
Abstract
To study the effects of selection marker, promoter type, and copy number on heterologous expression in Aspergillus nidulans, strains were constructed with single- and multicopy plasmid integrations bearing a reporter gene (lacZ) under the control of either an inducible (alcA) or constitutive (gpdA) promoter and one of three Aspergillus nutritional marker genes (argB, trpC, or niaD). β-Galactosidase activity in the transformants varied over three orders of magnitude, with the majority of levels in the range of 5×103–1×104 U/mg. Significant differences in mean expression levels were found when comparing single-copy transformants with the same promoter but a different marker. Transformants with the argB marker had the highest average expression, ∼threefold over the trpC or niaD clones. For each promoter, maximal expression for the set was seen in the range of the single-copy clones, implying that increasing the copy number does not reliably increase expression in Aspergillus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In filamentous fungal hosts transformed by chromosomal integration, increasing transgene dosage is a common strategy for improving heterologous expression. Although high-expressing clones can be isolated, expression levels are not known to be well correlated with gene copy number (van den Hondel et al. 1991). Several proposed explanations for this variation have been investigated. In the case of inducible Aspergillus promoters for which the conditions of induction and repression are well characterized, such as amdS (Kelly and Hynes 1987) or alcA (Davies 1991), the titration of endogenous transcription factors is shown when multiple integrated copies are present. Expression levels could be improved by adding copies of the activator genes; however, no clear relation between copy number or activator/promoter ratio and expression levels has emerged.
In Aspergillus, high-frequency transformation often results from the integration of transforming DNA at nonhomologous (ectopic) sites, as well as at the homologous region (Johnstone 1985). Apart from unforeseen pleiotropic effects due to gene disruption by randomly integrated transforming DNA, it has been suggested that certain chromosomal locations may be more favorable for heterologous expression than others, perhaps due to specific interaction with local regulatory elements, or more generally active native transcription in the neighborhood of normally highly expressed genes (Davis and Hynes 1991). Certain spatially or temporally regulated Aspergillus genes—e.g., the aflatoxin cluster (Chiou et al. 2002) and conidium-specific genes (Miller et al. 1987)—show dramatic changes in regulatory response when displaced from their original locus, and locus effects on heterologous expression have also been reported (Verdoes et al. 1995).
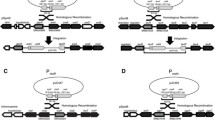
To study the effects of selection marker, promoter type, and copy number on heterologous expression, Aspergillus nidulans strains were constructed with single- and multicopy plasmid integrations bearing a lacZ expression cassette under the control of either an inducible (alcA) or constitutive (gpdA) Aspergillus promoter and one of three commonly used Aspergillus nutritional selection marker genes (argB, trpC, or niaD). To enhance homologous integration at the marker loci, a similar series of transformation vectors were constructed with engineered mutant alleles of the wild-type nutritional genes, complementary to those in the auxotrophic host strains, such that homologous integration would restore the functional gene (Fig. 1), whereas ectopic integration would result in a null phenotype (Hamer and Timberlake 1987). The transformants were assayed for transgene dosage and β-galactosidase expression levels.

Homologous recombination by a single crossover occurring between the engineered mutation of the selection marker in the Aspergillus transformation vectors and chromosomal mutation in the host reconstitutes the functional marker gene
Materials and methods
Fungal strains
Aspergillus nidulans strains A89 (biA1 argB2), A237 (pabaA1 yA2 trpC801), and A691 (biA1 niaD15) were obtained from the Fungal Genetics Stock Center (FGSC) of the University of Kansas. Aspergillus strains for spore and protoplast production were grown on glucose-nitrate minimal medium (Barratt et al. 1965) plus 4 mM arginine and 0.2 μg/L biotin or 4 mM tryptophan and 0.2 μg/L PABA, as required; NaNO3 was replaced at a 1:1 molar ratio by NH4Cl for strain A691. Transformed strains were grown on nitrate minimal medium plus vitamins.
Fungal transformation
Aspergillus transformation was performed as described by Dawe (Dawe et al. 2000), with the exception of replacing Novozym 234 and β-glucuronidase with Glucanex (Sigma L1412; at 10 mg/mL) and Driselase (Interspex 0456; at 2 mg/mL); additionally, the washed protoplast suspension was filtered through a 60-μM glass fritted funnel or nylon mesh (Small Parts) to remove undigested mycelia. Ten micrograms of transforming DNA was added to ∼105 protoplasts in 100 μL total volume for each transformation. Transformed protoplasts were plated onto minimal medium plus 1 M sucrose. Selected colonies were subcultured three times on minimal plates, and those displaying grossly abnormal growth phenotypes were discarded.
DNA manipulations and reagents
Plasmids were propagated in Escherichia coli strains DH5α, DH10b, and DM1 (Life Technologies), and XL1-Blue (Stratagene). PCR products for sequencing or ligation were cloned into the pCR4-TOPO vector (Invitrogen) and maintained in E. coli TOP10 (Invitrogen). Transformed E. coli hosts were cultivated at 37 °C on Luria-Bertani medium (Sigma) containing ampicillin (100 μg/mL) or kanamycin (50 μg/mL) from Fisher Scientific.
General recombinant DNA techniques were performed essentially as described by Sambrook (Sambrook and Russell 2001). Escherichia coli plasmid DNA was extracted using the Bio101 Mini-Prep Express kit (for colony screening), the Qiagen mini-spin kit (for sequencing and ligation), and the Qiagen Midi kit (for Aspergillus transformation). Aspergillus genomic DNA was extracted from colonies on agar, including spores, by a “quick prep” boiling lysis method (Chiou et al. 2002) for screening PCR, and from broth-cultured pellets by lyophilization, grinding and phenol-chloroform extraction Chow and Käfer (1993) for sequencing, quantitative PCR (QPCR), Southern analysis, and dot-blots. Restriction enzymes and T4 DNA ligase came from Roche and New England Biolabs and were used with the manufacturers’ buffers. Gibco DNA Taq polymerase was used for PCR-screening of transformants, the Roche Hi-Fidelity PCR kit for generating products for sequencing, and Stratagene Pfu Turbo polymerase for PCR mutagenesis. QuikChange PCR site-directed mutagenesis (Stratagene) was performed as per the manufacturer’s instructions using primers (and their complements) as noted (Table 1).
Sequencing of auxotrophic genes and construction of complementary mutant alleles
The sequencing of PCR products from genomic DNA prepared from strains A89, A237, and A691 revealed a single base deletion 248 bp downstream of the start codon of argB, causing a frameshift and premature stop codon [confirming previously published data (Ichioka et al. 2001)]; a single base deletion 349 bp downstream of the start codon in trpC; and a single base transversion at the last base pair of intron VI in niaD, respectively.
Complementary mutant alleles of the three genes (argB_1626, trpC_2716, and niaD_345; numbers correspond to nucleotide positions on their GenBank sequences M29819, X02390, and M58291, respectively) were made by QuikChange PCR site-directed mutagenesis. The argB_1626 mutation consists of two sequential stop codons at 925 bp downstream of the start codon, made using primer 1 and its complement (Table 1); trpC_2716 has a similar mutation 1,464 bp downstream of the trpC start codon, made using primer 2 and its complement. The niaD mutation in A691 was toward the 3′ end of the gene, so niaD_345 was designed with an altered start codon (nonsense mutation) via primer 3. NcoI restriction sites were removed from argB and trpC by the same method using primers 4 and 5, respectively, and their complements.
Vector construction
Plasmids pAST30 (Som and Kolaparthi 1994), pDC1 (Aramayo et al. 1989), and pDE3 (Ebbole 1990) came from the FGSC of the University of Kansas Medical Center, Kansas City, KS, USA. Plasmid pAN52-1 was a gift of Dr. P.J. Punt (Punt et al. 1990). Plasmid pCN2 was a gift from Nancy Keller, Department of Plant Pathology, University of Wisconsin-Madison. Plasmid pBluSK− was purchased from Stratagene.
Plasmid pDL3 consists of a 1.8-kb ClaI–EcoRI fragment from pDC1 containing the wild-type A. nidulansargB, first cloned into an EcoRI–NarI digest of pAST30 to create pDL1, followed by the insertion of a 380-bp PstI–SapI fragment from the polylinker region of pBluSK− into a SapI–PstI digest of pDL1 to create pDL3. Plasmid pDL6 was constructed from pDL3 by excision of the alcA promoter by BamHI, followed by the insertion of a 1.5-kb BamHI–BclI fragment of pAN52-1, containing the 3′ portion of the A. nidulansgpdA promoter. Plasmids pDL3c and pDL6c were then made via PCR mutagenesis of argB in pDL3 and pDL6, respectively, as detailed above (argB_1626).
Plasmid pDL5 contains the 3.8-kb trpC amplified from the A. nidulans genomic template, with primers 6 and 7 inserted into the 5-kb NotI fragment of pCN2; pDL7 was made by excision of the alcA promoter [alcA(p)]from pDL5 with BamHI and AvrII, and ligation of the 1.5-kb NheI–BamHI fragment of pAN52-1 containing the gpdA promoter [gpdA(p)]. Plasmids pDL5c and pDL7c were made by PCR mutagenesis of PDL5 and DL7 (trpC_2716).
Aspergillus integration vectors for lacZ expression contain the 3-kb E. colilacZ, with a 3′ terminal fusion to the 800-bp terminating region of the Aspergillus trpC [trpc(t)] from plasmid pDE3. The NcoI site in the trpC terminator was removed by PCR mutagenesis using primer 8 and its complement, and a 3.8-kb PCR product (primers 9 and 10) containing the lacZ::trpc(t) cassette plus a 5′ NcoI site was cloned into pCR4-TOPO, forming pTOPO-LZTC. A 3.8-kb NcoI–NotI fragment of pTOPO-LZTC was inserted into NotI–NcoI digests of pDL3, pDL3c, pDL6, and pDL6c, to create pDL11, pDL11c pDL12, and pDL12c, respectively. A 3.8-kb NcoI–HindIII fragment of pTOPO-LZTC was inserted into HindIII–NcoI digests of pDL5, pDL5c, pDL7, and pDL7c, to construct pDL13, pDL13c, pDL14, and pDL14c, respectively.
Plasmids pDL15 and pDL16 contain the 3.7-kb niaD flanked with XmaI and AvrII sites, amplified from A. nidulans genomic template with primers 11 and 12 inserted into SpeI–XmaI fragments of pDL13 and pDL14 to create pDL15 and pDL16, respectively; pDL15c and pDL16c were made by PCR mutagenesis (niaD_345). Constructs made entirely by cloning existing fragments were confirmed by restriction analysis only; those including PCR products were also sequenced.
Molecular genetic analysis of Aspergillus transformants
The PCR screening of transformants for integration of lacZ utilized primers 9 and 13, amplifying the 5′ 1.2-kb fragment of lacZ, along with primers 14 and 15, amplifying an 850-bp fragment starting in the 5′ untranslated region of argB as a positive control for genomic template.
Southern blotting and hybridization was performed as described by Sambrook, with minor modifications as suggested in the membrane manufacturer’s literature (NytranN+; Schleicer and Schuell). DNA probes were prepared by plasmid digestion and gel purification, and labeled with [α-32P] 2′-deoxycytidine 5′-triphosphate (New England Nuclear Life Sciences) using a (Roche) random-primed labeling kit. For the Southern blots, a [32P]-labeled 3-kb NcoI–EcoRI fragment of pDL11, containing the 5′ terminal region of the lacZ sequence, was used as a probe for the integration of lacZ, along with a [32P]-labeled 1-kb EcoRIargB fragment of pDC1, not contained in the argB vectors, as a control. Aspergillus genomic DNA (gDNA) was digested with StuI (with a single site in argB and unique on pDL11), BsaBI (with a single site in lacZ and also unique on pDL11), or both, and similar digests of pDL11 were added as control lanes. For the dot-blots, transformant gDNA was spotted directly onto a nylon membrane alongside a series of lacZ-bearing plasmid or host gDNA standards, and hybridized with [32P]-labeled lacZ or native single-copy gene probes; copy number was determined by interpolating the spot intensity of the samples from a fitted curve of the standard series. Hybridized blots were visualized by exposure on a Molecular Dynamics Phosphorimager screen, followed by imaging on the Amersham Typhoon 8600 scanner and analysis with their proprietary software, ImageQuant.
Real-time QPCR with the BioRad iCycler was performed essentially as per the manufacturer’s instructions (Boeckman et al. 2001). To determine lacZ copy number, primers 16 and 17 were used to amplify a 136-bp lacZ fragment, along with either primers 18 and 19 for a 95-bp argB fragment, or 20 and 21 for a 77-bp trpC fragment (each a native gene present at one copy per genome). For each trial, the following master mix was prepared and aliquotted at 50 μL into a 96-well plate: 37.75 μL H2O, 500 μL 10× PCR buffer (−Mg), 300 μL 50 mM MgCL2, 100 μL 10 mM mixed deoxyribonucleotide triphosphates (Invitrogen), 50 μL 1 mM fluorescein sodium (Sigma), 50 μL Synergy Brands (SYBR) Green I (Molecular Probes “10,000×Gel Stain” 1:1,000 in DMSO), 25 μL Platinum Taq polymerase (Invitrogen), 100 μL of each 10 mM forward and reverse primer. To each well, 1 μL genomic DNA template was added. PCR reaction conditions were 1 min at 94 °C, followed by 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 45 s; fluorescent data were acquired at the 60 °C step. Amplification cycling was followed by a “melt curve” (dissociation and renaturation); starting at 95 °C, the temperature was reduced by 1 °C at 10-s intervals to 35 °C, with fluorescent data taken at each decrement.
Under optimized reaction conditions yielding the same amplification efficiency for each, the single-copy native gene was amplified from a standard dilution series of A. nidulans genomic template, and lacZ from an identical genomic series spiked with known concentrations of a lacZ-bearing plasmid. Both genes were simultaneously amplified from genomic template samples from the transformants, using a sample of a known single-copy integrant as a control; samples and controls were each in triplicate. Product formation was monitored by the fluorescence of dsDNA-bound SYBR Green in each well vs time (cycle number). Using the bundled iCycler iQ software, a threshold fluorescence detection level was set, and standard curves were made for the amplification of each of the control dilution series (threshold cycle vs log starting template quantity). By comparing the points where the product curves of the unknowns crossed the threshold to those of the standard curves, the amount of starting template DNA, and hence lacZ copy number per genomic equivalent, was determined. A plot of the first derivative of the melt curve gave a single peak at the melting temperature of the target amplicon, indicating the absence of spurious products.
DNA sequencing was done at the University of California, Berkeley DNA Sequencing Facility and by Elim Biopharmaceutical, Hayward, CA, USA. Oligonucleotide synthesis services were provided by Operon Biotechnologies, Huntsville, AL, USA.
Enzyme activity assays
Assays were done on mycelial pellets grown for 72 h from minimal broth inoculated at ∼5×103 conidia per milliliter, with 50 mM methyl ethyl ketone added at 48 h for the alcA(p)::lacZ clones. β-Galactosidase activity was quantified using a modified version of the standard o-nitrophenyl-β-galactopyranoside method (Miller 1972) developed for Neurospora (Lee and Ebbole 1996). For each sample, 75–100 mg of (wet) tissue was homogenized in a 2-mL, screw-cap tube filled with 1 g of glass beads (0.5 mm in diameter) and 1 mL of ice-cold Z buffer (Sambrook and Russell 2001), plus 10 μL of 100 mM phenylmethanesulfonyl fluoride on a mini bead beater (Biospec Products, Bartlesville, OK, USA), twice, for 30 s each, with cooling on ice between bead-beatings; total protein content of the lysate was determined by the Bradford method (Bradford 1976). β-Galactosidase activity is reported as “Miller units” per milligram of total protein. Statistical analysis of expression data was performed with Analyse-it for Microsoft Excel, Leeds, UK (see http://www.analyse-it.com).
Results
Transformation and selection
Vectors bearing a lacZ expression cassette and either the wild-type or engineered mutant marker gene were used to transform the three auxotrophic hosts and the transformants examined for phenotype and genotype. Type I (homologous), Type II (ectopic), and Type III (gene conversion) (Hynes 1996) events were expected; an increased percentage of Type I integrants at a reduced overall transformation frequency was expected for the plasmids bearing complementary marker mutations. The observed transformation frequency for the mutated markers was <1 transformant per microgram of DNA per 105 protoplasts, a 100-fold reduction from the highest frequencies observed with the vectors bearing the corresponding wild-type markers. Transformants were subcultured three times on plates of minimal medium, and surviving clones, except those with grossly abnormal growth or morphology, were selected for genetic analysis and enzyme activity assays.
Molecular genetic analysis of transformants
Transformants were screened by PCR for integration of lacZ, and several positive clones were selected for Southern analysis; single- and multicopy integrants were identified (data not shown). To quantify transgene dosage for a large number of transformants, a rapid method utilizing QPCR was developed. A single-copy native gene (trpC or argB) and lacZ were simultaneously amplified from genomic template DNA from the host strains and transformants; by comparing the product curves to a series of standards, the amount of starting template DNA, and hence gene copy number, was determined (see “Materials and methods” for details).
The same samples were also tested by dot-blot: transformant gDNA was spotted directly onto a nylon membrane alongside a series of lacZ-bearing plasmid or host gDNA standards, and hybridized with [32P]-labeled lacZ or native single-copy gene probes; copy number was determined by interpolating the spot intensity of the samples from a fitted curve of the standard series (data not shown). The copy numbers determined using the two methods are in good agreement; the QPCR numbers are slightly lower than the blot numbers on average (Fig. 2). Each of these methods yielded a physically unlikely fractional copy number. Although first-generation transformants would be expected to harbor some untransformed nuclei (May 1992), subcultured clones should not; so, selected putative single-copy integrants were confirmed by Southern blot (data not shown) and further confirmed by PCR amplification of the regions of plasmid–genomic fusion (data not shown). QPCR results were normalized to a confirmed single-copy lacZ transformant.

Comparison of copy number by QPCR and dot-blot methods. The number of copies of lacZ integrated into the genome as determined by QPCR (y-axis) as a function of that as determined by dot blot is shown. Symbols indicate the mean copy number and error bars indicate the range of copy numbers for each clone
Of 205 transformants studied, 106, or 52%, were lacZ-positive by PCR; transformants without lacZ were presumed to be the result of Type III events (Table 2). Although a few transformants had nearly 40 copies, the large majority of transformants had five or less. The overall mean copy number was five copies of lacZ per genome, with a median of two; this varied only slightly among the transformants with different markers. Among the lacZ-positive clones, 46 or 43% had a calculated copy number of 1.0±0.50. In this group, of the 25 for which unambiguous Southern hybridizations were obtained, 20 were confirmed to be single-copy integrants, of which ten were integrated at the homologous (marker) locus.
β-Galactosidase expression
The variation of β-galactosidase expression levels in the transformants ranged from just above the native background activity of 0–100 U/mg protein to over three orders of magnitude greater. Plots of β-galactosidase activity vs lacZ copy number for the three markers show that there is no consistent gain in expression as copy number is increased (Fig. 3). Maximal expression for both alcA(p) and gpdA(p) strains occurred in several of the single-copy clones, and many of the higher-copy clones displayed lower levels. Those transformants with the argB marker had the highest average expression, roughly three times that of either the trpC or niaD clones (Fig. 4). Mean and median values for each group of clones with the same selection marker–promoter combination are similar to those of the corresponding subsets of multicopy and single-copy clones. In the smaller subset of single-copy clones confirmed by Southern blot, expression levels were in the range of 5×103–1×104 U/mg (Fig. 5), with the exception of the argB–gpdA clones at over 1×105 and several lower-expressing trpC–gpdA clones.

β-Galactosidase activity vs lacZ copy number. For each subset of transformants with the same promoter and lacZ copy number (shown on the x-axis, with the number of clones in each group in parentheses below), the range of β-galactosidase activities for those clones are shown on the y-axis, with boxes and whiskers indicating their relative distribution; clones that are the single transformants with a particular copy number are shown as a point for the mean activity. a The alcA clones; b the gpdA clones

Variation of β-galactosidase expression in transformant populations. For each subset of transformants with the same promoter and selection marker copy number (shown on the x-axis, with the number of clones in each group in parentheses) the range of β-galactosidase activities for those clones are shown on the y-axis. a Multiple-copy clones; b putative single-copy clones

β-Galactosidase expression in confirmed single-copy clones. Each bar on the x-axis represents β-galactosidase activity for a single clone, for which the single-copy lacZ integration was confirmed by Southern blot; the shaded bars indicate that the plasmid is integrated at the homologous marker locus
The expression data were tested for significance of differing means among marker–promoter groups by nonparametric analysis of variance using the Kruskal–Wallace H-test for all of the marker–promoter groups, as well as for the two subsets of the three different markers for each promoter, and were also tested pairwise by the Mann–Whitney U-test. The H- and U-statistics and corresponding P values are reported in Table 3.
Discussion
We examined expression differences among Aspergillus transformation vectors having the same expression cassette but different selection markers, promoters, copy numbers, and integration loci. The complementary mutant markers were designed to enhance the yield of single-copy homologous integrations; as expected, transformation efficiencies for the “targeting” vectors were markedly reduced as compared to the nontargeting vectors. On average, targeting vectors yielded <1 transformant colony per microgram of DNA per 1×105 protoplasts, approximately 100-fold lower than the corresponding wild-type markers.
From Table 2, the percentage of lacZ-positive integrations was 49% for the argB vectors, 44% for the trpC vectors, and 72% for the niaD vectors. The fractions of apparent Type III replacements for each of the three markers, calculated as [1−(lacZ-positive divided by total transformants)], were 51, 56, and 28% for the argB, trpC, and niaD vectors, respectively. The frequencies of single-copy integration were 45, 35, and 52% for the argB, trpC, and niaD vectors, respectively. Together, these results would suggest that integration targeting is more efficient at the niaD locus than at argB or trpC; Bird and Bradshaw determined that targeting was roughly five times more frequent at niaD than at the amdS locus (Bird and Bradshaw 1997). The trpC vectors yielded a higher percentage of single-copy integrants, implying that targeting is likelier here than at the other loci; however, this may, instead, be due to the use of the trpC terminator (on all the vectors). The niaD vectors yielded the highest fraction of confirmed single-copy integrants: five of seven. Based on the fraction of confirmed single-copy homologous integrations, overall, approximately 35% of the lacZ-positive transformants with the wild-type markers would be expected to be single-copy homologous integrants, whereas with the targeting vectors, five of the six transformants were single-copy and homologous.
Although the complementary mutation method is simpler than other targeting vector schemes utilizing Agrobacterium (Gouka et al. 1999; Michielse et al. 2005) or the λ-Red recombinase system (Chaveroche et al. 2000), the fractional improvement in single-copy homologous integrants is offset by the large loss in the overall transformation efficiency. The easiest route to single-copy homologous integrants may be transformation with wild-type markers followed by selection via Southern blot, PCR, or other means.
The β-galactosidase activity analyses showed that increasing transgene dosage does not reliably increase overall expression levels in A. nidulans. In the case of the strong constitutive gpdA promoter, intuitively, we might expect expression to increase linearly with copy number; for the alcA promoter it might be similar, but with a leveling-off at higher numbers due to the aforementioned titration effect. The differences in expression seen with clones bearing one or two copies, and the fact that maximal or near-maximal expression for the population occurs for several single-copy clones (seen in both promoter groups in Fig. 3), may be the result of variance in the physiological state of the transformants due to unknown, pleiotropic effects of random integration.
The highly variable expression in the multicopy integrants raises the question of whether a gene silencing effect is implicated. Several types of silencing related to duplicate gene copies have been observed in the filamentous fungi (Cogoni 2001). Evidence for transcriptional silencing by “RIP” methylation-transversion, first elucidated in N. crassa, was recently found in A. nidulans (Clutterbuck 2004). Posttranscriptional silencing, such as quelling in Neurospora, an RNA-interference mechanism related to cosuppression in plants with commonalities to systems in many higher organisms (Maine 2000), may in fact have been first described much earlier in A. flavus (Schmidt 2004). It may prove fruitful to study multicopy expression in A. nidulans with an RNA-silencing-deficient background, such as the recently discovered rsdA − genotype (Hammond and Keller 2005).
The populations show a correlation between marker association and expression, with the argB vectors the highest and niaD the lowest (Fig. 4). If this were related to the regulation of the native genes, especially when integrated at their native locus, then we might expect to see higher expression associated with natively up-regulated marker genes. Under the experimental conditions, argB and trpC should both be expressed at low constitutive levels (Yelton et al. 1983; Upshall et al. 1986), and niaD should be fully induced and derepressed (Johnstone et al. 1990), presenting no obvious explanation for the differences in expression levels.
The statistical results in Table 3, column A, show that there exist significant differences in mean expression among the single-copy groups, and the pairwise testing indicates that the argB and niaD groups are the most significantly different. For a given marker, mean expression under the two promoters differs insignificantly (Table 3, column C), with slightly more variable expression from gpdA(p) than from alcA(p). As the enzyme activity assay measures the accumulation of a fairly stable protein over time, this difference is likely due to constitutive expression from the gpdA promoter (soon after germination), whereas alcA is repressed until later, when the glucose is consumed and inducer is added (Felenbok 1991).
Expression variability among the putative single-copy clones follows the same trends as for the population as a whole. Expression of the confirmed single-copy clones shows some evidence for locus effects; single-copy homologous integrants give similar expressions from both promoters when located at niaD, whereas for gpdA(p), expression is markedly higher at the argB locus and lower at the trpC locus. In contrast, the two, confirmed, argB–gpdA single-copy integrants, one homologous and one ectopic, have similar expressions, suggesting that the locus is less important than the choice of selection marker and promoter. As there are also several cases where the same construct integrates singly at the same loci (Fig. 5; trpC–alcA and niaD–alcA), which have different activity levels; there may be other effects on expression specific to each clone, yet unrelated to the integration locus (perhaps related to mutagenesis during transformation). These transformant groups both use the inducible alcA promoter, so this may also be an artifact of induction conditions; i.e., because the experiments were performed in shaken flasks induced at the same time, the metabolic states of each clone may not have been identical, the glucose level in the medium may have been different, etc. The enyzme assays were normalized to total protein, rather than a measure of the overall metabolic state of the transformants; ideally, expression would be evaluated for a strain of interest in continuous culture, although this is impractical for a large number of samples.
The choice of nutritional marker when designing transformation vectors for A. nidulans can clearly influence the outcome in the transformants. For obtaining a high yield of homologous integrants, the niaD marker is preferred. The noted effects on expression associated with marker and promoter, along with the apparent saturation effects with increasing copy number, may find practical utility in the construction of A. nidulans strains for high-level heterologous expression; the argB–gpdA(p) combination, integrated singly at the homologous locus, would be a good choice for maximizing expression and avoiding unwanted pleiotropic effects.
References
Aramayo R, Adams TH et al (1989) A large cluster of highly expressed genes is dispensable for growth and development in Aspergillus-Nidulans. Genetics 122(1):65–71
Barratt RW, Johnson GB et al (1965) Wild-type and mutant stocks of Aspergillus Nidulans. Genetics 52(1):233–246
Bird D, Bradshaw R (1997) Gene targeting is locus dependent in the filamentous fungus Aspergillus nidulans. Mol Gen Genet 255(2):219–225
Boeckman F, Brisson M et al (2001) Real-time PCR: general considerations. Bulletin 2593. Bio-Rad Laboratories, Hercules
Bradford MM (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem 72(1–2):248–254
Chaveroche M-K, Ghigo J-M et al (2000) A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28(22):e97
Chiou CH, Miller M et al (2002) Chromosomal location plays a role in regulation of aflatoxin gene expression in Aspergillus parasiticus. Appl Environ Microbiol 68(1):306–315
Chow TYK, Käfer E (1993) A rapid method for isolation of total nucleic acids from Aspergillus nidulans. Fungal Genetics Newsletter 40:25–27
Clutterbuck (2004) MATE transposable elements in Aspergillus nidulans: evidence of repeat-induced point mutation. Fungal Genet Biol 41(3):308–316
Cogoni C (2001) Homology-dependent gene silencing mechanisms in fungi. Annu Rev Microbiol 55:381–406
Davies RW (1991) Molecular biology of a high-level recombinant protein production system in Aspergillus. In: Leong SA, Berka RM (eds) Molceular industrial mycology. Marcel Dekker, New York, pp 45–81
Davis MA, Hynes MJ (1991) Regulatory circuits in Aspergillus nidulans. In: Bennett JW (ed) More gene manipulations in fungi. Academic, New York, pp 151–189
Dawe AL, Willins DA et al (2000) Increased transformation efficiency of Aspergillus nidulans protoplasts in the presence of dithiothreitol. Anal Biochem 283(1):111–112
Ebbole DJ (1990) Vectors for construction of translational fusions to beta-galactosidase. Fungal Genetics Newsletter 37:15–16
Felenbok B (1991) The ethanol utilization regulon of Aspergillus-Nidulans — the Alca–Alcr system as a tool for the expression of recombinant proteins. J Biotechnol 17(1):11–17
Gouka RJ, Gerk C et al (1999) Transformation of Aspergillus awamori by Agrobacterium tumefaciens-mediated homologous recombination. Nat Biotechnol 17(6):598–601
Hamer JE, Timberlake WE (1987) Functional-organization of the Aspergillus-Nidulans Trpc promoter. Mol Cell Biol 7(7):2352–2359
Hammond TM, Keller NP (2005) RNA silencing in Aspergillus nidulans independent of RNA-dependent RNA polymerases. Genetics 169(2):607–617
Hynes MJ (1996) Genetic transformation of filamentous fungi. J Genet 75(3):297–311
Ichioka D, Itoh T et al (2001) An Aspergillus nidulans uvsC null mutant is deficient in homologous DNA integration. Mol Gen Genet 264(5):709–715
Johnstone IL (1985) Transformation of Aspergillus-Nidulans. Microbiol Sci 2(10):307–311
Johnstone IL, McCabe PC et al (1990) Isolation and characterization of the Crna–Niia–Niad Gene-Cluster for nitrate assimilation in Aspergillus-Nidulans. Gene 90(2):181–192
Kelly JM, Hynes MJ (1987) Multiple copies of the amdS gene of Aspergillus nidulans cause titration of trans-acting regulatory proteins. Curr Genet 12(1):21–31
Lee KW, Ebbole DJ (1996) A reliable microplate assay for determination of B-galactosidase activity in Neurospora crassa strains bearing lacZ fusions. Fungal Genetics Newsletter 43:35–36
Maine EM (2000) A conserved mechanism for post-transcriptional gene silencing? Genome Biol 1(3): reviews 1018.1–reviews 1018.4
May G (1992) Fungal technology. In: Kinghorn JR, Turner G (eds) Applied molecular genetics of filamentous fungi. Chapman & Hall, London
Michielse CB, Arentshorst M et al (2005) Agrobacterium-mediated transformation leads to improved gene replacement efficiency in Aspergillus awamori. Fungal Genet Biol 42(1):9–19
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Miller BL, Miller KY et al (1987) Position-dependent and position-independent mechanisms regulate cell-specific expression of the spoc1 gene-cluster of Aspergillus-Nidulans. Mol Cell Biol 7(1):427–434
Punt PJ, Dingemanse MA et al (1990) Functional elements in the promoter region of the Aspergillus-Nidulans Gpda gene encoding Glyceraldehyde-3-Phosphate Dehydrogenase. Gene 93(1):101–109
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schmidt FR (2004) RNA interference detected 20 years ago? Nat Biotechnol 22(3):267–268
Som T, Kolaparthi VSR (1994) Developmental decisions in Aspergillus-Nidulans are modulated by ras activity. Mol Cell Biol 14(8):5333–5348
Upshall A, Gilbert T et al (1986) Molecular analysis of the argb gene of Aspergillus-Nidulans. Mol Gen Genet 204(2):349–354
van den Hondel C, Punt P et al (1991) Heterologous gene expression in filamentous fungi. In: Bennett JW (ed) More gene manipulations in fungi. Academic, New York, pp 396–428
Verdoes JC, Punt PJ et al (1995) Molecular-genetic strain improvement for the overproduction of fungal proteins by filamentous fungi. Appl Microbiol Biotechnol 43(2):195–205
Yelton MM, Hamer JE et al (1983) Developmental regulation of the Aspergillus-Nidulans-Trpc gene. Proc Natl Acad Sci USA 80(24):7576–7580
Acknowledgement
This research was funded by a grant from Merck.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lubertozzi, D., Keasling, J.D. Marker and promoter effects on heterologous expression in Aspergillus nidulans . Appl Microbiol Biotechnol 72, 1014–1023 (2006). https://doi.org/10.1007/s00253-006-0368-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0368-8