
Abstract
Herein, flexible and porous Co3O4-carbon nanofibers (Co3O4-CNFs) were fabricated by electrospinning technique combining with the followed carbonization process. The effects of material composition and calcination temperature on morphology, pore structure, and electrochemical properties of the Co3O4-CNFs were systematically investigated. Results indicated that the obtained Co3O4-CNFs exhibited high porosity, high mechanical strength, and superior electrical conductivity. Electrochemical characterization results showed that the optimized Co3O4-CNFs as binder-free electrodes exhibited a specific capacitance of 369 F g−1 at the current density of 0.1 A g−1. Even at a high current density of 2 A g−1, the specific capacitance still remained at 181 F g−1, with the capacitance retention rate of 49%. Intriguingly, the prepared Co3O4-CNF film could recover to its original state easily after folding for three times, indicating good mechanical flexibility for free-standing electrodes. Coupled with the excellent mechanical flexibility, high specific capacitance, and simple fabrication process, the flexible and free-standing Co3O4-CNFs with hierarchical porous structure could be promising electrode materials for energy storage applications.
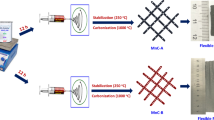
Flexible and porous Co3O4-carbon nanofibers were prepared by electrospinning and carbonization,which can be used as free-standing electrodes for supercapacitors.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Much attention has been focused to supercapacitors (SCs) because of their fascinating power density, long life cycles, fast charge and discharge rates, and high efficiency [1, 2]. However, the low energy-power ratio of SCs has been restricted to their applications. In recent years, numerous materials, such as carbonaceous materials [3, 4], metal oxides or hydroxides [5, 6], and conducting polymers [7, 8], have been widely used in SCs. Among the electroactive materials, Co3O4 has been extensively reported because of its high theoretical specific capacitance up to 3560 F g−1, high redox performance, controllable size and shape, relatively cost-effective, and widespread applications [9,10,11,12]. Recently, much progress has been made in the development of energy storage device [13,14,15,16,17]. For example, Pal et al. have demonstrated a single-step solvothermal method to synthesize self-supported ultra-small Co3O4 nanocubes over Ni foam, which exhibited pseudocapacitive performance of 1913 F g−1 at current density of 8 A g−1 [18]. Jiang et al. reported a technique combining hydrothermal and calcination for the synthesis of two-dimensional Co3O4 thin sheets, which showed a specific capacitance of 1500 F g−1 at 1 A g−1 [19]. Besides, the Co3O4 has also widely been used as wave-absorbing material, as they have good impedance matching [20,21,22,23,24]. For example, the hierarchically porous Co/C nanocomposites were fabricated by freezing dry and carbothermic reduction process with excellent absorption performance of its minimum reflection loss could reach to 34.2 dB, which was profit from the optimization of hierarchical porous microstructures and impedance matching [25]. However, the low electrical conductivity and inferior rate capacity of Co3O4 have greatly limited their practical application [26,27,28]. To address these drawbacks, the carbon-based Co3O4 composites for SCs electrodes have been investigated in various forms [29,30,31].
Carbon matrixes, such as carbon nanotube (CNT), graphene, and carbon nanofibers (CNFs), have been regarded as potential candidates for SC electrodes [32,33,34]. Recently, Zheng et al. constructed 3D hierarchical N-doped carbon-based Co3O4 nanopillar arrays as a binder-free electrode, which exhibited a maximum specific capacity of 978.9 F g−1 at 0.5 A g−1 [35]. Fan and co-workers reported the 2D thin Co3O4 nanosheets anchored on 3D porous graphene/nickel foam substrate through hydrothermal synthesis [36]. Because of the synergy of thin Co3O4 nanosheets and conductive grapheme layer, the composites displayed a high specific capacitance of 3533 F g−1 at a current of 1 A g−1. Besides, Su et al. prepared two-ply yarn SCs by electrochemical deposition of MnOx or Co3O4 materials on CNT/stainless steel (SS), the Co3O4/CNT/SS yarn electrode exhibited volumetric capacitance of 82.94 F/cm3 at 0.02 V/s [37]. Nevertheless, the complicated synthesis process and high cost of graphene and CNT may prevent their widespread application in SCs [38]. Fortunately, CNFs have been scaled up to industrial production by electrospinning technique, with a much lower cost than graphene and CNT, which have high mechanical strength, excellent chemical resistance, and superior electrical conductivity [39,40,41,42].
Traditional CNFs are mostly made from polyacrylonitrile (PAN), in which the high cost and environmental concerns limit its extensive applications. As a green and sustainable material, lignin is regarded as one of the most attractive precursors for CNFs because of its high carbon content (up to 60%) and aromatic monomers [43,44,45,46,47,48,49,50,51,52,53]. However, most of the lignin-based CNFs show lower mechanical strength as compared to PAN-based CNFs [54,55,56,57,58]. Although lignin-based CNFs may not meet the high requirement standard for construction materials, they can be promising electrode materials for energy storage applications. In addition, it is reported that the flexibility and electrochemical performances of lignin-based CNFs can be improved by rational design of the porous structure [59,60,61]. For example, Liu et al. reported the fabrication of free-standing hierarchical porous CNFs films by a metal ion-assistant acid corrosion process [62]. Results indicated that the porous structure could increase ion transportation path and facilitate the accessibility between CNFs and electrolyte. Samuel and co-workers demonstrated the preparation of flexible core–shell SnOx/CNF composites using poly (methyl methacrylate) as pore-forming agent. It was found that the SnOx/CNF-based symmetric SCs exhibited a specific capacitance of 289 F g−1 at a scan rate of 10 mV s−1 [63].
Inspired by the above-mentioned studies, we propose to synthesize lignin-derived porous Co3O4-CNFs by using electrospinning technique and the subsequent carbonization, in which the advantages of porous structure of CNFs with good mechanical performance, high theoretical specific capacitance of Co3O4, and the low cost of lignin can be integrated. The surface chemistry, crystalline structure, and morphology of the as-produced Co3O4-CNFs were characterized by FTIR, XPS, XRD, EDS, SEM, and TEM analyses. Moreover, the electrochemical behaviors of the Co3O4-CNFs electrodes were measured by cyclic voltammetry (CV), galvanostatic charging-discharging (GCD), and electrochemical impedance spectroscopy (EIS) tests. This work will provide a facile and versatile strategy to prepare porous CNF-based composite electrode materials for high-performance supercapacitors.
2 Experimental section
2.1 Materials
Cobalt nitrate hexahydrate, acetic acid, and N,N-dimethylformamide (DMF) were purchased from Beijing Chemical Works. PAN (Mw = 150 000) and terephthalic acid (TPA) were bought from Macklin. Lignin (Mw = 10,000) was purchased from Sigma-Aldrich. All chemicals used were analytical grades and utilized without further purification.
2.2 Synthesis of porous Co3O4-CNFs
A total of 0.50 g of PAN and 0.30 g of TPA were mixed and dissolved in 5 mL DMF at 60 °C forming a homogeneous colloidal solution. Meanwhile, 0.20 g lignin and 0.291 g cobalt nitrate hexahydrate were dissolved in the mixed solution of 0.5 mL acetic acid and 1 mL DMF. Then, the above two solutions were mixed together by stirring. The mixed solution was transferred into a plastic syringe (10 mL) equipped with capillary needle (0.42 mm in diameter). The solution was electrospun at a feeding rate of 0.5 mL h−1 under high voltage of 20 kV. Then, the fibers were collected with an aluminum foil sheet, which distance between the needle and collector was 18 cm. The fibers were then dried in fume hood. Then, the dried nanofibers were thermostabilized in tube furnace to 280 °C at 1 °C min−1 and kept at 280 °C for 2 h in air environment. Subsequently, they underwent a pyrolysis process in tube furnace at three different temperatures, 700, 800, and 900 °C for 2 h in N2 atmosphere with a heating rate of 3 °C min−1. Finally, the carbonization nanofibers were annealed at 300 °C for 60 min in air to obtain Co3O4 nanoparticles (NPs). The schematic illustration of the experimental process is shown in Scheme 1. The corresponding samples were labeled as Co3O4-CNFs-700, Co3O4-CNFs-800, and Co3O4-CNFs-900, respectively.

Schematic illustration of porous Co3O4-CNFs prepared by electrospinning and carbonization
2.3 Characterization
X-ray powder diffraction (XRD) detection was analyzed in the 2θ range from 10 to 80° on a Rigaku DMax-RB 91–0459 diffractometer with Cu Ka radiation at a scan rate of 4° min−1. The morphology of nanofibers was investigated by the high-resolution images, which were measured by FESEM (SU8010, Japan). Besides, the structures of CNFs were conducted by transmission electron microscopy (TEM, JEOL, JEM-1010). The X-ray photoelectron spectroscopy (XPS) date was performed on an ESCALAB 250Xi X-ray photoelectron spectroscope with a monochromatic Al Ka radiation. And the spectral elemental analysis was examined by EDS (Thermo Scientific attached with Hitachi S-4800 operated at 15 kV). The pore structure of Co3O4-CNFs was tested by N2 adsorption/desorption isotherms using Belsorp-max surface area detecting instrument. Thermogravimetric analysis (TGA) of the precursors was measured (SDT Q600) in air atmosphere with the temperature range of 30–600 ℃ at a heating rate of 10 ℃ min−1. FTIR spectra of lignin, precursor, and CNFs were collected on Thermo Nicolet (Nicolet iN10) with a resolution of 4 cm−1. Raman spectroscopy was recorded from 100 to 2000 cm−1 using a LabRAM HR Evolution by wavelength of 523 nm under ambient conditions.
2.4 Electrochemical measurement
Electrochemical properties were performed using CHI 660D electrochemistry workstation (Shanghai Chenhua Instrument Co., China) in three-electrode setup, where CNFs were served as working electrodes without any additional binders or conductive additives, Hg/HgO as reference electrode and platinum mesh as counter electrode, respectively. Electrochemical tests including CV, GCD, and EIS were performed in 3 M KOH aqueous electrolyte at room temperature.
3 Results and discussion
Figure 1a presents the electrospinning precursor before calcination, in which the obtained nanofibers are smooth and randomly ordered with a network structure and the average nanofiber diameter of 600 nm. After carbonization and oxidation process, the TPA component sublimated, generating many pores in the carbonized fibers. Furthermore, the appearance of Co3O4-CNFs became rough, and Co3O4 NPs were also found on the surface of the CNFs uniformly. It is probably because of the formation of porosity and Co3O4 NPs, the mean diameter of Co3O4-CNFs-700 shows little change after carbonization (Fig. 1b). While the calcination temperature increased to 800 ℃, the mean diameter of Co3O4-CNFs-800 reduced to 450 nm (Fig. 1c), which could be due to the further decomposition of polymer during the carbonization process. Additionally, the average diameter of Co3O4-CNFs-900 continues to reduce to 250 nm, and in the meantime, the quantity of cobalt nanoparticles decreases with the increase of particle size (Fig. 1d). Moreover, the element mappings in Fig. 2 demonstrate the existence and uniform dispersion of Co3O4 NPs. The energy-dispersive X-ray spectroscopy (EDS) analysis also proves the existence of C, N, O, and Co in the samples, which could be attributed to carbon, Co3O4, and the retention of N from PAN.

SEM images of a PAN-cobalt salt precursors, b Co3O4-CNFs-700, c Co3O4-CNFs-800, and d Co3O4-CNFs-900, respectively

The EDS element mappings of Co3O4-CNFs-700: a C; b N; c O; d Co
The transmission electron microscopy (TEM) images (Fig. 3) further reveal the detailed morphologies and structures of Co3O4-CNFs. Figure 3a, b show that the Co3O4 NPs exhibit a hollow structure with diameters ranging from 120 to 600 nm. As displayed, the hollow Co3O4 NPs are formed by Kirkendall effect, which refers to higher diffusion rate of outward Co than that of inward O [64]. In consequence, it enabled hollow Co3O4 NPs to form. When the calcining temperature was up to 900 °C, the hollow Co3O4 NPs turned into solid ones, and the size of these nanoparticles became large. It is suspected that Co/CoO is not transformed into Co3O4 crystals completely after oxidation process. In the meantime, the amorphous carbon transformed to graphitic carbon partially (Fig. 3c, d). More detailed structures of Co3O4-CNFs are demonstrated through the high-resolution TEM (HRTEM) and selected area electron diffraction (SAED). The measured d-spacings distance of carbon layers is 0.35 nm, assigning to the (002) plane of carbon (Fig. 3d). And the lattice fringe d-spacings of 0.285 and 0.243 nm related to the (220) and (311) planes of Co3O4 (Fig. 3e). The corresponding selected-area electronic diffraction (SAED) pattern shows a polycrystalline nature of the Co3O4, and the diffraction rings can be indexed to (220), (311), (400), (511), and (440) crystallographic planes of Co3O4 crystal (Fig. 3f).

TEM images of a Co3O4-CNFs-700, b Co3O4-CNFs-800, and c Co3O4-CNFs-900, respectively; d, e HRTEM images of Co3O4-CNFs-800, and f the corresponding SAED pattern
The formation of Co3O4 and graphitic carbon could be confirmed by X-ray diffraction (XRD), Raman, and X-ray photoelectron spectroscopy (XPS). Figure 4a shows the X-ray diffraction (XRD) pattern of Co3O4-CNF products. As shown, the six characteristic peaks of Co3O4-CNFs-900 at 31.6, 36.8, 45.2, 55.4, 59.5, and 65.1° can be corresponded to the lattice planes of (220), (311), (400), (422), (511), and (440), respectively, which belong to the JCPDS card no. 42–1467 of Co3O4. The broad diffraction peak located at 24.7° index to the (002) diffraction plane of carbon fibers. The diffraction peaks of Co3O4-CNFs-700 are not apparent, which may be due to the trace amount of Co3O4. From the XRD pattern of three different calcination temperatures, one can see that with the increase of temperature, the carbon and Co3O4 diffraction peak intensity increases. The XRD results reveal that Co3O4-CNFs include an amorphous carbon and Co3O4, which could greatly affect their electrochemical performance.

a XRD patterns of Co3O4-CNFs samples and b Raman spectrum.
Raman spectroscopy is the standard technique to investigate the structure and graphitic degree of carbon materials (Fig. 4b). Raman spectra of Co3O4-CNFs exhibit two visible peaks at around 1330 and 1590 cm−1, corresponding to the D (ascribed to defects and disordered carbon at the edges of the sp2 domain) and G (related to the E2g in-plane vibration of the graphite lattice of the C sp2 atom) bands, respectively [65,66,67,68]. The degree of graphitization could be calculated by the intensity ratio of D and G band (ID/IG). As we know, the higher the ratio is, the more disordered the carbon material will be. For Co3O4-CNFs-700, the ID/IG value is 1.13. With increase of calcination temperature, the ID/IG of Co3O4-CNFs-800 decreased to 1.05, indicating the increase content of graphite carbon. Furthermore, the Raman spectra of Co3O4-CNFs display characteristic peaks at 189, 466, 517, and 673 cm−1, corresponding to F12g, E2g, F22g, and A1g vibration modes of Co3O4, respectively, which is in good agreement with XRD results [69, 70].
The chemical composition of Co3O4-CNFs was further examined by X-ray photoelectron spectroscopy (XPS) (Fig. 5). Three typical peaks corresponding to the binding energies of C 1 s, N 1 s, and Co 2p are observed. As shown in Fig. 5, the XPS spectrum of C 1 s can be deconvoluted into four peaks with binding energies at 284.7, 285.4, 286.6, and 289.5 eV, corresponding to sp2 graphitic carbon, C–C, C–O, and O–C = O, respectively [71, 72]. The graphitic carbon was increased with the calcination temperature, which could enhance the electron transfer efficiency of CNFs. And the import of doped nitrogen from pyrolysis of PAN is also conducive to promote the conductivity of CNFs, resulting in the improvement of electrochemical performance [73,74,75]. The high-resolution N 1 s spectrum can be further fitted into three individual peaks, which are centered at 398.6, 400.2, and 401.4 eV, corresponding to the pyridinic N, pyrrolic N, and graphitic N, respectively [76,77,78]. At the same time, the peak at 401.4 eV of Co3O4-CNFs-900 is the strongest, and the corresponding graphitic N can improve the conductivity of CNFs, which indirectly indicates that the electrochemical performance of Co3O4-CNFs-900 sample is optimal. What is more, the high-resolution spectra of Co 2p of the three samples displayed two similar XPS peaks with two weaker satellite peaks. The spectra of Co 2p3/2 peak reveal binding energy at 795.6 and 780.0 eV, which is in good agreement with the reported data on Co3O4, further confirming the presence of Co3O4 in Co3O4-CNFs composites [79]. The principle peaks can be deconvoluted into four sub peaks, which the fitting peaks located at 781.3 and 796.9 eV are assigned to Co2+, and the 779.9 and 795.6 eV peaks are indexed to Co3+ [80]. The integrated intensity ratio of Co2+ to Co3+ of Co3O4-CNFs decreased at first and then increased with the increasing calcining temperature, which indicated that more Co2+ ions are generated accompanying with the generation of oxygen vacancies [81]. Benefiting from the advantage of high calcination temperature, high graphitic carbon and more oxygen vacancies will improve the specific capacity of the samples. Therefore, TEM, XRD, and XPS demonstrated that the homogeneously dispersed nanocrystalline Co3O4 and conductive carbon species existed in Co3O4-CNFs, which is desired for high-performance electrochemical materials.

XPS spectrum of Co3O4-CNFs, high-resolution C1s, N1s, and Co2p spectra
The nitrogen adsorption desorption measurement has been taken as represented in Fig. 6. The Co3O4-CNFs present a type-IV isotherm with a distinct hysteresis loop, showing the characteristics of a mesoporous material. Furthermore, the isotherm shows a sharp increase at high relative pressures (p/p0 > 0.9), demonstrating the macroporous characteristics [82,83,84,85]. Meanwhile, the corresponding pore size distribution curves (inset figures) also show the hierarchical pore structure of the Co3O4-CNFs. The total BET surface areas can be determined to be 86.31 m2 g−1 and 347.62 m2 g−1, respectively. The specific surface areas increase with the increasing calcination temperature, which can be ascribed to the high calcination temperature that offered more specific surface and pore structure by sublimation of the TPA component and degradation of lignin. That could be beneficial for achieving higher electrochemical performance due to the moderate BET surface areas by providing passageways for the ion to pass through with the hierarchical pore structure. TGA measurement was performed in air from 30 to 600 °C to confirm the content of Co3O4 in Co3O4-CNF sample (Fig. 7). The TGA curve of the Co3O4-CNFs-900 sample presented a sharp weight loss at 260–510 °C, which corresponds to the burning of carbon matrix [86,87,88,89]. Correspondingly, the DTG curve showed the main peaks at 395 °C and 468 °C. Therefore, the TGA-DTG analysis determined that the content percentage of Co3O4 was 54.9% in the sample.

N2 absorption and desorption isotherms and pore size distribution of the Co3O4-CNFs (inset figures)

TGA and DTG curves of the Co3O4-CNFs-900 sample in air

a The digital photographs of Co3O4-CNFs sample under 3-folded and could recover its initial state. b Digital photo images of the flexible Co3O4-CNFs sample. c The paper cutting cut by Co3O4-CNFs sample.
At the same time, the flexibility and foldability of the prepared Co3O4-CNFs were studied. As shown in Fig. 8, after three folds, Co3O4-CNFs could fully recover to their initial state, indicating their excellent flexibility and high mechanical strength. Such outstanding flexibility and bending of Co3O4-CNFs give them a superior benefit for applications in flexible devices. The results also revealed that the porous structure of as-obtained CNFs could conduce to improve the flexibility of CNFs by using TPA as a soft template, because the sublimation of TPA in carbonization process resulted in the formation of porous structure. The porous morphology of obtained CNFs could also be observed in TEM images (Fig. 3).When Co3O4-CNFs were bent, the flexibility of CNFs can be enhanced. Because the pore structure can expand stress distribution, thus reducing the stress, then its flexural performance can be optimized. At the same time, the existence of micro- and mesoporous can enhance ion transport rate and strengthen the charge accommodation, resulting in excellent electrochemical properties of the samples.

a FTIR spectra of lignin, PAN, PAN-cobalt salt precursors, and Co3O4-CNFs-700 and b possible lignin/PAN interaction.
The FTIR spectra of lignin PAN, PAN/cobalt salt precursors, and Co3O4-CNFs-700 fibers are shown in Fig.9a. In lignin, the characteristic peaks at 1607 cm−1 and 1411–1587 cm−1 correspond to C–C = C in aromatic ring, which is prevalent in lignin structure [90,91,92,93,94]. The band at 1037 cm−1 represents the absorption peak of carbonyl group [95,96,97,98]. The peak at 1456 cm−1 can be assigned to –CH2 bending vibration, and peak at 2245 cm−1 is ascribed to C≡N stretching vibration in PAN chain. As for the spectra of PAN-cobalt precursor composite fibers, the C–C = C peak in aromatic ring is still observed. Compared with lignin spectrum, the peak strength of carbonyl group at 1037 cm−1 is significantly reduced in lignin, which indirectly indicates that lignin interacts with the nitrile group in PAN. The peaks at 1594 and 1296 cm−1 can be attributed to N–O stretching vibration of PAN. Compared with PAN, the C≡N peak strength of PAN/cobalt precursor at 2245 cm−1 was also significantly reduced, indicating that with the introduction of lignin, part of C≡N bond is converted to C = N in PAN, and the N–O bond is formed with –O–H of lignin [99,100,101,102,103,104,105,106]. The possible connections between PAN and lignin are shown in Fig. 9b.
The obtained flexible Co3O4-CNFs were directly used as working electrode without adding binder or conducting agent, and the electrochemical properties were evaluated by CV, GCD, and EIS. Figure 10 shows the CV curves of different Co3O4-CNFs samples at scan rates ranging from 5 to 500 mV/s. As can be seen, the CV curves of three samples maintain a rectangular shape in the operating voltage range of − 0.2–0.8 V, indicating their ideal electrochemical performance with a fast charging-discharging process. Among them, Co3O4-CNFs-900 has the largest CV curve area, indicating its maximum capacitance. At the same time, Co3O4-CNFs have an arch curve at a scanning rate of 5 mV/s, indicating that it also has pseudocapacitive properties (Fig. 10d). The specific capacitance of Co3O4-CNFs-900 is the largest, possibly because the graphitization degree of carbon fiber increases with the increase of calcination temperature [107,108,109,110,111,112,113,114,115,116,117]. Besides, Co3O4 particles increase and their size becomes larger, which may be caused by the increase in the number of micropores. The CV curves of Co3O4-CNFs-700 and Co3O4-CNFs-800 have larger deformation than Co3O4-CNFs-900 at high scanning rates, indicating that their impedance is larger, and there is a charge transfer resistance, which mainly comes from the resistivity of samples and the ion diffusion resistance in micropores [111, 112].

CV curves of a Co3O4-CNFs-700, b Co3O4-CNFs-800, and c Co3O4-CNFs-900 at different scan rates, i.e., 5, 50, 100, 200, and 500 mV s−1; d CV curves of different Co3O4-CNFs samples at scan rates of 5 mV s−1
In addition, Fig. 11 shows the GCD curves of Co3O4-CNFs at various current densities ranging from 0.1 to 2 A g−1. It can be observed that the GCD curves exhibit a similar triangle shape in the range of − 0.2–0.8 V, indicating that the double-layer capacitance is reversible at wide range of current density. Meanwhile, the GCD curves show a small deviation, which also indicates that the Co3O4 has pseudocapacitance. At higher calcination temperature, Co3O4-CNFs-900 has higher graphitization and more micro- and mesopores. The unique 3D Co3O4-CNFs network pore structure could reduce the electron pathways between electrode and electrolyte, and the pores could serve as electrolyte reservoir. Thus, it shows the longest discharge time, indicating that they have the highest specific capacitance, which is in good agreement with the CV results. Figure 12a compares the capacitance values of different samples at different current densities. When the current density is 0.1 A g−1, the specific capacitance of Co3O4-CNFs-900, Co3O4-CNFs-800, and Co3O4-CNFs-700 is 369, 125, and 119 F g−1, respectively. When the current density is 2 A g−1, the specific capacitance of Co3O4-CNFs-900 is 181 F g−1, and its capacitance retention rate is 49%, exceeding most other flexible or self-standing similar carbon electrodes (Table 1). Besides, after the cycling measurements, the hierarchical porous CNFs network, as well as the morphology of Co3O4, was almost maintained, indicating the excellent structural stability of the electrode during the electrochemical process (Fig. 12a). Its good rate performance is mainly due to the fact that the porous CNF network structure can shorten the electronic pathway between electrode and electrolyte, and the doping of N and Co3O4 can further improve its electrical conductivity. However, when the current density increases, the specific capacitance of Co3O4–CNFs also tends to decrease gradually, which is due to the space limitation of CNF itself, so that only part of ions can penetrate the micropores.

GCD curves of a Co3O4-CNFs-700, b Co3O4-CNFs-800, and c Co3O4-CNFs-900 under different current densities; d GCD curves of different Co3O4-CNFs samples at the current density of 1 A g−1

a Specific capacity diagram at different current densities (the inset is the SEM image after cycling), and b alternating current impedance graph of Co3O4-CNF samples.
Figure 12b shows the Nyquist plot of Co3O4-CNFs in the frequency range of 100 kHz to 0.01 Hz. Obviously, the Nyquist diagram of Co3O4-CNFs-900 consists of semi-circular arc in the high-frequency region and double-layer capacitance response diagonal in the low-frequency region. The equivalent series resistance (Rs) of Co3O4-CNFs samples is 3.9, 2.53, and 2.63 Ω, respectively, indicating that the three samples have good conductivity. The interfacial charge transfer resistance (Rct) of Co3O4-CNFs-900 can be obtained from the diameter of solid axis semicircle in the high-frequency region, which is 6.93 Ω, while the Rct of Co3O4-CNFs-800 and Co3O4-CNFs-700 is higher. The results further indicate that the improvement of graphitization degree, the increase of Co3O4 particles, and the hierarchical pore numbers can improve the electronic conductivity of CNFs. In addition, the Warburg diffusion line of Co3O4-CNFs-900 is smaller, while the Warburg resistance region of Co3O4-CNFs-800 and Co3O4-CNFs-700 is more obvious. Warburg resistance region is short, indicating a high ion adsorption efficiency, and electrolyte ions can effectively diffuse at the electrode interface [122,123,124,125,126,127,128]. At lower frequency, Co3O4-CNFs-900 also shows a higher slope, demonstrating good double-layer capacitance behavior. It can also be concluded that Co3O4-CNFs-900 has high specific capacitance performance from EIS analysis.
4 Conclusions
In summary, we have successfully fabricated the flexible, free-standing, and porous Co3O4-CNFs by electrospinning technique and the followed carbonization process. TPA was used as a soft template during the synthesis process to form hierarchical pore structure, which significantly improved the porosity and flexibility of the as-prepared Co3O4-CNFs. The calcination temperatures played an important role in the elctrochemical properties of the obtained Co3O4-CNFs. For example, the Co3O4-CNFs-900 showed the highest specific capacitance of 369 F g−1 at the current density of 0.1 A g−1, which is about three times higher than that of the Co3O4-CNFs-700 at the same testing condition. Also, the Co3O4-CNFs-900 electrode exhibited good rate capability, i.e., the specific capacitance retained to 181 F g−1 at a high current density of 2 A g−1. The superior electrochemical performance was mainly attributed to the high electrical conductivity, the extra pseudocapacitance contributed by Co3O4 and N heteroatom, and the hierarchical pore structure (providing fast transport channels for ions). With the advantages of excellent mechanical flexibility, high conductivity, and high specific capacitance, the free-standing Co3O4-CNFs could be a promising candidate for the development of high-performance flexible energy storage devices.
References
Wang G, Zhang L, Zhang J (2012) A review of electrode materials for electrochemical supercapacitors. Chem Soc Rev 41:797–828. https://doi.org/10.1039/C1CS15060J
Ma C, Ma MG, Si C, Ji XX, Wan P (2021) Flexible Mxene-based composites for wearable devices. Adv Funct Mater 31:2009524. https://doi.org/10.1002/adfm.202009524
Xu T, Du H, Liu H, Liu W, Zhang X, Si C, Liu P, Zhang K (2021) Advanced nanocellulose-based composites for flexible functional energy storage devices. Adv Mater 33:202101368. https://doi.org/10.1002/adma.202101368
Liu H, Du H, Zheng T, Liu K, Ji X, Xu T, Zhang X, Si C (2021) Cellulose based composite foams and aerogels for advanced energy storage devices. Chem Eng J 426:130817. https://doi.org/10.1016/j.cej.2021.130817
Zhu Q, Huang Y, Li Y, Zhou M, Xu S, Liu X, Liu C, Yuan B, Guo Z (2021) Aluminum dihydric tripolyphosphate/polypyrrole-functionalized graphene oxide waterborne epoxy composite coatings for impermeability and corrosion protection performance of metals. Adv Compos Hybrid Mater. https://doi.org/10.1007/s42114-021-00265-6
Zygmuntowicz J, Łoś J, Kurowski B, Piotrkiewicz P, Kaszuwara W (2021) Investigation of microstructure and selected properties of Al2O3-Cu and Al2O3-Cu-Mo composites. Adv Compos Hybrid Mater 4:212–222. https://doi.org/10.1007/s42114-020-00188-8
Wei H, Li A, Kong D, Li Z, Cui D, Li T, Dong B, Guo Z (2021) Polypyrrole/reduced graphene aerogel film for wearable piezoresisitic sensors with high sensing performances. Adv Compos Hybrid Mater 4:86–95. https://doi.org/10.1007/s42114-020-00201-0
Li Z, Ma G, Ge R, Qin F, Dong X, Meng W, Liu T, Tong J, Jiang F, Zhou Y, Li K, Min X, Huo K, Zhou Y (2016) Free-standing conducting polymer films for high-performance energy devices. Angew Chem Int Ed Engl 55:979–982. https://doi.org/10.1002/anie.201509033
Huang J, Xu Y, Xiao Y, Zhu H, Wei J, Chen Y (2017) Mussel-inspired, biomimetics-assisted self-assembly of Co3O4 on carbon fibers for flexible supercapacitors. ChemElectroChem 4:2269–2277. https://doi.org/10.1002/celc.201700369
Zhang Y, Li Xi, Zhu T, Ma S, Li H, Sun G (2018) Facile fabrication hierarchical pore structure Li1.2Mn0.54Ni0.13Co0.13-xSrxO2 nanofiber for high-performance cathode materials in Li-ion battery. ES Mater Manuf 3:38–46. https://doi.org/10.30919/esmm5f201
Hou C, Wang B, Murugadoss V, Vupputuri S, Chao Y, Guo Z, Wang C, Du W (2020) Recent advances in Co3O4 as anode materials for high-performance Lithium-ion batteries. Eng Sci 11:19–30. https://doi.org/10.30919/es8d1128
Patil S, Bhat T, Teli A, Beknalkar S, Dhavale S, Faras M, Karanjkar M, Patil P (2020) Hybrid solid state supercapacitors (HSSC’s) for high energy & power density: An overview. Eng Sci 12:38–51. https://doi.org/10.30919/es8d1140
Tian Y, Yang X, Nautiyal A, Zheng Y, Guo Q, Luo J, Zhang X (2019) One-step microwave synthesis of MoS2/MoO3@graphite nanocomposite as an excellent electrode material for supercapacitors. Adv Compos Hybrid Mater 2:151–161. https://doi.org/10.1007/s42114-019-0075-4
Hou C, Fan G, Xie X, Zhang X, Sun X, Zhang Y, Wang B, Du W, Fan R (2021) TiN/Al2O3 binary ceramics for negative permittivity metacomposites at kHz frequencies. J Alloy Compd 855:157499. https://doi.org/10.1016/j.jallcom.2020.157499
Cai J, Xu W, Liu Y, Zhu Z, Liu G, Ding W, Wang G, Wang H, Luo Y (2019) Robust construction of flexible bacterial cellulose@Ni(OH)2 paper: toward high capacitance and sensitive H2O2 detection. Eng Sci 5:21–29. https://doi.org/10.30919/es8d669
Dong H, Li Y, Chai H, Cao Y, Chen X (2019) Hydrothermal synthesis of CuCo2S4 nano-structure and N-doped graphene for high-performance aqueous asymmetric supercapacitors. ES Energy Environ 4:19–26. https://doi.org/10.30919/esee8c221
Liu K, Du H, Zheng T, Liu W, Zhang M, Liu H, Zhang X, Si C (2021) Lignin-containing cellulose nanomaterials: preparation and applications. Green Chem. https://doi.org/10.1039/D1GC02841C
Pal M, Rakshit R, Singh AK, Mandal K (2016) Ultra high supercapacitance of ultra small Co3O4 nanocubes. Energy 103:481–486. https://doi.org/10.1016/j.energy.2016.02.139
Jiang Y, Chen L, Zhang H, Zhang Q, Chen W, Zhu J, Song D (2016) Two-dimensional Co3O4 thin sheets assembled by 3D interconnected nanoflake array framework structures with enhanced supercapacitor performance derived from coordination complexes. Chem Eng J 292:1–12. https://doi.org/10.1016/j.cej.2016.02.009
Li G, Ji Y, Zuo D, Xu J, Zhang H (2019) Carbon electrodes with double conductive networks for high-performance electrical double-layer capacitors. Adv Compos Hybrid Mater 2:456–461. https://doi.org/10.1007/s42114-019-00109-4
Du H, Parit M, Liu K, Zhang M, Jiang Z, Huang TS, Zhang X, Si C (2021) Multifunctional cellulose nanopaper with superior water-resistant, conductive, and antibacterial properties functionalized with chitosan and polypyrrole. ACS Appl Mater Interface 13(27):32115–32125. https://doi.org/10.1021/acsami.1c06647
Lu JS, Han X, Dai L, Li CY, Wang JF, Zhong YD, Yu FX, Si CL (2020) Conductive cellulose nanofibrils-reinforced hydrogels with synergetic strength, toughness, self-adhesion, flexibility and adjustable strain responsiveness. Carbohyd Polym 250:117010. https://doi.org/10.1016/j.carbpol.2020.117010
Du HS, Parit M, LiuK ZMM, Jiang ZH, Huang TS, Zhang XY, Si CL (2021) Engineering cellulose nanopaper with water resistant, antibacterial, and improved barrier properties by impregnation of chitosan and the followed halogenation. Carbohyd Polym 270:118372. https://doi.org/10.1016/j.carbpol.2021.118372
Lu Y, Yu G, Wei X, Zhan C, Jeon J, Wang X, Jeffryes C, Guo Z, Wei S, Wujcik E (2019) Fabric/multi-walled carbon nanotube sensor for portable on-site copper detection in water. Adv Compos Hybrid Mater 2:711–719. https://doi.org/10.1007/s42114-019-00122-7
Xie P, Liu Y, Feng M, Niu M, Liu C, Wu N, Sui K, Patil R, Pan D, Guo Z, Fan R (2021) Hierarchically porous Co/C nanocomposites for ultralight high-performance microwave absorption. Adv Compos Hybrid Mater 4:173–185. https://doi.org/10.1007/s42114-020-00202-z
Pang H, Li X, Zhao Q, Xue H, Lai W, Hu Z, Huang W (2017) One-pot synthesis of heterogeneous Co3O4-nanocube/Co(OH)2-nanosheet hybrids for high-performance flexible asymmetric all-solid-state supercapacitors. Nano Energy 35:138–145. https://doi.org/10.1016/j.nanoen.2017.02.044
Wang J, Zhang X, Wei Q, Lv H, Tian Y, Tong Z, Liu X, Hao J, Qu H, Zhao J, Li Y, Mai L (2016) 3D self-supported nanopine forest-like Co3O4@CoMoO4 core-shell architectures for high-energy solid state supercapacitors. Nano Energy 19:222–233. https://doi.org/10.1016/j.nanoen.2015.10.036
Liu W, Du HS, Zheng T, Si CL (2021) Recent insights on biomedical applications of bacterial cellulose based composite hydrogels. Curr Med Chem 33845720. https://doi.org/10.2174/0929867328666210412124444
Zhao Y, Liu C, Yi R, Li Z, Chen Y, Li Y, Mitrovic I, Taylor S, Chalker P, Yang L, Zhao C (2020) Facile preparation of Co3O4 nanoparticles incorporating with highly conductive MXene nanosheets as high-performance anodes for Lithium-ion batteries. Electrochim Acta 345:136203. https://doi.org/10.1016/j.electacta.2020.136203
Hu LQ, Du HS, Liu C, Zhang YD, Yu G, Zhang XY, Si CL, Li B, Peng H (2019) Comparative evaluation of the efficient conversion of corn husk filament and corn husk powder to valuable materials via a sustainable and clean biorefinery process. ACS Sustain Chem Eng 7:1327–1336. https://doi.org/10.1021/acssuschemeng.8b05017
Zhang M, Du H, Liu K, Nie S, Xu T, Zhang X, Si C (2021) Fabrication and applications of cellulose-based nanogenerators. Adv Compos Hybrid Mater 4. https://doi.org/10.1007/s42114-021-00312-2
Ma C, Yuan Q, Du H, Ma M, Si C, Wan P (2020) Multiresponsive MXene (Ti3C2Tx)-decorated textiles for wearable thermal management and human motion monitoring. ACS Appl Mater Inter 12:34226–34234. https://doi.org/10.1021/acsami.0c10750
Xu R, Liu K, Du H, Liu H, Cao X, Zhao X, Qu G, Li X, Li B, Si CL (2020) Falling leaves return to their roots: a review on the preparation of γ-valerolactone from lignocellulose and its application in the conversion of lignocellulose. ChemSusChem 13:6461–6476. https://doi.org/10.1002/cssc.202002008
Dong H, Li M, Jin Y, Wu Y, Huang C, Yang J (2020) Preparation of graphene-like porous carbons with enhanced thermal conductivities from lignin nano-particles by combining hydrothermal carbonization and pyrolysis. Front Energy Res 8:148. https://doi.org/10.3389/fenrg.2020.00148
Zheng Y, Li Z, Xu J, Wang T, Liu X, Duan X, Ma Y, Zhou Y, Pei C (2016) Multi-channeled hierarchical porous carbon incorporated Co3O4 nanopillar arrays as 3D binder-free electrode for high performance supercapacitors. Nano Energy 20:94–107. https://doi.org/10.1016/j.nanoen.2015.11.038
Fan H, Quan L, Yuan M, Zhu S, Wang K, Zhong Y, Chang L, Shao H, Wang J, Zhang J, Cao C (2016) Thin Co3O4 nanosheet array on 3D porous graphene/nickel foam as a binder-free electrode for high-performance supercapacitors. Electrochim Acta 188:222–229. https://doi.org/10.1016/j.electacta.2015.12.011
Su F, Lyu X, Liu C, Miao M (2016) Flexible two-ply yarn supercapacitors based on carbon nanotube/stainless steel core spun yarns decorated with Co3O4 nanoparticles and MnOx composites. Electrochim Acta 215:535–542. https://doi.org/10.1016/j.electacta.2016.08.140
Liu W, Du H, Zhang M, Liu K, Liu H, Xie H, Zhang X, Si C (2020) Bacterial cellulose-based composite scaffolds for biomedical applications: A review. ACS Sustain Chem Eng 8:7536–7562. https://doi.org/10.1021/acssuschemeng.0c00125
Liu Q, Zhong L, Zhao Q, Frear C, Zheng Y (2015) Synthesis of Fe3O4/polyacrylonitrile composite electrospun nanofiber mat for effective adsorption of tetracycline. ACS Appl Mater Inter 7:14573–14583. https://doi.org/10.1021/acsami.5b04598
Yang JE, Jang I, Kim M, Baeck SH, Hwang S, Shim SE (2013) Electrochemically polymerized vine-like nanostructured polyaniline on activated carbon nanofibers for supercapacitor. Electrochim Acta 111:136–143. https://doi.org/10.1016/j.electacta.2013.07.183
Lu J, Zhu W, Dai L, Si C, Ni Y (2019) Fabrication of thermo- and pH-sensitive cellulose nanofibrils-reinforced hydrogel with biomass nanoparticles. Carbohyd Polym 215:289–295. https://doi.org/10.1016/j.carbpol.2019.03.100
Du HS, Liu C, Zhang YD, Yu G, Si CL, Li B (2016) Preparation and characterization of functional cellulose nanofibrils via formic acid hydrolysis pretreatment and the followed high-pressure homogenization. Ind Crop Prod 94:736–745. https://doi.org/10.1016/j.indcrop.2016.09.059
An L, Si C, Wang G, Sui W, Tao Z (2019) Enhancing the solubility and antioxidant activity of high-molecular-weight lignin by moderate depolymerization via in situ ethanol/acid catalysis. Ind Crop Prod 128:177–185. https://doi.org/10.1016/j.indcrop.2018.11.009
Xu R, Du H, Liu C, Liu H, Wu M, Zhang X, Si C, Li B (2021) An efficient and magnetic adsorbent prepared in a dry process with enzymatic hydrolysis residues for wastewater treatment. J Clean Prod 313:127834. https://doi.org/10.1016/j.jclepro.2021.127834
Zhang H, Zhong J, Liu Z, Mai J, Liu H, Mai X (2021) Dyed bamboo composite materials with excellent anti-microbial corrosion. Adv Compos Hybrid Mater 4:294–305. https://doi.org/10.1007/s42114-020-00196-8
Li X, Xu R, Yang J, Nie S, Liu D, Liu Y, Si C (2019) Production of 5-hydroxymethylfurfural and levulinic acid from lignocellulosic biomass and catalytic upgradation. Ind Crop Prod 130:184–197. https://doi.org/10.1016/j.indcrop.2018.12.082
Chen SL, Wang GH, Sui WJ, Parvezc AM, Dai L, Si CL (2020) Novel lignin-based phenolic nanosphere supported palladium nanoparticles with highly efficient catalytic performance and good reusability. Ind Crop Prod 145:112164. https://doi.org/10.1016/j.indcrop.2020.112164
Lin W, Yang J, Zheng Y, Huang C, Yong Q (2021) Understanding the effects of different residual lignin fractions in acid-pretreated bamboo residues on its enzymatic digestibility. Biotechnol Biofuels 14(1):1–15. https://doi.org/10.1186/s13068-021-01994-y
Pei W, Chen ZS, Chan HYE, Zheng L, Liang C, Huang C (2020) Isolation and identification of a novel anti-protein aggregation activity of lignin-carbohydrate complex from chionanthus retusus leaves. Front Bioeng Biotechnol 8:573991. https://doi.org/10.3389/fbioe.2020.573991
Dong H, Zheng L, Yu P, Jiang Q, Wu Y, Huang C, Yin B (2020) Characterization and application of lignin-carbohydrate complexes from lignocellulosic materials as antioxidants for scavenging in vitro and in vivo reactive oxygen species. ACS Sustain Chem Eng 8(1):256–266. https://doi.org/10.1021/acssuschemeng.9b05290
Huang C, Tang S, Zhang W, Tao Y, Lai C, Li X, Yong Q (2018) Unveiling the structural properties of lignin-carbohydrate complexes in bamboo residues and its functionality as antioxidants and immunostimulants. ACS Sustain Chem Eng 6(9):12522–12531. https://doi.org/10.1021/acssuschemeng.8b03262
Huang C, Zheng Y, Lin W, Shi Y, Huang G, Yong Q (2020) Removal of fermentation inhibitors from pre-hydrolysis liquor using polystyrene divinylbenzene resin. Biotechnol Biofuels 13:188. https://doi.org/10.1186/s13068-020-01828-3
Pei W, Shang W, Liang C, Jiang X, Huang C, Yong Q (2020) Using lignin as the precursor to synthesize Fe3O4@lignin composite for preparing electromagnetic wave absorbing lignin-phenol-formaldehyde adhesive. Ind Crop Prod 154:112638. https://doi.org/10.1016/j.indcrop.2020.112638
Li Q, Xie S, Serem W, Naik M, Liu L, Yuan J (2017) Quality carbon fibers from fractionated lignin. Green Chem 19:1628–1634. https://doi.org/10.1039/C6GC03555H
Xu JY, Li CY, Dai L, Xu C, Zhong YD, Yu FX, Si CL (2020) Biomass fractionation and lignin fractionation towards lignin valorization. ChemSusChem 13(17):4284–4295. https://doi.org/10.1002/cssc.202001491
Wang X, Tang S, Chai S, Wang P, Qin J, Pei W, Bian H, Jiang Q, Huang C (2021) Preparing printable bacterial cellulose based gelatin gel to promote in vivo bone regeneration. Carbohydr Polym 270:118342. https://doi.org/10.1016/j.carbpol.2021.118342
Wang P, Yin B, Dong H, Zhang Y, Zhang Y, Chen R, Yang Z, Huang C, Jiang Q (2020) Coupling Biocompatible Au Nanoclusters and Cellulose Nanofibrils to Prepare the Antibacterial Nanocomposite Films. Front Bioeng Biotechnol 8:986. https://doi.org/10.3389/fbioe.2020.00986
Huang C, Dong H, Zhang Z, Bian H, Yong Q (2020) Procuring the nano-scale lignin in prehydrolyzate as ingredient to prepare cellulose nanofibril composite film with multiple functions. Cellulose 27(16):9355–9370. https://doi.org/10.1007/s10570-020-03427-9
Liu W, Du H, Liu H, Xie H, Xu T, Zhao X, Liu Y, Zhang X, Si C (2020) Highly efficient and sustainable preparation of carboxylic and thermostable cellulose nanocrystals via FeCl3-catalyzed innocuous citric acid hydrolysis. ACS Sustain Chem Eng 8:16691–16700. https://doi.org/10.1021/acssuschemeng.0c06561
Yang X, Xie H, Du H, Zhang X, Zou Z, Zou Y, Liu W, Lan H, Zhang X, Si C (2019) Facile extraction of thermally stable and dispersible cellulose nanocrystals with high yield via a green and recyclable FeCl3-catalyzed deep eutectic solvent system. ACS Sustain Chem Eng 7:7200–7208. https://doi.org/10.1021/acssuschemeng.9b00209
Liu HC, Chien A, Newcomb BA, Liu Y, Kumar S (2015) Processing, structure, and properties of lignin- and cnt-incorporated polyacrylonitrile-based carbon fibers. ACS Sustain Chem Eng 3:1943–1954. https://doi.org/10.1021/acssuschemeng.5b00562
Liu Y, Zhou J, Chen L, Zhang P, Fu W, Zhao H, Ma Y, Pan X, Zhang Z, Han W, Xie E (2015) Highly flexible freestanding porous carbon nanofibers for electrodes materials of high-performance all-carbon supercapacitors. ACS Appl Mater Inter 7:23515–23520. https://doi.org/10.1021/acsami.5b06107
Samuel E, Joshi B, Jo H, Kim Y, Swihart M, Yun J, Kim K, Yoon S (2017) Flexible and freestanding core-shell SnO/carbon nanofiber mats for high-performance supercapacitors. J Alloy Compd 728:1362–1371. https://doi.org/10.1016/j.jallcom.2017.09.103
Liao R, Wang H, Zhang W, Shi J, Huang M, Shi Z, Wei W, Li X, Liu S (2021) High-rate sodium storage performance enabled using hollow Co3O4 nanoparticles anchored in porous carbon nanofibers anode. J Alloy Compd 868:159262. https://doi.org/10.1016/j.jallcom.2021.159262
Lu X, Zhu D, Li X, Li M, Chen Q, Qing Y (2021) Gelatin-derived N-doped hybrid carbon nanospheres with an adjustable porous structure for enhanced electromagnetic wave absorption. Adv Compos Hybrid Mater. https://doi.org/10.1007/s42114-021-00258-5
Wang Z, Li X, Wang L, Li Y, Qin J, Xie P, Qu Y, Sun K, Fan R (2020) Flexible multi-walled carbon nanotubes/polydimethylsiloxane membranous composites toward high-permittivity performance. Adv Compos Hybrid Mater 3:1–7. https://doi.org/10.1007/s42114-020-00144-6
Yang W, Hou L, Xu X, Li Z, Ma X, Yang F, Li Y (2018) Carbon nitride template-directed fabrication of nitrogen-rich porous graphene-like carbon for high performance supercapacitors. Carbon 130:325–332. https://doi.org/10.1016/j.carbon.2018.01.032
Lin W, Xing S, Jin Y, Lu X, Huang C, Yong Q (2020) Insight into understanding the performance of deep eutectic solvent pretreatment on improving enzymatic digestibility of bamboo residues. Bioresour Technol 306:123163. https://doi.org/10.1016/j.biortech.2020.123163
Dou Y, Xu J, Ruan B, Liu Q, Pan Y, Sun Z, Dou SX (2016) Atomic layer-by-layer Co3O4/graphene composite for high performance lithium-ion batteries. Adv Energy Mater 6:1501835. https://doi.org/10.1002/aenm.201501835
Zhai T, Wan L, Sun S, Chen Q, Sun J, Xia Q, Xia H (2017) Phosphate ion functionalized Co3O4 ultrathin nanosheets with greatly improved surface reactivity for high performance pseudocapacitors. Adv Mater 29:1604167. https://doi.org/10.1002/adma.201604167
Huang C, Dong H, Su Y, Wu Y, Narron R, Yong Q (2019) Synthesis of carbon quantum dot nanoparticles derived from byproducts in bio-refinery process for cell imaging and in vivo bioimaging. Nanomaterials 9(3):387. https://doi.org/10.3390/nano9030387
Huang C, Lin W, Lai C, Li X, Jin Y, Yong Q (2019) Coupling the post-extraction process to remove residual lignin and alter the recalcitrant structures for improving the enzymatic digestibility of acid-pretreated bamboo residues. Bioresour Technol 285:121355. https://doi.org/10.1016/j.biortech.2019.121355
Liu H, Xu T, Liu K, Zhang M, Liu W, Li H, Du H, Si C (2021) Lignin-based electrodes for energy storage application. Ind Crop Prod 165:113425. https://doi.org/10.1016/j.indcrop.2021.113425
Wang Y, Hu Y, Hao X, Peng P, Shi J, Peng F, Sun RC (2020) Hydrothermal synthesis and applications of advanced carbonaceous materials from biomass: a review. Adv Compos Hybrid Mater 3:267–284. https://doi.org/10.1007/s42114-020-00158-0
Liu L, Jiang Z, Fang L, Xu H, Zhang H, Gu X, Wang Y (2017) Probing the crystal plane effect of Co3O4 for enhanced electrocatalytic performance toward efficient overall water splitting. ACS Appl Mater Inter 9:27736–27744. https://doi.org/10.1021/acsami.7b07793
Ma C, Cao W, Xin W, Bian J, Ma M (2019) Flexible and free-standing reduced graphene oxide and polypyrrole coated air-laid paper-based supercapacitor electrodes. Ind Eng Chem Res 58:12018–12027. https://doi.org/10.1021/acs.iecr.9b02088
Hu Q, Zhou J, Qiu B, Wang Q, Song G, Guo Z (2021) Synergistically improved methane production from anaerobic wastewater treatment by iron/polyaniline composite. Adv Compos Hybrid Mater 4:265–273. https://doi.org/10.1007/s42114-021-00236-x
Song X, Wang S, Bao Y, Liu G, Sun W, Ding L, Liu H, Wang H (2017) A high strength, free-standing cathode constructed by regulating graphitization and the pore structure in nitrogen-doped carbon nanofibers for flexible lithium–sulfur batteries. J Mater Chem A 5:6832–6839. https://doi.org/10.1039/C7TA01171G
Yan X, Tian L, He M, Chen X (2015) Three-dimensional crystalline/amorphous Co/Co3O4 core/shell nanosheets as efficient electrocatalysts for the hydrogen evolution reaction. Nano Lett 15:6015–6021. https://doi.org/10.1021/acs.nanolett.5b02205
Ma TY, Dai S, Jaroniec M, Qiao SZ (2014) Metal-organic framework derived hybrid Co3O4-carbon porous nanowire arrays as reversible oxygen evolution electrodes. J Am Chem Soc 136:13925–13931. https://doi.org/10.1021/ja5082553
Wei R, Fang M, Dong G, Lan C, Shu L, Zhang H, Bu X, Ho JC (2018) High-index faceted porous Co3O4 nanosheets with oxygen vacancies for highly efficient water oxidation. ACS Appl Mater Inter 10:7079–7086. https://doi.org/10.1021/acsami.7b18208
Gu Y, Pan Z, Zhang H, Zhu J, Yuan B, Pan D, Wu C, Dong B, Guo Z (2020) Synthesis of high performance diesel oxidation catalyst using novel mesoporous AlLaZrTiOx mixed oxides by a modified sol-gel method. Adv Compos Hybrid Mater 3:583–593. https://doi.org/10.1007/s42114-020-00193-x
Rehman S, Ahmed R, Ma K, Xu S, Tao T, Aslam M, Amir M, Wang J (2021) Composite of strip-shaped ZIF-67 with polypyrrole: a conductive polymer-MOF electrode system for stable and high specific capacitance. Eng Sci 13:71–78. https://doi.org/10.30919/es8d1263
Li X, Zhao W, Yin R, Huang X, Qian L (2018) A highly porous polyaniline-graphene composite used for electrochemical supercapacitors. Eng Sci 3:89–95. https://doi.org/10.30919/es8d743
Liu W, Du HS, Liu K, Liu HY, Xie HX, Si CL, Pang B, Zhang XY (2021) Sustainable preparation of cellulose nanofibrils via choline chloride-citric acid deep eutectic solvent pretreatment combined with high-pressure homogenization. Carbohyd Polym 267:118220. https://doi.org/10.1016/j.carbpol.2021.118220
Hou C, Yang W, Xie X, Sun X, Wang J, Naik N, Pan D, Mai X, Guo Z, Dang F, Du W (2021) Agaric-like anodes of porous carbon decorated with MoO2 nanoparticles for stable ultralong cycling lifespan and high-rate lithium/sodium storage. J Colloid Interf Sci 59:396–407. https://doi.org/10.1016/j.jcis.2021.03.149
Wang X, Zeng X, Cao D (2018) Biomass-derived nitrogen-doped porous carbons (NPC) and NPC/polyaniline composites as high performance supercapacitor materials. Eng Sci 1:55–63. https://doi.org/10.30919/es.180325
Xie HX, Zou ZF, Du HS, Zhang XY, Wang XM, Yang XH, Wang H, Li GB, Li L, Si CL (2019) Preparation of thermally stable and surface-functionalized cellulose nanocrystals via mixed H2SO4/oxalic acid hydrolysis. Carbohyd Polym 223:115116. https://doi.org/10.1016/j.carbpol.2019.115116
Zheng Y, Yu Y, Lin W, Jin Y, Yong Q, Huang C (2021) Enhancing the enzymatic digestibility of bamboo residues by biphasic phenoxyethanol-acid pretreatment. Bioresour Technol 325:124691. https://doi.org/10.1016/j.biortech.2021.124691
Xu R, Du H, Wang H, Zhang M, Wu M, Liu C, Yu G, Zhang X, Si C, Choi S, Li B (2021) Valorization of enzymatic hydrolysis residues from corncob into lignin-containing cellulose nanofibrils and lignin nanoparticles. Front Bioeng Biotech 9:677963. https://doi.org/10.3389/fbioe.2021.677963
Dai L, Lu J, Kong F, Liu K, Wei H, Si C (2019) Reversible photo-controlled release of bovine serum albumin by azobenzene-containing cellulose nanofibrils-based hydrogel. Adv Compos Hybrid Mater 2:462–470. https://doi.org/10.1007/s42114-019-00112-9
Wang H, Du HS, Liu K, Liu HY, Xu T, Zhang SY, Chen XQ, Zhang R, Li HM, Xie HX, Zhang XY, Si CL (2021) Sustainable preparation of bifunctional cellulose nanocrystals via mixed H2SO4/formic acid hydrolysis. Carbohyd Polym 266:118107. https://doi.org/10.1016/j.carbpol.2021.118107
Du HS, Liu C, Mu XD, Gong WB, Lv D, Hong YM, Si CL, Li B (2016) Preparation and characterization of thermally stable cellulose nanocrystals via a sustainable approach of FeCl3-catalyzed formic acid hydrolysis. Cellulose 23:2389–2407. https://doi.org/10.1007/s10570-016-0963-5
Zheng L, Yu P, Zhang Y, Wang P, Yan W, Guo B, Huang C, Jiang Q (2021) Evaluating the bio-application of biomacromolecule of lignin-carbohydrate complexes (LCC) from wheat straw in bone metabolism via ROS scavenging. Int J Biol Macromol 176:13–25. https://doi.org/10.1016/j.ijbiomac.2021.01.103
Du H, Liu W, Zhang M, Si C, Zhang X, Li B (2019) Cellulose nanocrystals and cellulose nanofibrils based hydrogels for biomedical applications. Carbohyd Polym 209:130–144. https://doi.org/10.1016/j.carbpol.2019.01.020
Liu K, Du H, Zheng T, Liu H, Zhang M, Zhang R, Li H, Xie H, Zhang X, Ma M, Si C (2021) Recent advances in cellulose and its derivatives for oilfield applications. Carbohyd Polym 259:117740. https://doi.org/10.1016/j.carbpol.2021.117740
Wang H, Xie HX, Du HS, Wang XM, Liu W, Duan YX, Zhang XY, Sun L, Zhang XY, Si CL (2020) Highly efficient preparation of functional and thermostable cellulose nanocrystals via H2SO4 intensified acetic acid hydrolysis. Carbohyd Polym 239:116233. https://doi.org/10.1016/j.carbpol.2020.116233
Si CL, Liu Z, Kim JK, Bae YS (2008) Structure elucidation of phenylethanoid glycosides from Paulownia tomentosa Steud. var. tomentosa wood. Holzforschung 62(2):197–200. https://doi.org/10.1515/HF.2008.047
Aqeel SM, Huang Z, Walton J, Baker C, Falkner D, Liu Z, Wang Z (2018) Polyvinylidene fluoride (PVDF)/polyacrylonitrile (PAN)/carbon nanotube nanocomposites for energy storage and conversion. Adv Compos Hybrid Mater 1:185–192. https://doi.org/10.1007/s42114-017-0002-5
Dai L, Zhu W, Lu J, Kong F, Si C, Ni Y (2019) A lignin-containing cellulose hydrogel for lignin fractionation. Green Chem 21(19):5222–5230. https://doi.org/10.1039/c9gc01975h
Das TK, Ghosh P, Das N (2019) Preparation, development, outcomes, and application versatility of carbon fiber-based polymer composites: A review. Adv Compos Hybrid Mater 2:214–233. https://doi.org/10.1007/s42114-018-0072-z
Hu W, Wang X, Wu L, Shen T, Ji L, Zhao X, Si C, Jiang Y, Wang G (2016) Pigenin-7-O-beta-D-glucuronide inhibits LPS-induced inflammation through the inactivation of AP-1 and MAPK signaling pathways in RAW 264.7 macrophages and protects mice against endotoxin shock. Food Func 7(2):1002–1013. https://doi.org/10.1039/c5fo01212k
Si CL, Kim JK, Bae YS, Li SM (2009) Phenolic compounds in the leaves of populus ussuriensis and their antioxidant activities. Planta Med 75(10):1165–1167. https://doi.org/10.1055/s-0029-1185476
Lin W, Cheng D, Yong Q, Huang C, Huang S (2019) Improving enzymatic hydrolysis of acid-pretreated bamboo residues using amphiphilic surfactant derived from dehydroabietic acid. Bioresour Technol 293:122055. https://doi.org/10.1016/j.biortech.2019.122055
Su Y, Dong H, Li M, Lai C, Huang C, Yong Q (2019) Isolation of the flavonoid from bamboo residues and its application as metal ion sensor in vitro. Polymers 11(9):1377. https://doi.org/10.3390/polym11091377
Huang C, Su Y, Shi J, Yuan C, Zhai S, Yong Q (2019) Revealing the effects of centuries of ageing on the chemical structural features of lignin in archaeological fir woods. New J Chem 43(8):3520–3528. https://doi.org/10.1039/c9nj00026g
Mirabootalebi SO (2020) A new method for preparing buckypaper by pressing a mixture of multi-walled carbon nanotubes and amorphous carbon. Adv Compos Hybrid Mater 3:336–343. https://doi.org/10.1007/s42114-020-00167-z
Dai L, Li Y, Kong F, Liu K, Si C, Ni Y (2019) Lignin-based nanoparticles stabilized pickering emulsion for stability improvement and thermal-controlled release of trans-resveratrol. ACS Sustain Chem Eng 7(15):13497–13504. https://doi.org/10.1021/acssuschemeng.9b02966
Xie H, Du H, Yang X, Si C (2018) Recent strategies in preparation of cellulose nanocrystals and cellulose nanofibrils derived from raw cellulose materials. Int J Polym Sci 7923068. https://doi.org/10.1155/2018/7923068
Huang C, Sun R, Chang H, Yong Q, Jameel H, Phillips R (2019) Production of dissolving grade pulp from tobacco stalk through SO2-ethanol-water fractionation, alkaline extraction, and bleaching processes. Bioresoures 14(3):5544–5558. https://doi.org/10.15376/biores.14.3.5544-5558
He J, Huang C, Lai C, Jin Y, Ragauskas A, Yong Q (2020) Investigation of the effect of lignin/pseudo-lignin on enzymatic hydrolysis by Quartz Crystal Microbalance. Ind Crop Prod 157:112927. https://doi.org/10.1016/j.indcrop.2020.112927
Huang C, Wang X, Liang C, Jia X, Yang G, Xu J, Yong Q (2019) A sustainable process for procuring. Biotechnol Biofuels 12:189. https://doi.org/10.1186/s13068-019-1527-3
Si C, Jiang J, Liu S, Hu H, Ren XD, Yu GJ, Yu GH (2013) A new lignan glycoside and phenolics from the branch wood of Pinus banksiana Lambert. Holzforschung 67(4):357–363. https://doi.org/10.1515/hf-2012-0137
Tian Y, Du H, Zhang M, Zheng Y, Guo Q, Zhang H, Luo J, Zhang X (2019) Microwave synthesis of MoS2/MoO2@CNT nanocomposites with excellent cycling stability for supercapacitor electrodes. J Mater Chem C 7:9545–9555. https://doi.org/10.1039/C9TC02391G
Zhang M, Nautiyal A, Du H, Wei Z, Zhang X, Wang R (2021) Electropolymerization of polyaniline as high-performance binder free electrodes for flexible supercapacitor. Electrochim Acta 376:138037. https://doi.org/10.1016/j.electacta.2021.138037
Sun D, He L, Chen R, Liu Y, Lv B, Lin S, Lin B (2019) Biomorphic composites composed of octahedral Co3O4 nanocrystals and mesoporous carbon microtubes templated from cotton for excellent supercapacitor electrodes. Appl Surf Sci 465:232–240. https://doi.org/10.1016/j.apsusc.2018.09.178
Xiao L, Qi H, Qu K, Shi C, Cheng Y, Sun Z, Yuan B, Huang Z, Pan D, Guo Z (2021) Layer-by-layer assembled free-standing and flexible nanocellulose/porous Co3O4 polyhedron hybrid film as supercapacitor electrodes. Adv Compos Hybrid Mater 4:306–316. https://doi.org/10.1007/s42114-021-00223-2
Li S, Yang K, Ye P, Ma K, Zhang Z, Huang Q (2020) Three-dimensional porous carbon/Co3O4 composites derived from graphene/Co-MOF for high performance supercapacitor electrodes. Appl Surf Sci 503:144090. https://doi.org/10.1016/j.apsusc.2019.144090
Tai Z, Lang J, Yan X, Xue Q (2012) Mutually enhanced capacitances in carbon nanofiber/cobalt hydroxide composite paper for supercapacitor. J Electrochem Soc 159(4):485–491. https://doi.org/10.1149/2.110204jes
Guan Q, Cheng J, Wang B, Ni W, Gu G, Li X, Huang L, Yang G, Nie F (2014) Needle-like Co3O4 anchored on the graphene with enhanced electrochemical performance for aqueous supercapacitors. ACS Appl Mater Interfaces 6:7626–7632. https://doi.org/10.1021/am5009369
Tummala R, Guduru R, Mohanty P (2012) Nanostructured Co3O4 electrodes for supercapacitor applications from plasma spray technique. J Power Sources 209: 44–51. https://doi.org/10.1016/j.jpowsour.2012.02.071
Huang C, He J, Min D, Lai C, Yong Q (2016) Understanding the nonproductive enzyme adsorption and physicochemical properties of residual lignins in moso bamboo pretreated with sulfuric acid and kraft pulping. Appl Biochem Biotech 180:1508–1523. https://doi.org/10.1007/s12010-016-2183-8
Zhang M, Nautiyal A, Du H, Li J, Liu Z, Zhang X, Wang R (2020) Polypyrrole film based flexible supercapacitor: mechanistic insight into influence of acid dopants on electrochemical performance. Electrochim Acta 357:136877. https://doi.org/10.1016/j.electacta.2020.136877
Huang C, He J, Wang Y, Min D, Yong Q (2015) Associating cooking additives with sodium hydroxide to pretreat bamboo residues for improving the enzymatic saccharification and monosaccharides production. Bioresour Technol 193:142–149. https://doi.org/10.1016/j.biortech.2015.06.073
Huang C, He J, Li X, Min D, Yong Q (2015) Facilitating the enzymatic saccharification of pulped bamboo residues by degrading the remained xylan and lignin-carbohydrates complexes. Bioresour Technol 192:471–477. https://doi.org/10.1016/j.biortech.2015.06.008
Tang W, Wu X, Huang C, Lin Z, Lai C, Yong Q (2021) Revealing migration discipline of lignin during producing fermentable sugars from wheat straw through autohydrolysis. Ind Crop Prod 171:113849. https://doi.org/10.1016/j.indcrop.2021.113849
Li X, Lu X, Nie S, Wang M, Yu Z, Duan B, Yang J, Xu R, Lu L, Si C (2020) Efficient catalytic production of biomass-derived levulinic acid over phosphotungstic acid in deep eutectic solvent. Ind Crop Prod 145:112154. https://doi.org/10.1016/j.indcrop.2020.112154
Qu K, Sun Z, Shi C, Wang W, Xiao L, Tian J, Huang Z, Guo Z (2021) Dual-acting cellulose nanocomposites filled with carbon nanotubes and zeolitic imidazolate framework-67 (ZIF-67) derived polyhedral porous Co3O4 for symmetric supercapacitors. Adv Compos Hybrid Mater. https://doi.org/10.1007/s42114-021-00293-2
Funding
This work was financially supported by the Foundation of State Key Laboratory of Biobased Material and Green Papermaking, Qilu University of Technology, Shandong Academy of Sciences (No. GZKF202001), and the Foundation of Key Laboratory of Pulp and Paper Science and Technology of Ministry of Education of China (No. ZR201901). The authors also gratefully acknowledge the “Beyond Research Innovation & Development for Good Enterprises + ” Project, supported by the Ministry of Education (MOE), the Technology Development Program (S3030198) funded by the Ministry of SMEs and Startups (MSS, Korea), 2021 Research Grant from Kangwon National University and this work was also partially supported by 2020 work was also partially supported by 2020 Jeollannam-do (“Industry-University-Institute collaboration agricultural industrial complex R&D supporting program” operated by Jeonnam Technopark) to S.E.C.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, S., Du, H., Liu, K. et al. Flexible and porous Co3O4-carbon nanofibers as binder-free electrodes for supercapacitors. Adv Compos Hybrid Mater 4, 1367–1383 (2021). https://doi.org/10.1007/s42114-021-00344-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42114-021-00344-8