
Abstract
The filamentous fungi Trichoderma reesei and Penicillium funiculosum produce highly effective enzyme mixtures that degrade the cellulose and hemicellulose components of plant cell walls. Many fungal species produce a glycoside hydrolase family 7 (Cel7A) cellobiohydrolase, a class of enzymes that catalytically process from the reducing end of cellulose. A direct amino acid comparison of these two enzymes shows that they not only have high amino acid homology, but also contain analogous N-linked glycosylation sites on the catalytic domain. We have previously shown (Jeoh et al. in Biotechnol Biofuels, 1:10, 2008) that expression of T. reesei cellobiohydrolase I in a commonly used industrial expression host, Aspergillus niger var. awamori, results in an increase in the amount of N-linked glycosylation of the enzyme, which negatively affects crystalline cellulose degradation activity as well as thermal stability. This complementary study examines the significance of individual N-linked glycans on the surface of the catalytic domain of Cel7A cellobiohydrolases from T. reesei and P. funiculosum by genetically adding or removing N-linked glycosylation motifs using site directed mutagenesis. Modified enzymes, expressed in A. niger var. awamori, were tested for activity and thermal stability. It was concluded that N-linked glycans in peptide loops that form part of the active site tunnel have the greatest impact on both thermal stability and enzymatic activity on crystalline cellulose for both the T. reesei and P. funiculosum Cel7A enzymes. Specifically, for the Cel7A T. reesei enzyme expressed in A. niger var. awamori, removal of the N384 glycosylation site yields a mutant with 70% greater activity after 120 h compared to the heterologously expressed wild type T. reesei enzyme. In addition, similar activity improvements were found to be associated with the addition of a new glycosylation motif at N194 in P. funiculosum. This mutant also exhibits 70% greater activity after 120 h compared to the wild type P. funiculosum enzyme expressed in A. niger var. awamori. Overall, this study demonstrates that “tuning” enzyme glycosylation for expression from heterologous expression hosts is essential for generating engineered enzymes with optimal stability and activity.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Glycosylation of proteins during synthesis is a common and functionally important modification that occurs during the secretion of proteins (Taylor and Drickamer 2003), resulting in the covalent linkage of carbohydrates to asparagine (N-linked) or to serine/threonine (O-linked) residues. N-linked glycans are typically bonded via β,1-4 linkages to one (or more) N-acetylglucosamine (GlcNAc) groups, which in turn is covalently linked to the amido nitrogen of asparagine via β,1-N bonds. Post-secretory exposure to extracellular endoglycosidic activities is responsible for some post-translational modification of many fungal proteins. Coupled with the initial peptide glycosylation events occurring in the cytosol and endoplasmic reticulum, and finishing in the Golgi, these extracellular processes form an integral component of the complex secretory machinery of fungi and are thought to be essential to the intrinsic function and stability of many fungal enzymes (Eriksson et al. 2004). Because protein glycosylation is inherently complex and possibly micro-heterogeneous, the impact on the structure and function of proteins is sometimes overlooked, however, the effects of glycosylation can be critically important, especially when heterologous hosts are used for enzyme production.
The filamentous fungi Trichoderma reesei and Penicillium funiculosum produce a diversity of glycoside hydrolase enzymes that are considered critical for the saccharification of both the hemicellulose and the cellulosic components of plant cell walls (Chen et al. 1987; Elshafei et al. 1991; Foreman et al. 2003; Gaur and Mishra 1990; Lachke et al. 1987; Manchanda et al. 1982). These two fungal species of commercial importance produce cellobiohydrolase enzymes in abundant quantities. Furthermore, the cellobiohydrolases produced by these fungal species are similar in primary sequence (60% overall identity), but differ markedly in their rates and extents of cellulose hydrolysis, with P. funiculosum having approximately twice the activity on crystalline cellulose compared to the T. reesei enzyme (Adney et al. 2003). In T. reesei, additional modification of the original glycan content is also thought to occur extracellularly from the action of an endogenous endoglycosidase-H enzyme (Stals et al. 2004).
Cellobiohydrolases, such as those produced by T. reesei and P. funiculosum, generally have a multi-domain structure characterized by a catalytic domain and a carbohydrate-binding module (CBM) separated by a linker peptide. Cellobiohydrolases are believed to act processively from either the reducing or the non-reducing ends of a cellulose chain, releasing successive cellobiose (and in some cases, glucose) molecules. The T. reesei and P. funiculosum cellobiohydrolases used in this study both process from the reducing end and have been classified according to the CAZY database as belonging to the glycoside hydrolase family 7 (Adney et al. 2003). The original Genbank submission of the P. funiculosum sequence described this enzyme as a xylanase/cellobiohydrolase and was designated XynA. For simplicity, we refer to this protein as the Cel7A enzyme from P. funiculosum.
Both O-linked and N-linked glycosylation are prevalent in fungal cellobiohydrolases: O-linked glycosylation occurs at the hydroxyl groups of serine (S) and threonine (T) residues in the linker region and N-linked glycosylation occurs at the asparagine (N) residues in the consensus sequence motif of N-X-S/T in the catalytic domain. In T. reesei, the cellobiohydrolase appears in culture filtrates as several isoforms with similar reported catalytic and adsorption properties (Medve et al. 1994, 1998). In studies of expressed and secreted T. reesei cellobiohydrolases, the considerable heterogeneity observed for the linked glycans can been attributed to both the strain and growth conditions (Eriksson et al. 2004). Recently, the predominant N-linked glycans associated with T. reesei Cel7A have been reported to be (ManP)0–1GlcMan7–8GlcNAc2 for the hyper-producing strains and (Man1–2P)0–1–2Man5–6–7GlcNAc2 for the wild type [WT] strain (Eriksson et al. 2004). Isoforms observed for T. reesei Cel7A are thought to be due, in part, to the random substitution of glycans on the N-glycosylation sites (Stals et al. 2004; Medve et al. 1998). Because of its complexity, the importance of N-linked glycosylation for secretion and stability of extracellular enzymes from filamentous fungi has not been clearly established (Adney et al. 2003; Chen et al. 1993; Eriksen et al. 1998). The widespread occurrence of the highly O-glycosylated linker, however, seems to indicate an essential stabilization function for this peptide and may also define limits of the conformational diversity of this linker (Receveur et al. 2002). It is clear that when Trichoderma is used for the heterologous expression of foreign genes, or when Trichoderma genes are expressed in heterologous hosts, a detailed understanding of the endogenous glycosylation pathways becomes essential.
The initial adsorption of cellobiohydrolases on solid substrates is undoubtedly an important initial step in processivity and hydrolysis. Deviations from the optimal initial binding conformation of the enzyme to substrate will likely occur if the enzymes are not properly processed during secretion, whether from homologous or heterologous hosts. Alternatively, the engineering of new N-linked sites on the protein surface may offer an opportunity to improve the binding or activity characteristics of these enzymes. Having a clear understanding of the structure and complexity of glycans linked to cellobiohydrolases should provide insight into the design of more stable and active enzyme mutants.
To this end, we presented a study of the biophysical properties of the T. reesei Cel7A cellobiohydrolase expressed in Aspergillus niger var. awamori (Jeoh et al. 2008). We reported that the recombinant T. reesei Cel7A enzyme exhibits six times the amount of N-linked glycosylation compared to the native T. reesei enzyme purified from a commercial preparation, as well as reduced activity and increased non-productive binding on both crystalline and acid-swollen cellulose (Jeoh et al. 2008).
As a complement to our previous work, we present here a study of the stability and activity of the Family 7 cellobiohydrolases from T. reesei and P. funiculosum expressed in A. niger var. awamori. In this study, these enzymes are referred to as T. reesei rCel7A and P. funiculosum rCel7A, respectively. We systematically evaluate the role of N-linked glycosylation by genetically eliminating or adding glycosylation sites and then analyzing the resulting enzyme for thermal stability and activity on crystalline cellulose.
Materials and methods
Acquisition of the cellobiohydrolase genes
Acquisition of the cel7a gene from T. reesei and expression of the gene product in A. niger var. awamori has been described in our previous study (Jeoh et al. 2008). The cel7a gene from Penicillium funiculosum (ATCC62998) was acquired from genomic DNA using PCR with primers based on the published GenBank sequence for P. funiculosum xylanase/cellobiohydrolase (AJ312295) (Adney et al. 2003) as previously described. The resulting PCR product was cloned into the vector pGEM so that it contained the entire cel7a coding sequence including the WT signal sequence. This plasmid was then used to construct the pFE2 expression vector for A. niger var. awamori and to perform site-directed mutations of N-linked glycosylation sites. Using this approach, primers were designed that isolated the translated portion of the cel7a gene sequence and added convenient restriction enzyme sites for cloning. Pfu polymerase, a polymerase with proofreading ability and high fidelity, was obtained from Stratagene (San Diego, CA) and used for all PCR reactions. Inserts containing the cel7a gene were prepared by gel purification of the PCR products followed by restriction digestion and directional cloning into the appropriate expression vector. Confirmation of the cel7a gene and the resultant gene product was based on DNA sequencing. The cel7a gene from T. reesei was acquired from cDNA as previously described (Laymon et al. 1996).
Modified proteins were produced using genes cloned from either cDNA or genomic DNA and expressed in A. niger var. awamori under the control of the A. niger glucoamylase promoter using the Escherichia coli/Aspergillus shuttle vector pFE2. Using the ProtParam tool available through ExPASy, the mature P. funiculosum enzyme consists of a 504-residue glycoprotein with a calculated molecular weight of 52,436 Daltons, whereas the mature T. reesei enzyme consists of a 497-residue glycoprotein with a calculated molecular weight of 52,247 Daltons. The sequences of the Cel7A enzymes are shown in Fig. 1. The T. reesei enzyme has three N-linked glycosylation sites (N45, N270 and N384) with an unlinked motif at N64. Similarly, the P. funiculosum enzyme has three N-linked sites at positions N45, N387 and N430, with a potential motif at N194 (e.g., structurally analogous to the N64 in T. reesei). In order to evaluate the role of N-linked glycosylation on the activity and stability of these enzymes, we established a library of mutants that were systematically modified by the elimination (or addition) of N-linked glycosylation motifs. Following the expression and purification of each mutant, we conducted an examination of the thermal stability and kinetic performance on bacterial crystalline cellulose to establish the relative importance of each N-linked glycosylation site.

Amino acid sequence of Cel7A enzymes from P. funiculosum and T. reesei. The sequence is derived from DNA sequencing with the CBM underlined, the linker domains in italics, and the signal sequence designated in lower case letters. Putative N-linked sites modified in this study are in bold and underlined on the respective enzymes
Culture and growth conditions
Escherichia coli DH5α was cultured in LB (Luria Broth) at 37 °C and 250 rpm whereas A. niger var. awamori ATCC22342 and P. funiculosum ATCC62998 were grown in CM media (per liter: glucose 10 g, yeast extract 5 g, tryptone 5 g, and 50 mL Clutterbuck’s salts solution, pH 7.5) at 29 °C and 250 rpm. Clutterbuck’s salts contain 120 g/L NaNO3, 10.4 g/L KCl, 10.4 g/L MgSO4 · 7H2O and 30.4 g/L KH2PO4. For the selection and maintenance of pFE2 and its derivatives, antibiotics were supplemented to the media at the following concentrations: Zeocin (Z or Zeo), 170 μg/mL for the initial selection of A. niger var. awamori transformants, 300 μg/mL for the growth of transformants for cellulase production, and Ampicillin (Amp), 100 μg/mL for E. coli. For the agar media, Bacto Agar (Difco) was added to CM at 2% and LB at 1.5%.
Modification of N-linked glycosylation sites
Mutants with targeted amino acid substitutions were generated following the protocol supplied with the QuickChange™ Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA). Mutagenic primers containing the desired mutation were designed and synthesized at Macromolecular Resources (Colorado State University, Fort Collins, CO). Mutations were confirmed by DNA sequencing. The primer sequences used for all enzymes in this study are shown in Table 1.
Cellobiohydrolase purification
The purification of all of the Cel7A enzymes was performed as previously described (Adney et al. 2003). Fungal hyphae were removed from 1-L shake flask growths by passing the broth first through Miracloth and then through glass fiber filters. The broth was concentrated and extensively diafiltered with 20 mM Bis-Tris, pH 5.8 buffer and applied to a HiPrep 16/10 DEAE FF column (Amersham Biosciences) equilibrated with 50 mM Bis-Tris, pH 5.8 buffer with a flow rate of 10 mL/min. The column was washed extensively with equilibration buffer and the bound fraction eluted with a linear gradient of 0.0–0.5 M NaCl in the same equilibration buffer. Fractions containing the Cel7A enzyme were pooled, concentrated and subjected to size exclusion chromatography using a Superose 12 Prep grade 35/600 column in 20 mM acetate, 100 mM NaCl, pH 5 buffer. The purity was confirmed as a single band using a NuPage 4–12% Bis-Tris gradient gel and MOPS–SDS buffer (Invitrogen) following the manufactures recommended conditions.
Production of bacterial cellulose
For the cellulose hydrolysis experiments, bacterial cellulose (BC), a highly recalcitrant, pure cellulose substrate was used because it represents a chemically clean form of crystalline cellulose. BC was produced as previously described (Jeoh et al. 2008) in static cultures of Gluconacetobacter xylinus sbsp sucrofermentans (ATCC 700178) in Hestrin Schramm medium (Hestrin and Schram 1954) with 1% (v/v) ethanol (Krystynowicz et al. 2002). The initial inoculum was prepared by growing frozen G. xylinus culture in 50 mL of the same medium (HS + 1% EtOH) at 26 °C for 3 days under static conditions. At the end of the 3 days, the culture flask was shaken vigorously to remove the cells from the pellicle. The cells in the supernatant were pelletized and used to inoculate 75 mL media in 750 mL rectangular tissue culture flasks. Production cultures were incubated at 26 °C for 5–7 days without agitation. At the end of the production period, the cells were re-pelletized and used in fresh media for growing subsequent batches of BC. The BC pellicles were washed according to a protocol outlined by Helbert and coworkers (Helbert et al. 2003) with the following modifications: following neutralization from the alkali wash, the cellulose pellicles were incubated in a 0.3% bleach solution (in 4 mM sodium acetate buffer) for 2 h at 70 °C. The pellicles were rinsed three times with distilled water to remove the bleach solution. Following the final water rinse, the pellicles were re-suspended in 5 mM sodium acetate, pH 5.0 buffer with 0.04% NaN3 and homogenized in a food processor. A final concentration of 1.9 mg/mL (standard deviation of 0.12 mg/mL) was determined from triplicate oven dry weights of 3 mL suspensions. The stock BC suspension was stored at 4 °C.
rCel7A stability by differential scanning calorimetry
Overall protein stability was measured by differential scanning microcalorimetry using a Microcal model VP-DSC calorimeter (Microcal, Inc., Northampton, MA), with data analysis by means of Origin for DSC software (Microcal). Thermograms were collected for samples containing 50 μg/mL protein at pH 5.0 in 20 mM sodium acetate with 100 mM NaCl. The calorimeter scan rate was 60 °C/h.
rCel7A N-glycan analysis by mass spectrometry
Oligosaccharide heterogeneity in the rCel7A enzymes was determined using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). For the MALDI-MS experiments, 1 μL of purified sample was mixed with 1 μL of sinapinic acid (10 mg/mL in 50% acetonitrile, 0.1% trifluoroacetic acid). The mixture was spotted on the MALDI-MS target and allowed to dry in the open air. The sample was analyzed by an Ultraflex-TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA) in positive ion, linear mode using a 25 kV accelerating voltage. External calibration was performed using a six-protein calibration mixture on a spot adjacent to the sample.
Cellulose hydrolysis time course experiments
Reactions containing 1.0 μM cellulase and 1.0 mg/mL BC in 0.25 mL reaction volumes were conducted at 38 °C. Triplicate reactions were incubated for each of 0.25, 0.5, 1, 2 and 4 h durations. Each reaction was setup by preparing the appropriate dilution of cellulase in 1.5 mL microcentrifuge tubes. BC was pre-incubated at 38 °C for a minimum of 30 minutes. The reactions were initiated by addition of the pre-incubated substrate and terminated by separating the liquid and solid phase by filtration using a manifold filtration system equipped with a 96-well 1.0 μM glass fiber filter frit (Innovative Microplate, Chicopee, MA). The liquid phase was assayed for reducing sugar concentration by the disodium-2,2′-bicinchoninate method (Doner and Irwin 1992) using cellobiose for the standard curve.
Results
Expression of rCel7A genes
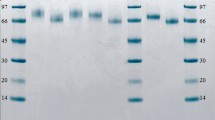
Table 2 lists the mutants examined in this study: three mutants of T. reesei rCel7A, four mutants of P. funiculosum rCel7A, and the recombinant WT enzymes of both species. The glucoamylase promoter and the TrpC terminator from A. niger var. awamori were used to promote and terminate transcription. Using this construct, the recombinant proteins were secreted into the culture medium and were purified using column chromatography. The purity of the enzyme preparation and the molecular weights were determined using SDS-PAGE, as shown in Fig. 2 and by MALDI-MS, as listed in Table 2. The replacement of asparagine with alanine resulted in a lower apparent molecular weight attributable to the removal of glycosylation at the glycosylation sites. In the case of the P. funiculosum enzyme, the replacement of the alanine at site 196 with either threonine or serine resulted in a new N-linked motif at N194. For the T. reesei enzyme, replacement of asparagine with alanine at site 45 resulted in an unstable protein, whereas the analogous site for the P. funiculosum produced a stable mutant protein.

Invitrogen 4–12% gradient NuPage gel with MOPS buffer. Lane 1 Mark 12 Std with weight markers included in kDa, 2 T. reesei rCel7A, 3 T. reesei N270A, 4 T. reesei N384A, 5 Mark 12 Std, 6 P. funiculosum rCel7A, 7 P. funiculosum N45A, 8 P. funiculosum N388A, 9 P. funiculosum N430A, 10 P. funiculosum A196S
Structural comparisons of the rCel7A enzymes
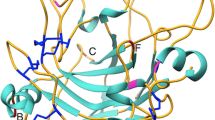
Structural examination of the glycosylation motifs found in the sequences of these two proteins shows similar placement of the N-linked glycosylation sites on the surfaces of the two proteins as illustrated by a homology model of the P. funiculosum enzyme and the published structure for T. reesei Cel7A (1cel) (Divne et al. 1994). The 3-D model for the P. funiculosum Cel7A enzyme was created using the software package Modeler (Accelrys, Inc. San Diego, CA) using the PDB template of T. reesei (1cel) as a model structure (Divne et al. 1994). The recommended values in the Modeler software were used to build the homology models. Similarities between the potential glycosylation sites for these two enzymes are illustrated in Fig. 3.

aP. funiculosum Cel7A enzyme with labeled glycosylation sites. The cellulose chain in the catalytic domain is taken from the T. reesei crystal structure. bT. reesei Cel7A enzyme with labeled glycosylation sites (Divne et al. 1994)
MALDI-MS results
The MALDI-MS results for the rCel7A enzymes are shown in Table 2. The two T. reesei rCel7A mutants exhibit a lower molecular weight, as expected. For the P. funiculosum rCel7A mutants, the N388A and N430A mutants exhibit lower molecular weights, whereas the A196S addition motif shows a significantly higher molecular mass. The P. funiculosum N45A mutant, however, shows roughly equivalent molecular weight to the rCel7A WT enzyme. This finding indicates little or no glycosylation at the N45A site, which has been observed earlier (Jeoh et al., unpublished results).
Differential scanning calorimetry analysis
DSC was used to evaluate the thermal stability of the rCel7A enzymes. The P. funiculosum rCel7A and T. reesei rCel7A exhibit a 1.8 °C difference in thermal transition temperature, as shown in Table 2. The higher thermal transition temperature for the P. funiculosum enzyme likely indicates either differences in the peptide secondary structure or differences in the extent and nature of the glycosylation of these two enzymes (or a combination of both).
Results from DSC studies of the recombinant N-linked glycosylation mutants of T. reesei rCel7A and P. funiculosum rCel7A are shown in Table 2. These results illustrate the conserved sensitivity associated with sites N384 and N388 in T. reesei and P. funiculosum, respectively, compared to the proteins expressed using the WT genes and the other N-linked mutants. For the P. funiculosum enzyme, the addition of the N-linked motif at N194 results in an approximately 1 °C reduction in the thermal denaturation temperature.
The role of N-glycosylation on T. reesei rCel7A activity
In the current study, two glycosylation mutants of T. reesei Cel7A, N270A and N384A, were successfully expressed and produced in A. niger var. awamori. The digestion curves for these mutants and the T. reesei rCel7A WT enzyme acting on bacterial cellulose (BC) are shown in Fig. 4.

Extent of hydrolysis time course for the T. reesei rCel7A WT and two mutant enzymes: N270A and N384A
The loss of the glycosylation site at position 270 leads to marginal improvements in activity over that of the rCel7A WT enzyme. A profound effect on the hydrolysis of BC was observed when the glycosylation site at position 384 was eliminated. The N384A mutant continued to digest BC for 120 h after the activities of both the rCel7A WT and the N270A mutant enzymes reached a plateau.
The role of N-glycosylation on P. funiculosum rCel7A activity
The activity of the P. funiculosum rCel7A WT enzyme was approximately equal to that of T. reesei rCel7A WT, as shown in Fig. 5. The low hydrolysis extents due to rapid drops in hydrolysis rates on BC indicate that both cellobiohydrolases were limited by the crystallinity of the substrate. Four single glycosylation mutants of the P. funiculosum enzyme (N45A, N388A, N430A, and A196S) were successfully expressed and purified from A. niger var. awamori. All mutations resulted in higher extents of BC hydrolysis. In each case, the mutants maintain higher hydrolysis rates in the 120-h period compared to the P. funiculosum rCel7A WT enzyme. Consistent with the previous observation for the case of the T. reesei rCel7A N384A mutant (analogous to N388 in P. funiculosum Cel7A), eliminating the glycan at N388 on P. funiculosum rCel7A resulted in significantly improved activity over the heterologously expressed P. funiculosum rCel7A WT enzyme. However, the N388A mutant exhibits nearly identical cellulose digestion activity to the N45A and N430A mutants. The A196S mutant, with the additional glycosylation site at N194, sustained higher hydrolysis rates longer than any of the other mutants tested or any of the P. funiculosum rCel7A WT enzymes (by 70% after 120 h).

Extent of hydrolysis time course for P. funiculosum rCel7A WT and the four single mutants: N45A, N388A, N430A, and A196S
Discussion
To determine the contribution of N-linked glycosylation to the stability and activity of family 7 glycoside hydrolases, we compared homologous N-linked sites of two closely related enzymes, the Cel7A enzymes from P. funiculosum and T. reesei expressed in an industrially important expression host, A. niger var. awamori. These enzymes have greater than 60% sequence homology and have structurally conserved N-linked glycosylation sites as shown in Fig. 3.
The effects of T. reesei Cel7A glycosylation on the digestion of BC can be predicted in part by the location of the N-linked motifs on the enzyme surface (Fig. 3b). There are three N-glycosylated sites on the catalytic domain of T. reesei Cel7A: N45 is located near the entrance to the catalytic site tunnel, N270 is located near the distal end (cellobiose-releasing) of the tunnel, and N384 is located at the “bottom” or cellulose-crystal-proximal surface of the enzyme on a peptide loop (amino acids 367–403), which partially forms the catalytic tunnel. Only two of the three mutant enzymes, rCel7A N270A and N384A, were successfully expressed for characterization, suggesting that the glycan at N45 is involved in stabilization of the protein. Analyses of the glycans on Cel7A from various strains of T. reesei have shown that N45 and N384 are typically glycosylated with an N-acetyl glucosamine residue (GlnNAc), whereas the glycan at N270 can vary from a single GlnNAc residue to large mannose structures such as GlnNAc2Man8 (Hui et al. 2001).
We find that the glycan at N384 has the greatest effect on the activity of T. reesei rCel7A on BC substrates (Fig. 4). N384 is situated on the bottom surface of the catalytic domain, on a loop (amino acids 367–403) that partially defines the tunnel shape at the distal end of the enzyme as illustrated in Fig. 3b. The absence of a glycan at N384 resulted in the improvement of the activity of this mutant by 70% after 120 h compared to the WT rCel7A. One possible explanation for the increased hydrolytic effectiveness of the N384A mutant enzyme is that the host-applied glycosylation at N384, which is considerably more extensive than that present at this position in the WT enzyme expressed from T. reesei, may function as a “spacer”, limiting the ability of the catalytic domain to approach closely to the cellulose surface. This glycan chain may thus perturb the ability of the catalytic domain to attain optimal alignment with cellulose chains or chain ends. If this steric hindrance exists, it is possible that for the rCel7A enzymes bound by their CBMs to the cellulose surface, the process of threading the cellulose chain productively into the active-site tunnel may be significantly hindered by the greater distance of the catalytic domain from the surface. This hypothesis suggests that the catalytic domain of the rCel7A WT enzyme eventually stalls on the substrate surface, with the threading of a cellulose chain into the active-site tunnel arrested at a point short of productive engagement. Alternatively, the absence of a large glycan at position 384 (as seen in mutant N384A) may in fact allow the enzyme to acquire a “tighter hold” on the substrate within its active site tunnel, thus improving the processivity of the enzyme. This concept is especially consistent with the model proposed in references (Varrot et al. 1999; Armand et al. 1997), where the loops of the active site tunnel are thought to slightly separate to accommodate new substrate. A large glycan at N384 may act as a wedge that impedes this mechanism by hindering the closing of the loops or as a cross linker, which interacts with surface residues on both sides of the cleft.
The crystal structure of the catalytic domain of P. funiculosum Cel7A is not currently available. However, functional implications of the structure can still be examined by backbone threading of the P. funiculosum sequence to that of its homologue, Talaromyces emersonii (1q9h; Grassick et al. 2004; Grassick et al. 2003). The modeled structure of P. funiculosum Cel7A (Fig. 3) is similar to the structure of T. reesei Cel7A. For example, the glycosylation sites at N45 and N388 are analogous to N45 and N384 on the T. reesei catalytic domain, respectively. The second analogous glycosylation site for the two cellobiohydrolases (N384 on T. reesei and N388 on P. funiculosum), have similar effects on the activity on BC, as shown in Fig. 5. In addition, whereas T. reesei Cel7A has a third glycosylation site near the distal end of the catalytic tunnel (N270), P. funiculosum Cel7A has a third site near the linker peptide attachment site (N430A). This linker peptide begins at G435. At this position, the glycan can potentially interact with the linker peptide. The absence of a glycan chain at this position (e.g., T. reesei Cel7A and the P. funiculosum mutant N430A) may in fact increase the linker peptide’s degree of freedom on the catalytic domain and allow the enzyme to attach to a recalcitrant substrate more effectively, thus increasing total bound concentration. The increased association of the enzyme to the substrate may also increase the probability of locating a reducing end on the substrate, thereby resulting in a higher rate of hydrolysis extents. Interestingly, whereas the N45A mutant of T. reesei rCel7A could not be expressed in active form, the analogous mutant in P. funiculosum rCel7A was readily expressed. The location of this glycosylation site appears to be similar for the two enzymes, which suggests that differences in the size and composition of the glycan may be a factor in its stability. Overall, the individual elimination of any of the three N-linked glycosylation sites at N45, N388, and N430 in the P. funiculosum enzyme yields three mutant enzymes with improved activity compared to the WT recombinant enzymes, as shown in Fig. 5.
Whereas the three elimination mutants of P. funiculosum yielded similar activities, the addition of a new glycosylation motif around N194 in the P. funiculosum enzyme proved especially interesting in terms of cellulose degradation activity. In the WT sequence, N194 is not part of an N-glycosylation motif (N-X-S/T) and thus not glycosylated (Fig. 1). Glycosylation of this site may be expected to impart steric hindrance between the enzyme and the substrate surface, however. In contrast to the N388 glycan, which is located on the “bottom” (cellulose-crystal-facing side) but nearer the exit to the active-site tunnel, the N194 glycan (A196S mutant) is located in a loop near the proximal end (entrance) of the active site tunnel (amino acids 192–198). Furthermore, addition of a glycan to this loop could affect the geometry of the active site tunnel. Figure 5 shows that the introduction of a glycan chain at N194 actually improves rCel7A enzyme activity by approximately 70% after 120 h compared to the recombinant WT enzyme. We speculate that the additional glycan introduced into the space between the catalytic domain and the cellulose surface may deter non-specific interaction with the substrate surface. This mutant (A196S) exhibited significantly higher hydrolysis rates in the latter (50-h onward) time course of the 120-h digestion on BC, although this effect was not as prevalent on phosphoric acid swollen cellulose (data not shown). The A196S mutation, therefore may help the enzyme overcome late-stage cellulose recalcitrance.
Conclusions
This study was intended to examine the effects of glycosylation on heterologously expressed T. reesei and P. funiculosum Cel7A enzymes, which are industrially relevant enzymes for the conversion of lignocellulosic biomass. A. niger var. awamori was used as the expression host. We created a library of mutant T. reesei and P. funiculosum rCel7A enzymes by the removal and/or the addition of specific N-linked glycosylation motifs using site-directed mutagenesis to probe the effect of individual N-linked glycosylation sites on enzyme stability and activity on crystalline cellulose.
It was found that when the two analogous glycosylation sites, N384 on the T. reesei enzyme and N388 on the P. funiculosum enzyme, were modified by the replacement of the asparagine with alanine, both enzymes showed higher rates and extents of hydrolysis compared to their respective rCel7A WT enzymes. Specifically for the T. reesei rCel7A enzyme, eliminating the N384 site imparts up to 70% greater activity compared to the rCel7A WT enzyme. In addition, sustained activity on the crystalline cellulose (BC) suggests that these enzymes have improved processivity. These findings suggest that the presence of covalently linked glycans at the base of the active site tunnel (defined by seven peptide loops in the enzyme) may inhibit loop flexibility by interacting with the charged surface amino acid residues in the vicinity of N384 and N388. This conclusion is consistent with our postulate that Cel7A enzymes may be described as “hinge proteins”, with a clamshell configuration (oriented with the hinge opposite the side interacting with cellulose). Thus, the interaction of loops and perhaps glycans at the surface helps to limit the motion of the hinge. In the case of P. funiculosum rCel7A, a glycoslylation motif at N194 exhibited increased activity over the heterologously expressed WT enzyme.
In this study, we established the contribution of glycan attachment to the intrinsic biological functions and stability of these enzymes. We present compelling evidence that glycans on the peptide loop near the active site entrance of these enzymes influence the activity of the enzymes and may be a target for improvement of specific enzyme performance. The fundamental principles and findings described in this study are directed toward understanding cellulase enzymes, specifically family 7 cellobiohydrolases, but are likely to apply to the heterologous fungal expression of other important biomass degrading enzymes, such as hemicellulases and accessory enzymes.
References
Adney WS et al (2003) Heterologous expression of Trichoderma reesei 1, 4-beta-d-glucan cellobiohydrolase (Cel 7A), in applications of enzymes to lignocellulosics. Amer Chemical Soc: Washington 40:3–437
Armand S et al (1997) A bifunctionalized fluorogenic tetrasaccharide as a substrate to study cellulases. J Biol Chem 272(5):2709
Chen CM et al (1987) Toward improved cellulases—targeted modifications of Trichoderma reesei exocellobiohydrolase-II using site specific mutagenesis. Abstracts of Papers of the American Chemical Society 194: 188–MBTD
Chen HZ, Hayn M, Esterbauer H (1993) Three forms of cellobiohydrolase-I from Trichoderma reesei. Biochem Mol Biol Int 30(5):901–910
Divne C et al (1994) The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 265(5171):524–528
Doner LW, Irwin PL (1992) Assay of reducing end-groups in oligosaccharide homologues with 2,2′-bicinchoninate. Anal Biochem 202(1):50–53
Elshafei AM et al (1991) The Saccharification of corn stover by cellulase from Penicillium funiculosum. Bioresour Technol 35(1):73–80
Eriksen SH, Jensen B, Olsen J (1998) Effect of N-linked glycosylation on secretion, activity, and stability of alpha-amylase from Aspergillus oryzae. Current Microbiology 37(2):117–122
Eriksson T et al (2004) Heterogeneity of homologously expressed Hypocrea jecorina (Trichoderma reesei) Cel7B catalytic module. Eur J Biochem 271(7):1266–1276
Foreman PK et al (2003) Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J Biol Chem 278(34):31988–31997
Gaur R, Mishra S (1990) Cellulase activity at different sites in two fungal species, Trichoderma harzianum and Penicillium funiculosum. Acta botanica Indica 18(1):141–143
Grassick A et al (2003) Crystallization and preliminary crystallographic analysis of the catalytic domain cellobiohydrolase I from Talaromyces emersonii. Acta Crystallographica Section D-Biological Crystallography 59:1283–1284
Grassick A et al (2004) Three-dimensional structure of a thermostable native cellobiohydrolase, CBHIB, and molecular characterization of the cel7 gene from the filamentous fungus, Talaromyces emersonii. Eur J Biochem 271(22):4495–4506
Helbert W et al (2003) Fluorescent cellulose microfibrils as substrate for the detection of cellulase activity. Biomacromolecules 4(3):481–487
Hestrin S, Schram M (1954) Synthesis of cellulose by Acetobacter xylinum. Biochem J 58:345–352
Hui JPM et al (2001) Characterization of cellobiohydrolase I (Cel7A) glycoforms from extracts of Trichoderma reesei using capillary isoelectric focusing and electrospray mass spectrometry. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences 752(2):349–368
Jeoh T et al (2008) Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnology for Biofuels 1(1):10
Krystynowicz A et al (2002) Factors affecting the yield and properties of bacterial cellulose. J Ind Microbiol Biotechnol 29(4):189–195
Lachke AH et al (1987) Strain selection criteria for Penicillium funiculosum in enzymatic hydrolysis of ligncellulosics. Biotechnol Lett 9(2):147–150
Laymon RA et al (1996) Cloning and expression of full-length Trichoderma reesei cellobiohydrolase I cDNAs in Escherichia coli. Appl Biochem Biotechnol 57–8:389–397
Manchanda AC, Jogdand VV, Karanth NG (1982) Studies on fermentation broth rheology of a Penicillium strain with cellulose as substrate. Journal of Chemical Technology and Biotechnology 32(6):660–665
Medve J, Stahlberg J, Tjerneld F (1994) Adsorption and synergism of cellobiohydrolase I and II of Trichoderma reesei during hydrolysis of microcrystalline cellulose. Biotechnol Bioeng 44:1064–1073
Medve J, Lee D, Tjerneld F (1998) Ion-exchange chromatographic purification and quantitative analysis of Trichoderma reesei cellulases cellobiohydrolase I, II and endoglucanase II by fast protein liquid chromatography. J Chromatogr A 808(1-2):153–165
Receveur V et al (2002) Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle X-ray scattering. J Biol Chem 277(43):40887–40892
Stals I et al (2004) Factors influencing glycosylation of Trichoderma reesei cellulases. I: postsecretorial changes of the O- and N-glycosylation pattern of Cel7A. Glycobiology 14(8):713–724
Taylor ME, Drickamer K (2003) Introduction to glycobiology. Oxford Universtiy Press, New York
Varrot A, Schulein M, Davies GJ (1999) Structural changes of the active site tunnel of Humicola insolens cellobiohydrolase, Cel6A, upon oligosaccharide binding. Biochemistry 38(28):8884–8891
Acknowledgments
This work was funded by the DOE Office of the Biomass Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Adney, W.S., Jeoh, T., Beckham, G.T. et al. Probing the role of N-linked glycans in the stability and activity of fungal cellobiohydrolases by mutational analysis. Cellulose 16, 699–709 (2009). https://doi.org/10.1007/s10570-009-9305-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-009-9305-1