baumgartner K, Henricot B, Bailey AM, Foster GD (2016) A native promoter and inclusion of an intron is necessary for efficient expression of GFP or mRFP in Armillaria mellea. Sci Rep 6:1–10
Foster AJ, Martin-Urdiroz M, Yan X, Wright HS, Soanes DM, Talbot NJ (2018) CRISPR-Cas9 ribonucleoprotein-mediated co-editing and counterselection in the rice blast fungus. Sci Rep 8:1–12
Foureau E, Clastre M, Millerioux Y, Simkin AJ, Cornet L, Dutilleul C, Besseau S, Marais E, Melin C, Guillard J, Crèche J, Giglioli-Guivarc’h N, Courdavault V, Papon N (2012) A TRP5/5-fluoroanthranilic acid counter-selection system for gene disruption in Candida guilliermondii. Curr Genet 58:245–254
Frandsen RJ, Khorsand-Jamal P, Kongstad KT, Nafisi M, Kannangara RM, Staerk D, Okkels FT, Binderup K, Madsen B, Lindberg Møller B, Thrane U, Mortensen UH (2018) Heterologous production of the widely used natural food colorant carminic acid in Aspergillus nidulans. Nat Sci Rep 8:1–10
Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA 85:8998–9002
Fujii T, Yamaoka H, Gomi K, Kitamoto K, Kumaga C (1995) Cloning and nucleotide sequence of the ribonuclease T1 gene (rntA) from Aspergillus oryzae and its expression in Saccharomyces cerevisiae and Aspergillus oryzae. Biosci Biotechnol Biochem 59:1869–1874
Fujii I, Ono Y, Tada H, Gomi K, Ebizuka Y, Sankawa U (1996) Cloning of the polyketide synthase gene atX from Aspergillus terreus and its identification as the 6-methylsalicylic acid synthase gene by heterologous expression. Mol Gen Genomics 253:1–10
Fujii I, Yoshida N, Shimomaki S, Oikawa H, Ebizuka Y (2005) An iterative type I polyketide synthase PKSN catalyzes synthesis of the decaketide alternapyrone with regio-specific octa-methylation. Chem Biol 12:1301–1309
Gao F, Hao Z, Sun X, Qin L, Zhao T, Liu W, Luo H, Yao B, Su X (2018) A versatile system for fast screening and isolation of Trichoderma reesei cellulase hyperproducers based on DsRed and fluorescence-assisted cell sorting. Biotechnol Biofuels 11:1–13
Gardiner DM, Jarvis RS, Howlett BJ (2005) The ABC transporter gene in the sirodesmin biosynthetic gene cluster of Leptosphaeria maculans is not essential for sirodesmin production but facilitates self-protection. Fungal Genet Biol 42:257–263
Garneau-Tsodikova S, Labby KJ (2016) Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives. Medchemcomm 7:11–27
Gems D, Johnstone IL, Clutterbuck AJ (1991) An autonomously replicating plasmid transforms Aspergillus nidulans at high frequency. Gene 98:61–67
Gibson DG, Young L, Chuang RY, Craig Venter J, Hutchison CA III, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–347
Giles NH, Case ME, Baum J, Geever R, Huiet L, Patel V, Tyler B (1985) Gene organization and regulation in the qa (quinic acid) gene cluster of Neurospora crassa. Microbiol Rev 49:338–358
Gomi K, Akeno T, Minetoki T, Ozeki K, Kumagai C, Okazaki N, Iimura Y (2000) Molecular cloning and characterization of a transcriptional activator gene, amyR, involved in the amylolytic gene expression in Aspergillus oryzae. Biosci Biotechnol Biochem 64:816–827
Goosen T, Bloemheuvel G, Gysler C, de Bie DA, van den Broek HWJ, Swart K (1987) Transformation of Aspergillus niger using the homologous orotidine-5′-phosphate-decarboxylase gene. Curr Genet 11:499–503
Goosen T, van Engelenburg F, Debets F, Swart K, Bos K, van den Broek H (1989) Tryptophan auxotrophic mutants in Aspergillus niger: inactivation of the trpC gene by cotransformation mutagenesis. Mol Gen Genet 219:282–288
Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89:5547–5551
Gossen M, Freundlieb S, Bender G, Müller G, Hillen W, Bujardt H (1995) Transcriptional activation by tetracyclines in mammalian cells. Science 268:1766–1769
Gouka RJ, Punt PJ, Van Den Hondel CAMJJ (1997) Glucoamylase gene fusions alleviate limitations for protein production in Aspergillus awamori at the transcriptional and (post)translational levels. Appl Environ Microbiol 63:488–497
Gould N, Hendy O, Papamichail D (2014) Computational tools and algorithms for designing customized synthetic genes. Front Bioeng Biotechnol 2:1–14
Gressler M, Hortschansky P, Geib E, Brock M (2015) A new high-performance heterologous fungal expression system based on regulatory elements from the Aspergillus terreus terrein gene cluster. Front Microbiol 6:1–16
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42:D699–D704
Gupta SK, Shukla P (2018) Glycosylation control technologies for recombinant therapeutic proteins. Appl Microbiol Biotechnol 102:10457–10468
Haas H, Redl B, Friedlin E, Stöffler G (1992) Isolation and analysis of the Penicillium chrysogenum phoA gene encoding a secreted phosphate-repressible acid phosphatase. Gene 113:129–133
Halo LM, Marshall JW, Yakasai AA, Song Z, Butts CP, Crump MP, Heneghan M, Bailey AM, Simpson TJ, Lazarus CM, Cox RJ (2008) Authentic heterologous expression of the tenellin iterative polyketide synthase nonribosomal peptide synthetase requires coexpression with an enoyl reductase. Chembiochem 9:585–594
Hansen BG, Genee HJ, Kaas CS, Nielsen JB, Regueira TB, Mortensen UH, Frisvad JC, Patil KR (2011a) A new class of IMP dehydrogenase with a role in self-resistance of mycophenolic acid producing fungi. BMC Microbiol 11:1–7
Hansen BG, Salomonsen B, Nielsen MT, Nielsen JB, Hansen NB, Nielsen KF, Regueira TB, Nielsen J, Patil KR, Mortensen UH (2011b) Versatile enzyme expression and characterization system for Aspergillus nidulans, with the Penicillium brevicompactum polyketide synthase gene from the mycophenolic acid gene cluster as a test case. Appl Environ Microbiol 77:3044–3051
Hanson G, Coller J (2018) Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol 19:20–30
He Y, Cox RJ (2016) The molecular steps of citrinin biosynthesis in fungi. Chem Sci 7:2119–2127
Herr A, Fischer R (2014) Improvement of Aspergillus nidulans penicillin production by targeting AcvA to peroxisomes. Metab Eng 25:131–139
Hess GT, Frésard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, Bassik MC (2016) Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods 13:1036–1042
Hoang HD, Maruyama JI, Kitamoto K (2015) Modulating endoplasmic reticulum-Golgi cargo receptors for improving secretion of carrier-fused heterologous proteins in the filamentous fungus Aspergillus oryzae. Appl Environ Microbiol 81:533–543
Hoefgen S, Lin J, Fricke J, Stroe MC, Mattern DJ, Kufs JE, Hortschansky P, Brakhage AA, Hoffmeister D, Valiante V (2018) Facile assembly and fluorescence-based screening method for heterologous expression of biosynthetic pathways in fungi. Metab Eng 48:44–51
Holm DMK (2013) Development and implementation of novel genetic tools for investigation of fungal secondary metabolism. PhD thesis, Technical University of Denmark
Holm DK, Petersen LM, Klitgaard A, Knudsen PB, Jarczynska ZD, Nielsen KF, Gotfredsen CH, Larsen TO, Mortensen UH (2014) Molecular and chemical characterization of the biosynthesis of the 6-MSA-derived meroterpenoid yanuthone D in Aspergillus niger. Chem Biol 21:519–529
Hoover DM, Lubkowski J (2002) DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res 30:1–7
Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cerretti DP, Urdal DL, Conlon PJ (1988) A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology 6:1204–1210
Hoshida H, Fujita T, Murata K, Kubo K, Akada R (2005) Copper-dependent production of a Pycnoporus coccineus extracellular laccase in Aspergillus oryzae and Saccharomyces cerevisiae. Biosci Biotechnol Biochem 69:1090–1097
Hoskins I, Roberts C (1994) Expression of the 3-phosphoglycerate kinase gene (pgkA) of Penicillium chrysogenum. Mol Gen Genet 243:270–276
Hurley JM, Chen CH, Loros JJ, Dunlap JC (2012) Light-inducible system for tunable protein expression in Neurospora crassa. G3 2:1207–1212
Hynes MJ, Pateman JAJ (1970) The genetic analysis of regulation of amidase synthesis in Aspergillus nidulans. Mol Gen Genet 108:107–116
Ilmén M, Onnela ML, Klemsdal S, Keränen S, Penttilä M (1998) Functional analysis of the cellobiohydrolase I promoter of the filamentous fungus Trichoderma reesei. Mol Gen Genet 253:303–314
Iriarte A, Sanguinetti M, Fernández-Calero T, Naya H, Ramón A, Musto H (2012) Translational selection on codon usage in the genus Aspergillus. Gene 506:98–105
Ishida H, Hata Y, Kawato A, Abe Y, Kashiwagi Y (2004) Isolation of a novel promoter for efficient protein production in Aspergillus oryzae. Biosci Biotechnol Biochem 68:1849–1857
Ishii N, Minami N, Chen EY, Medina AL, Chico MM, Kasamatsu H (1996) Analysis of a nuclear localization signal of simian virus 40 major capsid protein Vp1. J Virol 70:1317–1322
Jackson DC, Tang XL, Brown LE, Murray JM, White DO, Tregear GW (1986) Antigenic determinants of influenza virus hemagglutinin XII. Virology 155:625–632
Jalving R, Van De Vondervoort PJI, Visser J, Schaap PJ (2000) Characterization of the kexin-like maturase of Aspergillus niger. Appl Environ Microbiol 66:363–368
Jensen ED, Ferreira R, Jakočiunas T, Arsovska D, Zhang J, Ding L, Smith JD, David F, Nielsen J, Jensen MK, Keasling JD (2017) Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb Cell Factories 16:1–16
Jin FJ, Maruyama J, Juvvadi PR, Arioka M, Kitamoto K (2004) Adenine auxotrophic mutants of Aspergillus oryzae: development of a novel transformation system with triple auxotrophic hosts. Biosci Biotechnol Biochem 68:656–662
Jin FJ, Watanabe T, Juvvadi PR, Maruyama JI, Arioka M, Kitamoto K (2007) Double disruption of the proteinase genes, tppA and pepE, increases the production level of human lysozyme by Aspergillus oryzae. Appl Microbiol Biotechnol 76:1059–1068
Johnstone IL, Hughes SG, Clutterbuck AJ (1985) Cloning an Aspergillus nidulans developmental gene by transformation. EMBO J 4:1307–1311
Jørgensen TR, Park J, Arentshorst M, van Welzen AM, Lamers G, VanKuyk PA, Damveld RA, van den Hondel CAM, Nielsen KF, Frisvad JC, Ram AFJ (2011) The molecular and genetic basis of conidial pigmentation in Aspergillus niger. Fungal Genet Biol 48:544–553
Kamaruddin N, Storms R, Mahadi NM, Illias RM, Bakar FDA, Murad AMA (2018) Reduction of extracellular proteases increased activity and stability of heterologous protein in Aspergillus niger. Arab J Sci Eng 43:3327–3338
Keller NP (2019) Fungal secondary metabolism: regulation, function and drug discovery. Nat Rev Microbiol 17:167–180
Keller NP, Turner G, Bennett JW (2005) Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol 3:937–947
Kelly JM, Hynes MJ (1985) Transformation of Aspergillus niger by the amdS gene of Aspergillus nidulans. EMBO J 4:475–479
Kempken F (2013) Alternative splicing in ascomycetes. Appl Microbiol Biotechnol 97:4235–4241
Kempken F, Windhofer F (2004) Alternative splicing of transcripts of the transposon Restless is maintained in the foreign host Neurospora crassa and can be modified by introducing mutations at the 5′ and 3′ splice sites. Curr Genet 46:59–65
Kipriyanov SM, Kupriyanova OA, Little M, Moldenhauer G (1996) Rapid detection of recombinant antibody fragments directed against cell-surface antigens by flow cytometry. J Immunol Methods 196:51–62
Kistler HC, Broz K (2015) Cellular compartmentalization of secondary metabolism. Front Microbiol 6:68
Kitamoto N, Matsui J, Kawai Y, Kato A, Yoshino S, Ohmiya K, Tsukagoshi N (1998) Utilization of the TEF1-α gene (TEF1) promoter for expression of polygalacturonase genes, pgaA and pgaB, in Aspergillus oryzae. Appl Microbiol Biotechnol 50:85–92
Kitamoto N, Ono N, Yoshino Yasuda S (2015) Construction of quintuple protease and double amylase gene deletant for heterologous protein production in Aspergillus oryzae KBN616. Food Sci Technol Res 21:297–307
Kluge J, Terfehr D, Kück U (2018) Inducible promoters and functional genomic approaches for the genetic engineering of filamentous fungi. Appl Microbiol Biotechnol 102:6357–6372
Knudsen PB (2015) Development of scalable high throughput fermentation approaches for physiological characterisation of yeast and filamentous fungi. PhD thesis, Technical University of Denmark (DTU)
Krappmann S (2007) Gene targeting in filamentous fungi: the benefits of impaired repair. Fungal Biol Rev 21:25–29
Krishnan K, Askew DS (2014) The fungal UPR: a regulatory hub for virulence traits in the mold pathogen Aspergillus fumigatus. Virulence 5:334–340
Krügel H, Fiedler G, Smith C, Baumberg S (1993) Sequence and transcriptional analysis of the nourseothricin acetyltransferase-encoding gene nat1 from Streptomyces noursei. Gene 127:127–131
Kubodera T, Yamashita N, Nishimura A (2002) Transformation of Aspergillus sp. and Trichoderma reesei using the pyrithiamine resistance gene (ptrA) of Aspergillus oryzae. Biosci Biotechnol Biochem 66:404–406
Kupfer DM, Drabenstot SD, Buchanan KL, Hongshing L, Zhu H, Dyer DW, Roe BA, Murphy JW (2004) Introns and splicing elements of five diverse fungi. Eukaryot Cell 3:1088–1100
Kurzatkowski W, Törrönen A, Filipek J, Mach RL, Herzog P, Sowka S, Kubicek CP (1996) Glucose-induced secretion of Trichoderma reesei xylanases. Appl Environ Microbiol 62:2859–2865
Kwon MJ, Arentshorst M, Roos ED, Van Den Hondel CAMJJ, Meyer V, Ram AFJ (2011) Functional characterization of Rho GTPases in Aspergillus niger uncovers conserved and diverged roles of Rho proteins within filamentous fungi. Mol Microbiol 79:1151–1167
Lamb TM, Vickery J, Bell-Pedersen D (2013) Regulation of gene expression in Neurospora crassa with a copper responsive promoter. G3 3:2273–2280
Landowski CP, Huuskonen A, Wahl R, Westerholm-Parvinen A, Kanerva A, Hänninen AL, Salovuori N, Penttilä M, Natunen J, Ostermeier C, Helk B, Saarinen J, Saloheimo M (2015) Enabling low cost biopharmaceuticals: a systematic approach to delete proteases from a well-known protein production host Trichoderma reesei. PLoS One 10:1–28
Landowski CP, Mustalahti E, Wahl R, Croute L, Sivasiddarthan D, Westerholm-Parvinen A, Sommer B, Ostermeier C, Helk B, Saarinen J, Saloheimo M (2016) Enabling low cost biopharmaceuticals: high level interferon alpha-2b production in Trichoderma reesei. Microb Cell Factories 15:1–15
Leach DRF (1994) Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. BioEssays 16:893–900
Ley A, Coumou HC, Frandsen RJN (2015) Heterologous expression of MlcE in Saccharomyces cerevisiae provides resistance to natural and semi-synthetic statins. Metab Eng Commun 2:117–123
Li H, d’Anjou M (2009) Pharmacological significance of glycosylation in therapeutic proteins. Curr Opin Biotechnol 20:678–684
Li J, Wang J, Wang S, Xing M, Yu S, Liu G (2012) Achieving efficient protein expression in Trichoderma reesei by using strong constitutive promoters. Microb Cell Factories 11:1–10
Li W, Yu J, Li Z, Yin WB (2018) Rational design for fungal laccase production in the model host Aspergillus nidulans. Sci China Life Sci 61:1–11
Lin C, Baldwin T, Estell D, Fung J, Lin C, Pucci J, Razo J, Rider B, Throndset W, Trimbur D, Gastel F Van Ward M (2006) Develop systems for manufacturing 100,000,000 doses of an emergency pharmaceutical (e.g. vaccine or monoclonal antibody) within 2 months of product identification. Progress report, Genencor International, pp 1–21
Linder MB, Qiao M, Laumen F, Selber K, Hyytiä T, Nakari-Setälä T, Penttilä ME (2004) Efficient purification of recombinant proteins using hydrophobins as tags in surfactant-based two-phase systems. Biochemistry 43:11873–11882
Liu R, Chen L, Jiang Y, Zhou Z, Zou G (2015) Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov 1:1–11
Longley DB, Harkin DP, Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3:330–338
Lorang JM, Tuori RP, Martinez JP, Sawyer TL, Redman RS, Rollins JA, Wolpert TJ, Johnson KB, Rodriguez RJ, Dickman MB, Ciuffetti LM (2001) Green fluorescent protein is lighting up fungal biology. Appl Environ Microbiol 67:1987–1994
Lubertozzi D, Keasling JD (2006) Marker and promoter effects on heterologous expression in Aspergillus nidulans. Appl Microbiol Biotechnol 72:1014–1023
Lupton SD, Brunton LL, Kalberg VA, Overell RW (1991) Dominant positive and negative selection using a hygromycin phosphotransferase-thymidine kinase fusion gene. Mol Cell Biol 11:3374–3378
Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X (2016) Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods 13:1029–1035
Maruyama J, Nakajima H, Kitamoto K (2001) Visualization of nuclei in Aspergillus oryzae with EGFP and analysis of the number of nuclei in each conidium by FACS. Biosci Biotechnol Biochem 65:1504–1510
Marx F, Haas H, Reindl M, Stöffler G, Lottspeich F, Redl B (1995) Cloning, structural organization and regulation of expression of the Penicillium chrysogenum paf gene encoding an abundantly secreted protein with antifungal activity. Gene 167:167–171
Masai K, Marumaya J, Nakajima H, Kitamoto K (2003) In vivo visualization of the distribution of a secretory protein in Aspergillus oryzae hyphae using the RntA-EGFP fusion protein. Biosci Biotechnol Biochem 67:455–459
Matsuda Y, Mitsuhashi T, Lee S, Hoshino M, Mori T, Okada M, Zhang H, Hayashi F, Fujita M, Abe I (2016) Astellifadiene: structure determination by NMR spectroscopy and crystalline sponge method, and elucidation of its biosynthesis. Angew Chem 128:5879–5882
McNally MT, Free SJ (1988) Isolation and characterization of a Neurospora glucose-repressible gene. Curr Genet 14:545–551
Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, van den Hondel CAMJJ, Ram AFJ (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128:770–775
Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CAMJJ, Ram AFJ (2011) Fungal gene expression on demand: an inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl Environ Microbiol 77:2975–2983
Meyer V, Andersen MR, Brakhage AA, Braus GH, Caddick MX, Cairns TC, de Vries RP, Haarmann T, Hansen K, Hertz-Fowler C, Krappmann S, Mortensen UH, Peñalva MA, Ram AFJ, Head RM (2016) Current challenges of research on filamentous fungi in relation to human welfare and a sustainable bio-economy: a white paper. Fungal Biol Biotechnol 3:1–17
Miyauchi S, Te’o VS, Bergquist PL, Nevalainen KMH (2013) Expression of a bacterial xylanase in Trichoderma reesei under the egl2 and cbh2 glycosyl hydrolase gene promoters. New Biotechnol 30:523–530
Moralejo FJ, Watson AJ, Jeenes DJ, Archer DB, Martín JF (2001) A defined level of protein disulfide isomerase expression is required for optimal secretion of thaumatin by Aspergillus awamori. Mol Gen Genomics 266:246–253
Morris MC (2010) Fluorescent biosensors of intracellular targets from genetically encoded reporters to modular polypeptide probes. Cell Biochem Biophys 56:19–37
Morse NJ, Gopal MR, Wagner JM, Alper HS (2017) Yeast terminator function can be modulated and designed on the basis of predictions of nucleosome occupancy. ACS Synth Biol 6:2086–2095
Muller C, Hjort CM, Hansen K, Nielsen J (2002) Altering the expression of two chitin synthase genes differentially affects the growth and morphology of Aspergillus oryzae. Microbiology 148:4025–4033
Munawar A, Marshall JW, Cox RJ, Bailey AM, Lazarus CM (2013) Isolation and characterisation of a ferrirhodin synthetase gene from the sugarcane pathogen Fusarium sacchari. Chembiochem 14:388–394
Nagamine S, Liu C, Nishishita J, Kozaki T, Sogahata K, Sato Y, Minami A, Ozaki T, Schmidt-Dannert C, Maruyama J, Oikawa H (2019) Ascomycota Aspergillus oryzae is an efficient expression host for production of basidiomycota terpenes using genomic DNA sequences. Appl Environ Microbiol 85:1–21
Nakajima KI, Asakura T, Maruyama JI, Morita Y, Oike H, Shimizu-Ibuka A, Misaka T, Sorimachi H, Arai S, Kitamoto K, Abe K (2006) Extracellular production of neoculin, a sweet-tasting heterodimeric protein with taste-modifying activity, by Aspergillus oryzae. Appl Environ Microbiol 72:3716–3723
Nakano ET, Fox RD, Clements DE, Koo K, Stuart WD, Ivy JM (1993) Expression vectors for Neurospora crassa and expression of a bovine preprochymosin cDNA. Fungal Genet Rep 40:54–56
Nakari-Setälä T, Penttilä M (1995) Production of Trichoderma reesei cellulases glucose-containing media. Appl Environ Microbiol 61:3650–3655
Nakazato L, Dutra V, Broetto L, Staats CC, Vainstein MH, Schrank A (2006) Development of an expression vector for Metarhizium anisopliae based on the tef-1α homologous promoter. Appl Microbiol Biotechnol 72:521–528
Nielsen ML, Albertsen L, Lettier G, Nielsen JB, Mortensen UH (2006) Efficient PCR-based gene targeting with a recyclable marker for Aspergillus nidulans. Fungal Genet Biol 43:54–64
Nielsen MT, Nielsen JB, Anyaogu DC, Holm DK, Nielsen KF, Larsen TO, Mortensen UH (2013) Heterologous reconstitution of the intact geodin gene cluster in Aspergillus nidulans through a simple and versatile PCR based approach. PLoS One 8:e72871
Nielsen ML, Isbrandt T, Rasmussen KB, Thrane U, Hoof JB, Larsen TO, Mortensen UH (2017) Genes linked to production of secondary metabolites in Talaromyces atroroseus revealed using CRISPR-Cas9. PLoS One 12:1–9
Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH (2015) A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One 10:1–18
Nødvig CS, Hoof JB, Kogle ME, Jarczynska ZD, Lehmbeck J, Klitgaard DK, Mortensen UH (2018) Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in Aspergilli. Fungal Genet Biol 115:78–89
Oda K, Terado S, Toyoura R, Fukuda H, Kawauchi M, Iwashita K (2016) Development of a promoter shutoff system in Aspergillus oryzae using a sorbitol-sensitive promoter. Biosci Biotechnol Biochem 80:1792–1801
Ohno A, Maruyama JI, Nemoto T, Arioka M, Kitamoto K (2011) A carrier fusion significantly induces unfolded protein response in heterologous protein production by Aspergillus oryzae. Appl Microbiol Biotechnol 92:1197–1206
Okada M, Saito K, Wong CP, Li C, Wang D, Iijima M, Taura F, Kurosaki F, Awakawa T, Abe I (2017) Combinatorial biosynthesis of (+)-daurichromenic acid and its halogenated analogue. Org Lett 19:3183–3186
Osmani AH, May GS, Osmani SA (1999) The extremely conserved pyroA gene of Aspergillus nidulans is required for pyridoxine synthesis and is required indirectly for resistance to photosensitizers. J Biol Chem 274:23565–23569
Ouedraogo JP, Arentshorst M, Nikolaev I, Barends S, Ram AFJ (2016) I-SceI enzyme mediated integration (SEMI) for fast and efficient gene targeting in Trichoderma reesei. J Biotechnol 222:25–28
Pahirulzaman KAK, Williams K, Lazarus CM (2012) A toolkit for heterologous expression of metabolic pathways in Aspergillus oryzae. In: Hopwood D (ed) Methods in enzymology, vol 517. Academic, San Diego, pp 241–260
Paloheimo M, Haarmann T, Mäkinen S, Vehmaanperä J (2016) Production of industrial enzymes in Trichoderma reesei. In: Schmoll M, Dattenböck C (eds) Gene expression systems in fungi: advancements and applications, vol 1. Springer International Publishing, Cham, pp 23–57
Pantazopoulou A, Pinar M, Xiang X, Peñalva MA (2014) Maturation of late Golgi cisternae into RabERAB11 exocytic post-Golgi carriers visualized in vivo. Mol Biol Cell 25:2428–2443
Perez-Pinera P, Ousterout DG, Brunger JM, Farin AM, Glass KA, Guilak F, Crawford GE, Hartemink AJ, Gersbach CA (2013) Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods 10:239–244
Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA (2016) A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165:1493–1506
Petersen LM, Holm DK, Gotfredsen CH, Mortensen UH, Larsen TO (2015) Investigation of a 6-MSA synthase gene cluster in Aspergillus aculeatus reveals 6-MSA-derived aculinic acid, aculins A-B and epi-aculin A. Chembiochem 16:2200–2204
Petes T (1988) Recombination between repeated genes in microorganisms. Annu Rev Genet 22:147–168
Pohl C, Kiel J, Driessen A, Bovenberg R, Nygård Y (2016) CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synth Biol 5:754–764
Polli F, Meijrink B, Bovenberg RAL, Driessen AJM (2016) New promoters for strain engineering of Penicillium chrysogenum. Fungal Genet Biol 89:62–71
Pop C, Rouskin S, Ingolia NT, Han L, Phizicky EM, Weissman JS, Koller D (2014) Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol Syst Biol 10:1–15
Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ (1992) Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111:229–233
Pretorius IS, Boeke JD (2018) Yeast 2.0-connecting the dots in the construction of the world’s first functional synthetic eukaryotic genome. FEMS Yeast Res 18:1–15
Punt PJ, van den Hondel CAMJJ (1992) Transformation of filamentous fungi based on hygromycin b and phleomycin resistance markers. In: Wu R (ed) Methods in enzymology, vol 216. Academic, San Diego, pp 447–457
Punt PJ, Seiboth B, Weenink XO, Van Zeijl C, Lenders M, Konetschny C, Ram AFJ, Montijn R, Kubicek CP, Van Den Hondel CAMJJ (2001) Identification and characterization of a family of secretion-related small GTPase-encoding genes from the filamentous fungus Aspergillus niger: a putative SEC4 homologue is not essential for growth. Mol Microbiol 41:513–525
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183
Qin LN, Cai FR, Dong XR, Huang ZB, Tao Y, Huang JZ, Dong ZY (2012) Improved production of heterologous lipase in Trichoderma reesei by RNAi mediated gene silencing of an endogenic highly expressed gene. Bioresour Technol 109:116–122
Qin H, Xiao H, Zou G, Zhou Z, Zhong J (2017) CRISPR-Cas9 assisted gene disruption in the higher fungus Ganoderma species. Process Biochem 56:57–61
Rahman Z, Shida Y, Furukawa T, Suzuki Y, Okada H, Ogasawara W, Morikawa Y (2009) Evaluation and characterization of Trichoderma reesei cellulase and xylanase promoters. Appl Microbiol Biotechnol 82:899–908
Rantasalo A, Landowski CP, Kuivanen J, Korppoo A, Reuter L, Koivistoinen O, Valkonen M, Penttilä M, Jäntti J, Mojzita D (2018) A universal gene expression system for fungi. Nucleic Acids Res 1:1–15
Rantasalo A, Vitikainen M, Paasikallio T, Jäntti J, Landowski CP, Mojzita D (2019) Novel genetic tools that enable highly pure protein production in Trichoderma reesei. Sci Rep 9:1–12
Read G, Vining LC (1968) The biosynthesis of terreic acid. Chem Commun 16:935–937
Record E, Punt PJ, Chamkha M, Labat M, Van Den Hondel CAMJJ, Asther M (2002) Expression of the Pycnoporus cinnabarinus laccase gene in Aspergillus niger and characterization of the recombinant enzyme. Eur J Biochem 269:602–609
Rendsvig JKH, Workman CT, Hoof JB (2019) Bidirectional histone-gene promoters in Aspergillus: characterization and application for multi-gene expression. Fungal Biol Biotechnol 6:1–14
Riquelme M, Sánchez-León E (2014) The Spitzenkörper: a choreographer of fungal growth and morphogenesis. Curr Opin Microbiol 20:27–33
Rogers JK, Taylor ND, Church GM (2016) Biosensor-based engineering of biosynthetic pathways. Curr Opin Biotechnol 42:84–91
Rokas A, Wisecaver JH, Lind AL (2018) The birth, evolution and death of metabolic gene clusters in fungi. Nat Rev Microbiol 16:731–744
Roller S, Covill N (1999) The antifungal properties of chitosan in laboratory media and apple juice. Int J Food Microbiol 47:67–77
Romero B, Turner G, Olivas I, Laborda F, Ramón De Lucas J (2003) The Aspergillus nidulans alcA promoter drives tightly regulated conditional gene expression in Aspergillus fumigatus permitting validation of essential genes in this human pathogen. Fungal Genet Biol 40:103–114
Roze LV, Chanda A, Linz JE (2011) Compartmentalization and molecular traffic in secondary metabolism: a new understanding of established cellular processes. Fungal Genet Biol 48:35–48
Ryan MD, King AMQ, Thomas GP (1991) Cleavage of FMDV polyprotein is mediated by residues located within a 19 amino acid sequence. J Gen Virol 72:2727–2732
Sakai K, Kinoshita H, Shimizu T, Nihira T (2008) Construction of a citrinin gene cluster expression system in heterologous Aspergillus oryzae. J Biosci Bioeng 106:466–472
Sandermann H Jr (1992) Plant metabolism of xenobiotics. Trends Biochem Sci 17:82–84
Schalén M, Anyaogu DC, Hoof JB, Workman M (2016) Effect of secretory pathway gene overexpression on secretion of a fluorescent reporter protein in Aspergillus nidulans. Fungal Biol Biotechnol 3:1–14
Schäpe P, Kwon MJ, Baumann B, Gutschmann B, Jung S, Lenz S, Nitsche B, Paege N, Schütze T, Cairns TC, Meyer V (2019) Updating genome annotation for the microbial cell factory Aspergillus niger using gene co-expression networks. Nucleic Acids Res 47:559–569
Schuetze T, Meyer V (2017) Polycistronic gene expression in Aspergillus niger. Microb Cell Factories 16:1–8
Shi TQ, Gao J, Wang WJ, Wang KF, Xu GQ, Huang H, Ji XJ (2019) CRISPR/Cas9-based genome editing in the filamentous fungus Fusarium fujikuroi and its application in strain engineering for gibberellic acid production. ACS Synth Biol 8:445–454
Shimizu T, Ito T, Kanematsu S (2012) Transient and multivariate system for transformation of a fungal plant pathogen, Rosellinia necatrix, using autonomously replicating vectors. Curr Genet 58:129–138
Shoji J, Maruyama J, Arioka M, Kitamoto K (2005) Development of Aspergillus oryzae thiA promoter as a tool for molecular biological studies. FEMS Microbiol Lett 244:41–46
Sibthorp C, Wu H, Cowley G, Wong PWH, Palaima P, Morozov IY, Weedall GD, Caddick MX (2013) Transcriptome analysis of the filamentous fungus Aspergillus nidulans directed to the global identification of promoters. BMC Genomics 14:1–18
Song Z, Bakeer W, Marshall JW, Yakasai AA, Khalid RM, Collemare J, Skellam E, Tharreau D, Lebrun M-H, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ (2015) Heterologous expression of the avirulence gene ACE1 from the fungal rice pathogen Magnaporthe oryzae. Chem Sci 6:4837–4845
Stajich JE, Dietrich FS, Roy SW (2007) Comparative genomic analysis of fungal genomes reveals intron-rich ancestors. Genome Biol 8:1–13
Steinberg G, Peñalva MA, Riquelme M, Wösten HA, Harris SD (2017) Cell biology of hyphal growth. Microbiol Spectr 5:231–265
Sticha KRK, Sieg CA, Bergstrom CP, Hanna PE, Wagner CR (1997) Overexpression and large-scale purification of recombinant hamster polymorphic arylamine N-acetyltransferase as a dihydrofolate reductase fusion protein. Protein Expr Purif 10:141–153
Stone PJ, Makoff AJ, Parish JH, Radford A (1993) Cloning and sequence analysis of the glucoamylase gene of Neurospora crassa. Curr Genet 24:205–211
Stricker AR, Grosstessner-Hain K, Würleitner E, Mach RL (2006) Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and d-xylose metabolism in Hypocrea jecorina. Eukaryot Cell 5:2128–2137
Sun A, Peterson R, Te’o J, Nevalainen H (2016) Expression of the mammalian peptide hormone obestatin in Trichoderma reesei. New Biotechnol 33:99–106
Tada S, Gomi K, Kitamoto K, Takahashi K, Tamura G, Hara S (1991) Construction of a fusion gene comprising the Taka-amylase a promoter and the Escherichia coli β-glucuronidase gene and analysis of its expression in Aspergillus oryzae. Mol Gen Genet 229:301–306
Tanaka M, Tokuoka M, Gomi K (2014) Effects of codon optimization on the mRNA levels of heterologous genes in filamentous fungi. Appl Microbiol Biotechnol 98:3859–3867
Taylor DL, Hollingsworth TN, McFarland JW, Lennon NJ, Nusbaum C, Ruess RW (2014) A first comprehensive census of fungi in soil reveals both hyperdiversity and fine-scale niche partitioning. Ecol Monogr 84:3–20
Throndset W, Kim S, Bower B, Lantz S, Kelemen B, Pepsin M, Chow N, Mitchinson C, Ward M (2010) Flow cytometric sorting of the filamentous fungus Trichoderma reesei for improved strains. Enzym Microb Technol 47:335–341
Tian Y, Tan Y, Liu N, Yan Z, Liao Y, Chen J, De Saeger S, Yang H, Zhang Q, Wu A (2016) Detoxification of deoxynivalenol via glycosylation represents novel insights on antagonistic activities of Trichoderma when confronted with Fusarium graminearum. Toxins 8:1–15
Till P, Pucher ME, Mach RL, Mach-Aigner AR (2018) A long noncoding RNA promotes cellulase expression in Trichoderma reesei. Biotechnol Biofuels 11:1–16
Unkles SE, Campbell EI, Carrez D, Grieve C, Contreras R, Fiers W, van den Hondel CAMJJ, Kinghorn JR (1989) Transformation of Aspergillus niger with the homologous nitrate reductase gene. Gene 78:157–166
Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W (2000) Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci USA 97:7963–7968
Valkonen M, Ward M, Wang H, Penttilä M, Saloheimo M (2003) Improvement of foreign-protein production in Aspergillus niger var. awamori by constitutive induction of the unfolded-protein response. Appl Environ Microbiol 69:6979–6986
van den Brink HJM, Petersen SG, Rahbek-Nielsen H, Hellmuth K, Harboe M (2006) Increased production of chymosin by glycosylation. J Biotechnol 125:304–310
Vanegas KG, Lehka BJ, Mortensen UH (2017) SWITCH: a dynamic CRISPR tool for genome engineering and metabolic pathway control for cell factory construction in Saccharomyces cerevisiae. Microb Cell Factories 16:1–12
Vesth TC, Nybo JL, Theobald S, Frisvad JC, Larsen TO, Nielsen KF, Hoof JB, Brandl J, Salamov A, Riley R, Gladden JM, Phatale P, Nielsen MT, Lyhne EK, Kogle ME, Strasser K, McDonnell E, Barry K, Clum A, Chen C, LaButti K, Haridas S, Nolan M, Sandor L, Kuo A, Lipzen A, Hainaut M, Drula E, Tsang A, Magnuson JK, Henrissat B, Wiebenga A, Simmons BA, Mäkelä MR, de Vries RP, Grigoriev IV, Mortensen UH, Baker SE, Andersen MR (2018) Investigation of inter- and intraspecies variation through genome sequencing of Aspergillus section Nigri. Nat Genet 50:1688–1695
Vlaardingerbroek I, Beerens B, Shahi S, Rep M (2015) Fluorescence assisted selection of Transformants (FAST): using flow cytometry to select fungal transformants. Fungal Genet Biol 76:104–109
Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L (2010) Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res 38:5075–5087
Wanka F, Cairns T, Boecker S, Berens C, Happel A, Zheng X, Sun J, Krappmann S, Meyer V (2016) Tet-on, or Tet-off, that is the question: advanced conditional gene expression in Aspergillus. Fungal Genet Biol 89:72–83
Ward M, Wilson LJ, Carmona CL, Turner G (1988) The oliC3 gene of Aspergillus niger: isolation, sequence and use as a selectable marker for transformation. Curr Genet 14:37–42
Waring RB, May GS, Morris NR (1989) Characterization of an inducible expression system in Aspergillus nidulans using alcA and tubulin coding genes. Gene 79:119–130
Watanabe A, Ono Y, Fujii I, Sankawa U, Mayorga ME, Timberlake WE, Ebizuka Y (1998) Product identification of polyketide synthase coded by Aspergillus nidulans wA gene. Tetrahedron Lett 39:7733–7736
Watanabe A, Fujii I, Sankawa U, Mayorga ME, Timberlake WE, Ebizuka Y (1999) Re-identification of Aspergillus nidulans wA gene to code for a polyketide synthase of naphthopyrone. Tetrahedron Lett 40:91–94
Wei W, Chen L, Zou G, Wang Q, Yan X, Zhang J, Wang C, Zhou Z (2013) N-glycosylation affects the proper folding, enzymatic characteristics and production of a fungal β-glucosidase. Biotechnol Bioeng 110:3075–3084
Wernars K, Goosen T, Wennekes LMJ, Visser J, Bos CJ, van den Broek HWJ, van Gorcom RFM, van den Hondel CAMJJ, Pouwels PH (1985) Gene amplification in Aspergillus nidulans by transformation with vectors containing the amdS gene. Curr Genet 9:361–368
Wiemann P, Soukup AA, Folz JS, Wang PM, Noack A, Keller NP (2018) CoIN:co-inducible nitrate expression system for secondary metabolites in Aspergillus nidulans. Fungal Biol Biotechnol 5:1–10
Willis KJ (ed) (2018) State of the world’s fungi 2018. Report. Royal Botanic Gardens, Kew
Wu Y, Sun X, Xue X, Luo H, Yao B, Xie X, Su X (2017) Overexpressing key component genes of the secretion pathway for enhanced secretion of an Aspergillus niger glucose oxidase in Trichoderma reesei. Enzym Microb Technol 106:83–87
Xie X, Meehan MJ, Xu W, Dorrestein PC, Tang Y (2009) Acyltransferase mediated polyketide release from a fungal megasynthase. J Am Chem Soc 131:8388–8389
Xu J, Gong ZZ (2003) Intron requirement for AFP gene expression in Trichoderma viride. Microbiology 149:3093–3097
Xu W, Chooi Y-H, Choi JW, Li S, Vederas JC, Da Silva NA, Tang Y (2013) LovG: the thioesterase required for dihydromonacolin L release and lovastatin nonaketide synthase turnover in lovastatin biosynthesis. Angew Chem Int Ed 52:6472–6475
Yang S, Kuang Y, Li H, Liu Y, Hui X, Li P, Jiang Z, Zhou Y, Wang Y, Xu A, Li S, Liu P, Wu D (2013) Enhanced production of recombinant secretory proteins in Pichia pastoris by optimizing Kex2 P1’ site. PLoS One 8:1–11
Yoon J, Aishan T, Maruyama JI, Kitamoto K (2010) Enhanced production and secretion of heterologous proteins by the filamentous fungus Aspergillus oryzae via disruption of vacuolar protein sorting receptor gene AoVps10. Appl Environ Microbiol 76:5718–5727
Yoshino-Yasuda S, Karita S, Kato M, Kitamoto N (2012) Sequence analysis and heterologous expression of rhamnogalacturonan lyase A gene (AsrglA) from Shoyu koji mold, Aspergillus sojae KBN1340. Food Sci Technol Res 18:901–909
Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y (2015) Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell 59:744–754
Yuill E (1950) The numbers of nuclei in conidia of Aspergilli. Trans Br Mycol Soc 33:324–331
Zadra I, Abt B, Parson W, Haas H (2000) xylP promoter-based expression system and its use for antisense downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Appl Environ Microbiol 66:4810–4816
Zeilinger S, Mach RL, Schindler M, Herzog P, Kubicek CP (1996) Different inducibility of expression of the two xylanase genes xyn1 and xyn2 in Trichoderma reesei. J Biol Chem 271:25624–25629
Zeilinger S, Mach R, Kubicek C (1998) Two adjacent protein binding motifs in the cbh2 (Cellobiohydrolase II-encoding) promoter of the fungus Hypocrea jecorina (Trichoderma reesei) cooperate in the induction by cellulose. J Biol Chem 273:34463–34471
Zhang F, Keasling J (2011) Biosensors and their applications in microbial metabolic engineering. Trends Microbiol 19:323–329
Zhang DX, Lu HL, Liao X, St Leger RJ, Nuss DL (2013) Simple and efficient recycling of fungal selectable marker genes with the Cre-loxP recombination system via anastomosis. Fungal Genet Biol 61:1–9
Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH (2015) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4:1–8
Zhang C, Meng X, Wei X, Lu L (2016a) Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet Biol 86:47–57
Zhang H, Wang S, Zhang XX, Ji W, Song F, Zhao Y, Li J (2016b) The amyR-deletion strain of Aspergillus niger CICC2462 is a suitable host strain to express secreted protein with a low background. Microb Cell Factories 15:1–11
Zhang J, Zhang G, Wang W, Wei D (2018) Enhanced cellulase production in Trichoderma reesei RUT C30 via constitution of minimal transcriptional activators. Microb Cell Factories 17:1–14
Zhao C, Waalwijk C, de Wit PJGM, Tang D, van der Lee T (2013) RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genomics 14:1–16
Zheng YM, Lin FL, Gao H, Zou G, Zhang JW, Wang GQ, Chen GD, Zhou ZH, Yao XS, Hu D (2017) Development of a versatile and conventional technique for gene disruption in filamentous fungi based on CRISPR-Cas9 technology. Sci Rep 7:1–10
Zhou M, Wang T, Fu J, Xiao G, Liu Y (2015) Nonoptimal codon usage influences protein structure in intrinsically disordered regions. Mol Microbiol 97:974–987
Zhou Z, Dang Y, Zhou M, Li L, Yu C, Fu J, Chen S, Liu Y (2016) Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci USA 113:E6117–E6125
Zhou Z, Dang Y, Zhou M, Yuan H, Liu Y (2018) Codon usage biases co-evolve with transcription termination machinery to suppress premature cleavage and polyadenylation. Elife 7:1–29
Zhu H, Choi S, Johnston AK, Wing RA, Dean RA (1997) A large-insert (130 kbp) bacterial artificial chromosome library of the rice blast fungus Magnaporthe grisea: genome analysis, contig assembly, and gene cloning. Fungal Genet Biol 21:337–347
Zhu B, Cai G, Hall EO, Freeman GJ (2007) In-FusionTM assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. BioTechniques 43:354–359
Zubieta MP, Contesini FJ, Rubio MV, Gonçalves AEDSS, Gerhardt JA, Prade RA, Damasio ARDL (2018) Protein profile in Aspergillus nidulans recombinant strains overproducing heterologous enzymes. Microb Biotechnol 11:346–358
Acknowledgments
We are grateful to Michael Lynge Nielsen for carefully reading and editing this book chapter. This work has been supported by Innovation Fund Denmark, grant no.: 6150-00031B.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rendsvig, J.K.H., Futyma, M.E., Jarczynska, Z.D., Mortensen, U.H. (2020). Filamentous Fungi as Hosts for Heterologous Production of Proteins and Secondary Metabolites in the Post-Genomic Era. In: Benz, J.P., Schipper, K. (eds) Genetics and Biotechnology. The Mycota, vol 2. Springer, Cham. https://doi.org/10.1007/978-3-030-49924-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-49924-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-49923-5
Online ISBN: 978-3-030-49924-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)