
Abstract
Genetic tools for the fine-tuning of gene expression levels are a prerequisite for rational strain optimization through metabolic engineering. While Aspergillus niger is an industrially important fungus, widely used for production of organic acids and heterologous proteins, the available genetic tool box for this organism is still rather limited. Here, we characterize six novel constitutive promoters of A. niger providing different expression levels. The selection of the promoters was based on published transcription data of A. niger. The promoter strength was determined with the β-glucuronidase (gusA) reporter gene of Escherichia coli. The six promoters covered a GUS activity range of two to three orders of magnitude depending on the strain background. In order to demonstrate the power of the newly characterized promoters for metabolic engineering, they were used for heterologous expression of the cis-aconitate decarboxylase (cad1) gene of Aspergillus terreus, allowing the production of the building block chemical itaconic acid with A. niger. The CAD activity, dependent on the choice of promoter, showed a positive correlation with the specific productivity of itaconic acid. Product titers from the detection limit to up to 570 mg/L proved that the set of constitutive promoters is a powerful tool for the fine-tuning of metabolic pathways for the improvement of industrial production processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aspergillus niger is a widely used host organism for the industrial production of food processing enzymes and metabolites such as organic acids or antibiotics (Sauer et al. 2008; Tevz et al. 2010; Dashtban et al. 2011; Frisvad et al. 2011). Rational strain design becomes increasingly important for this microbial cell factory. To this end analysis and modelling of the cellular metabolism is followed by design of the metabolic pathways using genetic engineering (Andersen and Nielsen 2009; Kim et al. 2012; Sauer and Mattanovich 2012). A prerequisite for such rational engineering approaches is an adequate genetic tool box including markers, vectors, and promoters among others. The set of promoters, which have been described for heterologous gene expression in A. niger is currently quite focused on promoters providing a high expression level of the target gene (Fleissner and Dersch 2010). For fine-tuning of the gene expression, some promoters have been described which are inducible, thereby allowing for low and high expression from the same construct. However, these promoters require usually strictly defined conditions for high or low gene expression, respectively. Many of them are dependent on the carbon or nitrogen source in the culture medium such as the glucoamylase promoter PglaA of A. niger (Ganzlin and Rinas 2008), the alcohol dehydrogenase promoter PalcA of Aspergillus nidulans (Waring et al. 1989), or the Taka-amylase A promoter PamyB of Aspergillus oryzae (Kanemori et al. 1999). Other promoters are induced only in the presence of certain substances, like thiamine (thiamine promoter PthiA of A. oryzae) (Shoji et al. 2005) or estrogen (human estrogen receptor hERα) (Pachlinger et al. 2005). Recently, Meyer et al. (2011) have described a very tight, tuneable Tet-on (induced by tetracycline) expression system for A. niger, that is dependent on the doxycycline concentration. The advantage of these systems is the possible fine regulation of gene expression depending on the concentration of the inducer, which is very useful for scientific purposes. However, in an industrial production context the dependence on the lack or addition of a specific compound or specific other medium constraints are a disadvantage. Construction of a metabolic network, which constitutively gives rise to the required combination of enzyme activities allows for flexibility for bioprocess design and is not dependent on special media (components) and is therefore the preferred solution.
Among the constitutive promoters, which have been described for filamentous fungi are the alcohol dehydrogenase promoter PadhA of A. niger, the pyruvate kinase promoter Ppki of Trichoderma reesei (Limón et al. 1999) or the glyceraldehyde-3-phosphate dehydrogenase promoter PgpdA of A. nidulans (Hunter et al. 1992). While PgpdA is by far the most frequently used promoter for genetic engineering of Aspergillus ssp. all of them are useful for high-level gene expression.
However, the optimization of complex networks of metabolic pathways does not always require the highest activity of all genes of interest. In a lot of cases, it is more a question of fine-tuning and balancing of the involved activities. Notwithstanding, constitutive promoters providing different levels of expression strength are hardly characterized and not available with a sufficient degree of variability for filamentous fungi.
In this study, we cloned and characterized six novel constitutive promoters of the industrially important fungus A. niger, providing different levels from low to high expression of the target genes. In order to prove their functionality as a useful tool for metabolic engineering, we applied them on the metabolic pathway leading to the production of itaconic acid. Since A. niger lacks cis-aconitate decarboxylase activity, this organism is not naturally able to produce itaconic acid (Kubicek and Karaffa 2006). Heterologous expression of the cad1 gene of Aspergillus terreus in A. niger results in strains producing low amounts of itaconic acid in the culture medium (Li et al. 2011). In our study, we transformed the cad1 coding sequence of A. terreus under the control of six constitutive promoters to A. niger and confirmed their functionality as novel tool for metabolic engineering.
Materials and methods
Strains and media
Escherichia coli TOP10 was used as host for recombinant DNA manipulation. A. niger CBS 513.88 and ATCC 1015 were used as parental strains for A. niger transformation.
E. coli transformants were grown on LB (Sambrook and Russell 2001) supplemented with 75 μg/mL hygromycin. A. niger was cultured in liquid ME or solid MEA (3 % malt extract, 0.5 % peptone, and ±1.5 % agar), supplemented with 150 μg/mL hygromycin, or in Vogel’s minimal medium (2.5 g/L Na3-citrate 2 × H2O, 5 g/L KH2PO4, 2 g/L NH4NO3, 0.2 g/L MgSO4 7 × H2O, 0.1 g/L CaCl2 2 × H2O, 0.5 mg/L citric acid 1 × H2O, 0.5 mg/L ZnSO4 7 × H2O, 0.1 mg/L Fe(NH4)2 (SO4)2 6 × H2O, 0.025 mg/L CuSO4 5 × H2O, 0.005 mg/L MnSO4 1 × H2O, 0.005 mg/L H3BO3, 0.005 mg/L Na2MoO4 2 × H2O, and 0.005 mg/L biotin) (Vogel 1956) without MnSO4 and supplemented with 10 % glucose as carbon source. For the osmotic stabilization, 1.2 M sorbitol was added to the medium after transformation. Top agar was composed of osmotically stabilized MEA containing 0.75 % agar. Bottom agar was composed of osmotically stabilized MEA supplemented with 180 μg/mL hygromycin. For the detection of β-glucuronidase activity, additional 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid (X-gluc, 70 mg /L) was added to the plates.
Selection of constitutive promoters of A. niger using microarray expression data
The Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) was used to retrieve five gene expression data sets (GSE11405, GSE17329, GSE14285 (van den Berg et al. 2010), GSE17641 (Meyer et al. 2009), and GSE21752 (Jørgensen et al. 2010) of A. niger based on the Affymetrix chip platform GPL6758. For each gene, the average signal intensity was determined by calculation of the arithmetic mean from all data sets. Then the genes were grouped into two arbitrary subgroups (A and B): the first (A) containing all highly expressed genes with an average signal intensity >1,000 (463 genes) and a second group (B) containing weaker expressed genes with an average signal intensity between 230 and 299 (500 genes). In both subgroups, the most stably expressed genes were determined using the software NormFinder (Andersen et al. 2004). This software uses a model-based approach for the estimation of expression variation and can deal with heterogeneous data sets subdivided in distinct groups. With regard to this ability the expression data sets were subdivided into five sample groups according to the five gene expression data sets used for the analysis. The stability value was determined and, out of the first ten genes with the highest stability, three genes of each gene subgroup were randomly selected (Table 1). 1.5 kb fragments located upstream of the coding sequences of the six selected genes listed in Table 1 were PCR amplified from genomic DNA of A. niger ATCC 1015 and used as promoter sequences in this study.
Construction of gusA and cad1 expression cassettes
Plasmids were constructed using a previously published enzymatic assembly method (Gibson et al. 2009). Target DNA fragments were obtained by touchdown PCR using Phusion® High-Fidelity DNA Polymerase (New England Biolabs) under the following conditions: 98 °C for 10 s, 14 cycles of 98 °C for 10 s, 70 °C minus 1 °C/cycle for 30 s, 72 °C for 90–150 s, 25 cycles of 98 °C for 10 s, 55 °C for 30 s, 72 °C for 90–150 s, and a final extension at 72 °C for 10 min. The primers are specified in Table S1 in the Electronic supplementary material (ESM). Two series of plasmids A and B containing (A) gusA of E. coli (plasmids pGUS_mbfA, pGUS_coxA, pGUS_srpB, pGUS_tvdA, pGUS_mdhA, pGUS_manB) or (B) cad1 of A. terreus (plasmids pCAD_mbfA, pCAD_coxA, pCAD_srpB, pCAD_tvdA, pCAD_mdhA, pCAD_manB) were constructed for homologous integration of the expression cassette into the mus53 locus. The plasmids consisted of the following 11 fragments (in order): (1) one of the A. niger promoters amplified from genomic DNA of strain ATCC 1015 (Table 1), followed by the gusA coding sequence (2A) of E. coli amplified from genomic DNA of strain TOP10, or the cad1 coding sequence of A. terreus (2B) amplified from genomic DNA of strain DSM 62071 (Kanamasa et al. 2008); (3) the TtrpC terminator amplified from pAN52-1Not (GeneBank Accession No. Z32524); (4) the hph gene under the Ppki promoter and the Tcbh2 terminator of Hypocrea jecorina amplified from pRLMex30 (Mach et al. 1994) suitable for selection of both, E. coli and filamentous fungi (Steiger et al. 2011); (5) the TtrpC terminator amplified from pAN52-1Not (GeneBank Accession No. Z32524) suitable for selection marker recovery; (6) 1 kb of the 3′ flanking region of mus53 of A. niger amplified from genomic DNA of strain ATCC 1015; (7) PmeI restriction site; (8A/8B) AMA1 fungal origin of replication amplified in two pieces from pRG3-AMA1-NotI (Osherov et al. 2000) kindly provided by Joseph Strauss, University of Natural Resources and Life Sciences, Vienna (BOKU); (9) origin of replication for E. coli amplified from pUC19 (GenBank Accession No. M77789); (10) PmeI restriction site; (11) 1 kb of the 5′ flanking region of mus53 of A. niger amplified from genomic DNA of strain ATCC 1015. The map of the plasmids is included in the online resource (Fig. S1 in the ESM). Two or three overlapping PCR products were fused by secondary PCR. Four secondary fragments were enzymatically assembled for each plasmid. Obtained plasmids were cloned into E. coli and isolated according to standard methods (Sambrook and Russell 2001). In order to obtain integrative expression cassettes, the AMA1 and ori for E. coli were cut out with the PmeI restriction enzyme.
Transformation of A. niger
The preparation of protoplasts and the transformation of A. niger were performed based on Yelton et al. (1984). Conidia (1 × 108) were cultivated in 250 mL ME for 16–18 h at 30 °C. The mycelium was harvested by filtration through Miracloth (Calbiochem) and washed with deionized water. Protoplastation was achieved in the presence of 5 g/L of lysing enzymes from Trichoderma harzianum (Sigma®, L1412) in SMC (1.2 M sorbitol, 50 mM CaCl2, 20 mM Mes/NaOH, pH 5.8), for 4 h at 37 °C, 120 rpm. The protoplasts were collected by filtration through Miracloth and centrifugation at 2,000×g at 4 °C for 5 min. After a washing step with cold STC (1.2 M sorbitol, 50 mM CaCl2, 10 mM Tris/HCl, pH 7.5), the protoplasts were resuspended in STC and directly used for transformation. Five micrograms of linear DNA (30 μL) was mixed with 100 μL of at least 108 protoplasts and 330 μL freshly prepared PEG solution (25 % PEG 6000, 50 mM CaCl2, 10 mM Tris/HCl, pH 7.5), and incubated for 20 min on ice. After the addition of 2 mL PEG solution and 10 min incubation at room temperature, the protoplasts mixture was diluted with 4 mL of STC. The aliquots were mixed with 4 mL of liquid top agar, spread on bottom agar containing the antibiotic and incubated at 30 °C for 4–6 days. All transformants were three times purified by single colony isolation on the selection medium (Daud et al. 1985). The correct integration was verified by PCR analysis using specific genomic primers on both sides of the expression cassette. Only clones were further analyzed, which showed positive PCR results for both, the 5′and 3′ end of the expression cassette.
Preparation of cell-free extracts
To obtain cell-free extracts, fungi were cultivated on MEA for 26–28 h at 30 °C (inoculated with conidia). The mycelium was harvested into tubes containing either (for GUS activity) 2× GUS reaction buffer (×1 reaction buffer contains, 50 mM sodium phosphate, pH 7; 1 mM EDTA; 0.001 % Triton X-100; 5 mM DTT) or (for CAD activity) 0.2 M sodium phosphate buffer (pH 6.2), and disrupted with 0.4–0.6 mm acid washed glass beads (five times, 20 s, 4 m/s). The protein concentration was determined based on Bradford’s protocol (Bradford 1976) using the Bio-Rad Protein Assay according to the manufacturer’s instructions.
β-glucuronidase activity assay
For the measurement of the β-glucuronidase (GUS) activity, 10 μL of the cell-free extract in eight different dilutions was mixed with 40 μL of ×2 GUS reaction buffer and diluted with 50 μL of 4 mM 4-methylumbelliferyl-β-d-glucuronide (MUG). The reaction was carried out at 37 °C. The fluorescence was measured at excitation/emission of 355/460 nm with the time interval of 80 s. The linear range of fluorescence increase was used for the calculation of the GUS activity. One GUS unit is defined as the amount of the enzyme that catalyzes the conversion of 1 μM MUG/min.
Cis-aconitate decarboxylase activity assay
The cis-aconitate decarboxylase (CAD) activity was measured according to Bentley and Thiessen (1957). The reaction containing 8 mM freshly prepared cis-aconitic acid in 0.2 M sodium phosphate buffer (pH 6.2) and 200 μL cell-free extract, was carried out for 10 min at 37 °C and heat inactivated at 80 °C for 20 min. The CAD activity was measured by direct detection of itaconic acid as a reaction product by HPLC (as described below). One CAD unit is defined as the amount of the enzyme that catalyzes the conversion of 1 μM CAD/min.
Shake flask cultivation of cad1 transformants of A. niger
Conidia (5 × 107 or 5 × 108) were cultivated for 432 h at 33 °C, shaking (120 rpm) in 1 L baffled shake flasks containing 100 mL Vogel’s medium. Final concentration of the organic acids produced in the medium and the remaining glucose was determined by HPLC. The biomass was harvested by filtration through Miracloth. The dry weight was determined by drying the biomass in the oven at 110 °C for 2 days.
HPLC measurements
Nine hundred microliters of the HPLC samples were mixed with 100 μL of 0.04 M H2SO4, filtrated through 0.20 μm RC membrane filters and measured by HPLC (Shimadzu) with a Phenomenex Rezex ROA column (300 × 7.8 mm). A refraction index detector (RID-10A, Shimadzu) was used for detection of glucose, while a PDA detector (SPD-M20A, Shimadzu) at 200 nm was used for itaconic acid. The column was operated at 60 °C temperature, 1 mL/min flow rate, and 0.004 M H2SO4 as mobile phase.
Results
Selection of constitutive promoters of A. niger
A constitutive promoter, unlike an induced promoter, should have a constant transcription level under as many as possible different conditions. In order to find constitutive promoters spanning a wide range of expression levels, we reinvestigated all microarray expression data sets which were publicly available at the time being, in order to have a high variability in the cultivation conditions (e.g., different carbon and nitrogen sources, bioreactor and shake flask cultures). These data sets were analyzed for constitutively expressed genes with the software NormFinder. This software was originally developed to find stably expressed reference genes for quantitative PCR analysis. The underlying algorithm calculates which gene shows the lowest variance in expression level throughout a data set. The selected genes from this analysis are shown in Table 1.
To estimate the transcription strength of the promoters, the transcription of the genes controlled by the six promoters was analyzed in batch cultures of A. niger strain ATCC 1015 (Fig. 1). According to the transcriptional analysis with DNA microarrays, the six promoters cover a range of one order of magnitude in transcription level with promoter PmanB being the weakest and PmbfA the strongest promoter. Remarkably, the PmbfA promoter is significantly stronger than the characterized and widely used promoter of the gpdA gene. The gpdA transcription signal is in the range of the cox4 transcription signal which is the second strongest promoter of the selected six.

Relative expression intensities measured with a custom DNA microarray (Agilent). The expression intensities were normalized to the expression value of manB, which was arbitrarily set to 1. The axis has a logarithmic scaling to the base 10. Strain A. niger ATCC 1015 was cultivated in a glucose mineral medium and RNA was extracted in the late exponential growth phase
Promoter characterization using a gusA reporter system
Of the fragments located upstream of the coding sequences of the selected genes, 1.5 kb were PCR amplified and used as promoter regions in this study. To characterize the selected promoters for heterologous gene expression, cassettes with the GUS reporter gene under the control of the respective promoter were constructed and integrated into the genome of A. niger strains CBS 513.88 and ATCC 1015. Thus, the gene activity was directly observable on the agar plates for fungal growth (supplemented with X-Gluc; Fig. 2a). The blue color formation can be clearly seen for all promoter constructs except for PmanB, which is indistinguishable from the parental strain.

Clones of A. niger expressing the gusA gene of E. coli under control of the six analyzed promoters. a Clones of A. niger CBS 513.88 growing on with agar solidified Vogel´s minimal medium supplemented with X-gluc. (1) front and (2) back side of the plate. ps parental strain CBS 513.88. b GUS activity (U (mg protein)−1) in cell extracts of A. niger CBS 513.88 and ATCC 1015 clones containing the gusA gene of E. coli under the specified promoters. Strains were grown on MEA plates. Activity values are shown as arithmetic mean values with the standard deviation of at least two different clones, each measured in triplicate. The axis has a logarithmic scaling to the base 10
A fluorimetric assay based on the conversion of the fluorogenic substrate 4-methylumbelliferyl-β-d-glucuronide allowed the precise quantification of the enzyme activity. The obtained values are shown in Fig. 2b. In all of the transformants carrying one of the six different promoter constructs, the GUS activity was detectable and significantly above the basal level of the parental strains, which were used as a negative control in all experiments. In case of ATCC 1015 transformants, the GUS activity correlates well with the transcription data and the order of the promoters is the same in both data sets being PmbfA > PcoxA > PmdhA > PtvdA > PsrpB > PmanB. The six promoters cover an activity range of two orders of magnitude. Interestingly, in case of CBS 513.88, the order of the promoter strength changes slightly, being: PmbfA > PcoxA > PsrpB > PtvdA > PmdhA > PmanB (Fig. 2b). With three orders of magnitude, the range of activity between the strongest and the weakest promoter is even more pronounced in CBS 513.88. The GUS activity of a PgpdA construct showed a comparable activity as the Pcox4 construct, thus confirming the initial transcription data (data not shown).
Application of the constitutive promoters in a metabolic pathway
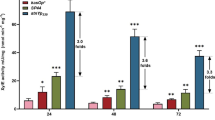
A. niger is known to be a powerful producer of citric acid, however, it is naturally not able to produce itaconic acid, an industrially promising building block chemical. However, this organic acid can be produced in A. niger by expressing cis-aconitate decarboxylase (cad1) from A. terreus, as it was previously shown (Li et al. 2011). We took advantage of this system to demonstrate the usability of the characterized promoters in a metabolic pathway. For this purpose, the cad1 gene of A. terreus was alternately put under control of one of the six promoters and the respective cassettes were integrated into the genome of the parental strain CBS 513.88. From each construct three randomly chosen colonies were tested for CAD activity (Fig. 3). CAD activity was found in all transformants tested and again the highest activity was found for strains carrying the PmbfA promoter construct whereas the lowest was detected using PmanB promoter. The rank of the other 4 promoters is comparable to the results of the GUS activity assay. Only the construct with PsrpB exhibits a lower CAD activity than expected, having this time slightly lower activity than the PtvdA construct.

CAD activity (in mU (mg protein)−1) in cell extracts of A. niger CBS 513.88 clones containing the cad1 coding sequence of A. terreus under the specified promoters. Strains were grown on MEA plates. Activity values are shown as arithmetic mean values with the standard deviation of three different clones, each measured in triplicate. The axis has a logarithmic scaling to the base 10. ps parental strain
The strains (at least three different clones of each promoter construct) were cultivated on Vogel’s minimal medium to produce itaconic acid in baffled shake flasks. Itaconic acid was obtained for all cultivations except for the transformants carrying the cad1 gene under the control of the very weak PmanB promoter (Fig. 4). A maximal titer of 567 mg/L was obtained using the PmbfA construct which is comparable to previously published data (∼700 mg/L) (Li et al. 2011).

Specific itaconic acid productivity q P (in mg itaconate (g cell dry weight)−1 (day)−1)) of A. niger CBS 513.88 clones containing the cad1 coding sequence of A. terreus under the specified promoter. The strains were cultivated in shake flasks on Vogel’s minimal medium. Specific productivity values are shown as arithmetic mean values with the standard deviation of at least three different clones. The concentrations above the bars represent the maximal itaconic acid titer (in milligrams per liter) obtained with the respective construct. ps parental strain
In order to compare the CAD activity with the produced amount of itaconic acid, the specific productivity was calculated for each culture and plotted against the respective CAD activity value (Fig. 5). Figure 5 clearly shows the correlation of the CAD activity, which is dependent on the promoter, with the specific productivity of itaconic acid. The high deviation of the single measurements can be explained by the natural variance between the transformants as well as the inherently high variability of cultivation conditions in baffled shake flasks.

Correlation between CAD activity (in mU (mg protein)−1) and specific itaconic acid productivity (in g itaconate (g cell dry weight)−1 (day)−1)) of A. niger CBS 513.88 clones containing the cad1 coding sequence of A. terreus under the control of the six analyzed promoters PmbfA (squares), PcoxA (x sign), PsrpB (plus sign), PtvdA (triangles), PmdhA (diamonds), and PmanB (circles). Each data point represents the mean value of activity and specific productivity of measurements done in triplicates
Discussion
For metabolic engineering purposes, it is crucial to have a powerful toolbox for the respective organism at hand. One important tool is well-characterized promoters in regard of expression strength and regulation. In this work we focused on the characterization of constitutive promoters with different expression strength. The NormFinder algorithm allowed us to explore all published transcriptomic data for the most stably expressed genes. Six promoters were chosen, namely PmbfA, PcoxA, PmdhA, PtvdA, PsrpB, and PmanB, which gave rise to a wide range of expression levels in their natural genetic contexts as shown with transcription analysis with DNA microarrays. Subcloning these sequences in front of the gusA reporter gene confirmed their applicability also in a heterologous context. Consistently, the mbfA promoter led to the highest expression level, even significantly exceeding the activity of the gpdA promoter, which is currently the promoter of choice for high heterologous gene expression in Aspergillus spp. (Flaherty and Payne 1997; Flipphi et al. 2001; Lombraña et al. 2004; Lubertozzi and Keasling 2008). Interestingly, the protein MbfA, encoded by mbfA, was not found in the intracellular proteome measured by Lu et al. (2010), whereas GpdA was found. On the one hand, this discrepancy can be explained by the measurement method used, which consisted of a 2D gel electrophoresis followed by a MALDI-TOF or Q-TOF detection. Not necessarily all proteins can be separated by this technique and detected and to our knowledge it was not shown that MbfA can be detected by this method. On the other hand, it can be speculated that the MbfA protein is very unstable and the actual concentration in the cell is low because of a massive turn-over cycle. However, further investigations are necessary to clarify this hypothesis.
The manB promoter showed consistently the lowest gene expression level, in all strains and construct combinations tested. The expression level of PmanB was two or three orders of magnitude lower than that of PmbfA. The other four promoters gave rise to transcription and activity levels between these two. However, the order of the strength varied with different strains and different constructs.
Interestingly, the absolute expression values and obtained activities are generally and consistently higher for A. niger CBS 513.88 as compared with A. niger ATCC 1015. This finding is plausible considering the background, where these strains are used and derived from: A. niger CBS 513.88 is known to be a good protein producing host and is the ancestor of industrially important enzyme production strains (Pel et al. 2007). A. niger ATCC 1015 is known as the ancestor of acidogenic strains used for the production of organic acids (Andersen et al. 2011). Selection and laboratory evolution of these strains have therefore been quite distinct. While for CBS 513.88 strong transcription and translation is a key feature as the desired products are proteins, the constraints which have been important for ATCC 1015 have been different. Whatever makes a good acid producer might not be determined by a generally optimal protein expression.
The functionality of the new promoter set for metabolic engineering purposes was demonstrated in a case study using the target metabolite itaconic acid. Heterologous expression of the cad1 gene of A. terreus in A. niger led to strains with an enzyme activity variation of three orders of magnitude. These strains accumulated itaconic acid in the supernatant to various degrees: While constructs with the strong PmbfA promoter produced up to 567 mg/L of itaconic acid under the tested conditions, no itaconic acid was detectable for PmanB constructs. Intermediate concentrations for the other constructs proved the correlation of the promoter strength with the final titer. This correlation could be further shown by representing the specific productivity of different strains versus the enzymatic activity.
The highest achieved titer of 567 mg/L is in the same order of magnitude as the previously published value of ∼700 mg/L (Li et al. 2011), which underlines the usefulness of the new promoters. It has to be stressed in this context that the production conditions have not been optimized in our case, as we just wanted to verify the correlation of the promoter strength with the production of a useful metabolite. The slightly lower values compared with Li et al. (2011), which we reached can be explained by the different parental strain (AB 1.13) and the cultivation conditions (bioreactor system) used.
In line with our findings, that protein expression was generally more efficient in strain CBS 513.88 as compared with ATCC 1015, strain CBS 513.88 produced higher itaconic acid titers than strain ATCC 1015 (data not shown). Since we could prove that the productivity of itaconic acid strongly correlates with the cad1 activity it does not come surprising that CBS 513.88 is the better producer under our tested conditions.
The presented constitutive promoters can be a valuable extension to a recently published inducible and tuneable expression system developed for A. niger (Meyer et al. 2011). With such a tuneable system (depending in this case on the inducer doxycycline) the optimal expression level for an enzyme could be determined. Subsequently, a constitutive promoter delivering the tailor-made expression intensity is applied for stable and robust gene expression. This has the advantage that the inducible system can be used for further rounds of experiments without interference of the already optimized enzyme activity and no additional, potentially harmful and/or expensive inducers are needed for industrial production processes.
Summarizing, in this paper, we characterized six novel constitutive promoters of A. niger and demonstrate their suitability as promoters for metabolic engineering purposes using the example of itaconic acid production.
References
Andersen MR, Nielsen J (2009) Current status of systems biology in Aspergilli. Fungal Genet Biol 46:S180–S190
Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJ, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, van Dijck PW, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, van Ooyen AJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A, van den Brink JM, Atkins A, Aerts A, Shapiro H, Pangilinan J, Salamov A, Lou Y, Lindquist E, Lucas S, Grimwood J, Grigoriev IV, Kubicek CP, Martinez D, van Peij NN, Roubos JA, Nielsen J, Baker SE (2011) Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21:885–897
Bentley R, Thiessen CP (1957) Biosynthesis of itaconic acid in Aspergillus terreus III The properties and reaction mechanism of cis-aconitic acid decarboxylase. J Biol Chem 226:703–720
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Dashtban M, Buchkowski R, Qin W (2011) Effect of different carbon sources on cellulase production by Hypocrea jecorina (Trichoderma reesei) strains. Int J Biochem Mol Biol 2:274–286
Daud F, Ortori GS, Roper JA (1985) Spontaneous IR duplications generated at mitosis in Aspergillus nidulans: further evidence of a preferential site of transposed attachment. Genetics 110:229–245
Flaherty JE, Payne GA (1997) Overexpression of aflR leads to upregulation of pathway gene transcription and increased aflatoxin production in Aspergillus flavus. Appl Environ Microbiol 63:3995–4000
Fleissner A, Dersch P (2010) Expression and export: recombinant protein production systems for Aspergillus. Appl Microbiol Biotechnol 87:1255–1270
Flipphi M, Mathieu M, Cirpus I, Panozzo C, Felenbok B (2001) Regulation of the aldehyde dehydrogenase gene (aldA) and its role in the control of the coinducer level necessary for induction of the ethanol utilization pathway in Aspergillus nidulans. J Biol Chem 276:6950–6958
Frisvad JC, Larsen TO, Thrane U, Meijer M, Varga J, Samson RA, Nielsen KF (2011) Fumonisin and ochratoxin production in industrial Aspergillus niger strains. PLoS One 6:e23496
Ganzlin M, Rinas U (2008) In-depth analysis of the Aspergillus niger glucoamylase (glaA) promoter performance using high-throughput screening and controlled bioreactor cultivation techniques. J Biotechnol 135:266–271
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345
Hunter GD, Bailey CR, Arst HN (1992) Expression of a bacterial aspartase gene in Aspergillus nidulans: an efficient system for selecting multicopy transformants. Curr Genet 22:377–383
Jørgensen TR, Nitsche BM, Lamers GE, Arentshorst M, van den Hondel CA, Ram AF (2010) Transcriptomic insights into the physiology of Aspergillus niger approaching a specific growth rate of zero. Appl Environ Microbiol 76:5344–5355
Kanamasa S, Dwiarti L, Okabe M, Park EY (2008) Cloning and functional characterization of the cis-aconitic acid decarboxylase (CAD) gene from Aspergillus terreus. Appl Microbiol Biotechnol 80:223–229
Kanemori Y, Gomi K, Kitamoto K, Kumagai C, Tamura G (1999) Insertion analysis of putative functional elements in the promoter region of the Aspergillus oryzae Taka-amylase A gene (amyB) using a heterologous Aspergillus nidulans amdS-lacZ fusion gene system. Biosci Biotechnol Biochem 63:180–183
Kim IK, Roldão A, Siewers V, Nielsen J (2012) A systems-level approach for metabolic engineering of yeast cell factories. FEMS Yeast Res 12(2):228–248
Kubicek CP, Karaffa L (2006) Organic acids. In: Ratledge C, Kristiansen B (eds) Basic biotechnology, 3rd edn. Cambridge University Press, Cambridge, pp 359–380
Li A, van Luijk N, ter Beek M, Caspers M, Punt P, van der Werf M (2011) A clone-based transcriptomics approach for the identification of genes relevant for itaconic acid production in Aspergillus. Fungal Genet Biol 48:602–611
Limón MC, Pintor-Toro JA, Benítez T (1999) Increased antifungal activity of Trichoderma harzianum transformants that overexpress a 33-kDa chitinase. Phytopathology 89:254–261
Lombraña M, Moralejo FJ, Pinto R, Martín JF (2004) Modulation of Aspergillus awamori thaumatin secretion by modification of bipA gene expression. Appl Environ Microbiol 70:5145–5152
Lu X, Sun J, Nimtz M, Wissing J, Zeng AP, Rinas U (2010) The intra- and extracellular proteome of Aspergillus niger growing on defined medium with xylose or maltose as carbon substrate. Microb Cell Factories 9:23
Lubertozzi D, Keasling JD (2008) Expression of a synthetic Artemesia annua amorphadiene synthase in Aspergillus nidulans yields altered product distribution. J Ind Microbiol Biotechnol 35:1191–1198
Mach RL, Schindler M, Kubicek CP (1994) Transformation of Trichoderma reesei based on hygromycin B resistance using homologous expression signals. Curr Genet 25:567–570
Meyer V, Arentshorst M, Flitter SJ, Nitsche BM, Kwon MJ, Reynaga-Peña CG, Bartnicki-Garcia S, van den Hondel CAMJJ, Ram AFJ (2009) Reconstruction of signaling networks regulating fungal morphogenesis by transcriptomics. Eukaryot Cell 8:1677–1691
Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CA, Ram AF (2011) Fungal gene expression on demand: an inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl Environ Microbiol 77:2975–2983
Osherov N, Mathew J, May GS (2000) Polarity-defective mutants of Aspergillus nidulans. Fungal Genet Biol 31:181–188
Pachlinger R, Mitterbauer R, Adam G, Strauss J (2005) Metabolically independent and accurately adjustable Aspergillus sp. expression system. Appl Environ Microbiol 71:672–678
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JaE, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EGJ, Debets AJM, Dekker P, van Dijck PWM, van Dijk A, Dijkhuizen L, Driessen AJM, d’Enfert C, Geysens S, Goosen C, Groot GSP, de Groot PWJ, Guillemette T, Henrissat B, Herweijer M, van den Hombergh JPTW, van den Hondel CaMJJ, van der Heijden RTJM, van der Kaaij RM, Klis FM, Kools HJ, Kubicek CP, van Kuyk Pa, Lauber J, Lu X, van der Maarel MJEC, Meulenberg R, Menke H, Mortimer Ma, Nielsen J, Oliver SG, Olsthoorn M, Pal K, van Peij NNME, Ram AFJ, Rinas U, Roubos JA, Sagt CMJ, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort PJJ, Wedler H, Wösten HAB, Zeng A-P, van Ooyen AJJ, Visser J, Stam H (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotech 25:221–231
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York, pp 6.4–6.12
Sauer M, Mattanovich D (2012) Construction of microbial cell factories for industrial bioprocesses. J Chem Technol Biotechnol 87:445–450
Sauer M, Porro D, Mattanovich D, Branduardi P (2008) Microbial production of organic acids: expanding the markets. Trends Biotechnol 26:100–108
Shoji JY, Maruyama J, Arioka M, Kitamoto K (2005) Development of Aspergillus oryzae thiA promoter as a tool for molecular biological studies. FEMS Microbiol Lett 244:41–46
Steiger MG, Vitikainen M, Uskonen P, Brunner K, Adam G, Pakula T, Penttilä M, Saloheimo M, Mach RL, Mach-Aigner AR (2011) Transformation system for Hypocrea jecorina (Trichoderma reesei) that favors homologous integration and employs reusable bidirectionally selectable markers. Appl Environ Microbiol 77:114–121
Tevz G, Bencina M, Legisa M (2010) Enhancing itaconic acid production by Aspergillus terreus. Appl Microbiol Biotechnol 87:1657–1664
van den Berg RA, Braaksma M, van der Veen D, van der Werf MJ, Punt PJ, van der Oost J, de Graaff LH (2010) Identification of modules in Aspergillus niger by gene co-expression network analysis. Fungal Genet Biol 47:539–550
Vogel HJ (1956) A convenient growth medium for Neurospora (Medium N). Microb Genet Bull 13:42–43
Waring RB, May GS, Morris NR (1989) Characterization of an inducible expression system in Aspergillus nidulans using alcA and tubulin-coding genes. Gene 79:119–130
Yelton MM, Hamer JE, Timberlake WE (1984) Transformation of Aspergillus nidulans by using a trpC plasmid. Proc Natl Acad Sci U S A 81:1470–1474
Acknowledgments
We acknowledge Karina Schindler for her contribution in plasmid construction and fungal transformation and Stefanie Müller for excellent technical assistance throughout the project. This work has been financially supported by the FHplus Program of the Austrian Research Promotion Agency FFG, Project METORGANIC. Furthermore this work has been supported by the Federal Ministry of Economy, Family and Youth (BMWFJ), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol, and ZIT—Technology Agency of the City of Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 104 kb)
Rights and permissions
About this article
Cite this article
Blumhoff, M., Steiger, M.G., Marx, H. et al. Six novel constitutive promoters for metabolic engineering of Aspergillus niger . Appl Microbiol Biotechnol 97, 259–267 (2013). https://doi.org/10.1007/s00253-012-4207-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4207-9