
Abstract
This is a continuation of a series focused on providing a stable platform for the taxonomy of phytopathogenic fungi and organisms. This paper focuses on 25 phytopathogenic genera: Alternaria, Capnodium, Chaetothyrina, Cytospora, Cyphellophora, Cyttaria, Dactylonectria, Diplodia, Dothiorella, Entoleuca, Eutiarosporella, Fusarium, Ilyonectria, Lasiodiplodia, Macrophomina, Medeolaria, Neonectria, Neopestalotiopsis, Pestalotiopsis, Plasmopara, Pseudopestalotiopsis, Rosellinia, Sphaeropsis, Stagonosporopsis and Verticillium. Each genus is provided with a taxonomic background, distribution, hosts, disease symptoms, and updated backbone trees. A new database (Onestopshopfungi) is established to enhance the current understanding of plant pathogenic genera among plant pathologists.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Contents and contributors (main contributors underlined)
51. Capnodium–P Chomnunti, RS Jayawardena
52. Chaetothyrina–S Hongsanan, RS Jayawardena
53. Cytospora–C Norphanphoun, RS Jayawardena, KD Hyde
54. Cyphellophora–Q Tian, RS Jayawardena
55. Cyttaria–AH Ekanayake
56. Dactylonectria–RH Perera
57. Entoleuca–MC Samarakoon
58. Eutiarosporella–RS Brahmanage, AJL Phillips, RS Jayawardena
59. Ilyonectria–RH Perera
60. Macrophomina–DS Tennakoon, AJL Phillips
61. Medeolaria–AH Ekanayake
62. Neonectria–RH Perera, F. Halleen
63. Neopestalotiopsis–NI de Silva, SSN Maharachchikumbura, RS Jayawardena
64. Plasmopara–IS Manawasinghe, EHC McKenzie
65. Pseudopestalotiopsis–NI de Silva, SSN Maharachchikumbura, RS Jayawardena
66. Rosellinia–MC Samarakoon
67. Sphaeropsis–DS Tennakoon, AJL Phillips
Updated genera
68. Alternaria–RS Jayawardena, KD Hyde
69. Diplodia–AJ Dissanayake, AJL Phillips
70. Dothiorella–RS Jayawardena, AJL Philips
71. Fusarium–RH Perera
72. Lasiodiplodia–AJL Phillips, RS Jayawardena
73. Pestalotiopsis–NI de Silva, SSN Maharachchikumbura
74. Stagonosporopsis–SC Jayasiri, RS Jayawardena
75. Verticillium–CG Lin
Introduction
One stop shop (OSS) is a series of papers focused on providing a stable platform for the taxonomy of plant pathogenic fungi and organisms. Genera included in these paper series are associated with plant diseases. However, some may not be well-known plant pathogens and Kochs’ postulates might have not been conducted in order to establish their pathogenicity. When this series was launched in 2014, its specific aims were mentioned (Hyde et al. 2014). Two issues of OSS have been published in which 50 genera were treated (Hyde et al. 2014; Jayawardena et al. 2019). In this study we treat 25 genera of plant pathogens as well as establish a new website, www.onestopshopfungi.org, to host a database for plant pathogenic fungi and organisms. This fungal database allows mycologists and plant pathologists to understand disease symptoms, host distribution, classification, morphology and provides an updated phylogeny which will enhance current understanding of plant pathogens and gain better insights into the current fungal classification system. The Onestopshopfungi webpage is an output funded by the Mushroom Research Foundation, Thailand, which is a non-government and non-profit organization. We invite all mycologists to contribute to make this a success. The outcome of this series provides a stable taxonomy and phylogeny for plant pathogens that can provide a reliable platform for mycologists and plant pathologists to accurately identify causal organisms.
Material and methods
Photo plates of the symptoms of the disease and morphological characters are given, when available. Classification follows Wijayawardene et al. (2018).
Sequence data from ex-type, ex-epitype or authentic strains for each species were retrieved from GenBank. Sequence data from single gene regions were aligned using Clustal X1.81 (Thompson et al. 1997) and further alignment of the sequences were carried out by using the default settings of MAFFT v.7 (Katoh and Toh 2008; http://mafft.cbrc.jp/alignment/server/), and manual adjustment was conducted using BioEdit where necessary. Gene regions were also combined using BioEdit v.7.0.9.0 (Hall 1999). Primers for each gene locus can be found in the bibliography related to the phylogeny presented for each genus. Phylogenetic analyses consisted of maximum likelihood (ML), maximum parsimony (MP) and Bayesian inference (BYPP). Maximum parsimony analysis was performed using PAUP (Phylogenetic Analysis Using Parsimony) v. 4.0b10 (Swofford 2002) to obtain the most parsimonious trees. Maximum likelihood analyses were also performed in raxmlGUIv.0.9b2 (Silvestro and Michalak 2012) or RAxML-HPC2 on XSEDE (8.2.8) in the CIPRES science gateway platform (Miller et al. 2010) using GTR+I+G model of evolution. Bayesian inference was used to construct the phylogenies using Mr. Bayes v.3.1.2 (Ronquist and Huelsenbeck 2003). MrModeltest v. 2.3 (Nylander et al. 2008) was used for statistical selection of best-fit model of nucleotide substitution and was incorporated into the analyses.
Results
Capnodium Mont., Annls Sci. Nat., Bot, sér. 3, 11: 233 (1849)
The genus Capnodium was introduced by Montagne (1849) to accommodate C. salicinum. Capnodium is one of the most commonly found sooty moulds in gardens and landscapes (Laemmlen 2011). Capnodium has a saprobic association with sap-feeding insects in the Order Homoptera, which includes aphids, whiteflies, soft scale, mealy bugs, leafhoppers and psyllids (Barr 1987). Gavrilov-Zimin (2017) reported that the larvae and female of a new species and a new monotypic genus of legless mealybug, Orbuspedum machinator, from bamboo twigs in southern Thailand are covered with densely packed fungal hyphae of the sooty mould Capnodium sp. Herath et al. (2012) reported that a tropical sooty mould (Capnodium sp.) is known to produce antibiotics such as tetramic acid, methiosetin and epicorazin A.
Capnodium species grow on honeydew, gradually covering the surface of the plant part affected by insects, colouring it with various shades of black. These fungi do not colonize the plant tissues or trigger symptoms. However, they alter the ability of the plant to perform photosynthesis and exchange of gases with the atmosphere. Severely affected leaves may die and fall, thereby affecting plant growth and survival. Therefore, we treat Capnodium as a main plant pathogenic group.
Classification—Dothideomycetes, Dothideomycetidae, Capnodiales, Capnodiaceae
Type species—Capnodium salicinum Mont., Annls Sci. Nat., Bot, sér. 3 11:234 (1849)
Distribution—Species of Capnodium have a wide distribution but are most common in tropical and subtropical regions (Chomnunti et al. 2014). They can be found on plants that have been previously fed upon by insects.
Disease symptoms—Dark mycelium coating surface of host can cause chlorosis and reduce photosynthetic ability of plants, which effects plant growth, reduces yield, and leading to marketability problems (Chomnunti et al. 2014; Fig 1). In higher latitudes, Capnodium spp. are scarce during the winter; the most common being C. salicinum in the UK (Cannon et al. 1985; Royal Botanic Gardens, Kew, UK National Collection of Dried Fungi, unpublished data). Warm-temperate climates in Australia and the Mediterranean countries provide an abundance of perennial foliage on which sooty moulds are able to establish themselves during the winter, and so persist from one season to the next (Fraser 1935; Reynolds and Gilbert 2005). In northern Thailand, most of the sooty mould infections are caused by Capnodium species (Chomnunti et al. 2014).

Sooty moulds on various host plants associated with insects. a On a hardwood tree, b on guava, c on coffee, d–f on mango
Hosts—Many plants when colonised by insects that produce honeydew. Species of Annona, Camellia, Citrus, Coffea, Chrysophyllum, Ficus, Malus, Mangifera, Olea, Populus, Prunus, Psidium, Rhododendron and Salix (Farr and Rossman 2019)
Morphological based identification and diversity
The asexual morph forms elongated pycnidia that develop from a superficial mycelium on living plant surfaces and produce tiny, hyaline conidia on top of the pycnidia (Chomnunti et al. 2011). Persoon (1822) mention that Fumago citri is the sooty mould but it was not well described and completed; therefore it was transferred to genus Polychaeton by Léveillé (1847). Later, Berkeley-Desmazieres (1849) transferred all species once known in the genus Fumago to Capnodium. Molecular evidence revealed that Polychaeton is an asexual stage of Capnodium, therefore, both are the same organism. According to “one fungus one name” and the Melbourne Code under Art. 57.2, Capnodium was considered for conservation as it has a larger number of epithets and is more widely used in this group of fungi, even though Polychaeton is the older name (Chomnunti et al 2011, 2014; McNeill et al. 2012; Hyde et al. 2013; Wijayawardene et al. 2014, 2017, 2018; Liu et al. 2015; Hongsanan et al. 2015).
The morphology of Capnodium species can be recognised by black mycelial growth spreading on the host surface, which produces superficial colonies with septate, dark brown hyphae and cylindrical and bitunicate asci. On host surface Capnodium species share the same ecological niche and are similar in appearance to other genera and families of sooty moulds; often found with sexual and asexual states growing together and living in complex communities (Faull et al. 2002; Hughes 2003; Hughes and Seifert 2012; Chomnunti et al. 2014; Hongsanan et al. 2015).
Molecular based identification and diversity
DNA sequencing data of Capnodium coffeae, C. coartatum, C. salicinum, C. coffeicola and C. dematum and eleven unidentified Capnodium spp. are available in GenBank, including sequence data for LSU, SSU and ITS (4/7/2019). Hongsanan et al. (2015) introduced a new species Capnodium coffeicola. It differs from other Capnodium species in having pycnidia with short and black stalks at the base and is swollen at the central part, and it has cylindrical to oblong conidia, but its placement is supported with phylogenetic analysis using LSU and ITS sequence data.
Sooty moulds often grow in colonies of more than one species, and taxonomic descriptions thus often unknowingly combine elements of different genera and species. Identification based on morphology only is difficult as there are overlapping morphological characters among many taxa (Chomnunti et al. 2011, 2014). To achieve accurate generic and species identification and taxonomic placements, phylogenetic studies using large subunit ribosomal RNA (LSU rRNA) gene sequences and the internal transcribed spacer regions and 5.8S nrDNA gene (ITS) were performed (Crous et al. 2009; Chomnunti et al. 2011, 2014; Liu et al. 2015; Hongsanan et al. 2015).
This study reconstructs the phylogeny of Capnodium based on analyses of ITS sequence data (Table 1, Fig. 2) and corresponds with previous studies (Chomnunti et al. 2011, 2014; Hongsanan et al. 2015). This can be used as a backbone tree in the identification of Capnodium species (Fig. 3).

Morphology of Capnodium sp. a pycnidia on the host. b, d, e, f stalked pycnidia c mycelium network, g conical pycnidium and pycnidial wall, h ostiole surrounded by hyaline hyphae, i conidia. Scale bars: b, d–f = 100 μm, c, g = 50 μm, h, = 20 μm, i = 10 μm

Phylogenetic tree generated by maximum likelihood analysis of ITS sequence data of Capnodium species. Related sequences were obtained from GenBank. Thirty-eight strains are included in the analyses, which comprise 512 characters including gaps. The tree was rooted with Scoria leucadendri (CBS 131318). Tree topology of the ML analysis was similar to the MP analysis. The best scoring RAxML tree with a final likelihood value of − 2398.970137 is presented. The matrix had 236 distinct alignment patterns, with 16.01% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.285338, C = 0.285338, G = 0.241464, T = 0.238894; substitution rates AC = 1.204962, AG = 1.857184, AT = 2.642904, CG = 1.319860, CT = 4.051266, GT = 1.000000; gamma distribution shape parameter α = 0.287147. The maximum parsimonious dataset consisted of constant 327, 125 parsimony-informative and 60 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of two equally most parsimonious trees with a length of 358 steps (CI = 0.735, RI = 0.875, RC = 0.630, HI = 0.265) in the first tree. RAxML and maximum parsimony bootstrap support value ≥ 50% are shown, respectively, near the nodes. Ex-type strains are in bold
Recommended genetic marker (genus level)—LSU
Recommended genetic markers (species level)—LSU, ITS
Sequence data of LSU, SSU and ITS are available for five species of Capnodium in GenBank but none of them has complete sequence data. LSU is useful for preliminary identification at the generic level (Chomnunti et al 2011, 2014; Quaedvlieg et al 2014). Hongsanan et al. (2015) recommended the use of combined LSU and ITS sequence data to identify the species. More protein-coding gene loci should be sequenced to clarify the taxonomic problems in this genus. In the current analyses, C. cortatum was not included due to lack of ITS sequences in GenBank. A revision of this genus is needed as it may reveal many new species. Re-sequencing of species as well as designating epitypes or representative species is also important.
Accepted number of species: There are 140 epithets in Index Fungorum (2019), however only four species have DNA sequence data.
References: Chomnunti et al 2011, 2014; Quaedvlieg et al 2014; Hongsanan et al. 2015 (morphology, phylogeny).
Chaetothyrina Theiss., Annls mycol. 11(6):495 (1913)
The genus Chaetothyrina was established by Theissen (1913), with C. musarum (Speg.) Theiss as the type species. Chaetothyrina was placed in Micropeltidaceae based on its superficial, flattened base, poorly developed thyriothecium and irregular meandering arrangement of compact hyphae of walled cells. Singtripop et al. (2016) provided molecular data of one reference specimen and one new species. Hongsanan et al. (2017) established a new species of Chaetothyrina and introduced a new family Phaeothecoidiellaceae to accommodate species of Chaetothyrina, Houjia and Phaeothecoidiella in Capnodiales. Based on its placement in phylogenetic trees and the morphological uniqueness, Micropeltidaceae was excluded from Microthyriales and treated as family incertae sedis in Lecanoromycetes (Hongsanan et al. 2017; Zeng et al. 2019) (Fig. 4).

Disease symptoms caused by Chaetothyrina spp. a on mango, b, d appearance of thyriothecia on hosts, c on a banana, e on mango leaves
Classification—Dothideomycetes, incertae sedis, Capnodiales, Phaeothecoidiellaceae
Type species—Chaetothyrina musarum (Speg.) Theiss., Annls mycol. 11(6):495 (1913)
Distribution—Known from Brazil, Cook Islands, Dominican Republic, India, Mexico, Pakistan, Panama, Thailand, US
Disease symptoms—Sooty blotch and flyspeck
Species in this genus cause flyspeck disease on various plants, such as C. musarum on Musa sp. and C. panamensis (F. Stevens & Dorman) Arx on Oncoba laurina. Sooty blotch and flyspeck (SBFS) is a disease complex caused by nearly 80 fungal species (Singtripop et al. 2016) that are epiphytes which blemish the epicuticular wax layer of several fruit crops, such as apple, pear, orange, persimmon, banana and grape worldwide (Gleason et al. 2011; Gao et al. 2014), cutting sale price and limiting the growth rate of fruit production (Williamson and Sutton 2000; Gao et al. 2014). ‘Sooty blotch’ is characterized by colonies produced on host tissues from superficial, spreading, dark irregular blotches of mycelium with or without sclerotium-like structures or fruiting bodies. On the other hand, ‘flyspeck’ defines clusters of shiny, small, black sclerotium-like structures or fruiting bodies, lacking visible intercalary mycelium (Gleason et al. 2011; Mayfield et al. 2012; Singtripop et al. 2016).
Hosts—Species of Anacardium, Anodendron, Anogeissus, Carallia, Cassia, Chonemorpha, Dalbergia, Dianella, Euonymus, Hevea, Iiana, Magnifera, Magnolia, Mammea, Maytenus, Memecylon, Mitragyna, Musa, Myrcia, Ochrocarpos, Olea, Oncoba, Phoebe, Similax, Streblus and Vochysia.
Morphological based identification and diversity
Chaetothyrina is characterized by superficial, flattened thyriothecia, with base poorly developed, with thyriothecial setae and 1-septate ascospores (Reynolds and Gilbert 2005; Singtripop et al. 2016; Hongsanan et al. 2017). Chaetothyrina can be distinguished from other species in Micropeltidaceae on the basis of thyriothecial setae appearance, shape and septation of the ascospores (Singtripop et al. 2016; Hongsanan et al. 2017). Twenty-three species of Chaetothyrina epithets are listed in Index Fungorum (2019), but sequence data are available for only two species (4/7/2019). Chaetothyrina is a poorly studied genus. Fresh collections and sequence data are needed for this genus. The disease cycle of this genus is yet to be established (Fig. 5).

Chaetothyrina guttulataa Thyriothecium when viewed in squash mount. b Surface of thyriothecium. c Section through thyriothecium. d Ascus when immature. e Asci at maturity. f Ascospores. Scale bars: a = 50 µm, b, d, e = 10 µm, c = 100 µm, f = 5 µm
Molecular based identification and diversity
Singtripop et al. (2016) provided a reference type specimen of C. musarum with sequence data. Using combined LSU, SSU and ITS sequence data, Chaetothyrina clustered as a sister genus to Houjia and Phaeothecoidiella within Capnodiales (Hongsanan et al. 2017; Table 2, Fig. 6).

Phylogenetic tree generated by maximum parsimony analysis of combined LSU, SSU and ITS sequence data. Thirty-nine strains are included in the analyses, which comprised 2225 characters including gaps. The tree was rooted with Myriangium duriaei (CBS 260.36) and M. hispanicum (CBS 247.33). The maximum parsimonious dataset consisted of 1645 constant, 461 parsimony-informative and 119 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of ten equally most parsimonious trees with a length of 1637 steps (CI = 0.549, RI 0.736, RC = 0.404, HI = 0.451) in the first tree. MP and ML bootstrap values ≥ 50% and bayesian posterior probabilities ≥ 0.90 (BYPP) are shown respectively near the nodes. Ex-type strains are in bold
Recommended genetic markers (genus level)—LSU and SSU
Recommended genetic markers (species level)—ITS and RPB2
Accepted number of species: There are 23 epithets in Index Fungorum (2019). However, only two species have molecular data.
References: Reynolds and Gilbert 2005; Singtripop et al. 2016; Hongsanan et al. 2017 (morphology, phylogeny)
Cytospora Ehrenb., Sylv. mycol. berol.: 28 (1818)
Cytospora was introduced by Ehrenberg (1818) as the type genus of the family Cytosporaceae in Diaporthales (Wehmeyer 1975; Barr 1978; Eriksson et al. 2001; Castlebury et al. 2002). The genus is an important pathogenic fungus, causing canker and dieback on branches of a wide range of hosts with a wide distribution (Adams et al. 2005, 2006; Hyde et al. 2017, 2018; Norphanphoun et al. 2017, 2018).
Classification—Sordariomycetes, Diaporthomycetidae, Diaporthales, Valsaceae
Type species—Cytospora chrysosperma (Pers.) Fr. 1823
Distribution—Worldwide
Disease symptoms—Canker and dieback disease on branches
Hosts—Species of Abies, Acer, Berberis, Betula, Ceratonia, Cornus, Cotinus, Crataegus, Elaeagnus, Eriobotrya, Eucalyptus, Juniperus, Lumnitzera, Malus, Picea, Pinus, Platanus, Platycladus, Populus, Prunus, Pyrus, Quercus, Rosa, Salix, Sequoia, Sibiraea, Sorbaronia, Sorbus, Spiraea, Styphnolobium, Syringa, Syzygium, Tibouchina, Ulmus, Vitis and Xylocarpus (Norphanphoun et al. 2018).
Morphological based identification and diversity
Cytospora is characterized by multi-loculate conidiomata with ostiolar necks and unicellular, elongate-allantoid to subcylindrical, hyaline conidia (Fan et al. 2015a, b; Norphanphoun et al. 2017, 2018; Fig. 7). The genus which was reported as causing canker diseases in many woody plants was established in 1818 and studied in detail by taxonomists (Fries 1823; Saccardo 1884). Valsa Fr. was reported as the sexual stage of this genus and therefore, Valsa was treated as a synonym of Cytospora (1818) based on The International Code of Nomenclature for Algae, Fungi, and Plants (ICN, McNeill et al. 2012), with Cytospora being the oldest and most widely used name (Adams et al. 2005; Fotouhifar et al. 2010; Fan et al. 2014; Rossman et al. 2015). Previously, the conventional identification of species in Cytospora was based on their host association, often with vague morphological descriptions. Mycologists began to elucidate the relationships between Cytospora species and their hosts, with morphological observations combined with phylogenetic analyses using internal transcribed spacer (ITS) regions as an effective fungal DNA barcode (Adams et al. 2005, 2006; Fotouhifar et al. 2010; Schoch et al. 2012). The establishment of multi-gene analyses using ITS, LSU, ACT, RPB2, TUB2 has proved comprehensive for the species level (Fan et al. 2015a, b, 2020; Liu et al. 2015; Yang et al. 2015; Hyde et al. 2016; Li et al. 2016; Norphanphoun et al. 2017, 2018; Phookamsak et al. 2019).

Cytospora ampulliformisa stromatal habit in wood. b fruiting bodies on the substrate. c Surface of fruiting bodies. d Cross-section of the stroma showing conidiomata. e Peridium, f ostiolar neck. g–i Conidiogenous cells with attached conidia. j Mature conidia. k, l Colonies on MEA (k-from above, l-from below). Scale bars: a = 2 mm, b = 1 mm, c = 500 µm, d, f = 200 µm, e = 50 µm, g, h = 10 µm, i, j = 5 µm
Molecular based identification and diversity
Comprehensive multigene phylogenetic analyses for this genus were performed by Fan et al. (2015a, b, 2020) and Norphanphoun et al. (2017, 2018).
This study reconstructs the phylogeny of Cytospora based on analyses of a combined ITS, LSU, ACT and RPB2 sequence data (Table 3, Fig 8). The phylogenetic tree is updated with recently introduced Cytospora species and corresponds to previous studies (Norphanphoun et al. 2018).


Phylogenetic tree generated by maximum parsimony analysis of combined ITS, LSU, ACT and RPB2 sequence data of Cytospora species. Related sequences were obtained from GenBank. One hundred and eleven strains are included in the analyses, which comprised 2266 characters including gaps. The tree was rooted with Diaporthe vaccinii (CBS 160.32). The maximum parsimonious dataset consisted of 1358 constant, 596 parsimony-informative and 312 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of ten equally most parsimonious trees with a length of 3836 steps (CI = 0.364, RI 0.649, RC = 0.236, HI = 0.636) in the first tree. MP and ML bootstrap values ≥ 50% and Bayesian posterior probabilities ≥ 0.90 are shown respectively near the nodes. The scale bar indicates 10 changes per site. Ex-type strains are in bold
Recommended genetic markers (genus level)—LSU, ITS
Recommended genetic markers (species level)—ITS, ACT and RPB2
Accepted number of species: There are 630 species in Index Fungorum (2019) and 110 species have molecular data.
References: Fan et al. 2015a, b, Lawrence et al. 2016, Senanayake et al. 2017, 2018 (morphology), Norphanphoun et al. 2017, 2018 (morphology, phylogeny).
Cyphellophora G.A. de Vries, Mycopath. Mycol. appl. 16(1):47(1962)
Cyphellophora is cosmopolitan, comprising species distributed from a broad range of environmental sources as human and animal disease, saprobes, epiphytes and plant pathogens (de Hoog et al. 1999, 2000; Jacob and Bhat 2000; Decock et al. 2003; Crous et al. 2007; Zhuang et al. 2010; Feng et al. 2014; Mayfield et al. 2012; Gao et al. 2014; Phookamsak et al. 2019). Most species, including the type species, C. laciniata, were isolated from nails or skin of humans, resulting in clinical symptoms (Feng et al. 2014). Phylogenetically, C. phyllostachysdis clustered with C. europaea, a human or mammal infection of hyperkeratosis (de Hoog et al. 2000). In contrast, C. phyllostachysdis causes sooty blotch and flyspeck (SBFS) of bamboo and is not found on humans (Gao et al. 2014). The sooty mould species C. jingdongensis was introduced with a sexual morph; it reduces plant photosynthesis but does not damage or cause disease of the plant (Chomnunti et al. 2014; Yang et al. 2018).
Classification—Eurotiomycetes, Chaetothyriomycetidae, Chaetothyriales, Cyphellophoraceae
Type species—Cyphellophora laciniata G.A. de Vries, Mycopath. Mycol. appl. 16(1):47(1962)
Distribution—Australia, Brazil, China, Germany, India, Israel, Korea, Taiwan
Disease symptoms—Sooty blotch and flyspeck (main symptoms of this disease are given under Chaetothyrina).
To date, C. artocarpi, C. guyanensis, C. jingdongensis, C. musae, C. olivacea, C. oxyspora, C. phyllostachydis and C. sessilis have been isolated from plant materials (Gams and Holubová-Jechová 1976; de Hoog et al. 1999; Decock et al. 2003; Gao et al. 2014; Yang et al. 2018). Cyphellophora artocarpi, C. musae, C. phyllostachydis and C. sessilis were reported to cause sooty blotch and flyspeck from apple, jackfruit (Artocarpus heterophyllus) and bamboo (Phyllostachys heterocycla, Sinobambusa tootsik), resulting in significant economic damage (Zhuang et al. 2010; Mayfield et al. 2012; Gao et al. 2014).
Hosts—Artocarpus heterophyllus, Dendrocalamus strictus, Eucalyptus sp., Helomeco velane, Hylomecon verlance, Malus domestica, Musa sp., Phyllostachys sp., Sinobasmbusa tootsik and Stenocalyx uniflorus.
Morphological based identification and diversity
It is difficult to identify this black yeast-like genus based solely on morphological characters since the characters are very similar to those of other black yeast-like fungi, such as Phialophora and Pseudomicrodochium. Species of Cyphellophora resemble those of Phialophora in having melanized thalli with intercalary or terminal phialides bearing collarettes, but Phialophora has aseptate conidia whereas Cyphellophora produces larger, fusiform to sigmoid, aseptate to multi-septate conidia (Réblová et al. 2013). Cyphellophora can also be compared to Pseudomicrodochium, the former having melanized thalli while they are hyaline in Pseudomicrodochium (Decock et al. 2003; de Hoog et al. 2000, 2011). Yang et al. (2018) introduced C. jingdongensis as the first sexual morph, which is characterized by subglobose to globose, non-ostiolate ascomata, ellipsoidal to cylindrical asci and fusoid, 1–3 septate ascospores. However, the asexual morph of C. jingdongensis was difficult to observe in culture to compare with other species in Cyphellophora (Yang et al. 2018). There are 26 epithets of Cyphellophora in Index Fungorum (2019). Yang et al. (2018) clarified 23 species in this genus. To properly delineate these species, phylogenetic studies using multi-loci sequences (ITS, LSU, RPB1 and TUB2) and the secondary structures of ITS analyses are needed (Réblová et al. 2013; Feng et al. 2014; Gao et al. 2014; Yang et al. 2018).
Molecular based identification and diversity
Based on SSU and LSU sequence data, Cyphellophora clustered in a well-supported clade within the Chaetothyriales (Feng et al. 2014). Generic and species delimitation with morphological characters, ecological traits, host distribution and phylogenetic analyses using the internal transcribed spacer region (ITS), the partial β-tubulin gene (TUB2), the nuclear large subunit rDNA gene (LSU) and the DNA dependent RNA polymerase II largest subunit (RPB1) were recently performed (Feng et al. 2014; Gao et al. 2014). The present study reconstructs the phylogeny of Cyphellophora based on analyses of a combined ITS, TUB2, LSU and RPB1 sequence data (Table 4, Fig. 9). The phylogenetic tree in this study is updated with recently introduced Cyphellophora species and corresponds to previous studies (Feng et al. 2014; Gao et al. 2014). Cyphellophoroa indica and C. taiwanensis lack sequences in GenBank (4/7/2019). Cyphellophoroa hylomeconis was synonymized as Camptophora hylomeconis and C. eugeniae was synonymized as Aphanophora eugeniae (Réblová et al. 2013). Cyphellophoroa eucalypti were synonymized as C. guyanensis (Feng et al. 2014). Therefore, these species were not included in the present phylogenetic analyses (Fig. 9).

Phylogram generated from RAxML analysis based on combined sequences of ITS, LSU, RPB1 and TUB2 sequences of all accepted species of Cyphellophora. Forty-one strains are included in the analyses, which comprise 2514 characters including gaps. The tree was rooted with Cladophialophora immunda (CBS 834.96). Tree topology of the ML analysis was similar to the MP and BYPP analyses. The best scoring RAxML tree with a final likelihood value of − 5928.387430 is presented. The matrix had 337 distinct alignment patterns, with 12.44% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.234866, C = 0.250597, G = 0.284325, T = 0.230211; substitution rates AC = 1.492532, AG = 2.025910, AT = 2.769660, CG = 1.674732, CT = 8.545312, GT = 1.000000; gamma distribution shape parameter α = 0.136482. RAxML and maximum parsimony bootstrap support value ≥ 50% are shown respectively near the nodes. Bayesian posterior probabilities ≥ 0.95 (BYPP) indicated as thickened black branches. Ex-type strains are in bold
Recommended genetic markers (genus level)—LSU and SSU
Recommended genetic markers (species level)—ITS, LSU, TUB2, RPB1 and secondary (2D) structure of ITS analyses
LSU is useful for preliminary identification at the generic level (Feng et al. 2014). Réblová et al. (2013) resolved Cyphellophora and Phialophora as close relatives within the Chaetothyriales, although both genera were paraphyletic based on analysis of ITS, TUB2 and nuc28S rDNA sequence data. It is recommended to use a combination of ITS, LSU, TUB2, RPB1 and secondary (2D) structure of ITS analyses (Réblová et al. 2013; Feng et al. 2014; Gao et al. 2014) in order to identify to the species level.
Accepted number of species: 24 species
References: Vries 1962, 1986; Matsushima 1987; Walz and de Hoog 1987; Decock et al. 2003; Crous et al. 2013, 2016; Réblová et al. 2013; Feng et al. 2014; Gao et al. 2014; Madrid et al. 2016; Yang et al. 2018 (morphology, phylogeny)
Cyttaria Berk., Trans. Linn. Soc. London 19:40 (1842)
This genus is geographically restricted to South America (Argentina and Chile) and Southeastern Australasia (including Tasmania, and New Zealand) (Peterson and Pfister 2010). Cyttaria species are found in the secondary phloem and xylem, cambium and cortex of the hosts. They produce trunk and branch cankers that arise due to localized, stimulated cambial activity attributed to the presence of hyphae of Cyttaria (Wilson 1907; Gutierrez de Sanguinetti 1988). Cyttaria species are considered as weak parasites (Gamundí and De Lederkremer 1989).
Classification—Leotiomycetes, Leotiomycetidae, Cyttariales, Cyttariaceae
Type species—Cyttaria darwinii Berk., Trans. Linn. Soc. London 19:40 (1842)
Distribution—Argentina, Australia, Chile, New Zealand, Tasmania.
Disease symptoms—Canker, galls
These species are known to cause two types of cankers: globose and longitudinal. Globose cankers arise from growth mainly in the transverse axis of the branch while longitudinal cankers arise from growth mainly along the long axis (Rawlings 1956; Gamundi 1971). Development of perennial galls on branches and stems may lead to malformation and occasional death of branches (Gadgil 1985).
Hosts—Nothofagus spp.
Morphological based identification and diversity
Ascomata of Cyttaria species are orange, pitted apothecia similar to deeply dimpled golf balls. Each fruiting body is composed of 1–200 apothecia immersed in a sterile fleshy-gelatinous stroma. Asci are 8-spored, inoperculate and amyloid. Ascospores are uninucleate, subglobose to ovoid, smooth to rugulose, at first hyaline to yellowish but later becoming pigmented (Mengoni 1986; Peterson et al 2010).
Molecular based identification and diversity
The first phylogenetic analysis which included Cyttaria was done by Gargas and Taylor (1995) showing its relationship with other discomycetes. Wang et al. (2006) showed its placement within Leotiomycetes using combined analysis of SSU, LSU and 5.8S rDNA gene sequence data and then confirmed by Ekanayaka et al. (2017). Peterson and Pfister (2010) did large scale phylogeny for Cyttaria including all accepted 12 species in the genus using sequence data of partial nucSSU, nucLSU and mtSSU rRNA, as well as tef1. They found Cyttaria to be a strongly supported clade and suggested a close relationship between Cyttaria and some members of the Helotiales (Cordierites, Encoelia, Ionomidotis and Chlorociboria) (Peterson and Pfister 2010). The present study reconstructs the phylogeny of Cyttaria based on analyses of a combined LSU, SSU and mtSSU sequence data (Table 5, Fig. 10). The phylogenetic tree is updated with recently introduced Cyttaria species and corresponds to previous studies (Feng et al. 2014; Gao et al. 2014).

Phylogram generated from RAxML analysis based on combined sequences of LSU, SSU and mtSSU sequences of all the accepted species of Cyttaria. Related sequences were obtained from GenBank. Thirty-four strains are included in the analyses, which comprise 3480 characters including gaps. Single gene analyses were carried out and compared with each species, to compare the topology of the tree and clade stability. The tree was rooted with Chlorociboria cf. aeruginosa (OSC 100056). The best scoring RAxML tree with a final likelihood value of − 7505.900855 is presented. The matrix had 360 distinct alignment patterns, with 39.17% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.256, C = 0.214, G = 0.280, T = 0.250; substitution rates AC = 1.190197, AG = 1.207782, AT = 0.373130, CG = 0.681125, CT = 3.724394, GT = 1.000000; gamma distribution shape parameter α = 0.020000. RAxML and maximum parsimony bootstrap support value ≥ 50% (BT) are shown respectively near the nodes
Recommended genetic markers (genus level)—ITS, LSU
Recommended genetic markers (species level)—nucSSU, nucLSU, mitSSU rRNA, and tef1
Combined nucSSU, nucLSU, mitSSU rRNA, and tef1 can resolve almost all species of Cyttaria currently known from sequence data (Peterson et al 2010).
Accepted number of species: There are 21 epithets in Index Fungorum (2019). However, 12 species have molecular data and are treated as accepted.
References: Mengoni 1986, Peterson et al 2010 (morphology); Peterson and Pfister (2010), Ekanayaka et al. 2017 (morphology, phylogeny).
Dactylonectria L. Lombard & Crous, in Lombard et al., Phytopath. Mediterr. 53(3): 523 (2014)
The genus Dactylonectria was introduced by Lombard et al. (2014) for a group of species which were previously treated in Ilyonectria (Chaverri et al. 2011; Cabral et al. 2012a, b, c). In morphology, Dactylonectria resembles Ilyonectria and Neonectria but can be distinguished by their characteristic ovoid to obpyriform, smooth to finely warted, dark-red ascomata with a papillate ostiolar region at the apex (Lombard et al. 2014; Gordillo and Decock 2018). Species of this genus are mostly associated with Vitis sp., while some species are also recorded from other hosts (Farr and Rossman 2019).
Classification—Sordariomycetes, Hypocreomycetidae, Hypocreales, Nectriaceae
Type species—Dactylonectria macrodidyma (Hallen, Schroers & Crous) L. Lombard & Crous, in Lombard et al., Phytopath. Mediterr. 53(3): 527 (2014)
Distribution—Worldwide
Disease symptoms—Black foot disease, black root rot
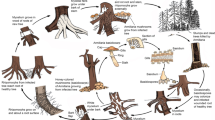
Characteristic symptoms of black foot disease include a reduction in root biomass and root hairs with sunken and necrotic lesions (Halleen et al. 2006). Severe necrosis of the root system results in stunting, wilting, leaf chlorosis, browning and leaf drop prior to death (Parkinson et al. 2017). Dactylonectria alcacerensis, D. estremocensis, D. macrodidyma, D. novozelandica, D. pauciseptata, D. pinicola, D. torresensis and D. vitis are associated with black foot disease of grapevine (Cabral et al. 2012a; Lombard et al. 2014) (Fig. 11).

Symptoms of black foot disease on Vitis spp. a, b dead plants and stunted growth of grapevine. c, e Infection of rootstock. d Blocked xylem vessels. f Black streak (Courtesy of Halleen)
Hosts—Abies sp., Annona cherimola, Anthrium sp., Arbutus unedo, Cistus albidus, Crataegus azalous, Erica melanthera, Eriobotrya japonica, Ficus sp., Fragaria sp., Hordeum vulgare, Ilex aquifolium, Juglans regia, Juniperus phoenicea, Lonicera sp., Myrtus communis, Persea americana, Picea glauca, Pinus sp., Pistacia lentiscus, Prunus domestica, Pyracantha sp., Quercus sp., Rosmarinus officinalis, Santolina chamaecyparissus and Vitis sp.
Morphological based identification and diversity
Lombard et al. (2014) accepted ten species in Dactylonectria based on ITS, LSU, TUB2 and tef1 sequence data and morphological characters. Later, Gordillo and Decock (2018) introduced another four species to the genus based on morphology and sequence data.
Fourteen Dactylonectria species have been described with DNA sequence data in GenBank. Dactylonectria species produce cylindrocarpon-like asexual morphs, several of which were previously treated in Ilyonectria (Lombard et al. 2014). Dactylonectria species were distinguished mainly by phylogenetic inference and using unique fixed single nucleotide polymorphisms (SNP’s) rather than morphological characters (Lombard et al. 2014; Gordillo and Decock 2018).
Molecular based identification and diversity
Lombard et al. (2014) re-evaluated genera with cylindrocarpon-like asexual morphs based on multi-gene phylogeny of ITS, LSU, TUB2 and tef1 genes. Gordillo and Decock (2018) analysed His3 together with latter gene regions, for delimiting the species in Dactylonectria. Lombard et al. (2015) also supported the fact that Dactylonectria is monophyletic and distinct from Ilyonectria. In this study, we reconstruct the phylogeny of Dactylonectria based on analyses of a combined ITS, LSU, TUB2 and tef1 sequence data (Table 6, Fig. 12). The phylogenetic tree is updated with recently introduced Dactylonectria species and corresponds to previous studies (Lombard et al. 2014, 2015; Gordillo and Decock 2018).

Phylogram generated from RAxML analysis based on combined sequences of ITS, LSU, TUB and tef1 sequences of all the accepted species of Dactylonectria. Related sequences were obtained from GenBank. Fifteen taxa are included in the analyses, which comprise 2460 characters including gaps. Single gene analyses were carried out and compared with each species, to compare the topology of the tree and clade stability. The tree was rooted with Campylocarpon fasciculare (CBS 112613). Tree topology of the ML analysis was similar to the BI. The best scoring RAxML tree with a final likelihood value of − 6772.195394 is presented. The matrix had 261 distinct alignment patterns, with 0.96% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.230657, C = 0.279364, G = 0.252128, T = 0.237852; substitution rates AC = 1.388608, AG = 2.845402, AT = 2.389715, CG = 0.838197, CT = 7.220493, GT = 1.000000; gamma distribution shape parameter α = 0.650385. RAxML and maximum parsimony bootstrap support value ≥ 50% are shown respectively near the nodes. Bayesian posterior probabilities ≥ 0.95 (BYPP) indicated as thickened black branches. Ex-type strains are in bold
Recommended genetic markers (genus level)—ITS, LSU, TUB2, tef1
Recommended genetic markers (species level)—TUB, tef1
Accepted number of species: 14 species
References: Cabral et al. 2012a, b; Halleen et al. 2004, Schroers et al. 2008; Lombard et al. 2014; Gordillo and Decock 2018 (morphology, phylogeny).
Entoleuca Syd., Annls mycol. 20(3/4):186 (1922)
The genus Entoleuca Syd. (Xylariaceae) consists of saprobic and plant pathogenic species distributed in Europe. Entoleuca mammata causes canker diseases (commonly known as Hypoxylon canker) on Malus sp. (Rosaceae), Populus sp., Salix sp. (Salicaceae) and Sorbus sp. (Rosaceae) (Shaw 1973; Callan 1998; Kasanen et al. 2004; Eriksson 2014) and also occurs as a saprobe on decaying tree trunks. The species are distributed in terrestrial habitats in temperate regions. The genus is characterized by its known sexual morph. It is characterized by partially embedded solitary or aggregated orbicular stroma, that has a whitish surface when young and dark surface at maturity, papillate ostiole; multiple, monostichous and embedded ascomata in stromata; 8-spored, unitunicate asci that are cylindrical, long pedicellate, with J+ apical ring bluing in Melzer’s reagent and uniseriate, unicellular, ellipsoidal inequilateral, brown, with straight to oblique germ slit ascospores (Rogers and Ju 1996; Daranagama et al. 2018). Daranagama et al. (2018) provided an identification key with emphasis on the coarsely papillate ostiole in Entoleuca.
Sydow and Petrak (1922) introduced the genus with E. callimorpha as the type species. Until 1994, Hypoxylon mammatum was considered a similar taxon to E. callimorpha. However, Læssøe and Spooner (1994) and Læssøe (1994) treated H. mammatum as a separate, synonym to Rosellinia. Based on these taxonomic confusions, Rogers and Ju (1996) revised the type, authentic and other specimens and re-established the genus Entoleuca.
Classification—Sordariomycetes, Xylariomycetidae, Xylariales, Xylariaceae
Type species—Entoleuca callimorpha Syd., in Sydow & Petrak, Annls mycol. 20(3/4):186 (1922)
Distribution—Austria, Canada, Poland, Sweden, USA
Disease symptoms—Canker
Symptoms may vary on the stage of disease development. Young cankers appear as slightly sunken, yellowish orange areas with irregular margins. Later, the outer-most bark within the canker breaks out in blisters exposing a powdery grey mat of fungal tissue and conidia. Then the patches of bark start to flake off making the canker rough and black in the centre. Advancing margins of the enlarging cankers become yellowish orange (Ostry 2013). Hosts—Known from Malus sylvestris, Populus sp., Salix sp. and Sorbus aucuparia.
Morphological based identification and diversity
Currently, the genus comprises three species: E. callimorpha, E. ellisii and E. mammata (Sydow and Petrak 1922; Rogers and Ju 1996; Ju et al. 2004; Index Fungorum 2019). Due to the presence of clear papillate ostioles, they have been distinguished from closely related genera such as Amphirosellinia, Nemania, and Rosellinia. Molecular data are only available for E. mammata, which is the most important species in the genus as a pathogen. Rogers and Ju (1996) observed that there are no distinguishing morphological differences among E. mammata isolates from different hosts. However, there is a high polymorphism, but no major phylogenetic differences among the isolates from Europe (Kasanen et al. 2004). Ju et al. (2004) introduced E. ellisii based on characterizations of ascospore and germ slit. Therefore, a combination of morphological and phylogenetic analyses are needed for species delimitation of Entoleuca.
Molecular based identification and diversity
Several recent studies have focused on the molecular phylogeny of Entoleuca, especially E. mammata. Phylogenetic based population studies revealed that higher polymorphism occurs in North American than in Europe (Kasanen et al. 2004). The sterile mycelia associated with Pinus tabulaeformis and its ITS-based phylogenetic analyses revealed that the genus Entoleuca clusters in Xylariaceae and is closely related to Nemania (Guo et al. 2003). Daranagama et al. (2018) revisited the family Xylariaceae and due to the morphological differences and conidial state characters, they suggested that it is useful to maintain the taxa as distinct genera. Daranagama et al. (2018) and Wendt et al. (2018) conducted multi-gene phylogenetic analyses using ITS, LSU, RPB2 and TUB2 and revealed that E. mammata clusters with Rosellinia corticium with high support. Due to the lack of molecular data from other species and other gene regions, it is difficult to place Entoleuca in an appropriate family.
In this study, we included the available sequences of Entoleuca in the analysis done for Rosellinia (Table 7, Fig 13).

Phylogenetic tree generated by maximum likelihood analysis of combined ITS, LSU and RPB2 sequence data of Entoleuca and Rosellinia species. Related sequences were obtained from GenBank. Forty strains are included in the analyses, which comprise 2336 characters including gaps. Single gene analyses were carried out and compared with each species, to compare the topology of the tree and clade stability. The tree was rooted with Xylaria bambusicola (WSP 205), X. grammica (HAST 479) and X. hypoxylon (CBS 122620). Tree topology of the ML analysis was similar to the MP. The best scoring RAxML tree with a final likelihood value of − 12521.202450 is presented. The matrix had 793 distinct alignment patterns, with 51.67% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.241655, C = 0.263843, G = 0.260086, T = 0.234416; substitution rates AC = 1.764265, AG = 4.237635, AT = 0.953946, CG = 1.541272, CT = 8.643978, GT = 1.000000; gamma distribution shape parameter α = 1.105097. The maximum parsimonious dataset consisted of constant 1567, 629 parsimony-informative and 140 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of two equally most parsimonious trees with a length of 2112 steps (CI = 0.545, RI = 0.668, RC = 0.364, HI = 0.455) in the first tree. RAxML and maximum parsimony bootstrap support value ≥ 50% are shown respectively near the nodes. Ex-type strains are in bold
Recommended genetic markers (genus level)—LSU and ITS
Recommended genetic markers (species level)—RPB2 and TUB2
Combined LSU, ITS, RPB2 and TUB2 provide a satisfactory resolution for resolving species.
Accepted number of species: Three species
References: Sydow and Petrak 1922; Rogers and Ju 1996; Ju et al. 2004 (morphology), Daranagama et al. 2018 (morphology, phylogeny).
Eutiarosporella Crous, in Crous et al., Phytotaxa 202(2): 85 (2015)
Eutiarosporella was introduced by Crous et al. (2015) and is typified by Eutiarosporella tritici (B. Sutton & Marasas) Crous on Triticum aestivum from South Africa. The genus was named on account of its similarity to Tiarosporella Höhn. (Crous et al. 2006). Eutiarosporella species are coelomycetes that are saprobes or pathogens which occur in terrestrial habitats (Crous et al. 2015; Thynne et al. 2015; Li et al. 2016). Eutiarosporella species have been reported from Celtis africana (Rosales), Triticum aestivum (Poales), Acacia karroo (Fabales) and Dactylis glomerata (Poales) (Thambugala et al. 2014; Crous et al. 2015). On wheat, it causes the economically important disease known as white grain disorder (Thynne et al. 2015). Several studies have reported this genus on woody hosts as a saprobe (Jami et al. 2012, 2014; Dissanayake et al. 2016).
Classification—Dothideomycetes, incertae sedis, Botryosphaeriales, Botryosphaeriaceae
Type species—Eutiarosporella tritici (B. Sutton & Marasas) Crous, in Crous et al., Phytotaxa
202(2):85 (2015)
Distribution—Worldwide
Disease symptoms—White grain disorder of wheats
White grain disorder shrivels and discolours (white to light grey) wheat grain (Thynne et al. 2015). Affected grains are more brittle and can break during harvesting. Infected spikelets of green heads may show bleaching appreance or grey discolouration. At first, the bleached florets may show blue-gray ‘highlights’. Rachis of affected heads and the upper peduncle may show a brownish discolouration (Thynne et al. 2015).
Even though species of this genus have been found to be associated with several hosts other than wheat, their diseases have not been described.
Hosts—Acacia karroo, Arrhenatherum elatius, Avenella flexuosa, Celtis africana, Dactylis glomerata, Triticum aestivum and Vachelloa karroo (Farr and Rossman 2019).
Morphological based identification and diversity
Eutiarosporella is characterized by hairy conidiomata with long necks, and holoblastic conidiogenesis, features which are clearly distinguishable from Tiarosporella (Höhnel 1919; Crous et al. 2015). This genus is morphologically similar to Marasasiomyces (long-necked, hairy conidiomata, and holoblastic conidiogenesis), except that it forms conidiomata in clusters, which are not found in Marasasiomyces (Crous et al. 2015). Li et al. (2016) reported the sexual morph of Eutiarosporella in E. dactylidis for the first time from Avenella flexuosa (Poales). The sexual morph is characterised by globose ascomata, with a central ostiole, a two-layered peridium, hyphae-like pseudoparaphyses and hyaline, aseptate, fusoid to ovoid ascospores, with a mucilaginous sheath (Thambugala et al. 2014).
Based on ITS and LSU sequence data, three species were initially included in this genus, E. africana (Jami et al.) Crous, E. tritici (B. Sutton & Marasas) Crous and E. urbis-rosarum (Jami et al.) Crous by Crous et al. (2015). Subsequently, E. darliae E. Thynne et al., E. tritici-australis E. Thynne, et al. and E. dactylidis (Thambug., Camporesi & K.D. Hyde) Dissan., Camporesi & K.D. Hyde were accommodated in the genus (Crous et al. 2015; Thynne et al. 2015; Li et al. 2016), which now comprises seven species (Dissanayake et al. 2016; Wijayawardene et al. 2017).
Colony and conidial morphology are the primary characters to identify species within this genus. Colonies on nutrient-rich media (PDA or V8-OMA) grow rapidly (Thynne et al. 2015). However, we consider morphological characters alone are inadequate to identify species due to plasticity and overlapping of conidial dimensions. Therefore, incorporation of molecular data together with morphology is recommended.
Molecular based identification and diversity
Taxonomy of Eutiarosporella is largely based on DNA sequence data to reveal the phylogenetic relationships between the species (Crous et al. 2015; Thynne et al. 2015; Dissanayake et al. 2016; Li et al. 2016). According to studies by Crous et al. (2015), Thynne et al. (2015) and Li et al. (2016), ITS and LSU are the most suitable loci for delineation of species within the genus. The phylogram generated with sequences available in GenBank including ex-epitype sequences is provided in Fig. 14 (Table 8). Our phylogenetic analyses are in accordance with previous studies by Crous et al. (2015), Thynne et al. (2015), Dissanayake et al. (2016) and Li et al. (2016).

Phylogram generated from maximum likelihood analysis based on combined LSU and ITS sequence data retrieved from GenBank. The tree is rooted in Tiarosporella paludosa (CPC 22701 and CBS 114650). Tree topology of the ML analysis was similar to the Bayesian analysis. The best scoring RAxML tree with a final likelihood value of − 7055.996836 is presented. The matrix had 380 distinct alignment patterns, with 40.12% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.229604, C = 0.264209, G = 0.282595, T = 0.223593; substitution rates AC = 1.357965, AG = 1.612491, AT = 0.913118, CG = 2.194420, CT = 5.121479, GT = 1.000000; gamma distribution shape parameter α = 0.137391. Maximum likelihood bootstrap support values greater than 60% are indicated above the nodes. Ex-type (ex-epitype) and voucher strains are in bold
Recommended genetic markers (genus level)—LSU and SSU
Recommended genetic markers (species level)—ITS and LSU
Accepted number of species:Seven species.
References: Crous et al. (2015), Thynne et al. (2015), Dissanayake et al. (2016), Li et al. 2016 (morphology, phylogeny).
Ilyonectria P. Chaverri & Salgado, in Chaverri et al., Stud. Mycol. 68:69 (2011)
Species of Ilyonectria (Nectriaceae, Hypocreales) are important soil-borne pathogens of various woody and herbaceous plant hosts. Ilyonectria species are cosmopolitan and are found on a wide range of hosts (Chaverri et al. 2011). They are mostly associated with root diseases and stem cankers (Seifert et al. 2003; Halleen et al. 2004, 2006; Chaverri et al. 2011; Cabral et al. 2012a, b, c; Vitale et al. 2012; Lombard et al. 2013; Aiello et al. 2014). There are 23 species of Ilyonectria, all associated with disease symptoms of their original plant hosts (Chaverri et al. 2011; Cabral et al. 2012a,c; Lombard et al. 2013, 2014; Aiello et al. 2014). Ilyonectria is a well-known genus causing black foot rot of grapevines in various countries (Halleen et al. 2003, 2004, 2006; Chaverri et al. 2011; Cabral et al. 2012a, b, c; Lombard et al. 2014).
Classification—Sordariomycetes, Hypocreomycetidae, Hypocreales, Nectriaceae
Type species—Ilonectria radicicola (Gerlach & L. Nilsson) P. Chaverri & Salgado, in Chaverri et al., Stud. Mycol. 68:71 (2011)
Distribution—Worldwide
Disease symptoms—Black foot disease, black root rot
Symptoms are given under the genus Dactylonectria
Hosts—Wide host range including plant genera in Amaryllidaceae, Aracaceae, Araliaceae, Cupressaceae, Fagaceae, Liliaceae, Myrtaceae, Pinaceae, Proteaceae, Rosaceae, Strelitziaceae and Vitaceae (Farr and Rossman 2019).
Morphological based identification and diversity
The genus Ilyonectria was introduced based on I. radicicola as the type species, to accommodate Neonectria species belonging to the “N. radicicola” group (Booth 1959). This genus has asexual morphs and belonged to Booth’s Group 3 (chlamydospores and microconidia present, Booth 1966; Chaverri et al. 2011; Lombard et al. 2014). Molecular phylogenetic studies revealed that Ilyonectria, as originally conceived, was paraphyletic (Cabral et al. 2012a, c; Lombard et al. 2013, 2014). Conidial size, culture characters and molecular data enabled the separation of Ilyonectria species (Cabral et al. 2012a, c; Lombard et al. 2013, 2014).
Molecular based identification and diversity
Delineating between species of Ilyonectria can be achieved with histone (His3) gene region. The topologies of the phylogenetic tree (Fig. 15, Table 9) is similar to previous studies done on this genus (Cabral et al. 2012a; Lombard et al. 2014).

Phylogenetic tree generated by maximum likelihood analysis of combined ITS, TEF, TUB, and His3 sequence data of Ilyonectria species. Twenty-three strains are included in the analyses, which comprise 2242 characters including gaps. The tree is rooted in Campylocarpon fasciculare. Tree topology of the ML analysis was similar to the one generated from BI (Figure not shown). The best scoring RAxML tree with a final likelihood value of − 9669.617830 is presented. The matrix had 507 distinct alignment patterns, with 5.11% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.219321, C = 0.325516, G = 0.222910, T = 0.230587; substitution rates AC = 0.215721, AG = 0.328038, AT = 0.225653, CG = 0.615208, CT = 5.798530, GT = 1.000000; gamma distribution shape parameter α = 0.518017. Maximum likelihood bootstrap support values ≥ 60% and bayesian posterior probabilities ≥ 95 (BYPP) are indicated above or near the nodes. Ex-type strains are in bold
Recommended genetic markers (genus level)—ITS, LSU, tef1, TUB2
Recommended genetic markers (species level)—tef1, TUB2, His3
Accepted number of species:23 species
References: Booth 1959, 1966 (morphology), Cabral et al. 2012a, b, c; Lombard et al. 2013, 2014 (morphology, phylogeny).
Macrophomina Petr., Annls mycol. 21: 314 (1923)
Species of Macrophomina are mostly pathogens that cause damping-off, seedling blight, collar rot, stem rot, charcoal rot, basal stem rot and root rot in many plant species (Arora et al. 2001; Pal et al. 2001; Gupta et al. 2002; Sarr et al. 2014; Wijayawardene et al. 2017). The type species, Macrophomina phaseolina (Tassi) Goid., is a seed-borne polyphagous pathogen that affects more than 500 crop and non-crop species, including economically important crops, such as soybean, sunflower, common bean, peanut, corn, sorghum, cowpea and cotton (Gupta et al. 2002; Ndiaye et al. 2010; Sarr et al. 2014).
Classification—Dothideomycetes, incertae sedis, Botryosphaeriales, Botryosphaeriaceae
Type species—Macrophomina phaseolina (Tassi) Goid., Annali Sper. agr., N.S. 1(3): 457 (1947)
Distribution—Worldwide
Disease symptoms—Charcoal rot, collar rot, damping off, root rot, seedling blight, stem rot, wilt
Seedling damage can occur when infected seeds are planted. Infected plants may produce slightly smaller leaflets than healthy plants and have reduced vigour. As the disease advances, leaflets turn yellow, wilt and turn brown (Adorada et al. 2018). A grey/silver discolouration can be observed in the roots and lower stem when the plants split open (Romero Luna et al. 2017; Koehler and Shew 2018; Meena et al. 2018). In charcoal rot, the abundant production of minute black sclerotia by the fungus causes the rotted tissues to become blackened. Infections on soybean lead to early maturation and incomplete pod filling (ElAraby et al. 2003; Yang and Navi 2005; Sarr et al. 2014). In peanut, it causes seed and seedling rots, wilt, root and stem rots, leaf spot and rotting of developing pods and seeds (Gupta et al. 2002; Deshwal et al. 2003).
Hosts—This soil-borne fungus can infect more than 500 agricultural crops and weed species including, Fragaria, Glycine, Helianthus, Sorghum and Zea.
Morphological based identification and diversity
Eight species names are recorded in Index Fungorum (2019), however, sequences are available for only two species Macrophomina phaseolina and M. pseudophaseolina (Sarr et al. 2014). Morphological characteristics of M. phaseolina are mostly similar to M. pseudophaseolina, except that conidia of the latter are shorter.
Colony and conidial morphology are the primary characters used to identify species within this genus (Ellis 1971, 1976; Simmons 1992). However, the connectivity of sexual and asexual morphs is not proven, as no sexual morph has been obtained from nature or culture (Crous et al. 2006; Wijayawardene et al. 2017, 2018). According to the morphological identifications, Macrophomina phaseolina has conidia with apical mucoid appendages as found in Tiarosporella (Sutton and Marasas 1976). Nevertheless, it can be distinguished from Tiarosporella in having conidia with apical mucoid appendages, per currently proliferating conidiogenous cells and dark brown (at maturity) conidia (Crous et al. 2006; Phillips et al. 2013). Morphologically M. phaseolina is similar to M. pseudophaseolina, except that conidia of the latter are shorter.
Molecular based identification and diversity
Phillips et al. (2013) suggested that phylogenetic analysis of a combined SSU, LSU, ITS, tef1 and TUB2 genes provide better resolution. Sarr et al. (2014) used ITS, tef1, ACT, CAL and TUB2 sequence data representing a large sample of Macrophomina isolates from many hosts. According to the multi-gene analysis of SSU, LSU, ITS, tef1 and TUB2 genes in this study (Fig. 16, Table 10), the two species cluster in a well-supported clade with high bootstrap values (100% ML, 1.00 BYPP). The overall topology of our phylogeny tree is similar to previous studies.

Phylogenetic tree generated by maximum likelihood analysis of combined ITS, LSU, SSU, tef1 and TUB2 sequences. Related sequences were obtained from GenBank. Forty-four strains are included in the analyses, which comprise 3477 characters including gaps. Single gene analyses were carried out and compared with each species, to compare the topology of the tree and clade stability. The tree was rooted with Dothiorella iberica (CBS 113188 and CBS 115041). Tree topology of the ML analysis was similar to the MP and BI. The best scoring RAxML tree with a final likelihood value of − 12764.659013 is presented. The matrix had 898 distinct alignment patterns, with 28.31% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.238049, C = 0.253522, G = 0.272022, T = 0.236408; substitution rates AC = 1.121500, AG = 2.393284, AT = 1.053637, CG = 1.711098, CT = 4.682724, GT = 1.000000; gamma distribution shape parameter α = 0.545763. The maximum parsimonious dataset consisted of constant 2820, 575 parsimony-informative and 82 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of two equally most parsimonious trees with a length of 1490 steps (CI = 0.626, RI = 0.861, RC = 0.539, HI = 0.374) in the first tree. RAxML, maximum parsimony bootstrap support values ≥ 65% and Bayesian posterior probabilities ≥ 0.95 (BYPP) are shown respectively near the nodes. Ex-type strains are in bold
Recommended genetic markers (genus level)cosmopolitan genus, and—LSU and SSU
Recommended genetic markers (species level)—ITS, tef1, ACT, CAL and TUB2
Accepted number of species: There are eight epithets in Index Fungorum (2019) However, two species have molecular data.
References: Crous et al. 2006; Phillips et al. 2013; Sarr et al. 2014, Wijayawardene et al. 2017 (morphology, phylogeny).
Medeolaria Thaxt., Proc. Amer. Acad. Arts & Sci. 57(17): 432 (1922)
The genus Medeolaria belongs to the family Medeolariaceae (Medeolariales, Leotiomycetes, Ascomycota). Medeolaria was introduced by Thaxter (1922) and typified with Medeolaria farlowii. Medeolaria species are pathogens of Medeola virginiana (Liliaceae). Currently, the known distribution of this genus is only from America.
Classification—Leotiomycetes, Medeolariales, Medeolariaceae
Type species—Medeolaria farlowii Thaxt., Proc. Amer. Acad. Arts & Sci. 57(17): 432 (1922)
Distribution—America
Disease symptoms—This fungus causes gall-like deformations on thickened, hypertrophic 9 parts below leaf whorls of herbaceous stems of the host tissue, in autumn. However, they are present not only in stem lesions of the host plant but in uninfected leaves, stems and rhizomes (Pfister et al 2013). Pfister et al. (2013) also showed the long-term perpetuation of the fungus in populations of the plant. They suggested the fungus remains as a systemic infection of vegetative plant parts and when the plant reproduces clonally, this infection is carried in populations of the host plant (Pfister et al 2013).
Hosts—Magnolia spp.
Morphological based identification and diversity
This genus contains only a single species, Medeolaria farlowii Thaxter (1922), described from material collected from Magnolia. It produces erumpent, indefinite apothecia with a palisade layer of asci and paraphyses. An excipulum is absent or is a very thin layer. The hymenium layer forms fusiform swellings below and/or between the shortened internodes of the host plant. Ascospores are large, fusiform to naviculate, with a dark, striate outer wall. The asexual morph of this fungus is unknown (Korf 1973; Pfister and LoBuglio 2009; Ekanayaka et al 2017).
Molecular based identification and diversity
The first stable taxonomic placement for this genus was provided by Korf (1973) under the family Medeolariaceae, order Medeolariales within Leotiomycetes, according to its morphology. Recent phylogenetic studies (LoBuglio and Pfister 2010; Pfister et al 2013; Ekanayaka et al. 2017) confirmed its phylogenetic relationship with Leotiomycetes (Fig. 10), but the phylogenetic position within the class is unresolved.
Recommended genetic marker (genus level)—ITS
Recommended genetic marker (species level)—ITS
ITS is the best single genetic marker for the genus Medeolaria (Pfister et al 2013). Pfister et al. (2013) provided primers, designed to specifically amplify ITS rDNA regions of Medeolaria farlowii.
Accepted number of species: One species
References: Korf 1973; LoBuglio and Pfister 2010; Pfister et al. 2013(morphology, phylogeny).
Neonectria Wollenw., Annls mycol. 15(1/2):52 (1917)
Neonectria is a cosmopolitan genus, and their asexual morphs are common in tropical and temperate regions (Chaverri et al. 2011). Neonectria species can be found on the bark of recently dead woody plants and sometimes on decaying herbaceous material (Samuels and Brayford 1990; Samuels and Brayford 1990, 1993, 1994; Rossman et al. 1999; Castlebury et al. 2006; Chaverri et al. 2011). Some species of Neonectria are plant pathogens causing cankers and other diseases on hardwood and coniferous trees (Castlebury et al. 2006; Rossman and Palm-Hernández 2008; Crane et al. 2009; Chaverri et al. 2011; Schmitz et al. 2017; Wenneker et al. 2017). Neonectria neomacrospora has been added to the European and Mediterranean Plant Protection Organization (EPPO) alert list (EPPO, 2019).
Classification—Sordariomycetes, Hypocreomycetidae, Hypocreales, Nectriaceae
Type species—Neonectria ramulariae Wollenw., Annls mycol. 15(1/2):52 (1917)
Distribution—Worldwide
Disease symptoms—Canker
Dead shoots can be observed in the lower branches or all over the affected tree. Affected branches or trunks show canker and some may have abundant resin flow. When the canker girdles the affected area, part of the tree above the canker dies. Under humid conditions characteristic small, red fruiting bodies will be formed. Badly affected trees will eventually die (Castlebury et al. 2006).
Beech (Fagus) bark disease is caused by N. coccinea, N. ditissima, N. fuckeliana and N. faginata. Cankers of fruit trees are caused by N. rugulosa and N. ditissima. Shoot dieback of Abies species are caused by N. neomacrospora (Castlebury et al. 2006; Rossman et al. 2008; Crane et al. 2009; Chaverri et al. 2011; Schmitz et al. 2017; Wenneker et al. 2017).
Hosts—Wide host range including plant genera in Amaryllidaceae, Aracaceae, Araliaceae, Betulaceae, Ericaceae, Fagaceae, Lauraceae, Myrtaceae, Pinaceae, Proteaceae, Rosaceae, Sapindaceae and Vitaceae (Farr and Rossman 2019).
Morphological based identification and diversity
The genus Neonectria was established by Wollenweber (1917). The generic concept of Neonectria has been revised by different authors (Booth 1959; Samuels and Brayford 1994; Rossman et al. 1999). Rossman et al. (1999) accepted only three species (N. coccinia, N. galligena and N. ramulariae) in Neonectria. Subsequently, species were added to the genus based on morphology and/or phylogeny (Hirooka et al. 2005; Castlebury et al. 2006; Luo and Zhuang 2010a, b; Zhao et al. 2011; Lombard et al. 2014, 2015). However, some unrelated species were transferred to other genera based on molecular analyses and morphological data (Lombard et al. 2014, 2015). There are 31 species recognised in the genus, while 23 species have sequence data in GenBank (4/7/2019). Morphological characters (perithecial morphology, ascospore size, macroconidial morphology, presence or absence of microconidia and chlamydospores) along with DNA sequence analysis are appropriate for identification of Neonectria species (Brayford et al. 2004).
Molecular based identification and diversity
Since 2001, DNA sequence analysis has been used to clarify the taxonomy of Neonectria (Mantiri et al. 2001; Brayford et al. 2004; Halleen et al. 2004; Hirooka et al. 2005; Chaverri et al. 2011). Mantiri et al. (2001) and Brayford et al. (2004) used mtSSU rDNA sequence data to infer intrageneric relationships of some Neonectria and Cylindrocarpon species. Later, Halleen et al. (2004) used mtLSU rDNA, TUB2 and nrDNA ITS regions to separate some Cylindrocarpon species included in the N. mammoidea group. Chaverri et al. (2011) approached a comprehensive treatment of Cylindrocarpon and Neonectria based on combined loci analyses and morphological data. Chaverri et al. (2011) defined Neonectria sensu stricto within Nectriaceae with Cylindrocarpon sensu stricto based on multi-gene phylogeny of ITS, LSU, tef1, TUB2, ACT, and RPB1. The ITS, tef1 and TUB2 loci possess highly variable regions (Chaverri et al. 2011) and are important in species delimitation of Neonectria. Rossman et al. (2013) proposed to protect generic name Neonectria over Cylindrocarpon. Maharachchikumbura et al. (2015) considered Cylindrodendrum not to be congeneric with Neonectria and accepted Neonectria over Cylindrocarpon.
This study reconstructs the phylogeny of Neonectria based on analyses of a combined ITS, LSU, tef1 and TUB2 sequence data (Table 11, Fig. 17). The phylogenetic tree is updated with recently introduced Neonectria species and corresponds to previous studies (Chaverri et al. 2011; Lombard et al. 2014; Mantiri et al. 2001).

Phylogenetic tree generated by maximum likelihood analysis of combined ITS, LSU, tef1 and TUB sequence data of Neonectria species. Related sequences were obtained from GenBank. Twenty-three strains are included in the analyses, which comprise 2336 characters including gaps. Tree topology of the ML analysis was similar to the one generated from BI (figure not shown). The best scoring RAxML tree with a final likelihood value of − 7942.756270 is presented. The matrix had 525 distinct alignment patterns, with 18.13% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.223071, C = 0.285136, G = 0.261197, T = 0.230596; substitution rates AC = 1.213729, AG = 2.500008, AT = 1.727890, CG = 0.720430, CT = 6.191594, GT = 1.000000; gamma distribution shape parameter α = 0.749195. Maximum likelihood bootstrap support (≥ 55%) and posterior probabilities (BYPP ≥ 0.90) from Bayesian inference analysis are indicated respectively near the nodes. Ex-type strains are in bold. The tree is rooted in Thelonectria gongylodes
Recommended genetic markers (genus level)—LSU, ITS, tef1 and TUB2
Recommended genetic markers (species level)—ITS, tef1 and TUB2
Accepted number of species: 28 species
References: Rossman et al. 1999 (morphology), Brayford et al. 2004; Hirooka and Kobayashi 2007; Chaverri et al. 2011; Lombard et al. 2014 (morphology, phylogeny).
Neopestalotiopsis Maharachch., K.D. Hyde & Crous (2014), in Maharachchikumbura et al., Stud. Mycol. 79:147 (2014a)
Neopestalotiopsis is an important plant pathogenic, saprobic and endophytic genus commonly present in tropical and subtropical ecosystems. The genus was introduced by Maharachchikumbura et al. (2014b). Species of Neopestalotiopsis are appendage-bearing asexual coelomycetes in the family Sporocadaceae (Jayawardena et al. 2016).
Classification—Sordariomycetes, Xylariomycetidae, Amphisphaeriales, Sporocadaceae
Type species—Neopestalotiopsis protearum (Crous & L. Swart) Maharachch. et al., in Maharachchikumbura et al., Stud. Mycol. 79:147 (2014a)
Distribution—Worldwide
Disease symptoms—Canker, dieback, fruit rots, leaf spot
Pathogenic Neopestalotiopsis are recorded in post-harvest fruit rots of grapes, trunk diseases in grapevine in China, India and France, leaf spot disease of grapevine in China and leaf blights in many plant species worldwide (Hyde et al. 2014; Jayawardena et al. 2015, 2016; Maharachchikumbura et al. 2017).
Neopestalotiopsis species infect a variety of grapevine cultivars, causing diseases including grapevine dieback, fruit rot, postharvest disease and severe defoliation. Initial symptoms of fruit rot disease are mostly observed at the splits between the pedicel and the berry and at the wounds of the fruits and severely infected fruits become rotten and separate completely from the pedicel (Jayawardena et al. 2015). Neopestalotiopsis asiatica and N. javaensis are associated with grapevine trunk disease (Maharachchikumbura et al. 2017). Grapevine trunk diseases reduce the yield and quality of grapes, even leading to partial or total death of individual plants.
Neopestalotiopsis clavispora and N. surinamensis cause guava scab (Solarte et al. 2018). Neopestalotiopsis ellipsospora causes leaf spot on sweet potatoes (Maharachchikumbura et al. 2016). Neopestalotiopsis clavispora causes crown and root rot of strawberry worldwide while N. iranensis infects leaves and fruits of strawberry (Ayoubi and Soleimani 2016), with the pathogen initially developing circular, black, and slightly sunken spots that expand outwards on the surface. Droplets of spores are scattered over the white aerial mycelial area and later cause soft decay of the fruit flesh (Ayoubi and Soleimani 2016). Canker and dieback on blueberry in Chile and Uruguay are also caused by N. clavispora (Espinoza et al. 2008; González et al. 2012; Chamorro et al. 2016).
Neopestalotiopsis samarangensis has been described from wax apple fruit rot in Thailand (Maharachchikumbura et al. 2013a, b). In fruit rots, the initial symptom is small, circular, black, slightly sunken spots on fruits. Later, the spots enlarged rapidly, become sunken and result in a soft decay of the fruit flesh (Maharachchikumbura et al. 2013a, b).
Hosts—Species of Fragaria × ananassa, Ipomoea, Malus, Psidium, Vaccinium and Vitis
Morphological based identification and diversity
Neopestalotiopsis species can be differentiated using morphology and molecular phylogeny (Maharachchikumbura et al. 2014b). There are 36 species epithets listed in Index Fungorum (2019). Neopestalotiopsis species differ from Pestalotiopsis and Pseudopestalotiopsis in having somewhat versicolorous median cells (Maharachchikumbura et al. 2014b) whereas both Pestalotiopsis and Pseudopestalotiopsis have concolourous median cells (Maharachchikumbura et al. 2014b) as well as its conidiophores which are indistinct and often reduced to conidiogenous cells (Maharachchikumbura et al. 2014b).
Conidial morphology is widely used in taxonomy in pestalotioid fungi (Steyaert 1949; Guba 1961; Nag Raj 1993; Maharachchikumbura et al. 2012, 2014b). Species delimitation based on morphological characters is limited as these characters are plastic and vary between hosts and environments (Maharachchikumbura et al. 2011, 2016). Therefore, phylogenetic species recognition is an effective method to identify different pestalotioid species (Maharachchikumbura et al. 2016).
Molecular based identification and diversity
Neopestalotiopsis species can be roughly separated from Pestalotiopsis and Pseudopestalotiopsis based on the total number of base pairs in the ITS region (Maharachchikumbura et al. 2014b). However, the use of ITS sequences alone does not resolve Neopestalotiopsis species (Maharachchikumbura et al. 2012). Therefore, Maharachchikumbura et al. (2014b) suggested using combined ITS, TUB2 and tef1 genes to provide a better resolution in phylogenetic analyses. This study reconstructs the phylogeny of Neopestalotiopsis based on a combined ITS, TUB2 and tef1 sequence data (Fig 18, Table 12) and reveals similar phylogenetic relationships to previous studies by Maharachchikumbura et al. (2014b, 2016).

Phylogram generated from maximum likelihood analysis based on combined ITS, TUB2 and tef1 sequence data of Neopestalotiopsis species. Related sequences were obtained from GenBank. Forty-three strains are included in the combined sequence analyses, which comprise 1391 characters with gaps. Pestalotiopsis diversiseta (MFLUCC 12-0287) is used as the outgroup taxa. The best scoring RAxML tree with a final likelihood value of − 5457.035085 is presented. The matrix had 409 distinct alignment patterns, with 6.30% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.231067, C = 0.270889, G = 0.213946, T = 0.284098; substitution rates AC = 0.847461, AG = 2.876343, AT = 1.282349, CG = 0.723831, CT = 3.850003, GT = 1.000000; gamma distribution shape parameter α = 0.235476. The maximum parsimonious dataset consisted of 1026 constant, 177 parsimony-informative and 188 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of ten equally most parsimonious trees with a length of 650 steps (CI = 0.688, RI = 0.609, RC = 0.419, HI = 0.312) in the first tree. RAxML and maximum parsimony bootstrap support value ≥ 50% and posterior probabilities BYPP ≥ 0.90 from Bayesian inference analysis are indicated respectively near the nodes, are shown respectively near the nodes. Ex-type strains are in bold
Recommended genetic marker (genus level)—LSU
Recommended genetic markers (Species level)—ITS, TUB2 and tef1
Accepted number of species:41 species.
References: Maharachchukumbura 2012, 2014b (morphology, phylogeny); Maharachchukumbura et al. 2016 (morphology, phylogeny); Jayawardena et al. 2015, 2016 (morphology, phylogeny, pathogenicity)
Plasmopara J. Schröt., in Cohn, Krypt.-Fl. Schlesien 3.1(9–16): 236 (1886) [1889]
The genus Plasmopara belongs to the family Peronosporaceae of the Peronosporales in Oomycetes (Riethmüller et al. 2002; Görg et al. 2017). This genus is included in this study as it is an important plant pathogen on many economically important crops. Plasmopara was introduced by Schröter (1886), but no species was assigned as a type for the genus (Constantinescu et al. 2005). Plasmopara nivea is considered as the type species of this genus (Constantinescu et al. 2005). Plasmopara species are commonly known as downy mildew pathogens. There are 2064 records in USDA fungal database under genus Plasmopara (Farr and Rosman 2019). Downy mildew has become one of the most troublesome diseases in agriculture including P. viticola on grape, P. geranii on geranium and P. halstedii on sunflower (McTaggart et al. 2015). Kamoun et al. (2015) categorized P. viticola among the top ten Oomycetes pathogens in plant pathology. Current interest in this genus is to understand the co-evolution with the host and effective disease management (Thines and Kamoun 2010). Taxonomically useful morphological or ecological characters are few for the downy mildews and this makes identification of synapomorphic states impossible (Göker et al. 2003).
Classification—Oomycota incertae sedis, Peronosporea, Peronosporidae, Peronosporales, Peronosporaceae
Type species—Plasmopara nivea (Unger) J. Schröt.
Distribution—Worldwide
Disease symptoms—Downy mildew
Hosts—Species belonging to this genus are obligate biotrophs on a wide range of hosts including Acanthaceae, Asteraceae, Balsaminaceae, Geraniaceae, Malvaceae, Onagraceae, Orobanchaceae, Violaceae and Vitaceae (Voglmayr et al. 2004; Thines and Kamoun 2010; McTaggart et al. 2015).
Morphological based identification and diversity
Wilson (1907) was the first to consider P. pygmaea as the type of Plasmopara. However, even after a century of discussion of nomenclatural and taxonomic problems of this genus, Plasmopara was segregated in two different groups by morphology and phylogeny (Constantinescu et al. 2005). Constantinescu et al. (2005) proposed to introduce a new generic name for P. pygmaea and for six other related species and they have established the current classification for Plasmopara.
Species belonging to Plasmopara have the following characters. Hyphae are intercellular, haustoria are intracellular, as obpyriform, globose, or slightly elongated vesicles (Göker et al. 2003; Voglmayr et al. 2004; Constantinescu et al. 2005). A callose sheath often surrounds haustoria. Sporangiophores are mostly present on the under leaf surface of the host, but sometimes also on other parts of the plant (eg. P. viticola produces sporangiophores on inflorescences and young berries, Zhang et al. 2017). Sporangiophores are colourless, branched in the upper part. They branch monopodially, in two to more orders (Göker et al. 2003; Constantinescu et al. 2005). Branches are more or less divergent, ending in a number of elongated ultimate branchlets. The newly formed wall closing the tip after sporangium discharge (Constantinescu et al. 2005). Callose plugs usually present in trunk and/or branches. Sporangiogenesis is holoblastic. These species produce sporangia synchronously, which vary in shape, sporangia wall is colourless, appearing smooth in light microscopy but showing various types of ornamentations in the electron microscope (Constantinescu et al. 2005). There are 199 epithets listed in Index Fungorum (2019), however, 40 of them do not belong to Plasmopara based on phylogenetic evidence (Figs. 19, 20).

Grape downy mildew disease symptoms. a Infected grapevines, b–c appearance of oil spot on the upper leaf surface. d Sporulation on the lower leaf surface. e Young infected berries with sporulation. f Infection on fruits

Disease cycle of grape downy mildew. Redrawn from Kassemeyer (2017)
Oospores develop a single germ tube, terminating with sporangium, once a sporangium disseminated, by rain flash or wind, it releases zoospores (Ash 2000; Rossi et al. 2008; Carisse 2016; Kamoun et al. 2015; Wilcox et al. 2015).
Molecular based identification and diversity
Molecular and phylogenetic studies have shown that Plasmopara is polyphyletic (Riethmüller et al. 2002; Göker et al. 2003, 2007; Voglmayr et al. 2004; Voglmayr and Constantinescu 2008). Even though Wilson (1907) proposed P. pygmaea as the type species of the genus Plasmopara, it has a close relationship with Bremia, Paraperonospora and Basidiophora (Göker et al. 2003). With these morphological and phylogenetic aspects, many species that are traditionally included in Plasmopara have moved into new genera. The newly introduced genera are Viennotia (Göker et al. 2003), Protobremia (Voglmayr et al. 2004), and Plasmoverna (Constantinescu et al. 2005). Constantinescu et al. (2005) resolved Plasmopara phylogeny, introducing Plasmoverna as a new genus to accommodate the morphologically dissimilar and polyphyletic taxa belonging to previous classifications. Voglmayr and Constantinescu (2008) re-classified three species of Plasmopara into new genus Novotelnova Voglmayr & Constant. The species belonging to Novotelnova were identical in the analyses of the nuLSU and nuSSU-ITS1-5.8S datasets. Therefore, in the present study, we follow Voglmayr and Constantinescu (2008) to provide a backbone tree for Plasmopara using combined nuLSU sequence data (Fig. 21, Table 13).

Phylogenetic tree generated by maximum parsimony analysis of LSU sequence data of Plasmopara species. Related sequences were obtained from GenBank. Nineteen strains are included in the analyses, which comprise 1263 characters including gaps. The tree was rooted with Phytophthora arece (AR 234). Tree topology of the MP analysis was similar to the ML. The maximum parsimonious dataset consisted of constant 1053, 110 parsimony-informative and 100 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of two equally most parsimonious trees with a length of 365 steps (CI = 0.641, RI = 0.539, RC = 0.345, HI = 0.359) in the first tree. The best scoring RAxML tree with a final likelihood value of − 3660.849005 is presented. The matrix had 254 distinct alignment patterns, with 30.91% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.229156, C = 0.177763, G = 0.308486, T = 0.284595; substitution rates AC = 0.706031, AG = 4.368078, AT = 1.048734, CG = 0.141039, CT = 7.063364, GT = 1.000000; gamma distribution shape parameter α = 0.163551. RAxML and maximum parsimony bootstrap support value ≥ 50% (BT) are shown respectively near the nodes
The downy mildew pathogens have been studied extensively to understand their host specificity and co-evolution with the host plants. The grape downy mildew has been identified as a host-specific cryptic species (Rouxel et al 2013; Zhang et al. 2017). Rouxel et al. (2013) considered the cryptic species as formae speciales: P. viticola f. sp. riparia (lineage A occurring on V. riparia and some hybrids); P. viticola f. sp. aestivalis (lineage B found on V. aestivalis, V. labrusca, V. vinifera and some hybrids); P. viticola f. sp. vinifera (lineage C occurring on V. vinifera and some hybrids); P. viticola f. sp. quinquefolia (lineage D found on V. quinquefolia). To understand the cryptic lineages genealogical concordance phylogenetic species recognition (GCPSR) approach is currently accepted (Taylor et al. 2000; Rouxel et al. 2013). GCPSR facilitate the most convenient analysis for species that cannot be cultivated or mate in control conditions (O’Donnell et al. 2000; Steenkamp et al. 2002; Rouxel et al. 2013).
Recommended genetic marker (genus level)—LSU
Recommended genetic marker (species level)—LSU
The universal barcode for the Oomycetes, the cytochrome oxidase subunit 1 and 2 genes (cox 1 and cox 2) are used. However Choi et al. (2015) suggested cox2 is better suited to this because of its ease of amplification among oomycete lineages, better performance on herbarium specimens, higher discriminatory power at the species level and the availability of a large taxonomically diverse database that already includes many species of oomycetes, especially the downy mildew. In previous studies, nuLSU gene regions (D1–D3 and D7–D8 sequences) were widely used (Riethmüller et al. 2002; Göker et al. 2003, 2007; Voglmayr et al. 2004; Voglmayr and Constantinescu 2008). Branch supports of the backbone tree was often higher in the nuLSU data, which resulted in a larger data matrix and a higher number of parsimony-informative characters than when ITS was used (Voglmayr and Constantinescu 2008). No study has combined these gene regions or any other gene regions as the marker to understand the phylogenetic relationship within the genus.
Accepted number of species: There are 199 species in Index Fungorum (2019) and only 19 species have molecular data in this genus.
References: Riethmüller et al. 2002; Göker et al. 2003; 2007; Voglmayr et al. 2004; Voglmayr and Constantinescu 2008; Rouxel et al 2013; Zhang et al. 2017 (morphology, phylogeny).
Pseudopestalotiopsis Maharachch., K.D. Hyde & Crous (2014), in Marachchikumbura et al., in Maharachchikumbura et al., Stud. Mycol. 79:180 (2014a)
The genus was introduced by Maharachchikumbura et al. (2014b) with Pseudopestalotiopsis theae (Sawada) Maharachch., K.D. Hyde & Crous as the type species. Species of Pseudopestalotiopsis are appendage-bearing phenotypically diverse coelomycetes in the family Sporocadaceae and are commonly found in tropical and subtropical ecosystems (Jaklitsch et al. 2016; Maharachchikumbura et al. 2016). Pseudopestalotiopsis is characterized by brown to dark brown or olivaceous median cells and knobbed or not knobbed apical appendages (Maharachchikumbura et al. 2014b, 2016). The epitype of Pseudopestalotiopsis theae (Sawada) Steyaert was designated from fresh leaves of Camellia sinensis collected in Thailand (Maharachchikumbura et al. 2013a, b). Pseudopestalotiopsis has been studied for the production of various secondary metabolites with diverse structural features, with antitumour, antifungal, antimicrobial and other activities (Ding et al. 2008; Maharachchikumbura et al. 2011, 2016).
Pseudopestalotiopsis theae is economically significant as it has been identified as a pathogen in major tea-growing areas in the world (Maharachchikumbura et al. 2016). Pseudopestalotiopsis theae causes grey blight of tea and reduces yield (Maharachchikumbura et al. 2011, 2013a, b, 2016). Pseudopestalotiopsis theae was also isolated as an endophyte from different hosts (Camellia nitidissima, C. sinensis, Holarrhena antidysenterica, Podocarpus macrophyllus, Terminalia arjuna) or as a saprobe (seeds of Diospyros crassiflora) (Maharachchikumbura et al. 2011, 2013a, b, 2016).
Classification—Sordariomycetes, Xylariomycetidae, Amphisphaeriales, Sporocadaceae
Type species—Pseudopestalotiopsis theae (Sawada) Maharachch., in Maharachchikumbura et al., Stud. Mycol. 79:183 (2014a)
Distribution—China, India, Indonesia, Malaysia, Thailand (Maharachchikumbura et al. 2016)
Disease symptoms—Pseudopestalotiopsis theae causes grey blight in major tea growing areas in the world (Horikawa 1986, Maharachchikumbura et al. 2013a, b, 2016). The pathogen develops circular to irregular leaf spots initially and grey, brown margins when mature, covering up to half of the leaf with acervuli (Maharachchikumbura et al. 2016). Pseudopestalotiopsis ixorae and P. taiwanensis cause a leaf spot which initially develops small, circular, ash-coloured spots which later turn into brown spots (Tsai et al. 2018).
Hosts—Averrhoa carambola, Camellia sp., Cinnamomum sp., Cocos nucifera, Diospyros crassiflora, Fragaria sp., Hibiscus rosa-sinensis, Holarrhena antidysenterica, Ixora sp., Kandelia obovate, Macaranga sp., Pandanus odoratissimus, Podocarpus macrophyllus, Prunus sp., Terminalia arjuna and Thea sinensis
Morphological based identification and diversity
Pseudopestalotiopsis can be distinguished from Neopestalotiopsis and Pestalotiopsis by dark concolourous median cells with indistinct conidiophores (Maharachchikumbura et al. 2014b, 2016). However, there could be a wide host range for Pseudopestalotiopsis species and the actual number of species could be much higher than presently known (Maharachchikumbura et al. 2011, 2016).
Conidial morphology is widely used in taxonomy in pestalotioid fungi (Steyaert 1949; Guba 1961; Nag Raj 1993; Maharachchikumbura et al. 2011, 2012, 2014b). Species delimitation based on morphological characters is limited as these characters are plastic and vary between hosts and environments (Maharachchikumbura et al. 2011, 2016). Therefore, phylogenetic species recognition is an effective method to identify different pestalotioid species (Maharachchikumbura et al. 2016).
Molecular based identification and diversity
ITS sequence data alone is not sufficient for species delimitation of Pseudopestalotiopsis. Therefore, Maharachchikumbura et al. (2012) suggested a phylogenetic analysis of combined ITS, TUB2 and tef1 genes provide better resolution as compared to single gene phylogeny (Fig. 22, Table 14).

Phylogram generated from maximum likelihood analysis based on combined ITS, TUB2 and tef1 sequence data of Pseudopestalotiopsis species. Related sequences were obtained from GenBank. Twenty-five strains are included in the combined sequence analyses, which comprise 1404 characters with gaps. Neopestalotiopsis natalensis (CBS 138.41) was used as the outgroup taxa. The best scoring RAxML tree with a final likelihood value of − 4028.799660 is presented. The matrix had 274 distinct alignment patterns, with 6.34% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.235765, C = 0.270775, G = 0.213073, T = 0.280387; substitution rates AC = 1.242401, AG = 3.217138, AT = 1.272343, CG = 0.837226, CT = 4.463116, GT = 1.000000; gamma distribution shape parameter α = 0.229606. The maximum parsimonious dataset consisted of 1122 constant, 79 parsimony-informative and 203 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of four equally most parsimonious trees with a length of 386 steps (CI = 0.832, RI = 0.737, RC = 0.613, HI = 0.168) in the first tree. RAxML and maximum parsimony bootstrap support value ≥ 50% are shown respectively near the nodes. Bayesian posterior probabilities ≥ 0.95 (BYPP) indicated as thickened black branches. Ex-type strains are in bold
Recommended genetic markers (genus level)—LSU (as outlined in Maharachchikumbura et al. 2012)
Recommended genetic markers (species level)—ITS, TUB2 and tef1 (as outlined in Maharachchikumbura et al. 2012)
Accepted number of species: 20 species
References: Maharachchukumbura 2013a, b, 2014b, 2016b (morphology, phylogeny)
Rosellinia De Not., G. bot. ital.1 (1): 334(1844)
Rosellinia (Xylariaceae) species are characterized mainly as saprobes, some endophytes and occasionally as pathogens. They have a worldwide distribution and common in both temperate and tropical regions (Petrini 1993, 2013; ten Hoopen and Krauss 2006). Plant pathogenic Rosellinia species play a vital role in economically important crops, trees and ornamental plants. Rosellinia desmazieresii and R. necatrix are mostly known from temperate regions, while R. bunodes is known only from the tropics causing root rot on fruit trees and vines (Agrios 2005; ten Hoopen and Krauss 2006). Among the root diseases cause by Rosellinia species, R. bunodes is responsible for black root rot, R. necatrix for white root rot and R. pepo for stellate root rot (Castro et al. 2013). Species of this genus can survive as microslerotia in wood, roots and soil and the infection spreads through feeder roots when they contact hyphae or microsclerotia (Ploetz et al. 2003).
Rosellinia was introduced to accommodate species which are characterized by uniascomatal and carbonaceous stromata that develop within a subiculum. There have been different contradiction placements of Rosellinia. Miller (1928) placed it in the family Xylariaceae and this was confirmed in morphology and phylogeny-based studies later (Hsieh et al. 2010; Daranagama et al. 2015). Daranagama et al. (2018) and Wendt et al. (2018) revealed that Rosellinia is closely related to Entoleuca and Nemania.
Classification—Sordariomycetes, Xylariomycetidae, Xylariales, Xylariaceae
Type species—Rosellinia aquila (Fr.) Ces. & De Not., G. bot. ital.1 (1): 334(1844)
Distribution—Worldwide
Disease symptoms—Root rots
Black root rot is characterized by the occurrence in patches that extend in a circular pattern. Rosellinia bunodes the main causal agent of black root rot typically shows black branching strands that are firmly attached to the roots and may form condensed irregular knots and chlorotic leaves may shed gradually (Sivanesan and Holiday 1972; Oliverira et al. 2008).
The symptoms of white root rot caused by R. necatrix in the upper parts of the plants (such as yellow foliage, shrivelled fruits, no new growth) cannot be recognized in early stages of root infection. Cottony, white mycelia cover feeder roots of a tree and decay sets in. Mycelia grow into the soil and upward in the tree forming small, pale patches under or in the bark of major roots, root crown and lower trunk which eventually decay. A purple canker in wood at the root crown of young trees can also be caused by the fungus. Diseased trees will defoliate and premature death may occur (Pérez-Jiménez 2006; Pasini et al. 2016).
Hosts—This genus has a wide range of hosts including Adoxaceae, Annonaceae, Apiaceae, Asteraceae, Betulaceae, Celastraceae, Convolvulaceae, Euphorbiaceae, Fabaceae, Fagaceae, Grossulariaceae, Juglandaceae, Lauraceae, Moraceae, Myrtaceae, Oleaceae, Pinaceae, Poaceae, Rosaceae, Rutaceae, Salicaceae, Sapindaceae, Scrophulariaceae, Tamaricaceae, Verbenaceae, Vitaceae and Zingiberaceae.
Morphological based identification and diversity
The genus is characterized by globose-subglobose, uni- to multiloculate, often collapsed ascomata, mostly detached from the stroma wall,; septate, hyaline paraphyses, asci that are 8-spored, unitunicate, cylindrical to clavate, long pedicellate, rounded at the apex, with J + apical ring bluing in Melzer’s reagent, massive barrel-shaped with distinctive rings ascospores that are uniseriate, unicellular, elongated ellipsoidal-fusiform, light to dark brown, with germ slits, cellular appendages and/or may be slimy sheaths or caps and a dematophora-like or geniculosporium-like asexual morphs (Daranagama et al. 2018).
Rosellinia is a large genus with 483 epithets in Mycobank, 517 in Index Fungorum and 311 in Global Biodiversity Information Facility (GBIF); there are currently 158 accepted species (Petrini 2013; Li and Guo 2015; Li et al. 2015, 2016; Su et al. 2016; Crous et al. 2017; Fournier et al. 2017a, b; Tibpromma et al. 2017). Petrini (2013) found seven morphologically distinct groups with distinguishing morphological characters associated with the shape, size and orientations of stroma, ostiole, ascospores and germ slit, which can be used for species delimitation (Fig. 23).

Rosellinia bunodes (holotype, K(M) 62957, Sri Lanka, Peradeniya, on dead wood, November 1867, G.H.K. Thwaites) a, e Stromata (e ostiole white arrow). b–d Herbarium details. f Paraphyses. g–j Ascospores (g germ slit black arrow). Scale bars: e = 500 µm, g–j = 20 µm, f = 5 µm
Molecular based identification and diversity
Protein coding gene sequences are available for nine species of Rosellinia, mostly with only ITS and LSU sequence data. However, with the limited data, Daranagama et al. (2018) provided an updated backbone tree for genera in Xylariaceae, and Rossellinia clustered with Nemania and Entoleuca. Several phylogenetic studies have focused on pathogenic species such as R. bunodes and R. pepo. Castro et al. (2013) investigated R. bunodes and R. pepo isolated from Coffea arabica (Rubiaceae), Hevea brasiliensis (Euphorbiaceae), Macadamia integrifolia (Proteaceae), Psidium guajava (Myrtaceae) and Theobroma cacao (Malvaceae) using ITS based phylogenetic analyses from Colombia. Another ITS-based phylogenetic study identified R. necatrix, the pathogen responsible for white root disease on Aronia melanocarpa (Rosaceae) in Korea (Choi et al. 2017).
This study reconstructs the phylogeny of Rosellinia based on analyses of combined ITS, LSU and RPB2 sequence data (Table 7, Fig. 13). The phylogenetic tree is updated with recently introduced Rosellinia species and corresponds to previous studies (Li et al. 2015, 2016; Su et al. 2016; Crous et al. 2017; Fournier et al. 2017a, b; Tibpromma et al. 2017).
Recommended genetic markers (genus level)—LSU, ITS
Recommended genetic marker (species level)—ITS
Based on several studies and the availability of sequence data, ITS based phylogenetic studies are sufficient to identify Rosellinia to species level. There are few other studies carried out using LSU, ITS and RPB2 sequences. With a lack of sequence data for most species, there are some contradictions for the species and generic delimitat ion.
Accepted number of species: 158with only 26 species with molecular data
References: Petrini 2013; Li and Guo 2015; (morphology), Castro et al. 2013; Li et al. 2015, 2016; Crous et al. 2017; Fournier et al. 2017a, b; Tibpromma et al. 2017, Daranagama et al. 2018 (morphology, phylogeny), Shimizu et al. 2012; dos Santos et al. 2017; Arjona-Girona and López-Herrera 2018; Kleina et al. 2018 (pathogenicity).
Sphaeropsis Sacc., Michelia 2: 105. 1880.
The genus Sphaeropsis was introduced by Saccardo (1880) (for species of Diplodia with brown, aseptate conidia), with S. visci as the type species. Sphaeropsis is the asexual morph of Phaeobotryosphaeria (Phillips et al. 2008, 2013; Wijayawardene et al. 2017). Species in Sphaeropsis seem to be cosmopolitan in distribution since they have been recorded from both temperate and tropical countries (i.e. Germany, New Zealand, South Africa, Thailand (Phillips et al. 2013; Slippers et al. 2014; Farr and Rossman 2019). Host specificity of Sphaeropsis has not yet been clarified and species have been recorded from various plant families.
Classification—Dothideomycetes, incertae sedis, Botryosphaeriales, Botryosphaeriaceae
Type species—Sphaeropsis visci (Alb. & Schwein.) Sacc.
Distribution—Worldwide
Disease symptoms—calyx-end rot, stem end rot
The decayed tissues in rot diseases are firm or spongy and brown in colour. The skin of decayed areas generally remains brown or dark brown but may appear dark in aged areas (Kim et al. 2005).
Hosts—Broad range of hosts, including Myrtaceae, Rutaceae, Santalaceae and Vitaceae.
Morphological based identification and diversity
Over 600 species names are listed in Index Fungorum (2019), but few of them are currently in use and for most species cultures are not available except for S. citrigena (A.J.L. Phillips et al.) A.J.L. Phillips & A. Alves, S. eucalypti Berk. & Broome, S. porosa (Van Niekerk & Crous) A.J.L. Phillips & A. Alves, and S. visci (Alb. & Schwein.) Sacc. Pycnidial paraphyses in Sphaeropsis species distinguish this genus from Diplodia species, which do not have paraphyses. The aseptate, smooth-walled conidia of Sphaeropsis species differentiate them from Lasiodiplodia species, which have 1-septate, striate conidia. Recently, S. variabilis was transferred to a separate genus, Oblongocollomyces due to distinct morphological differences (Yang et al. 2017).
Colony and conidial morphology are the primary characters to identify species within this genus (Ellis 1971, 1976; Simmons 1992). The sexual and asexual morphs connection of Sphaeropsis was established by Phillips et al. (2008) who obtained coelomycetes with large, brown, aseptate conidia typical of Sphaeropsis from Phaeobotryosphaeria culture. Four Sphaeropsis species have been identified from culture. Sphaeropsis porosa differs from other species in having distinct pitted conidial walls. Sphaeropsis visci and S. citrigena can be distinguished from each other with their conidial pigmentation and swollen paraphyses tips (Phillips et al. 2013).
Molecular based identification and diversity
Phillips et al. (2013) suggested that phylogenetic analysis of combined SSU, LSU, ITS, tef1 and TUB2 genes provide better resolution compared to ITS alone. This study provides the phylogenetic analyses of combined ITS, LSU, SSU, tef1 and TUB2 sequence data (Table 10, Fig. 16). The topology of the Sphaeropsis species tree is identical to the phylogeny tree of Phillips et al. (2013).
Recommended genetic markers (genus level)—LSU and SSU
Recommended genetic markers (species level)—ITS, tef1 and TUB2
Accepted number of species: There are 624 species epithets in Index Fungorum (2019) under this genus. However, only four species have sequence data.
References: Phillips et al. 2013; Yang et al. 2017 (morphology, phylogeny).
Updates on important phytopathogens
Alternaria Nees, Syst. Pilze (Würzburg): 72 (1816)
Species of Alternaria are saprotrophs on dead vegetation and are frequently isolated from soil, air, dust and water-damaged buildings (Ellis 1971, 1976; De Hoog and Horré 2002; Runa et al. 2009; Woudenberg et al. 2013; Lawrence et al. 2016). The majority of species, however, are pathogens, infecting a vast array of host species (Jayawardena et al. 2019). A detailed background, diseases and the symptoms, morphological characters is discussed in Jayawardena et al. (2019). In this paper, we provide an update for the sections in Alternaria based on six gene combination analyses (Al Ghafri et al. 2019; Table 15, Fig. 24).

Phylogenetic tree generated by maximum parsimony analysis of combined SSU, LSU, ITS, GPDH, tef1 and RPB2 sequence data of Alternaria species. One hundred strains are included in the analyses, which comprised 4056 characters including gaps. The tree was rooted with Stemyphylium herbarium (CBS 191.86) and Pleospora tarda (CBS 714.68). The maximum parsimonious dataset consisted of 3091 constant, 852 parsimony-informative and 113 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of ten equally most parsimonious trees with a length of 4520 steps (CI = 0.335, RI 0.708, RC = 0.237, HI = 0.665) in the first tree. MP and ML bootstrap values ≥ 50% and Bayesian posterior probabilities ≥ 0.90 are shown respectively near the nodes. Ex-type strains are in bold
Diplodia Fr., in Montagne, Annls Sci. Nat., Bot., sér. 2 1: 302 (1834)
The genus Diplodia was introduced by Montagne (1834) and comprises species with hyaline or dark brown, aseptate or 1-septate, thick-walled conidia (Phillips et al. 2005). Diplodia is defined by having unilocular, solitary or aggregated conidiomata lined with conidiogenous cells that form conidia at their tips (Phillips et al. 2005). The type species of Diplodia is Diplodia mutila (Montagne 1834; Fries 1849), but there are no living cultures linked to the holotype of this species. As this has severely hampered studies on taxonomy and phylogeny of Diplodia, Alves et al. (2004) provided a detailed description of D. mutila based on an isolate from grapevines in Portugal (CBS 112553). Alves et al. (2014) designated an epitype for Diplodia mutila, with associated ex-epitype cultures. This epitype confirmed in all ways with the isotype of D. mutila and with the asexual morph on BPI 599153 as described by Alves et al. (2004). Diplodia mutila has hyaline conidia that become brown and one-septate after discharge from the pycnidia. Species of Diplodia can be differentiated on slight differences in conidial dimensions (Alves et al. 2014).
Classification—Dothideomycetes, incertae sedis, Botryosphaeriales, Botryosphaeriaceae
Type species—Diplodia mutila (Fr. : Fr.) Fr., Summa Veg. Scand. 2: 417 (1849)
Distribution—Worldwide
Disease symptoms—Diebacks, cankers, fruit rots.
Hosts—Plurivorous on woody hosts.
Morphological based identification and diversity
Diplodia is a large genus and a search in MycoBank (2019) revealed 1398 names while Index Fungorum (2019) has 1268 names. Cryptic speciation is common in the genus, which makes species identification difficult if based only on morphological characters (Phillips et al. 2012, 2013). Dissanayake et al. (2016) included 26 Diplodia species in their phylogeny. Recently, a novel species Diplodia eriobotryicola on Eriobotrya japonica from Spain was introduced by González-Domínguez et al. (2016). Yang et al. (2017) introduced D. pyri on Pyrus sp., the Netherlands, D. citricarpa on Citrus sp., Iran, and D. gallae on galls of Quercus sp. However, the ITS and tef1 of the novel species, D. citricarpa are not available in GenBank and hence we could not include this species in our phylogeny. The genus now comprises 30 species known from culture.
Molecular based identification and diversity
Earlier taxonomic studies on Diplodia using molecular data employed ITS rDNA, but this single marker can underestimate species diversity among closely related or cryptic species. Multiple gene sequence concordance phylogenies have therefore been applied to identify cryptic or previously overlooked species of Diplodia (Slippers et al. 2004a, b, c; Burgess et al. 2006; Phillips et al. 2005, 2012, 2013; Hyde et al. 2014; Dissanayake et al. 2016). As the tef1 gene is considerably more variable than the ITS rDNA region in these taxa, data from tef1 have been combined with ITS sequence data. Unfortunately, no single gene region is sufficient to distinguish all species in this genus. The present phylogenetic analysis was performed based on up to date ex-holotype or ex-epitype sequence data available in GenBank (Fig. 25, Table 16).

Phylogenetic tree generated by maximum likelihood analysis of combined ITS and tef1 sequence data of Diplodia species. Related sequences were obtained from GenBank. Forty nine strains are included in the analyses, which comprise 866 characters including gaps. The tree was rooted with Lasiodiplodia theobromae (CBS 164.96). Tree topology of the ML analysis was similar to the BYPP. The best scoring RAxML tree with a final likelihood value of − 3342.903931 is presented. The matrix had 278 distinct alignment patterns, with 9.02%. % of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.207781, C = 0.297582, G = 0.261770, T = 0.232868; substitution rates AC = 0.993193, AG = 3.566477, AT = 0.787748, CG = 1.607220, CT = 4.471399, GT = 1.000000; gamma distribution shape parameter α = 1.224079. RAxML bootstrap support values ≥ 80% (BT) are shown respectively near the nodes. Bayesian posterior probabilities ≥ 0.5 (PP) indicated as thickened black branches. Ex-type strains are in bold
Recommended genetic markers (genus level)—SSU and LSU
Recommended genetic markers (species level)—ITS, tef1, TUB2
Accepted number of species: 30 species
References: Phillips et al. 2013 (morphology, phylogeny, distribution, hosts); Dissanayake et al. 2016 (phylogeny).
Dothiorella Sacc., Michelia 2(6): 5 (1880)
Dothiorella was proposed by Saccardo to accommodate D. pyrenophora (Hyde et al. 2014). Members of this genus are pathogens, endophytes and saprobes (Phillips et al. 2013; Dissanayake et al. 2016). Taxonomy of this genus has been in a state of flux for decades (Phillips et al. 2013). Sivanesan (1984) treated D. pyrenophora as a synonym of Dothichiza sorbi (asexual morph of Dothiora pyrenophora). However, Sivanesan (1984) was referring to Dothiorella pyrenophora Sacc. (1884) which, according to Sutton (1977), is a later homonym of Dothiorella pyrenophora Sacc. (1880). Crous and Palm (1999) studied the holotype of D. pyrenophora and considered it a synonym of Diplodia. However, Phillips et al. (2005) based on both morphological and molecular data revived the genus Dothiorella for species in which the conidia become brown and 1-septate while attached to the conidiogenous cells.
Classification—Dothideomycetes, incertae sedis, Botryosphaeriales, Botryosphaeriaceae
Type species—Dothiorella pyrenophora Berk. ex Sacc., Michelia 2 (6): 5 (1880) (1909)
Distribution—Worldwide
Disease symptoms—Diebacks, cankers, fruit rots.
Hosts—Plurivorous on woody hosts.
Morphological based identification and diversity
Species of this genus were mostly described based on host association, which has led to the introduction of many species names and currently there are 393 epithets in Index Fungorum (2019). Slippers et al. (2013) suggested that host association cannot be considered as an important factor in species delimitation, many names are likely to be synonyms. Phillips et al. (2008) introduced a new genus Spencermartinsia to accommodate dothiorella-like species with apiculate ascospores. However, Yang et al. (2017) based on six-gene phylogeny and a broad taxon sampling considered that Spencermartinsia should be treated as a synonym of Dothiorella. Phillips et al. (2013) listed all cultures available for this genus and provided a phylogenetic tree and a key to the species. In that study 13 species names and 16 unnamed lineages were listed. Hyde et al. (2014), Dissanayake et al. (2016) and Yang et al. (2017) provided updates for the genus. Hyde et al. (2014) accepted 19 species and Dissanayake et al. (2016) accepted 30 species in the genus. After making Spencermartinsia a synonym of Dothiorella Yang et al. (2017) accepted 36 species in this genus.
Phillips et al. (2013) differentiated 13 Dothiorella species on the basis of conidiomata and conidial dimensions. However, the dimensions of these characters overlap between species. Therefore, using morphology alone without molecular data is not suitable to define species.
Molecular based identification and diversity
Recent studies have re-evaluated this genus based on multi-gene phylogeny of ITS, TUB2 and tef1 sequence data. We reconstruct the phylogeny of Dothiorella based on analyses of a combined ITS and tef1 sequence data (Table 17, Fig. 26). The phylogenetic tree is updated with recently introduced Dothiorella species and corresponds to previous studies (Dissanayake et al. 2016; Yang et al. 2017; Hyde et al. 2018; Phookamsak et al. 2019). In the analyses, it appears that several species are synonyms, such as D. parva/D. guttulata and D. rhamni/D. eriobotryae and possibly others. Therefore, a thorough revision of the genus is recommended to clarify the status of these dubious species.

Phylogenetic tree generated by maximum parsimony analysis of combined ITS and tef1 sequence data of Dothiorella species. Related sequences were obtained from GenBank. Forty eight strains are included in the analyses, which comprised 873 characters including gaps. The tree was rooted with Neofusicoccum parvum (CMW9081) and N. mangiferae (CMW7024). The maximum parsimonious dataset consisted of 572 constant, 204 parsimony-informative and 97 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of ten equally most parsimonious trees with a length of 891 steps (CI = 0.532, RI 0.738, RC = 0.393, HI = 0.468) in the first tree. MP and ML bootstrap values ≥ 50% and Bayesian posterior probabilities ≥ 0.90 are shown respectively near the nodes. The scale bar indicates 10 changes per site. Ex-type strains are in bold
Recommended genetic markers (genus level)—SSU and LSU
Recommended genetic markers (species level)—ITS and tef1
Accepted number of species: Currently, 393 species names are listed for Dothiorella in Index Fungorum (2019). Cultures and DNA sequences are available for 46 species, therefore 46 species are currently accepted inDothiorella.
References: Phillips et al. 2013; Dissanayake et al. 2016 (morphology, phylogeny, distribution, hosts), Yang et al. 2017 (morphology and phylogeny)
Fusarium Link, Mag. Gesell. naturf. Freunde, Berlin 3(1-2): 10 (1809)
Fusarium is a genus with 20 monophyletic species complexes (Rana et al. 2017).
Species formely belonged to F. solani species complex were transferred to genus Neocosmospora based on sexual morph characters and molecular phylogeny (Lombard et al. 2015; Sandoval-Denis and Crous (2018). Fusarium species are saprobes, parasites, endophytes, soil-borne or isolated from water (Rana et al. 2017). Species of Fusarium are economically important fungi as they are responsible for blights, cankers, rots, and wilts of horticultural, ornamental and forest crops in both agricultural and natural ecosystems, worldwide, and also human infections (Rana et al. 2017; Varela et al. 2013; Peraldi et al. 2014; Al-Hatmi et al. 2019; Maryani et al. 2019a, b). In nature, sexual morphs of Fusarium occur less commonly than the asexual morphs (Gräfenhan et al. 2011; Rossman et al. 1999).
Classification—Sordariomycetes, Hypocreomycetidae, Hypocreales, Nectriaceae
Type species—Fusarium sambucinum Fuckel, Hedwigia 2: 135.1863.
Distribution—Worldwide
Disease symptoms—blights, cankers, rots, and wilts
Plant pathogenic species of this genus have the capability to change their lifestyle to saprotrophic and can survive for long periods as chlamydospores in host tissues. Fusarium species damage their hosts by systemically colonizing and occluding the host xylem (Ploetz et al. 2003).
Hosts—Known from many host plant families.
Morphological based identification and diversity
Fusarium was also known from the sexual morphic fungus name Gibberella, which was suppressed in favour of Fusarium by Rossman et al. (2013). Variation and mutation in culture and lack of clear morphological characters for separating species are the main problems which make the species concept of Fusarium much broader (Geiser et al. 2004). It leads to the incorrect and confusing application of species names to toxigenic and pathogenic isolates (Geiser et al. 2004). Species boundaries have been inferred using multi-gene phylogenetic methods, reflecting the species diversity more than morphological treatments (Aoki and O’Donnell 1999; Geiser et al. 2004; O’Donnell 2000; O’Donnell et al. 1998a, b; Ward et al. 2002). A combined phylogenetic analysis of LSU, ITS, RPB2 and new phylogenetic marker acl1 by Gräfenhan et al. (2011), revealed that the early concept of Fusarium is not monophyletic. Fusarium sensu Wollenweber divided into two large groups, basal ‘Fusarium-like clades’, and the other one terminal ‘Fusarium clade’ in the Nectriaceae (Gräfenhan et al. 2011) (Fig. 27).

Sexual morph of a Fusarium sp. a Herbarium material. b Ascomata on the host. c Section of ascomata. d Section of the ostiolar region. e Peridium in face view. f–h Asci (h in Melzer’s reagent). i, j Ascospores. k Germinating ascospore. l, m Colony on MEA. Scale bars: c = 100 µm, d = 50 µm, e–k = 20 µm
Molecular based identification and diversity
ITS and LSU are least informative in species-level identification of Fusarium (O’Donnell et al. 1998a; Hyde et al. 2014). Moreover, non-orthologous copies of the ITS2, which can lead to wrong phylogenetic inferences, can be detected in many species of Fusarium (Geiser et al. 2004; O’Donnell et al. 1998a, b). Generally, for the species-level identification of fungi intron-rich regions of protein-coding genes are used as the markers (Geiser et al. 2004). The translation elongation factor 1-a (tef1), which lacks non-orthologous copies of the gene, is highly informative at the species level in Fusarium (Geiser et al. 2004). RPB1 and RPB2 are also very informative gene regions for species identification of Fusarium (O’Donnell et al. 2013; Hyde et al. 2014). Lombard et al. (2018) observed that tef1 and RPB2 genes provide better resolution of the species in the F. oxysporum complex than cmdA and tub2 (Fig. 28, Table 18).


Phylogram generated from RAxML analysis based on combined RPB1, RPB2 and tef1 sequences of accepted species of Fusarium. Related sequences were obtained from GenBank. One hundred sixty-five taxa are included in the analyses, which comprise 3980 characters including gaps. Single gene analyses were carried out and compared with each species, to compare the topology of the tree and clade stability. The tree was rooted in Fusicolla aquaeductuum (NRRL 20696). Tree topology of the ML analysis was similar to the BYPP. The best scoring RAxML tree with a final likelihood value of − 63956.723151 is presented. The matrix had 2254 distinct alignment patterns, with 25.13% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.257298, C = 0.251723, G = 0.248896, T = 0.242083; substitution rates AC = 1.366599, AG = 4.760724, AT = 1.266971, CG = 0.903914, CT = 9.564945, GT = 1.000000; gamma distribution shape parameter α = 1.089091. Maximum likelihood bootstrap support values ≥70% (BT) and Bayesian posterior probabilities ≥ 0.99 (PP) are given near the nodes respectively
Recommended genetic markers (genus level)—ATP citrate lyase (acl1), tef1 and ITS
Recommended genetic markers (species level)—calmodulin-encoding gene (cmdA), tub2, tef1, RPB1 and RPB2
Accepted number of species: There are 1552 species epithets in Index Fungorum (2019) under this genus. More than 175 species have DNA sequence data.
References: Booth 1971, Rossman et al. 1999 (morphology), Rana et al. 2017, Gräfenhan et al. 2011; Laurence et al. 2014; Lombard et al. 2018, 2019; Maryani et al. 2019a, b; Wang et al. 2019; Nalim et al. 2011 (morphology, phylogeny).
Lasiodiplodia Ellis & Everh., Bot. Gaz. 21:92 (1896)
According to Clendenin (1896), a fungus causing rot of sweet potatoes imported from Java was identified by Ellis in 1894 as a new genus and he named the fungus Lasiodiplodia tubericola. However, Ellis (1894) did not describe the fungus or publish the new genus. Clendenin (1896) provided a description of the genus and the species, attributing both to Ellis and Everhardt. Griffin and Maublanc (1909) considered that on account of the pycnidial paraphyses, Botryodiplodia theobromae, described by Patouillard and de Lagerheim (1892), was more suitably accommodated in Lasiodiplodia. Since the epithet theobromae (1892) is older than tubericola (1896), L. theobromae should be regarded as the type species of Lasiodiplodia. Neither Patouillard and de Lagerheim (1892) nor Clendenin (1896) referred to any type or other specimens of the genus or species. Pavlic et al. (2004) could not locate the types, and they could not find any specimens from the original hosts or origins. Phillips et al. (2013) designated CBS H-21411 as neotype with CBS 164.96 as culture ex-neotype.
The sexual morph has been reported for L. theobromae, but the connection with the asexual morph has not been confirmed (Phillips et al. 2013). Sexual morphs have also been reported for L. pseudotheobromae (Tennakoon et al. 2016), L. gonubiensis (Trakunyingcharoen et al. 2015) and L. lignicola (Phillips et al. 2013) with clear evidence that connects sexual with asexual morphs.
Classification—Dothideomycetes, incertae sedis, Botryosphaeriales, Botryosphaeriaceae
Type species—Lasiodiplodia theobromae (Pat.) Griffon & Maubl., Bull. Soc. mycol. Fr. 25: 57 (1909)
Distribution—Worldwide, mostly confined to tropical and sub-tropical regions, but becoming increasingly more common in warm temperate regions.
Disease symptoms—Diebacks, cankers, fruit rots.
Hosts—Plurivorous on woody hosts
Morphological based identification and diversity
The pigmented, 1-septate conidia with longitudinal striations together with the pycnidial paraphyses distinguish Lasiodiplodia from all other genera in Botryosphaeriaceae (Phillips et al. 2013). Striations on the conidia distinguish it from Diplodia, the conidiomata paraphyses distinguish it from Neodeightonia, which also has striate conidia. Although Barriopsis has striate conidia and paraphyses, Lasiodiplodia is unique in the Botryosphaeriaceae because striations are visible on immature, hyaline conidia. Although Phillips et al. (2013) differentiated 18 species in Lasiodiplodia on the basis of conidial morphology (especially dimensions) and morphology of the paraphyses, in reality, species in Lasiodiplodia cannot be identified with any confidence from their morphology and molecular data are necessary for definitive identifications.
Molecular based identification and diversity
Denman et al. (2000) suggested that Lasiodiplodia could be a synonym of Diplodia. When Crous et al. (2006) re-organized Botryosphaeria on the basis of LSU phylogeny they split the genus into 10 genera, but could not resolve the position of Lasiodiplodia or separate it from Diplodia. Following a multi-locus approach (SSU, ITS, LSU, tef1 and TUB2) Phillips et al. (2008) showed that Lasiodiplodia constitutes a clear phylogenetic lineage.
For many years, only the type species of Lasiodiplodia (L. theobromae) was mentioned in the phytopathological and mycological literature, and it was regarded as a cosmopolitan, plurivorous pathogen restricted mainly to tropical and sub-tropical regions (Punithalingam 1976, 1980). Soon after the widespread application of DNA-based phylogenies, Pavlic et al. (2004) introduced L. gonubiensis as a new species on the basis of conidial morphology and ITS sequence data. Soon after, Burgess et al. (2006) described three new species (L. crassispora, L. venezuelensis and L. rubropurpurea) from the tropics based on ITS and tef1 sequence data and morphological characters. Alves et al. (2008) also used ITS and tef1 sequence data to reveal two cryptic species in the L. theobromae complex. Over the years more species were introduced and Phillips et al. (2013) listed 18 species and Dissanayake et al. (2016) listed 31 species known from culture. Today the figure stands at 40 (Fig. 29). Apart from L. theobromae, all species have been introduced almost entirely on the basis of DNA sequence phylogenies. Although the phylogenies were derived from analysis of multiple loci (mostly ITS, tef1 and TUB2 and sometimes RPB2) the genealogical concordance phylogenetic species recognition concept (Taylor et al. 2000) has not always been strictly applied and species have been introduced on the basis of minor differences in only one locus. The result is that some species are not well separated phylogenetically (Fig. 29, Table 19), such as L. hyalina and L. thailandica, L. chinensis, L. sterculiae, L. pseudotheobromae, L. pyriformis and L. crassispora. In a detailed study of five loci of 19 Lasiodiplodia species, Cruywagen et al. (2017) concluded that several accepted species (L. viticola, L. missouriana, L. laeliocattleyae, L. brasiliense) may, in fact, be hybrids. There has been no such study of the 16 species introduced after the work of Cruywagen et al. (2017). In view of the questionable status of several species in Lasiodiplodia, there is an urgent need to re-assess all of the species currently accepted in this genus.

Phylogenetic tree generated by maximum likelihood analysis of combined ITS and tef1 sequence data of Lasiodiplodia species. Related sequences were obtained from GenBank. Forty nine strains are included in the analyses, which comprise 866 characters including gaps. The tree was rooted with Barriopsis tectonae and B. iraniana. Tree topology of the ML analysis was similar to MP and BYPP. The best scoring RAxML tree with a final likelihood value of − 3733.342990 is presented. The matrix had 253 distinct alignment patterns, with 4.41% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.211797, C = 0.285190, G = 0.260783, T = 0.242230; substitution rates AC = 0.983905, AG = 3.303939, AT = 1.281593, CG = 0.950258, CT = 5.553417, GT = 1.000000; gamma distribution shape parameter α = 0.221126. RAxML bootstrap support values ≥ 80% are shown respectively near the nodes. Bayesian posterior probabilities ≥ 0.5 (BYPP) indicated as thickened black branches. Ex-type strains are in bold
Recommended genetic markers (genus level)—SSU and LSU
Recommended genetic markers (species level)—ITS, tef1, TUB2
Accepted number of species: Currently, 51 species names are listed for Lasiodiplodia in MycoBank and Index Fungorum (2019). Cultures and DNA sequences are available for 43 species, three of which have been reduced to synonymy under existing names. Thus, 40 species are currently recognised inLasiodiplodia.
References: Phillips et al. 2013 (morphology, phylogeny, distribution, hosts); Dissanayake et al. 2016 (species).
Pestalotiopsis Steyaert, Bull. Jard. bot. État Brux. 19: 300 (1949)
Pestalotiopsis is an appendage-bearing, 5-celled conidia (asexual coelomycetes) in the family Sporocadaceae (Maharachchikumbura et al. 2014a, b; Jayawardena et al. 2016). The genus was introduced by Steyaert (1949). Pestalotiopsis species are widely distributed throughout tropical and temperate regions (Guba 1961; Maharachchikumbura et al. 2012, 2014a). Pestalotiopsis species have been isolated from dead leaves, bark, twigs, soil, polluted stream water, wood, paper, fabrics, and wool (Guba 1961; Maharachchikumbura et al. 2012, 2014a). Some species have been associated with human and animal infections, and others (e.g. P. guepinii and P. microspora) have also been isolated from extreme environments (Maharachchikumbura et al. 2014b).
Classification—Sordariomycetes, Xylariomycetidae, Amphisphaeriales, Sporocadaceae
Type species—Pestalotiopsis guepinii (Desm.) Steyaert [as ‘guepini’], Bull. Jard. bot. État Brux. 19(3): 312 (1949)
Distribution—Worldwide
Disease symptoms—Species of Pestalotiopsis cause a variety of diseases in plants including canker lesions, shoot dieback, leaf spots, needle blight, tip blight, grey blight, scabby canker, severe chlorosis, fruit rots and various post-harvest diseases (Maharachchikumbura et al. 2013a, b, 2014a, b). These pathogens reduce production and cause economic loss in apple, blueberry, coconut, chestnut, ginger, grapevine, guava, hazelnut, lychee, mango, orchid, peach, rambutan, tea and wax apple due to diseases (Maharachchikumbura et al. 2013a, b, 2014a, b). Grapevine trunk diseases are the most destructive diseases of grapevines that impact the economic production and longevity of vineyards and even leading to partial or total death of individual plants. Therefore, the initial identification of the causal agent is essential for early control of these diseases (Jayawardene et al. 2015; Maharachchikumbura et al. 2017). Pestalotoid fungi have been reported as pathogens on a variety of grapevine cultivars, causing diseases including grapevine dieback, fruit rot, postharvest disease and severe defoliation and they infect all plant parts including leaves, canes, wood, berries and flowers (Jayawardene et al. 2015; Maharachchikumbura et al. 2017). Pestalotiopsis menezesiana (Bres. & Torr.) Bissett. and P. uvicola (Spegazzini) Bissett, are the most common species recorded from grapevine around the world and especially P. biciliata are associated with trunk grapevine disease (Jayawardene et al. 2015; Maharachchikumbura et al. 2017).
Hosts—Broad range of hosts including members of Altingiaceae, Arecaceae, Bromeliaceae, Euphorbiaceae, Myrtaceae, Poaceae, Proteaceae, Rosaceae, Rutaceae, Theaceae and Vitaceae.
Morphological based identification and diversity
There are around 250 species, most of which were named according to their host associations (Maharachchikumbura et al. 2014a, b). However, Pestalotiopsis species are not hosted specific and are found on a wide range of plants and substrates (Jeewon et al. 2003; Lee et al. 2006; Maharachchikumbura et al. 2014a, b). They exhibit considerable diversity in phenotype, and group together based on similarities in conidial morphology (Jeewon et al. 2003; Maharachchikumbura et al. 2012, 2013a, b, 2014a, b). Considering morphology, conidial length, width, median cell length, the colour of median cells and length of the apical appendages appear to be stable characters within Pestalotiopsis (Jeewon et al. 2003; Maharachchikumbura et al. 2014b).
Pestalotiopsis guepinii was considered to be the type species of the genus described from stems and leaves of Camellia japonica collected in France, and is characterised by 5-celled conidia with three concolourous median cells, hyaline terminal cells and simple or unbranched appendages arising from the apex of the apical cell (Steyaert 1949; Maharachchikumbura et al. 2014b). Nag Raj (1985) regarded P. maculans as the type species of Pestalotiopsis with P. guepinii as a synonym. Jeewon et al. (2003) also accepted P. maculans clusters with species having concolourous median cells based on phylogenetic analysis of ITS sequence data and that P. karstenii might be a synonym of P. maculans (Maharachchikumbura et al. 2014b).
Most Pestalotiopsis species lack sexual morphs. The sexual morph of Pestalotiopsis was treated as Pestalosphaeria Barr, with the type species Pestalosphaeria concentrica collected from grey-brown spots on living leaves of Rhododendron maximum in North Carolina, USA (Maharachchikumbura et al. 2014b). Pestalosphaeria concentrica is characterised by immersed, subglobose ascomata and unitunicate, cylindrical asci with a J + apical ring; ascospores uniseriate in the ascus, ellipsoid, pale dull brown and 2-septate (Maharachchikumbura et al. 2014b).
Pestalotiopsis species have the ability to switch life-modes as endophytes, pathogens and saprobes (Hu et al. 2007; Maharachchikumbura et al. 2012). Therefore, many endophytic and plant pathogenic Pestalotiopsis species persist as saprobes and have been isolated from dead leaves, bark and twigs (Maharachchikumbura et al. 2012, 2013a, b, 2014b).
Pestalotiopsis species that were isolated as endophytes are important in the discovery of novel compounds with medicinal, agricultural and industrial applications (Maharachchikumbura et al. 2014b; Xu et al. 2010, 2014). Pestalotiopsis species are a rich source for bioprospecting compared to other fungal genera, and more than 100 compounds have been isolated from Pestalotiopsis (Maharachchikumbura et al. 2014b; Xu et al. 2010, 2014).
Molecular based identification and diversity
Maharachchikumbura et al. (2012) tested with 10 gene regions to resolve species boundaries in Pestalotiopsis (actin, calmodulin, glutamine synthase, glyceraldehyde-3- phosphate dehydrogenase, ITS, LSU, 18S nrDNA, RNA polymerase II, tef1 and TUB2). Maharachchikumbura et al. (2014b) used phylogenetic analysis of combined ITS, TUB2 and tef1 genes to successfully resolve Pestalotiopsis species (Fig. 30, Table 20).

Phylogram generated from RAxML analysis based on combined ITS, TUB2 and tef1 sequences of all the accepted species of Pestalotiopsis. Related sequences were obtained from GenBank. Seventy-nine taxa are included in the analyses, which comprise 1581 characters including gaps. The tree was rooted in Neopestalotiopsis cubana (CBS 600.96) and N. saprophytica (MFLUCC 12-0282). Tree topology of the ML analysis was similar to the BYPP and MP. The best scoring RAxML tree with a final likelihood value of − 12269.881063 is presented. The matrix had 763 distinct alignment patterns, with 15.79% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.234129, C = 0.293516, G = 0.211518, T = 0.260837; substitution rates AC = 1.189917, AG = 3.402399, AT = 1.153875, CG = 1.001451, CT = 4.301074, GT = 1.000000; gamma distribution shape parameter α = 0.283001. The maximum parsimonious dataset consisted of 948 constant, 450 parsimony-informative and 183 parsimony-uninformative characters. The parsimony analysis of the data matrix resulted in the maximum of ten equally most parsimonious trees with a length of 1962 steps (CI = 0.498, RI = 0.697, RC = 0.347, HI = 0.502) in the first tree. RAxML and maximum parsimony bootstrap support value ≥ 50% and BYPP ≥ 0.90 values are shown near the nodes. The scale bar indicates 10 changes per site. The ex-type strains are in bold
Recommended genetic marker (genus level)—LSU (as outlined in Maharachchikumbura et al. 2012)
Recommended genetic markers (species level)—ITS, TUB2 and tef1 (as outlined in Maharachchikumbura et al. 2012)
Accepted number of species: There are 360 epithets in Index Fungorum in this genus, however, 75 species with DNA sequence data are accepted.
References: Maharachchikumbura et al. 2013a, b, 2014b, 2016 (morphology, phylogeny)
Stagonosporopsis Died. emend. Aveskamp et al., Stud. Mycol. 65: 44. 2010
Stagonosporopsis is a coelomycetous genus in the family Didymellaceae (de Gruyter et al. 2013), accommodating several important phytopathogenic species. Some of the species have described sexual forms in Didymella (Diedicke 1912; Aveskamp et al. 2010). Some Stagonosporopsis species have quarantine importance. Stagonosporopsis andigena is listed in Annex IAI by European Union (EU), meaning its introduction to EU is prohibited. This pathogen is also listed in the A1 list by the European and Mediterranean Plant Protection Organization (EPPO 2019). Stagonosporopsis chrysanthemi is another species listed in A2 list by EPPO (EPPO 2019).
Classification—Dothideomycetes, Pleosporomycetidae, Pleosporales, Didymellaceae
Type species—Stagonosporopsis boltshauseri (Sacc.) Died. 1912
Distribution—Worldwide
Disease symptoms—the Black blight of potato, gummy stem blight, ray blight
All plant parts may be attacked by S. chrysanthemi and S. inoxydabilis, however, flowers and cuttings are highly susceptible. Death of flowers and buds, a necrotic lesion on leaves and peduncles of unopened buds, soft rot of cortex of roots and discolouration of bark are the main symptom of the disease. Eventually, plant death occurs (Fox 1998; Pethybridge et al. 2008). In gummy stem blight of cucurbits, symptoms can be observed on all above ground and reproductive parts. Leaf spots are the main diagnostic character of this disease. Most of the circular or triangular shaped spots start at the margin of the leaf or extend towards the margin. The centre of the leaf spot is a lighter shade of brown than the surrounding portion. As leaf spots coalesce leaf blights occur. Actively expanding lesions on leaves, petioles, and pedicels often appear as water-soaked. Cankers may form on crowns, main stems and vines (Keinath 2013). Stagonosporopsis andigena the black blight of potato causal agent affects leaves, petioles and stems causing lesions and premature leaf drop, but does not infect the underground parts. On leaves, the pathogen causes small, blackish concentric lesions. The initial symptoms can be observed on the lower leaves, however, as the disease progresses lesions may also develop in upper leaves as well. Lesions may coalesce and severely affected leaves may turn blackish giving a scorched appearance (EFSA panel on plant health 2019).
Hosts—Amaranthaceae, Asteraceae, Campanulaceae, Caryophyllaceae, Cucurbitaceae, Fabaceae, Lamiaceae, Pinaceae, Ranunculaceae, Solanaceae and Valerianaceae.
Morphological based identification and diversity
Stagonosporopsis is characterized by ellipsoidal to subglobose, aseptate to 3 septate conidia and sexual morph with ellipsoidal, fusiform or obovoid, 1 septate ascospore (Aveskamp et al. 2010; Chen et al. 2015). Stagonosporopsis was originally separated from Ascochyta by Diedicke (1912) based on the occasional formation of multi-septate (Stagonospora-like) conidia. In the phylogenetic reassessment of Didymellaceae (Aveskamp et al. 2010) based on the sequences LSU and ITS of the nrDNA and TUB2 region, multiple Phoma species, including P. ligulicola, were recovered in a high supported clade with the interpretive types of the genus Stagonosporopsis; S. actaeae (Boerema 1997, Boerema et al. 2004). In addition, S. tanaceti shows morphological similarity to S. inoxydabilis but can be differentiated by the faster growth rate, larger conidia, presence of chlamydospores, and lack of ascomata in culture (Vaghefi et al. 2012). Morphological characters overlap between the species in this genus and species are primarily separated based on molecular data.
Molecular based identification and diversity
Most comprehensive multigene phylogeny analyses for this genus were performed by Aveskamp et al. (2010), Vaghefi et al. (2012), Hyde et al. (2014), Chen et al. (2015, 2017) and Jayasiri et al. (2019). Five-marker phylogeny of the Stagonosporopsis spp. for which these DNA sequence data are available is shown (Table 21).
Identification of Stagonosporopsis species associated with ray blight of Asteraceae can be achieved through multi-locus sequence typing (Aveskamp et al. 2010; Vaghefi et al. 2012) and also with a species-specific multiplex PCR assay developed by Vaghefi et al. (2016). This assay is based on a four-primer PCR that targets the intergenic spacer of the nrDNA of the ray blight pathogens, producing species-specific amplicons of ~ 560 in S. chrysanthemi, ~ 630 bp in S. inoxydabilis and ~ 400 bp in S. tanaceti, which can be easily differentiated on an agarose gel (Vaghefi et al. 2016).
This study reconstructs the phylogeny of Stagonosporopsis based on analyses of a combined ITS, LSU, RPB2 and TUB2 sequence data (Fig. 31). The phylogenetic tree is updated with recently introduced Stagonosporopsis species and corresponds to previous studies (Chen et al. 2017, Jayasiri et al. 2019).

Phylogenetic tree generated by maximum likelihood analysis of combined ITS, LSU, RPB2 and TUB2 sequence data of Stagonosporopsis species. Related sequences were obtained from GenBank. Thirty-two are included in the analyses, which comprise 2756 characters including gaps. Single gene analyses were carried out and compared with each species, to compare the topology of the tree and clade stability. The tree was rooted with Heterophoma poolensis (CBS 113.20). Tree topology of the ML analysis was similar to the MP. The best scoring RAxML tree with a final likelihood value of − 8386.622374 is presented. The matrix had 388 distinct alignment patterns, with 11.83% of undetermined characters or gaps. Estimated base frequencies were as follows; A = 0.245798, C = 0.237676, G = 0.273178, T = 0.243348; substitution rates AC = 1.882600, AG = 4.123164, AT = 2.015920, CG = 0.907833, CT = 12.626910, GT = 1.000000; gamma distribution shape parameter α = 1.105097. RAxML bootstrap support values ≥ 50% and Bayesian posterior probabilities ≥ 0.95 (BYPP) are shown respectively near the nodes. Ex-type strains are in bold
Recommended genetic marker (genus level)—ITS
Recommended genetic markers (species level)—TUB2 and RPB2
Accepted number of species: There are 55 species in Index Fungorum (2019) and only 24 species are accepted/ have molecular data in this genus.
References: Chen et al. 2015, 2017, Jayasiri et al. 2019 (morphology, phylogeny).
Verticillium Nees, Syst. Pilze (Würzburg): 57 (1816) [1816-17]
The genus Verticillium Nees was introduced by Nees von Esenbeck (1816) for a single saprotrophic species, V. tenerum Nees, which was proposed as the type species. Zare et al. (2004) synonymized V. tenerum under Acrostalagmus luteoalbus (Link) Zare, W. Gams & Schroers, and Gams et al. (2005) proposed V. dahliae Kleb. as the conserved type of the genus Verticillium.
Verticillium includes several plant pathogenic species that infect trees, insects, mushrooms and, in particular, dicotyledonous plants: V. dahliae Kleb., V. albo-atrum Reinke et Berth., V. nigrescens Pethybr., V. nubilum Pethybr., V. tricorpus Isaac., V. theobromae (Turc.) Mas. & Hughes and V. fungicola (Preuss) Hassebrauk (Pegg and Brady 2002). Zare et al. (2007) assigned V. nigrescens to the genus Gibellulopsis and V. theobromae to Musicillium. Zare and Gams (2008) assigned V. fungicola to the genus Lecanillium. Verticillium dahliae and V. albo-atrum are the two most notorious species which cause Verticillium wilt diseases in a wide range of mainly dicotyledonous hosts and result in billions of dollars of damage annually in crop losses worldwide (Pegg and Brady 2002; Barbara and Clewes 2003; Inderbitzin et al. 2011).
Verticillium species, in particular, V. dahliae and V. albo-atrum, can infect a wide range of plant species. Many hosts of Verticillium species were given by different mycologists, e.g., Van der Meer (1925); Rudolph (1931); Engelhard and Carter (1956); Parker (1959); Stark (1961); Devaux and Sackston (1966); Himelick (1969), including high-value crop plants, e.g., cotton (Land et al. 2016), lettuce (Garibaldi et al. 2007; Powell et al. 2013), mango (Baeza-Montanez et al. 2010; Ahmed et al. 2014), gold kiwifruit (Auger et al. 2009), bean (Berbegal and Armengol 2009; Sun et al. 2016; Blomquist et al. 2017), watermelon (Bruton et al. 2007), olive tree (Lo Giudice et al. 2010; Kaliterna et al. 2016), potato (Pace-Lupi et al. 2006), pumpkin (Rampersad 2008). The most comprehensive hosts’ list was provided by Pegg and Brady (2002).
Classification—Sordariomycetes, Hypocreomycetidae, Glomerellales, Plectosphaerellaceae
Type species—Verticillium dahliae Kleb., Mykol. Zentbl. 3: 66 (1913)
Distribution—Worldwide
Disease symptoms—Wilt
Wilt caused by Verticillium species is a serious fungal disease that causes injury or death to many plant species. Symptoms of this disease vary according to the host species and to environmental conditions including sudden wilting of small branches, yellowing of foliage, stunt growth and premature defoliation. Olive- green to black streaks can be observed in the sapwood of infected branches. In cross sections, vascular tissue appears as a dark ring or pinpoint dark spots. Initial symptoms can occur on one side of the tree or the entire plant (Fradin and Thomma 2006; Blum et al. 2018).
Hosts—Species of this genus have a broad host range including members of Amaranthaceae, Amaryllidaceae, Asteraceae, Brassicaceae, Musaceae, Pinaceae, Rosaceae, Rubiaceae, Sapindaceae, Solanaceae, Theaceae, Vitaceae and Zingiberaceae (Farr and Rossman 2019).
Morphological based identification and diversity
There are 270 records in Index Fungorum (2019) and 288 records in MycoBank (Crous et al. 2004) in this genus. Most of the species have been transferred to other genera, e.g., Gibellulopsis (Zare et al. 2007), Haptocillium (Zare and Gams 2001b), Lecanicillium (Zare and Gams 2001a), Musicillium (Zare et al. 2007), Pochonia (Zare and Gams 2001a, b), Simplicillium (Zare and Gams 2001a). Only ten species, V. albo-atrum, V. alfalfae, V. dahliae, V. isaacii, V. klebahnii, V. longisporum, V. nonalfalfae, V. nubilum, V. tricorpus and V. zaregamsianum, were accepted within Verticillium sensu stricto (Inderbitzin et al. 2011). No sexual states are known. Descriptions and a key to these ten species were provided by Inderbitzin et al. (2011).
Molecular based identification and diversity
The first phylogenetic analysis of Verticillium species was made by Morton et al. (1995) to analysis the relationship between V. alboatrum and V. dahliae based on the internal transcribed spacer regions and intervening 5.8S rDNA (ITS) sequences data (Fig. 32, Table 22). Subsequently, SSU, LSU, RNA polymerase II largest subunit gene (RPB1), the cytochrome oxidase subunit III gene (cox3), the small ribosomal rRNA subunit (rns), NADH dehydrogenase subunit genes (nad1 and nad3) were used (Barbara and Clewes 2003; Pantou et al. 2005; Zare and Gams 2008, 2016).

Phylogenetic tree generated from Bayesian analysis based on combined ACT, tef1, GPD and TS sequence data for the genus Verticillium. Sixteen strains are included in the combined genes sequence analyses which comprise total of 2597 characters (609 characters for the ACT, 620 characters for tef1, 744 characters for GPD, 624 characters for TS) after alignment. For the Bayesian analysis, two parallel runs with six chains were run for 1,000,000 generations and trees were sampled every 100th generation, resulted in 20,002 trees from two runs of which 15,002 trees were used to calculate the posterior probabilities (each run resulted in 10,001 trees of which 7501 trees were sampled). Bootstrap support values for maximum likelihood (ML, first set) and maximum parsimony (MP, second set) greater than 75% and Bayesian posterior probabilities greater than 0.95 are indicated above or below the nodes. Ex-type strains are in bold. The tree is rooted with Gibellulopsis nigrescens (PD596)
Recommended genetic marker (genus level)—LSU
Recommended genetic marker (species level)—ITS
Accepted number of species:10 species.
References: Inderbitzin et al. 2011 (morphology and phylogeny); Barbara and Clewes 2003, Morton et al. 1995, Pantou et al. 2005, Zare and Gams 2008, 2016 (phylogeny); Fradin and Thomma 2006, Blum et al. 2018 (pathogenicity)
References
Adams GC, Wingfield MJ, Common R, Roux J (2005) Phylogenetic relationships and morphology of Cytospora species and related teleomorphs (Ascomycota, Diaporthales, Valsaceae) from Eucalyptus. Stud Mycol 52:1–144
Adams GC, Roux J, Wingfield MJ (2006) Cytospora species (Ascomycota, Diaporthales, Valsaceae): introduced and native pathogens of trees in South Africa. Australas Plant Pathol 35:521–548
Agrios GN (2005) Introduction to plant pathology. Elsevier Academic Press, Amsterdam
Ahmed Y, Cirvilleri G, D’Onghia AM, Yaseen T (2014) First report of Verticillium wilt of Mango (Mangifera indica) caused by Verticillium dahliae in Italy. Plant Dis 98:1156–1157
Aiello D, Guarnaccia V, Vitale A, Cirvilleri G, Granata G, Epifani F, Perrone G, Polizzi G, Groenewald JZ, Crous PW (2014) Ilyonectria palmarum sp. nov. causing dry basal stem rot of Arecaceae. Eur J Plant Pathol 138:347–359
Al Ghafri AA, Maharachchikumbura SS, Hyde KD, Al-Saady NA, Al-Sadi AM (2019) A new section and a new species of Alternaria from Oman. Phytotaxa 405:279–289
Al-Hatmi AMS, Meletiadis J, Curfs-Breuker I, Bonifaz A, Meis JF, De Hoog GS (2019) In vitro combinations of natamycin with voriconazole, itraconazole and micafungin against clinical Fusarium strains causing keratitis. J Antimicrob Chemo 71: 953–955
Alves A, Correia A, Luque J, Phillips AJL (2004) Botryosphaeria corticola sp. nov. on Quercus species, with notes and description of Botryosphaeria stevensii and its anamorph, Diplodia mutila. Mycologia 96:598–613
Alves A, Crous PW, Correia A, Phillips AJL (2008) Morphological and molecular data reveal cryptic speciation in Lasiodiplodia theobromae. Fungal Divers 28:1–13
Alves A, Linaldeddu BT, Deidda A, Scanu B, Phillips AJL (2014) The complex of Diplodia species associated with Fraxinus and some other woody hosts in Italy and Portugal. Fungal Divers 67:143–156
Aoki T, O’Donnell K (1999) Morphological characterization of Gibberella coronicola sp. nov., obtained through mating experiments of Fusarium pseudograminearum. Mycoscience 40:443–453
Arjona-Girona I, López-Herrera CJ (2018) Study of a new biocontrol fungal agent for avocado white root rot. Biol Control 117:6–12
Arora NK, Kang SC, Maheshwari DK (2001) Isolation of siderophore–producing strains of Rhizobium meliloti and their biocontrol potential against Macrophomina phaseolina that causes charcoal rot of groundnut. Current Sci 81:673–677
Ash G (2000) Downy mildew of grape. The Plant Health Instructor Auger J, Perez I, Fullerton RA, Esterio M (2009) First report of Verticillium wilt of gold kiwifruit, Actinidia chinensis cv. Hort 16A, caused by Verticillium albo–atrum in Chile. Plant Dis 93:553–553
Aveskamp MM, de Gruyter J, Woudenberg JHC, Verkley GJM, Crous PW (2010) Highlights of the Didymellaceae: a polyphasic approach to characterize Phoma and related pleosporalean genera. Stud Mycol 65:1–60
Ayoubi N, Soleimani MJ (2016) Strawberry fruit rot caused by Neopestalotiopsis iranensis sp. nov., and N. mesopotamica. Curr Microbiol 72(3):329–336
Baeza-Montanez L, Gomez-Cabrera R, Garcia-Pedrajas MD (2010) First report of Verticillium wilt caused by Verticillium dahliae on Mango Trees (Mangifera indica) in Southern Spain. Plant Dis 94:380–381
Barbara DJ, Clewes E (2003) Plant pathogenic Verticillium species: how many of them are there? Mol Plant Pathol 4:297–305
Barr ME (1978) The Diaporthales in North America: with emphasis on Gnomonia and its segregates. Mycol Mem 7:1–232
Barr ME (1987) New taxa and combinations in the Loculoascomycetes. Mycotaxon, USA
Berbegal M, Armengol J (2009) First report of Verticillium wilt of faba bean caused by Verticillium dahliae in Spain. Plant Dis 93:432–432
Berkeley MJ, Desmazières JBHJ (1849) On some moulds referred by authors to Fumago and to certain allied or analogous forms. J Hortic Soc Lond 4:3–19
Blomquist CL, Rooney-Latham S, Barrera N (2017) First report of Verticillium wilt caused by Verticillium dahliae on fava bean in the United States. Plant Dis 101:384–384
Blum A, Bressan M, Zahid A, Trinsoutrot-Gattin I, Driouich A, Laval K (2018) Verticillium wilt on fiber flax: symptoms and pathogen development in planta. Plant Dis 102:2421–2429
Boerema GH (1997) Contributions towards a monograph of Phoma (Coelomycetes). V. Subdivision of the genus in sections. Mycotaxon (USA)
Boerema GH, de Gruyter J, Noordeloos ME, Hamers MEC (2004) Phoma identification manual. CABI Publishing, Cambridge
Booth C (1959) Studies of Pyrenomycetes. IV. Nectria (part 1). Mycol Pap 73:1–115
Booth C (1966) The genus Cylindrocarpon. Mycol Pap 104:1–56
Booth C (1971) The genus Fusarium. Commonwealth Mycological Institute, Kew, Surrey, England
Brayford D, Honda BM, Mantiri FR, Samuels GJ (2004) Neonectria and Cylindrocarpon: the Nectria mammoidea group and species lacking microconidia. Mycologia 96:572–597
Bruton BD, Fish WW, Subbarao KV, Isakeit T (2007) First report of Verticillium wilt of Watermelon in the Texas high plains. Plant Dis 91(8):1053–1053
Burgess TI, Barber PA, Mohali S, Pegg G, de Beer W, Wingfield MJ (2006) Three new Lasiodiplodia spp. from the tropics, recognized based on DNA sequence comparisons and morphology. Mycologia 98:423–435
Cabral A, Groenewald JZ, Rego C, Oliveira H, Crous PW (2012a) Cylindrocarpon root rot: multi-gene analysis reveals novel species within the Ilyonectria radicicola species complex. Mycol Prog 11:655–688
Cabral A, Rego C, Crous PW, Oliveira H (2012b) Virulence and cross-infection potential of Ilyonectria spp. to grapevine. Phytopathol Mediterr 52:340–354
Cabral A, Rego C, Nascimento T, Oliveira H, Groenewald JZ, Crous PW (2012c) Multi-gene analysis and morphology reveal novel Ilyonectria species associated with black foot disease of grapevines. Fungal Biol 116:62–80
Callan BE (1998) Diseases of Populus in British Columbia: a diagnostic manual. Canadian Forest Service, Victoria, BC
Cannon PF, Hawksworth DL, Sherwood-Pike MA (1985) The British Ascomycotina, an annotated checklist. Commonwealth Agricultural Bureaux, Slough
Carisse O (2016) Development of grape downy mildew (Plasmopara viticola) under northern viticulture conditions: influence of fall disease incidence. Eur J Plant Pathol 144:773–783
Castlebury LA, Rossman AY, Jaklitsch WJ, Vasilyeva LN (2002) A preliminary overview of Diaporthales based on large subunit nuclear ribosomal DNA sequences. Mycologia 94:1017–1031
Castlebury LA, Rossman AY, Hyten AS (2006) Phylogenetic relationships of Neonectria/Cylindrocarpon on Fagus in North America. Canad J Bot 84:1417–1433
Castro BL, Carreño AJ, Galeano NF, Roux J, Wingfield MJ, Gaitán ÁL (2013) Identification and genetic diversity of Rosellinia spp. associated with root rot of coffee in Colombia. Australas Plant Pathol 42:515–523
Chamorro M, Aguado A, De los Santos B (2016) First report of root and crown rot caused by Pestalotiopsis clavispora (Neopestalotiopsis clavispora) on strawberry in Spain. Plant Dis 100:1495
Chaverri P, Gazis RO, Samuels GJ (2011) Trichoderma amazonicum, a new endophytic species on Hevea brasiliensis and H. guianensis from the Amazon basin. Mycologia 103:139–151
Chen Q, Jiang JR, Zhang GZ, Cai L, Crous PW (2015) Resolving the Phoma enigma. Stud Mycol 82:137–217
Chen Q, Hou LW, Duan WJ, Crous PW, Cai L (2017) Didymellaceae revisited. Stud Mycol 87:105–159
Choi Y-J, Beakes G, Glockling S, Kruse J, Nam B, Nigrelli L, Ploch S, Shin H-D, Shivas RG, Telle S, Voglmayr H, Thines M (2015) Towards a universal barcode of oomycetes—a comparison of the cox1 and cox2 loci. Mol Ecol Res 15:1275–1288
Choi IY, Oh HT, Lee WH, Cho SE, Shin HD (2017) First report of white root rot caused by Rosellinia necatrix on Aronia melanocarpa in Korea. Plant Dis 101(1):253–253
Chomnunti P, Schoch CL, Aguirre-Hudson B, Ko-Ko TW, Hongsanan S, Jones EBG, Kodsueb R, Phookamsak R, Chukeatirote E, Bahkali AH, Hyde KD (2011) Capnodiaceae. Fungal Divers 51:103–134
Chomnunti P, Hongsanan S, Aguirre-Hudson B, Tian Q, Peršoh D, Dhami MK, Alias AS, Xu J, Liu X, Stadler M, Hyde KD (2014) The sooty moulds. Fungal divers 66:1–36 Clendenin L (1896) Lasiodiplodia Ellis & Everh. n. gen. Bot Gaz 21:92–93
Clendenin L (1896) Lasiodiplodia E. & E., n. gen. Bot Gaz 21:92
Constantinescu O, Voglmayr H, Fatehi J, Thines M (2005) Plasmoverna gen. nov. and the taxonomy and nomenclature of Plasmopara (chromista, peronosporales). Taxon 54:813–821
Crane PE, Hopkins AJ, Dick MA, Bulman LS (2009) Behaviour of Neonectria fuckeliana causing a pine canker disease in New Zealand. Canad J Forest Res 39:2119–2128
Crous PW, Palm ME (1999) Reassessment of the anamorph genera Botryodiplodia, Dothiorella and Fusicoccum. Sydowia 51:167–175
Crous PW, Gams W, Stalpers JA, Robert V, Stegehuis G (2004) MycoBank: an online initiative to launch mycology into the 21st century. Stud Mycol 50:19–22
Crous PW, Slippers B, Wingfield MJ, Rheeder J, Marasas WF, Philips AJ, Alves A, Burgess T, Barber P, Groenewald JZ (2006) Phylogenetic lineages in the Botryosphaeriaceae. Stud Mycol 55:235–253
Crous PW, Schubert K, Braun U, De Hoog GS, Hocking AD, Shin HD, Groenewald JZ (2007) Opportunistic, human–pathogenic species in the Herpotrichiellaceae are phenotypically similar to saprobic or phytopathogenic species in the Venturiaceae. Stud Mycol 58:185–217
Crous PW, Braun U, Wingfield MJ, Wood AR, Shin HD, Summerell BA, Alfenas AC, Cumagun CJR, Groenewald JZ (2009) Phylogeny and taxonomy of obscure genera of microfungi. Persoonia 22:139–161
Crous PW, Wingfield MJ, Guarro J, Cheewangkoon R, van der Bank M, Swart WJ, Stchigel AM, Cano-Lira JF, Roux J, Madrid H, Damm U, Wood AR, Shuttleworth LA, Hodges CS, Munster M, de Jesús Yáñez-Morales M, Zúñiga-Estrada L, Cruywagen EM, de Hoog GS, Silvera C, Najafzadeh J, Davison EM, Davison PJN, Barrett MD, Barrett RL, Manamgoda DS, Minnis AM, Kleczewski NM, Flory SL, Castlebury LA, Clay K, Hyde KD, Maússe-Sitoe SND, Chen S, Lechat C, Hairaud M, Lesage-Meessen L, Pawlowska J, Wilk M, Sliwińska-Wyrzychowska A, Mętrak M, Wrzosek M, Pavlic-Zupanc D, Maleme HM, Slippers B, Mac Cormack WP, Archuby DI, Grünwald NJ, Tellería MT, Dueñas M, Martín MP, Marincowitz S, de Beer ZW, Perez CA, Gené J, Marin-Felix Y, Groenewald JZ (2013) Fungal planet description sheets: 154–213. Persoonia 31:188–296
Crous PW, Müller M, Sánchez RM, Giordano L, Bianchinotti M, Anderson FE, Groenewald JZ (2015) Resolving Tiarosporella spp. allied to Botryosphaeriaceae and Phacidiaceae. Phytotaxa 202:73–93
Crous PW, Wingfield MJ, Burgess TI, Hardy GE, Crane C, Barrett S, Canolira JF, Le Roux JJ, Thangavel R, Guarro J, Stchigel AM, Martín MP, Alfredo DS, Barber PA, Barreto RW, Baseia IG, Cano-Canals J, Cheewangkoon R, Ferreira RJ, Gené J, Lechat C, Moreno G, Roets F, Shivas RG, Sousa JO, Tan YP, Wiederhold NP, Abell SE, Accioly T, Albizu JL, Alves JL, Antoniolli ZI, Aplin N, Araújo J, Arzanlou M, Bezerra JDP, Bouchara JP, Carlavilla JR, Castillo A, Castroagudín VL, Ceresini PC, Claridge GF, Coelho G, Coimbra VRM, Costa LA, da Cunha KC, da Silva SS, Daniel R, de Beer ZW, Dueñas M, Edwards J, Enwistle P, Fiuza PO, Fournier J, García D, Gibertoni TB, Giraud S, Guevara-Suarez M, Gusmão LFP, Haituk S, Heykoop M, Hirooka Y, Hofmann TA, Houbraken J, Hughes DP, Kautmanová I, Koppel O, Koukol O, Larsson E, Latha KPD, Lee DH, Lisboa DO, Lisboa WS, López-Villalba Á, Maciel JLN, Manimohan P, Manjón JL, Marincowitz S, Marney TS, Meijer M, Miller AN, Olariaga I, Paiva LM, Piepenbring M, Poveda-Molero JC, Raj KNA, Raja HA, Rougeron A, Salcedo I, Samadi R, Santos TAB, Scarlett K, Seifert KA, Shuttleworth LA, Silva GA, Silva M, Siqueira JPZ, Souza-Motta CM, Stephenson SL, Sutton DA, Tamakeaw N, Telleria MT, Valenzuela-Lopez N, Viljoen A, Visagie CM, Vizzini A, Wartchow F, Wingfield BD, Yurchenko E, Zamora JC, Groenewald JZ (2016) Fungal planet description sheets: 469–557. Persoonia 37:218–403
Crous PW, Wingfield MJ, Burgess TI, Hardy GE, Barber PA, Alvarado P, Barnes CW, Buchanan PK, Heykoop M, Moreno G, Thangavel R, van der Spuy S, Barili A, Barrett S, Cacciola SO, Cano-Lira JF, Crane C, Decock C, Gibertoni TB, Guarro J, Guevara-Suarez M, Hubka V, Kolařík M, Lira CRS, Ordoñez ME, Padamsee M, Ryvarden L, Soares AM, Stchigel AM, Sutton DA, Vizzini A, Weir BS, Acharya K, Aloi F, Baseia IG, Blanchette RA, Bordallo JJ, Bratek Z, Butler T, Cano-Canals J, Carlavilla JR, Chander J, Cheewangkoon R, Cruz RHSF, da Silva M, Dutta AK, Ercole E, Escobio V, Esteve-Raventós F, Flores JA, Gené J, Góis JS, Haines L, Held BW, Jung MH, Hosaka K, Jung T, Jurjević Ž, Kautman V, Kautmanova I, Kiyashko AA, Kozanek M, Kubátová A, Lafourcade M, LaSpada F, Latha KPD, Madrid H, Malysheva EF, Manimohan P, Manjón JL, Martín MP, Mata M, Merényi Z, Morte A, Nagy I, Normand AC, Paloi S, Pattison N, Pawłowska J, Pereira OL, Petterson ME, Picillo B, Raj KNA, Roberts A, Rodríguez A, Rodríguez-Campo FJ, Romański M, Ruszkiewicz-Michalska M, Scanu B, Schena L, Semelbauer M, Sharma R, Shouche YS, Silva V, Staniaszek-Kik M, Stielow JB, Tapia C, Taylor PWJ, Toome-Heller M, Vabeikhokhei JMC, van Diepeningen AD, Van Hoa N, Van Tri M, Wiederhold NP, Wrzosek M, Zothanzama J, Groenewald JZ (2017) Fungal planet description sheets: 558–624. Persoonia 38:240–384
Cruywagen EM, Slippers B, Roux J, Wingfield MJ (2017) Phylogenetic species recognition and hybridisation in Lasiodiplodia: a case study on species from baobabs. Fungal Biol 121:420–436
Daranagama DA, Camporesi E, Tian Q, Liu X, Chamyuang S, Stadler M, Hyde KD (2015) Anthostomella is polyphyletic comprising several genera in Xylariaceae. Fungal Divers 73:203–238
Daranagama DA, Hyde KD, Sir EB, Thambugala KM, Tian Q, Samarakoon MC, McKenzie EH, Jayasiri SC, Tibpromma S, Bhat JD, Liu X, Stadler M (2018) Towards a natural classification and backbone tree for Graphostromataceae, Hypoxylaceae, Lopadostomataceae and Xylariaceae. Fungal Divers 88:1–165
de Gruyter J, Woudenberg JHC, Aveskamp MM, Verkley GJM, Groenewald JZ, Crous PW (2013) Redisposition of Phoma-like anamorphs in Pleosporales. Stud Mycol 75:1–36
de Hoog GS, Weenink XO, van den Ende AHGG (1999) Taxonomy of the Phialophora verrucosa complex with the description of two new species. Stud Mycol 43:107–122
de Hoog GS, Mayser P, Haase G, Horre R, Horrevorts AM (2000) A new species, Phialophora europaea, causing superficial infections in humans. Mycoses 43:409–416
de Hoog GS, Horré R (2002) Molecular taxonomy of the Alternaria and Ulocladium species from humans and their identification in the routine laboratory. Mycoses 45:259–276
de Hoog GS, Vicente VA, Najafzadeh MJ, Harrak MJ, Badali H, Seyedmousavi S (2011) Waterborne Exophiala species causing disease in cold-blooded animals. Persoonia 27:46–72
de Vries GA (1962) Cyphellophora laciniata nov. gen., nov. sp. and Dactylium fusarioides Fragoso et Ciferri. Mycopathol Mycol Appl 16:47–54
de Vries GA, Elders MCC, Luykx MHF (1986) Description of Cyphellophora pluriseptata sp. nov. Anton Leeuw Int J G 52:141–143
Decock C, Delgado-Rodríguez G, Buchet S, Seng JM (2003) A new species and three new combinations in Cyphellophora, with a note on the taxonomic affinities of the genus, and its relation to Kumbhamaya and Pseudomicrodochium. Antonie van Leeuwenhoek 84:209–216
Denman S, Crous PW, Taylor JE, Kang J-C, Pascoe I, Wingfield MJ (2000) An overview of the taxonomic history of Botryosphaeria, and a re–evaluation of its anamorphs based on morphology and ITS rDNA phylogeny. Stud Mycol 45:129–140
Deshwal VK, Dubey RC, Maheshwari DK (2003) Isolation of plant growth–promoting strains of Bradyrhizobium (Arachis) sp. with biocontrol potential against Macrophomina phaseolina causing charcoal rot of peanut. Curr Sci 84:443–448
Devaux AL, Sackston WE (1966) Taxonomy of Verticillium species causing wilt of horticultural crops in Quebec. Can J Bot 44:803–811
Diedicke H (1912) Die Abteilung Hyalodidymae der Spaerioideen. Ann Myc 10:135–152
Ding G, Jiang L, Guo L, Chen X, Zhang H, Che Y (2008) Pestalazines and pestalamides, bioactive metabolites from the plant pathogenic fungus Pestalotiopsis theae. J Nat Prod 71:1861–1865
Dissanayake AJ, Phillips AJL, Li XH, Hyde KD (2016) Botryosphaeriaceae: current status of genera and species. Mycosphere 7:1001–1073
dos Santos AF, Thomazi H, Duarte HSS, Machado EB, Silva CN, Tessmann DJ (2017) First report of root rot caused by Rosellinia bunodes on a poplar species (Populus deltoides) in Brazil. Plant Dis 101:632–632
Ehrenberg CG (1818) Sylvae mycologicae berolinenses. Formis Theophili Bruschcke, Berlin, Germany [In Latin]
Ekanayaka AH, Ariyawansa HA, Hyde KD, Jones EBG, Daranagama DA, Phillips AJL, Hongsanan SC, Jayasiri SC, Zhao Q (2017) Discomycetes: the apothecial representatives of the phylum Ascomycota. Fungal Divers 87:237–298
ElAraby ME, Kurle JE, Stetina SR (2003) First report of charcoal rot (Macrophomina phaseolina) on soybean in Minnesota. Plant Dis 87:202–202
Ellis MB (1971) Dematiaceous Hyphomycetes. Commonwealth Mycological Institute, Kew, Surrey, England
Ellis MB (1976) More Dematiaceous Hyphomycetes. Commonwealth Mycological Institute, Kew, Surrey, England
Ellis JB, Everhart BM (1984) New species of fungi from various localities. Proc Acad Nat Sci Phila 46:322–386
Engelhard AW, Carter JC (1956) Isolations of Verticillium albo-atrum from woody hosts in Illinois, 1945–1955. Plant Dis Rep 40:459–462
Eriksson OE (2014) Checklist of the non-lichenized ascomycetes of Sweden. Acta Universitatis Upsaliensis
Eriksson OE, Baral H-O, Currah RS, Hansen K, Kurtzman CP, Rambold G, Læssøe T (2001) Outline of Ascomycota. Myconet 6:1–27
Espinoza JG, Briceño EX, Keith LM, Latorre BA (2008) Canker and twig dieback of blueberry caused by Pestalotiopsis spp. and a Truncatella sp. in Chile. Plant Dis 92:1407–1414
European and Mediterranean Plant Protection Organization (2019) https://www.eppo.int/
Fan X, Liang YM, Ma R, Tian CM (2014) Morphological and phylogenetic studies of Cytospora (Valsaceae, Diaporthales) isolates from Chinese scholar tree, with description of a new species. Mycoscience 55:252–259
Fan X, Hyde KD, Liu M, Liang Y, Tian C (2015a) Cytospora species associated with walnut canker disease in China, with description of a new species C. gigalocus. Fungal Biol 119:310–319
Fan X, Hyde KD, Yang Q, Liang Y, Ma R, Tian C (2015b) Cytospora species associated with canker disease of three anti-desertification plants in northwestern China. Phytotaxa 197:227–244
Farr DF, Rossman AY (2019) Fungal databases, systematic mycology and microbiology laboratory, ARS, USDA. http://nt.ars-grin.gov/fungaldatabases/
Faull JL, Olejnik I, Ingrouille M, Reynolds D (2002) A reassessment of the taxonomy of some tropical sooty moulds. Trop Mycol 2:33–40
Feng P, Lu Q, Najafzadeh MJ, van den Ende AG, Sun J, Li R, Xi L, Vicente VA, Lai W, Lu C, de Hoog GS (2014) Cyphellophora and its relatives in Phialophora: biodiversity and possible role in human infection. Fung Diver 65:17–45
Filiz ÜNAL, Koca Ercan, Aşkin A, Kurbetli İ, Sarpkaya K (2018) Identification and virules of Sphaeropsis tip blight (Sphaeropsis sapinea) on Pinus spp. in Istanbul and Bursa parks. Acta Biol Turc 31:18–21
Fotouhifar KB, Hedjaroude GA, Leuchtmann A (2010) ITS rDNA phylogeny of Iranian strains of Cytospora and associated teleomorphs. Mycologia 102:1369–1382
Fournier J, Lechat C, Régis Courtecuisse R, Moreau P-A (2017a) The genus Rosellinia (Xylariaceae) in Guadeloupe and Martinique (French West Indies). Ascomycete.org 9:171–208.
Fournier J, Lechat C, Courtecuisse R (2017b) The genus Biscogniauxia (Xylariaceae) in Guadeloupe and Martinique (French West Indies). Ascomycete.org 9:67–99
Fox RTV (1998) Chrysanthemum ray blight. Mycologist 12:135–136
Fradin EF, Thomma BP (2006) Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol 7:71–86
Fraser L (1935) An investigation of the sooty mould of New South Wales IV the species of the Eucapnodieae. Proc Linn Soc NSW 40:159–178
Fries EM (1823) Systema mycologicum, vol 2. Greifswald, Germany
Fries EM (1849) Summa vegetabilium Scandinaviae, pp 1–572
Gadgil PD (1985) Cyttaria galls on silver beech. Forest Research Institute
Gams W, Holubová-Jechová V (1976) Chloridium and some other dematiaceous hyphomycetes growing on decaying wood. Stud Mycol 13:1–99
Gams W, Zare R, Summerbell RC (2005) (1654) Proposal to conserve the generic name Verticillium (anamorphic Ascomycetes) with a conserved type. Taxon 54:179–179
Gamundi IJ (1971) Las Cyttariales sudamericanas (Fungi–Ascomycetes). Darwiniana, pp 461–510
Gamundí IJ, De Lederkremer RM (1989) Los hongos andino-patagónicos del género Cyttaria. Sus hidratos de carbono. Cieñe Investig 43:4–13
Gao L, Sun G, Zhang R, Gleason ML (2014) Secondary spread of Zygophiala wisconsinensis on the surface of apple fruit. Eur J Plant Pathol 139:117–124
Gargas A, Taylor JW (1995) Phylogeny of discomycetes and early radiations of the apothecial ascomycotina inferred from SSu rDNA sequence data. Exp Mycol 19:7–15
Garibaldi A, Gilardi G, Gullino ML (2007) First report of Verticillium wilt caused by Verticillium dahliae on lettuce in Italy. Plant Dis 91:770–770
Gavrilov-Zimin IA (2017) A remarkable example of symbiosis between an animal and a fungus in a new species of legless mealybug (Insecta: Pseudococcidae). J Nat Hist 51:2211–2224
Geiser DM, del Mar Jiménez-Gasco M, Kang S, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, O’donnell K (2004) FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. Eur J Plant Pathol 110:473–479
Georgieva MI, Hlebarska S (2016) A review of Sphaeropsis sapinea occurrence on Pinus species in Bulgaria. J BioSci Biotechnol 5:247–250
Gleason ML, Batzer JC, Sun G, Zhang R, Arias MMD, Sutton TB, Crous PW, Ivanović M, McManus PS, Cooley DR, Mayr U, Weber RWS, Yoder KS, Del Ponte EM, Biggs AR, Oertel B (2011) A new view of sooty blotch and flyspeck. Plant Dis 95:368–383
Göker M, Voglmayr H, Riethmüller A, Weiß M, Oberwinkler F (2003) Taxonomic aspects of Peronosporaceae inferred from bayesian molecular phylogenetics. Can J Bot 81:672–683
Göker M, Voglmayr H, Riethmüller A, Oberwinkler F (2007) How do obligate parasites evolve? A multi–gene phylogenetic analysis of downy mildews. Fungal Genet Biol 44(2):105–122
González P, Alaniz S, Montelongo MJ, Rauduviniche L, Rebellato J, Silvera-Pérez E, Mondino P (2012) First report of Pestalotiopsis clavispora causing dieback on blueberry in Uruguay. Plant Dis 96:914–914
González-Domínguez E, Alves A, León M, Armengol J (2016) Characterization of Botryosphaeriaceae species associated with diseased loquat (Eriobotrya japonica) in Spain. Plant Pathol 66:77–89
Gordillo A, Decock C (2018) Cylindrocarpon-like (ascomycota, hypocreales) species from the Amazonian rain forests in Ecuador: additions to Campylocarpon and Dactylonectria. Cryptogam Mycol 38:409–434
Görg M, Ploch S, Kruse J, Kummer V, Runge F, Choi YJ, Thines M (2017) Revision of Plasmopara (Oomycota, Peronosporales) parasitic to impatiens. Mycol Prog 16:791–799
Gräfenhan T, Schroers HJ, Nirenberg HI, Seifert KA (2011) An overview of the taxonomy, phylogeny, and typification of nectriaceous fungi in Cosmospora, Acremonium, Fusarium, Stilbella and Volutella. Stud Mycol 68:79–113
Griffin WM, Maublanc A (1909) Sur une maladie du cacaoyer. Bull Soc Mycol Fr 25:51–58
Guba EF (1961) Monograph of pestalotia and monochaetia. Harvard University Press, Cambridge
Guo LD, Huang GR, Wang YU, He WH, Zheng WH, Hyde KD (2003) Molecular identification of white morphotype strains of endophytic fungi from Pinus tabulaeformis. Mycol Res 107:680–688
Gupta C, Dubey R, Maheshwari D (2002) Plant growth enhancement and suppression of Macrophomina phaseolina causing charcoal rot of peanut by fluorescent Pseudomonas. Biol Fertil Soils 35:399–405
Gutiérrez de Sanguinetti MM (1988) Exomorfología y anatomía de los tumores en Nothofagus antarctica (Fagaceae) atribuidos a Cyttaria harioti y Cyttaria hookeri (Cytariaceae)
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Halleen F, Crous PW, Petrini O (2003) Fungi associated with healthy grapevine cuttings in nurseries, with special reference to pathogens involved in the decline of young vines. Australas Plant Pathol 32:47–52
Halleen F, Schroers HJ, Groenewald JZ, Crous PW (2004) Novel species of Cylindrocarpon (Neonectria) and Campylocarpon gen. nov. associated with black foot disease of grapevines (Vitis spp.). Stud Mycol 50:431–455
Halleen F, Schroers HJ, Groenewald JZ, Rego C, Oliveira H, Crous PW (2006) Neonectria liriodendri sp. nov., the main causal agent of black foot disease of grapevines. Stud Mycol 55:227–234
Herath K, Jayasuriya H, Zink DL, Sigmund J, Vicente F, de la Cruz M, Basilio A, Bills GF, Polishook JD, Donald R, Phillips J, Goetz M, Singh SB (2012) Isolation, structure elucidation, and antibacterial activity of methiosetin, a tetramic acid from a tropical sooty mold (Capnodium sp.). J Nat Prod 75:420–424
Himelick EB (1969) Tree and shrub hosts of Verticillium albo–atrum. Biological notes no. 066
Hirooka Y, Kobayashi T, Natsuaki KT (2005) Neonectria castaneicola and Neo. rugulosa in Japan. Mycologia 97:1058–1066
Höhnel F (1919) Fünfte vorläufige Mitteilung mykologische Ergebnisse (Nr. 300–500). Ber Dtsch Botanischen Ges 37:153–161
Hongsanan S, Tian Q, Bahkali AH, Yang JB, Mckenzie EH, Chomnunti P, Hyde KD (2015) Zeloasperisporiales ord. nov., and two new species of Zeloasperisporium. Cryptogam Mycol 36:301–318
Hongsanan S, Zhao RL, Hyde KD (2017) A new species of Chaetothytina on branches of mango, and introducing Phaeothecoidiellaceae fam. nov. Mycosphere 8:137–146
Horikawa T (1986) Yield loss of new tea shoots due to tea gray blight caused by Pestalotia longiseta SPEGAZZINI. Bulletin of the Shizuoka Tea Experiment Station, Japan
Hsieh HM, Lin CR, Fang MJ, Rogers JD, Fournier J, Lechat C, Ju YM (2010) Phylogenetic status of Xylaria subgenus Pseudoxylaria among taxa of the subfamily Xylarioideae (Xylariaceae) and phylogeny of the taxa involved in the subfamily. Mol Phylogenet Evol 54:957–969
Hu H, Jeewon R, Zhou D, Zhou T, Hyde KD (2007) Phylogenetic diversity of endophytic Pestalotiopsis species in Pinus armandii and Ribes spp.: evidence from rDNA and β-tubulin gene phylogenies. Fungal Divers 24:1–22
Hughes SJ (2003) Capnofrasera dendryphioides, a new genus and species of sooty moulds. N Z J Bot 41:139–146
Hughes SJ, Seifert KA (2012) Taxonomic and nomenclatural notes on sooty mould name based on species mixtures: Hormiscium handelii and Torula lecheriana. Mycoscience 53:17–24
Hyde KD, Jones EBG, Liu JK, Ariyawansha H, Boehm E, Boonmee S, Braun U, Chomnunti P, Crous PW, Dai DQ, Diederich P, Dissanayake A, Doilom M, Doveri F, Hongsanan S, Jayawardena R, Lawrey JD, Li YM, Liu YX, Lücking R, Monkai J, Muggia L, Nelsen MP, Pang KL, Phookamsak R, Senanayake IC, Shearer CA, Suetrong S, Tanaka K, Thambugala KM, Wijayawardene NN, Wikee S, Wu HX, Zhang Y, Aguirre-Hudson B, Alias SA, Aptroot A, Bahkali AH, Bezerra JL, Bhat DJ, Camporesi E, Chukeatirote E, Gueidan C, Hawksworth DL, Hirayama K, De Hoog S, Kang JC, Knudsen K, Li WJ, Li XH, Liu ZY, Mapook A, McKenzie EHC, Miller AN, Mortimer PE, Phillips AJL, Raja HA, Scheuer C, Schumm F, Taylor JE, Tian Q, Tibpromma S, Wanasinghe DN, Wang Y, Xu JC, Yacharoen S, Yan JY, Zhang M (2013) Families of Dothideomycetes. Fungal Divers 63:1–313
Hyde KD, Nilsson RH, Alias SA, Ariyawansa HA, Blair JE, Cai L, de Cock AWAM, Dissanayake AJ, Glockling SL, Goonasekara ID, Gorczak M, Hahn M, Jayawardena RS, van Kan JAL, Laurence MH, Lévesque CA, Li X, Liu JK, Maharachchikumbura SSN, Manamgoda DS, Martin FN, McKenzie EHC, McTaggart AR, Mortimer PE, Nair PVR, Pawłowska J, Rintoul TL, Shivas RG, Spies CFJ, Summerell BA, Taylor PWJ, Terhem RB, Udayanga D, Vaghefi N, Walther G, Wilk M, Wrzosek M, Xu JC, Yan JY, Zhou N (2014) One stop shop:backbones trees for important phytopathogenic genera: I. Fungal Divers 67:21–125
Hyde KD, Hongsanan S, Jeewon R, Bhat DJ, McKenzie EHC, Jones EBG, Phookamsak R, Ariyawansa HA, Boonmee S, Zhao Q, Abdel-Aziz FA, Abdel-Wahab MA, Banmai S, Chomnunti P, Cui BK, Daranagama DA, Das K, Dayarathne MC, de Silva NI, Dissanayake AJ, Doilom M, Ekanayaka AH, Gibertoni TB, Góes-Neto A, Huang SK, Jayasiri SC, Jayawardena RS, Konta S, Lee HB, Li WJ, Lin CG, Liu JK, Lu YZ, Luo ZL, Manawasinghe IS, Manimohan P, Mapook A, Niskanen T, Norphanphoun C, Papizadeh M, Perera RH, Phukhamsakda C, Richter C, de Santiago ALCM, Drechsler-Santos ER, Senanayake IC, Tanaka K, Tennakoon TMDS, Thambugala KM, Tian Q, Tibpromma S, Thongbai B, Vizzini A, Wanasinghe DN, Wijayawardene NN, Wu HX, Yang J, Zeng XY, Zhang H, Zhang JF, Bulgakov TS, Camporesi E, Bahkali AH, Amoozegar MA, Araujo-Neta LS, Ammirati JF, Baghela A, Bhatt RP, Bojantchev D, Buyck B, da Silva GA, de Lima CLF, de Oliveira RJV, de Souza CAF, Dai YC, Dima B, Duong TT, Ercole E, Mafalda-Freire F, Ghosh A, Hashimoto A, Kamolhan S, Kang JC, Karunarathna SC, Kirk PM, Kytövuori I, Lantieri A, Liimatainen K, Liu ZY, Liu XZ, Lücking R, Medardi G, Mortimer PE, Nguyen TTT, Promputtha I, Raj KNA, Reck MA, Lumyong S, Shahzadeh-Fazeli SA, Stadler M, Soudi MR, Su HY, Takahashi T, Tangthirasunun N, Uniyal P, Wang Y, Wen TC, Xu JC, Zhang ZK, Zhao YC, Zhou JL, Zhu L (2016) Fungal diversity notes 367–490: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 80:1–270
Hyde KD, Norphanphoun C, Abreu VP, Bazzicalupo A, Chethana KWT, Clericuzio M, Dayarathne MC, Dissanayake AJ, Ekanayaka AH, He MQ, Hongsanan S, Huang SK, Jayasiri SC, Jayawardena RS, Karunarathna A, Konta S, Kušan I, Lee H, Li J, Lin CG, Liu NG, Lu YZ, Luo ZL, Manawasinghe IS, Mapook A, Perera RH, Phookamsak R, Phukhamsakda C, Siedlecki I, Soares AM, Tennakoon DS, Tian Q, Tibpromma S, Wanasinghe DN, Xiao YP, Yang J, Zeng XY, Abdel-Aziz FA, Li WJ, Senanayake IC, Shang QJ, Daranagama DA, de Silva NI, Thambugala KM, Abdel-Wahab MA, Bahkali AH, Berbee ML, Boonmee S, Bhat DJ, Bulgakov TS, Buyck B, Camporesi E, Castañeda-Ruiz RF, Chomnunti P, Doilom M, Dovana F, Gibertoni TB, Jadan M, Jeewon R, Jones EBG, Kang JC, Karunarathna SC, Lim YW, Liu JK, Liu ZY, Plautz HL Jr, Lumyong S, Maharachchikumbura SSN, Matočec N, McKenzie EHC, Mešić A, Miller D, Pawłowska J, Pereira OL, Promputtha I, Romero AI, Ryvarden L, Su HY, Suetrong S, Tkalčec Z, Vizzini A, Wen TC, Wisitrassameewong K, Wrzosek M, Xu JC, Zhao Q, Zhao RL, Mortimer PE (2017) Fungal diversity notes 603–708: taxonomic and phylogenetic notes on genera and species. Fungal Divers 87:1–235
Hyde KD, Chaiwan N, Norphanphoun C, Boonmee S, Camporesi E, Chethana KWT, Dayar-athne MC, de Silva NI, Dissanayake AJ, Ekanayaka AH, Hongsanan S, Huang SK, Jayasiri SC, Jayawardena RS, Jiang HB, Karunarathna A, Lin CG, Liu JK, Liu NG, Lu YZ, Luo ZL, Maharachchimbura SSN, Manawasinghe IS, Pem D, Perera RH, Phukhamsakda C, Samarakoon MC, Senwanna C, Shang QJ, Tennakoon DS, Thambugala KM, Tibpromma S, Wanasinghe DN, Xiao YP, Yang J, Zeng XY, Zhang JF, Zhang SN, Bulgakov TS, Bhat DJ, Cheewangkoon R, Goh TK, Jones EBG, Kang JC, Jeewon R, Liu ZY, Lumyong S, Kuo CH, McKenzie EHC, Wen TC, Yan JY, Zhao Q (2018) Mycosphere notes 169–224. Mycosphere 9:271–430
Inderbitzin P, Bostock RM, Davis RM, Usami T, Platt HW, Subbarao KV (2011) Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS ONE 6:e28341
Index Fungorum (2019) http://www.indexfungorum.org/names/Names.asp
Jacob M, Bhat DJ (2000) Two new endophytic fungi from India. Cryptogam Mycol 21:81–88
Jaklitsch WM, Gardiennet A, Voglmayr H (2016) Resolution of morphology-based taxonomic delusions: Acrocordiella, Basiseptospora, Blogiascospora, Clypeosphaeria, Hymenopleella, Lepteutypa, Pseudapiospora, Requienella, Seiridium and Strickeria. Persoonia 37:82–105
Jami F, Slippers B, Wingfield MJ, Gryzenhout M (2012) Five new species of the Botryosphaeriaceae from Acacia Karroo in South Africa. Cryptogam Mycol 33:245–267
Jami F, Slippers B, Wingfield MJ, Gryzenhout M (2014) Botryosphaeriaceae species overlap on four unrelated, native South African hosts. Fungal Biol 118:168–179
Jayasiri SC, Hyde KD, Jones EBG, McKenzie EHC, Jeewon R, Phillips AJL, Bhat DJ, Wanasinghe DN, Liu JK, Lu YZ, Kang JC, Xu J, Karunarathna SC (2019) Diversity, morphology and molecular phylogeny of Dothideomycetes on decaying wild seed pods and fruits. Mycosphere 10:1–186
Jayawardena RS, Zhang W, Liu M, Maharachchikumbura SS, Zhou Y, Huang J, Nilthong S, Wang Z, Li X, Yan J, Hyde KD (2015) Identification and characterization of Pestalotiopsis-like fungi related to grapevine diseases in China. Fungal Biol 119:348–361
Jayawardena RS, Liu M, Maharachchikumbura SS, Zhang W, Xing Q, Hyde KD, Nilthong S, Li X, Yan J (2016) Neopestalotiopsis vitis sp. nov. causing grapevine leaf spot in China. Phytotaxa 258:63–74
Jayawardena RS, Hyde KD, Jeewon R, Ghobad-Nejhad M, Wanasinghe DN, Liu N, Phillips AJL, Oliveira-Filho JRC, da Silva GA, Gibertoni TB, Abeywikrama P, Carris LM, Chethana KWT, Dissanayake AJ, Hongsanan S, Jayasiri SC, McTaggart AR, Perera RH, Phutthacharoen K, Savchenko KG, Shivas RG, Thongklang N, Dong W, Wei D, Wijayawardena NN, Kang JC (2019) One stop shop II: taxonomic update with molecular phylogeny for important phytopathogenic genera: 26–50. Fungal Divers 94(1):41–129
Jeewon R, Liew EC, Simpson JA, Hodgkiss IJ, Hyde KD (2003) Phylogenetic significance of morphological characters in the taxonomy of Pestalotiopsis species. Mol Phylogenet Evol 27:372–383
Ju YM, Rogers JD, Hsieh HM, Vasilyeva L (2004) Amphirosellinia gen. nov. and a new species of Entoleuca. Mycologia 96:1393–1402
Kaliterna J, Milicevic T, Bencic D, Mesic A (2016) First report of Verticillium wilt caused by Verticillium dahliae on Olive trees in Croatia. Plant Dis 100:2526–2526
Kamoun S, Furzer O, Jones JD, Judelson HS, Ali GS, Dalio RJ, Roy SG, Schena L, Zambounis A, Panabières F, Cahill D (2015) The Top 10 oomycete pathogens in molecular plant pathology. Mol Plant Pathol 16:413–434
Kasanen R, Hantula J, Ostry M, Pinon J, Kurkela T (2004) North American populations of Entoleuca mammata are genetically more variable than populations in Europe. Mycol Res 108:766–774
Kassemeyer HH (2017) Fungi of grapes. In: König H, Unden G, Fröhlich J (eds) Biology of microorganisms on grapes, in must and in wine. Springer, Cham, pp 103–132
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298
Keinath AP (2013) Diagnostic guide for gummy stem blight and black rot on cucurbits. Plant Health Prog 14:35
Kim YK, Xiao CL, Rogers JD (2005) Influence of culture media and environmental factors on mycelial growth and pycnidial production of Sphaeropsis pyriputrescens. Mycologia 97:25–32
Kirk PM, Cannon PF, Minter DW, Stalpers JA (2008) Dictionary of the fungi 10th edition. CAB International, Wallingford, UK
Kleina HT, Santos ÁFD, Duarte HDSS, Machado EB (2018) Physiological characterization of Rosellinia bunodes and symptomatology of Rosellinia root rot in poplar seedlings. Revista Árvore 42(1):e420111
Koehler AM, Shew HD (2018) First report of charcoal rot of stevia caused by Macrophomina phaseolina in North Carolina. Plant Dis 102:241–241
Korf RP (1973) Discomycetes and Tuberales. In: Ainsworth GC, Sparrow FK, Sussman AS (eds) The fungi: an advanced treatise, vol 4A. Academic press, London, pp 249–319
Laemmlen FF (2011) Sooty mold. Integrated pest management for home gardeners and landscape professionals. Pest notes. University of California, Agriculture and Natural Resources, California, USA
Læssøe T, Spooner BM (1994) Rosellinia and Astrocystis (Xylariaceae): new species and genetic concepts. Kew Bull 49:1–70
Læssøe T (1994) Index Ascomycetum 1. Xylariaceae. Syst Ascomycetum 13:43–112
Land CJ, Lawrence KS, Newman M (2016) First report of Verticillium dahliae on Cotton in Alabama. APS publications 100
Laurence MH, Summerell BA, Burgess LW, Liew EC (2014) Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biol 118:374–384
Lawrence DP, Rotondo F, Gannibal PB (2016) Biodiversity and taxonomy of the pleomorphic genus Alternaria. Mycol Prog 15:3
Lee S, Crous PW, Wingfield MJ (2006) Pestalotioid fungi from Restionaceae in the Cape Floral Kingdom. Stud mycol 55:175–187
Léveillé JH (1847) Mycologie, mycétologie. D’Orbigny Dictionnaire univ d’Hist nat 9:261–303
Li W, Guo L (2015) Rosellinia brunneola sp. nov. and R. beccariana new to China. Mycotaxon 130:233–236
Li Q, Kang J, Hyde KD (2015) Two new Rosellinia species from Southwest China. Mycotaxon 130(2):563–567
Li GJ, Hyde KD, Zhao RN, Hongsanan S, Abdel-Aziz FA, Abdel-Wahab MA, Alvarado P, Alves-Silva G, Ammirati JF, Ariyawansa HA, Baghela A, Bahkali AH, Beug M, Bhat DJ, Bojantchev D, Boonpratuang T, Bulgakov TS, Camporesi E, Boro MC, Ceska O, Chakraborty D, Chen JJ, Chethana KWT, Chomnunti P, Consiglio G, Cui BK, Dai DQ, Dai YC, Daranagama DA, Das K, Dayarathne MC, Crop ED, De Oliveira RJV, de Souza CAF, de Souza JI, Dentinger BTM, Dissanayake AJ, Doilom M, Drechsler-Santos ER, Ghobad-Nejhad M, Gilmore SP, Góes-Neto A, Gorczak M, Haitjema GH, Hapuarachchi KK, Hashimoto A, He MQ, Henske JK, Hirayama K, Iribarren MJ, Jayasiri SC, Jayawardena RS, Jeon SJ, Jesus AL, Jerônimo GH, Jones EBG, Kang JC, Karunarathna SC, Kirk PM, Konta S, Kuhnert E, Langer E, Lee HS, Lee HB, Li WJ, Li XH, Liimatainen K, Lima DX, Lin CG, Liu JK, Liu XZ, Liu ZY, Luangsaard JJ, Lücking R, Lumbsch HT, Lumyong S, Leaño EM, Marano AV, Matsumura M, McKenzie EHC, Mongkol-samrit S, Mortimer PE, Nguyen TTT, Niskanen T, Norphan-phoun C, O’Malley MA, Parnmen S, Pawłowska J, Perera RH, Phookamsak R, Phukhamsakda C, Pires-Zottarelli CLA, Raspé O, Reck MA, Rocha SCO, de Santiago ALCMA, Senanayake IC, Setti L, Shang QJ, Singh SK, Sir EB, Solomon KV, Song J, Srikitikulchai P, Stadler M, Suetrong S, Takahashi H, Takahashi T, Tanaka K, Tang LP, Thambugala KM, Thanakitpipattana D, Theodorou MK, Thongbai B, Thummarukcharoen T, Tian Q, Tibpromma S, Verbeken A, Vizzini A, Vlasák J, Voigt K, Wanasinghe DN, Wang Y, Weerakoon G, Wen HA, Wen TC, Wijayawardene NN, Wongkanoun S, Wrzosek M, Xiao YP, Xu JC, Yan JY, Yang J, Yang SD, Hu Y, Zhang JF, Zhao J, Zhou LW, Persoh D, Phillips AJL, Maharachchikumbura SSN (2016) Fungal diversity notes 253–366: taxonomic and phylogeneticcontributions to fungal taxa. Fungal Divers 78:1–237
Liu JK, Hyde KD, Jones EBG, Ariyawansa HA, Bhat DJ, Boonmee S, Maharachchikumbura SSN, McKenzie EHC, Phookamsak R, Phukhamsakda C, Shenoy BD, Abdel-Wahab MA, Buyck B, Chen J, Chethana KWT, Singtripop C, Dai DQ, Dai YC, Daranagama DA, Dissanayake AJ, Doliom M, Dsouza MJ, Fan XL, Goonasekara ID, Hirayama K, Hongsanan S, Jayasiri SC, Jayawardena RS, Karunarathna SC, Li WJ, Mapook A, Norphanphoun C, Pang KL, Perera RH, Peršoh D, Pinruan U, Senanayake IC, Somrithipol S, Suetrong S, Tanaka K, Thambugala KM, Tian Q, Tibpromma S, Udayanga D, Wijayawardene NN, Wanasinghe D, Wisitrassameewong K, Zeng XY, Abdel-Aziz FA, Adamčík S, Bahkali AH, Boonyuen N, Bulgakov T, Callac P, Chomnunti P, Greiner K, Hashimoto A, Hofstetter V, Kang JC, Lewis D, Li XH, Liu ZY, Liu ZY, Matumura M, Mortimer PE, Rambold R, Randrianjohany E, Sato G, Indrasutdhi VS, Tian CM, Verbeken A, Brackel W, Wang Y, Wen TC, Xu JC, Yan JY, Zhao RL, Camporesi E (2015) Fungal diversity notes 1-100: taxonomic and phylogenetic contributions to fungal species. Fungal Divers 72:1–197
LoBuglio KF, Pfister DH (2010) Placement of Medeolaria farlowii in the Leotiomycetes, and comments on sampling within the class. Mycol Prog 9(3):361–368
Lo Giudice V, Raudino F, di San Lio RM, Cacciola SO, Faedda R, Pane A (2010) First report of a decline and wilt of young Olive trees caused by simultaneous infections of Verticillium dahliae and Phytophthora palmivora in Sicily. Plant Dis 94:1372–1373
Lombard L, Bezuidenhout CM, Crous PW (2013) Ilyonectria black foot rot associated with Proteaceae. Australas Plant Pathol 42:337–349
Lombard L, Van Der Merwe A, Groenewald JZ, Crous PW (2014) Lineages in Nectriaceae: re-evaluating the generic status of Ilyonectria and allied genera. Phytopathol Mediterr 1:515–532
Lombard L, van der Merwe NA, Groenewald JZ, Crous PW (2015) Generic concepts in Nectriaceae. Stud Mycol 80:189–245
Lombard L, Sandoval-Denis M, Lamprecht SC, Crous PW (2018) Epitypification of Fusarium oxysporum – clearing the taxonomic chaos. Persoonia 43:1–47
Lombard L, Van Doorn R, Crouse PW (2019) Neotypification of Fusarium chlamydosporuma reappraisal of a clinically important species complex. Fungal Syst Evol 4:183–200
Luo J, Zhuang W-Y (2010a) Chaetopsinectria (Nectriaceae, Hypocreales), a new genus with Chaetopsina anamorphs. Mycologia 102:976–984
Luo J, Zhuang W-Y (2010b) Three new species of Neonectria (Nectriaceae, Hypocreales) with notes on their phylogenetic positions. Mycologia 102:142–152
Madrid H, Hernandez-Restrepo M, Gené J, Cano J, Guarro J, Silva V (2016) New and interesting chaetothyrialean fungi from Spain. Mycol Prog 15:1179–1201
Maharachchikumbura SS, Guo LD, Chukeatirote E, Chukeatirote AH, Hyde KD (2011) Pestalotiopsis-morphology, phylogeny, biochemistry and diversity. Fungal Divers 50:167
Maharachchikumbura SSN, Guo LD, Cai L, Chukeatirote E, Wu WP, Sun X, Crous PW, Bhat DJ, McKenzie EHC, Bahkali AH, Hyde KD (2012) A multi-locus backbone tree for Pestalotiopsis, with a polyphasic characterization of 14 new species. Fungal Divers 56:95–129
Maharachchikumbura SSN, Chukeatirote E, Guo LD, Crous PW, Mckenzie EHC, Hyde KD (2013a) Pestalotiopsis species associ-ated with Camellia sinensis(tea). Mycotaxon 123:47–61
Maharachchikumbura SSN, Guo LD, Chukeatirote E, McKenzie EHC, Hyde KD (2013b) A destructive new disease of Syzygium samarangense in Thailand caused by the new species Pestalotiopsis samarangensis. Trop Plant Pathol 38:227–235
Maharachchikumbura SS, Hyde KD, Groenewald JZ, Xu J, Crous PW (2014a) Pestalotiopsis revisited. Stud Mycol 79:121–186
Maharachchikumbura SS, Guo LD, Chukeatirote E, Hyde KD (2014b) Improving the backbone tree for the genus Pestalotiopsis; addition of P. steyaertii and P. magna sp. nov. Mycol Prog 13:617–624
Maharachchikumbura SS, Hyde KD, Jones EG, McKenzie EH, Huang SK, Abdel-Wahab MA, Daranagama DA, Dayarathne M, D’souza MJ, Goonasekara ID, Hongsanan S, Jayawardena RS, Kirk PM, Konta S, Liu JK, Lui ZY, Norphanphoun C, Pang KL, Perera RH, Senanayake IC, Shang Q, Shenoy BD, Xiao Y, Bahkali AH, Kang J, Somrothipol S, Suetrong S, Wen T, Xu J (2015) Towards a natural classification and backbone tree for Sordariomycetes. Fungal Divers 72:199–301
Maharachchikumbura SS, Guo LD, Liu ZY, Hyde KD (2016) Pseudopestalotiopsis ignota and Ps. camelliae spp. nov. associated with grey blight disease of tea in China. Mycol Prog 15:22
Maharachchikumbura SS, Larignon P, Al-Sadi AM, Zuo-Yi LIU (2017) Characterization of Neopestalotiopsis, Pestalotiopsis and Truncatella species associated with grapevine trunk diseases in France. Phytopathol Mediterr 55:380–390
Mantiri FR, Samuels GJ, Rahe JE, Honda BM (2001) Phylogenetic relationships in Neonectria species having Cylindrocarpon anamorphs inferred from mitochondrial ribosomal DNA sequences. Canad J Bot 79:334–340
Maryani N, Lombard L, Poerba YS, Subandiyah S, Crous PW, Kema GH (2019a) Phylogeny and genetic diversity of the banana Fusarium wilt pathogen Fusarium oxysporum f. sp. cubense in the Indonesian centre of origin. Stud Mycol 92:155–194
Maryani N, Sandoval-Denis M, Lombard L, Crous PW, Kema GHJ (2019b) New endemic Fusarium species hitch-hiking with pathogenic Fusarium strains causing Panama disease in small-holder banana plots in Indonesia. Persoonia 43:48–69
Matsushima T (1987) Matsushima. Mycol Mem 5:1–100
Mayfield DA, Karakaya A, Batzer JC, Blaser JM, Gleason ML (2012) Diversity of sooty blotch and flyspeck fungi from apples in northeastern Turkey. Eur J Plant Pathol 135:805–815
McNeill J, Barrie FR, Buck WR, Demoulin V, Greuter W, Hawksworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, van Reine WFP, Smith GE, Wiersema JH, Turland NJ (eds) (2012) International code of nomenclature for algae, fungi, and fungal diversity plants (Melbourne Code), Adopted by the Eighteenth International Botanical Congress Melbourne, Australia, July 2011
McTaggart AR, Shuey LS, McKenna SG, Davis RI, Shivas RG (2015) Plasmopara sphagneticolae sp. nov. (Peronosporales) on Sphagneticola (Asteraceae) in Australia. Australas Plant Pathol 44:81–85
Meena BR, Nagendran K, Tripathi AN, Kumari S, Sagar V, Gupta N, Rai AB, Singh B (2018) First report of charcoal rot caused by Macrophomina phaseolina in Basella alba in India. Plant Dis 102(8):1669
Mengoni T (1986) El aparato apical del asco de Cyttaria harioti (Ascomycetes, Cyttariales) con microscopia fotonica y electronica. Bol Soc Argent Bot 24:393–401
Miller JH (1928) Biologic studies in the Sphaeriales—I. Mycologia 20:187–213
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: 2010 gateway computing environments workshop (GCE), pp 1–8. IEEE
Montagne JFC (1834) Notice sur les plantes cryptogames récemment découvertes en France contenant aussi l’indication précis des localités de quelques espèces les plus rares de la flore française. Ann Sci Nat Bot Sér 2:295–307
Montagne C (1849) Capnodium, novum Fungorum genus
Morton A, Carder JH, Barbara DJ (1995) Sequences of the internal transcribed spacers of the ribosomal RNA genes and relationships between isolates of Verticillium alboatrum and V. dahliae. Plant Pathol 44:183–190
Nag Raj TR (1985) Redisposals and redescriptions in the Monochaetia-Seiridium, Pestalotia-Pestalotiopsis complexes. II. Pestalotiopsis besseyii (Guba) comb. nov. and Pestalosphaeria varia sp. nov. Mycotaxon 22:43–75
NagRaj TR (1993) Coelomycetous anamorphs with appendage-bearing conidia. Mycologue Publications, Waterloo, ON
Nalim FA, Samuels GJ, Wijesundera RL, Geiser DM (2011) New species from the Fusarium solani species complex derived from perithecia and soil in the old World tropics. Mycologia 103:1302–1330
Ndiaye M, Termorshuizen AJ, van Bruggen AHC (2010) Effects of compost amendment and the biocontrol agent Clonostachys rosea on the development of charcoal rot (Macrophomina phaseolina) on cowpea. J Plant Pathol 92:173–180
Nees von Esenbeck CDG (1816) System der Pilze und Schwämme. Stahelschen Buchhandlung, Würtzburg
Norphanphoun C, Doilom M, Daranagama DA, Phookamsak R, Wen TC, Bulgakov TS, Hyde KD (2017) Revisiting the genus Cytospora and allied species. Mycosphere 8:51–97
Norphanphoun C, Raspé O, Jeewon R, Wen TC, Hyde KD (2018) Morphological and phylogenetic characterisation of novel Cytospora species associated with mangroves. MycoKeys 38:93–120
Nylander JAA, Wilgenbusch JC, Warren DL, Swofford DL (2008) AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24:581–583
O’Donnell K (2000) Molecular phylogeny of the Nectria haematococca-Fusarium solani species complex. Mycologia 92:919–938
O’Donnell K, Kistler HC, Cigelnik E, Ploetz RC (1998a) Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc Natl Acad Sci USA 95:2044–2049
O’Donnell K, Cigelnik E, Casper HH (1998b) Molecular phylogenetic, morphological, and mycotoxin data support reidentification of the Quorn mycoprotein fungus as Fusarium venenatum. Fungal Genet Biol 23:57–67
O’Donnell K, Kistler HC, Tacke BK, Casper HH (2000) Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc Natl Acad Sci USA 97:7905–7910
O’Donnell K, Rooney AP, Proctor RH, Brown DW, McCormick SP, Ward TJ, Frandsen RJ, Lysøe E, Rehner SA, Aoki T, Robert VA (2013) Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Gen Biol 52:20–31
Oliveira ML, Melo GL, Niella ARR, Silva VR (2008) Black root rot caused by Rosellinia pepo, a new disease of the clove tree in Brazil. Tropical Plant Pathol 33:90–95
Ostry ME (2013) Hypoxylon canker. Infectious Forest Diseases, p 407
Pace-Lupi TG, Porta-Puglia A, Ippolito A, Nigro F (2006) First record of Verticillium dahliae on potato in Malta. Plant Dis 90:1108–1108
Pal KK, Tilak KVBR, Saxcna AK, Dey R, Singh CS (2001) Suppression of maize root diseases caused by Macrophomina phaseolina, Fusarium moniliforme and Fusarium graminearum by plant growth promoting rhizobacteria. Microbiol Res 156:209–223
Pantou MP, Strunnikova OK, Shakhnazarova VY, Vishnevskaya NA, Papalouka VG, Typas MA (2005) Molecular and immunochemical phylogeny of Verticillium species. Mycol Res 109:889–902
Parker KG (1959) Verticillium hadromycosis of deciduous tree fruits. Plant disease reporter, suppl 255
Parkinson LE, Shivas RG, Dann EK (2017) Pathogenicity of nectriaceous fungi on avocado in Australia. Phytopathology 107:1479–1485
Pasini L, Prodorutti D, Pastorelli S, Pertot I (2016) Genetic diversity and biocontrol of Rosellinia necatrix infecting apple in Northern Italy. Plant Dis 100:444–452
Patouillard N, de Lagerheim G (1892) Champignons de l’Équateur (Pugillus II). Bull Soc Mycol Fr 8:113–140
Pavlic D, Slippers B, Coutinho TA, Gryzenhout M, Wingfield MJ (2004) Lasiodiplodia gonubiensis sp. nov., a new Botryosphaeria anamorph from native Syzygium cordatum in South Africa. Stud Mycol 50:313–322
Pegg GF, Brady BL (2002) Verticillium wilts. CABI Publishing, Oxford, UK
Peraldi A, Griffe LL, Burt C, McGrann GRD, Nicholson P (2014) Brachypodium distachyon exhibits compatible interactions with Oculimacula spp. and Ramularia collocygni, providing the first pathosystem model to study eyespot and ramularia leaf spot diseases. Plant Pathol 63:554–562
Pérez-Jiménez RM (2006) A review of the biology and pathogenicity of Rosellinia necatrix—the cause of white root rot disease of fruit trees and other plants. J Phytopathol 154(5):257–266
Peterson KR, Pfister DH (2010) Phylogeny of Cyttaria inferred from nuclear and mitochondrial sequence and morphological data. Mycologia 102(6):1398–1416
Peterson KR, Pfister DH, Bell CD (2010) Cophylogeny and biogeography of the fungal parasite Cyttaria and its host Nothofagus, southern beech. Mycologia 102(6):1417–1425
Pethybridge SJ, Hay FS, Clarkson RA, Groom T, Wilson CR (2008) Host range of Australian Phoma ligulicola var inoxydablis isolates from pyrethrum. J Phytopathol 156(7–8):506–508
Petrini LE (1993) Rosellinia species of the temperate zones. Sydowia 44:169–281
Petrini LE (2013) Rosellinia-a world monograph
Pfister DH, LoBuglio KF (2009) Placement of Medeolaria farlowii in the Leotiomycetes, and comments on sampling within the class. Mycol Prog 9:361–368
Pfister DH, Agnello C, Lantieri A, LoBuglio KF (2013) The Caloscyphaceae (Pezizomycetes, Ascomycota), with a new genus. Mycol Progress 12(4):667–674
Phillips AJL, Alves A, Abdollahzadeh J, Slippers B, Wingfield MJ, Groenewald JZ, Crous PW (2013) The Botryosphaeriaceae: genera and species known from culture. Stud Mycol 76:51–167
Phillips AJL, Alves A, Correia A, Luque J (2005) Two new species of Botryosphaeria with brown, 1-septate ascospores and Dothiorella anamorphs. Mycologia 97:513–529
Phillips AJL, Alves A, Pennycook SR, Johnston PR, Ramaley A, Akulov A, Crous PW (2008) Resolving the phylogenetic and taxonomic status of dark-spored teleomorph genera in the Botryosphaeriaceae. Persoonia 21:29–55
Phillips AJL, Lopes J, Abdollahzadeh J, Bobev S, Alves A (2012) Resolving the Diplodia complex on apple and other Rosaceae hosts. Persoonia 29:29–38
Phookamsak R, Hyde KD, Jeewon R, Bhat DJ, Jones EBG, Maharachchikumbura SSN, Raspé O, Karunarathna SC, Wanas-inghe DN, Hongsanan S, Doilom M, Tennakoon DS, Firmino AL, Machado AR, Ghosh A, Karunarathna A, Mešsić A, Dutta AK, Thongbai B, Devadatha B, Norphanphoun C, Senwanna C, Wei D, Pem D, Ackah FK, Wang GN, Jiang HB, Madrid H, Lee HB, Goonasekara ID, Manawasinghe IS, Kušan Cano J, Gené J, Li J, Das K, Acharya K, Raj KNA, Latha KPD, Chethana KWT, He MQ, Duenas M, Jadan M, Martín MP, Dayarathne MC, Samarakoon MC, Raza M, Park MS, Telleria MT, Chaiwan N, Matočec N, de Silva NI, Pereira OL, Singh PN, Manimohan P, Uniyal P, Shang QJ, Bhatt RP, Perera RH, Alvarenga RLM, Nogal-Prata S, Singh SK, Vadthanarat S, Oh SY, Huang SK, Rana S, Konta S, Paloi S, Jayasiri SC, Jeon SJ, Mehmood T, Gibertoni TB, Nguyen TTT, Singh U, Thiyagaraja V, Sarma VV, Dong W, Yu XD, Lu YZ, Lim YW, Chen Y, Tkalčec Z, Zhang ZF, Luo ZL, Daranagama DA, Thambugala KM, Tibpromma S, Camporesi E, Bulgakov T, Dissanayake AJ, Senanayake IC, Dai DQ, Tang LZ, Khan S, Zhang H, Promputtha I, Cai L, Chomnunti P, Zhao RL, Lumyong S, Boonmee S, Wen TC, Mortimer PE, Xu J (2019) Fungal diversity notes 929–1036: taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers 95:1–273
Ploetz RC, Lim TK, Menge JA, Kenneth G, Rohrbach KG, Michaolides TJ (2003) Common pathogens of tropical fruit crops. Diseases of tropical fruit crops. CABI Publishing, Wallingford, pp 1–19
Powell M, Gundersen B, Miles C, Coats K, Inglis DA (2013) First report of Verticillium wilt on lettuce (Lactuca sativa) in Washington caused by Verticillium tricorpus. Plant Dis 97(7):996–997
Punithalingam E (1976) Botryodiplodia theobromae. CMI descriptions of pathogenic fungi and bacteria, No. 519. Commonwealth Mycological Institute, Kew, Surrey, England
Punithalingam E (1980) Plant diseases attributed to Botryodiplodia theobromae. Pat J Cramer, Vaduz
Quaedvlieg W, Binder M, Groenewald JZ, Summerell BA, Carnegie AJ, Burgess TI, Crous PW (2014) Introducing the consolidated species concept to resolve species in the Teratosphaeriaceae. Persoonia 33:1–40
Rampersad SN (2008) First report of Verticillium dahliae causing wilt in pumpkin in Trinidad. Plant Dis 92:1136–1136
Rana A, Sahgal M, Johri BN (2017) Fusarium oxysporum: genomics, diversity and plant–host interaction. In: Satyanarayana T, Deshmukh S, Johri B (eds) Developments in fungal biology and applied mycology. Springer, Singapore, pp 159–199
Rawlings GB (1956) Australasian Cyttariaceae. New Zealand Forest Service
Réblová M, Untereiner WA, Réblová K (2013) Novel evolutionary lineagesrevealed in the Chaetothyriales (Fungi) based on multigene phylogenetic analyses and comparison of ITS secondary structure. PLoS ONE 8:e63547. https://doi.org/10.1371/journal.pone.0063547
Rees AA, Webber JF (1988) Pathogenicity of Sphaeropsis sapinea to seed, seedlings and saplings of some Central American pines. Trans Br Mycol Soc 91(2):273–277
Riethmüller A, Voglmayr H, Göker M, Weiß M, Oberwinkler F (2002) Phylogenetic relationships of the downy mildews (peronosporales) and related groups based on nuclear large subunit ribosomal dna sequences. Mycologia 94:834–849
Reynolds DR, Gilbert GS (2005) Epifoliar fungi from Queensland, Australia. Aust Syst Bot 18(3):265–289
Rogers JD, Ju YM (1996) Entoleuca mammata comb. nov. for Hypoxylon mammatum and the genus Entoleuca. Mycotaxon 59:441
Romero Luna MP, Mueller D, Mengistu A, Singh AK, Hartman GL, Wise KA (2017) Advancing our understanding of charcoal rot in soybeans. J Integr Pest Manag 8(1):8
Ronquist F, Huelsenbeck J (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rossi V, Caffi T, Bugiani R, Spanna F, Valle DD (2008) Estimating the germination dynamics of Plasmopara viticola oospores using hydro-thermal time. Plant Pathol 57(2):216–226
Rossman AY, Palm-Hernández ME (2008) Systematics of plant pathogenic fungi: why it matters. Plant Dis 92:1376–1386
Rossman AY, Samuels GJ, Rogerson CT, Lowen R (1999) Genera of the Bionectriaceae, Hypocreaceae and Nectriaceae (Hypocreales, Ascomycetes). Stud Mycol 42:1–248
Rossman AY, Seifert KA, Samuels GJ, Minnis AM, Schroers HJ, Lombard L, Crous PW, Põldmaa K, Cannon PF, Summerbell RC, Geiser DM (2013) Genera in Bionectriaceae, Hypocreaceae, and Nectriaceae (Hypocreales) proposed for acceptance or rejection. IMA Fungus 4:41–51
Rossman AY, Adams GC, Cannon PF, Castlerbury LA, Crous PW, Gryzenhout M, Jaklitsch WM, Mejia LC, Stoykov D, Udayanga D, Voglmayr H, Walker DM (2015) Recommendations of generic names in Diaporthales competing for protection or use. IMA Fungus 6:145–154
Rouxel M, Mestre P, Comont G, Lehman BL, Schilder A, Delmotte F (2013) Phylogenetic and experimental evidence for host specialized cryptic species in a biotrophic oomycete. New Phytol 197:251–263
Rudolph BA (1931) Verticillium hadromycosis. Hilgardia 5:197–361
Runa F, Park MS, Pryor BM (2009) Ulocladium systematics revisited: phylogeny and taxonomic status. Mycol Prog 8:35
Saccardo PA (1884) Sylloge fungorum, vol 3. Typis Seminarii, Italy
Saccardo PA (1880) Fungi gallici, ser. II. Michelia 2:38–135
Samuels GJ, Brayford D (1990) Variation in Nectria radicicola and its anamorphs, Cylindrocarpon destructans. Mycol Res 94:433–442
Samuels GJ, Brayford D (1993) Phragmosporous Nectria species with Cylindrocarpon anamorphs. Sydowia 45:55–80
Samuels GJ, Brayford D (1994) Species of Nectria (sensulato) with red perithecia and striate ascospores. Sydowia 46:75–161
Sandoval-Denis M, Crous PW (2018) Removing chaos from confusion: assigning names to common human and animal pathogens in Neocosmospora. Persoonia 41:109–129
Sarr MP, Ndiaye MB, Groenewald JZ, Crous PW (2014) Genetic diversity in Macrophomina phaseolina, the causal agent of charcoal rot. Phytopathol Mediterr 53:250–268
Schmitz S, Charlier A, Chandelier A (2017) First report of Neonectria neomacrospora on Abies grandis in Belgium. New Dis Rep 36:17
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109:6241–6246
Schröter J (1886) Kryptogamen-Flora von Schlesien, vol 3(1). J. Cramer, Lehre, pp 129–256
Seifert KA, McMullen CR, Yee D, Reeleder RD, Dobinson KF (2003) Molecular differentiation and detection of ginseng-adapted isolates of the root rot fungus Cylindrocarpon destructans. Phytopathology 93:1533–1542
Senanayake IC, Crous PW, Groenewald JZ, Maharachchikumbura SSN, Jeewon R, Phillips AJL, Bhat JD, Perera RH, Li QR, Li WJ, Tangthirasunun N, Norphanphoun C, Karunarathna SC, Camporesi, E, Manawasighe IS, Al-Sadi AM, Hyde KD (2017) Families of Diaporthales based on morphological and phylogenetic evidence. Stud Mycol 86:217–296
Senanayake IC, Jeewon R, Camporesi E, Hyde KD, Zeng YJ, Tian SL, Xie N (2018) Sulcisporasupratumida sp. nov. (Phaeosphaeriaceae, Pleosporales) on Anthoxanthum odoratum from Italy. MycoKeys 38:35–46
Shaw CG (1973) Host fungus index for the Pacific Northwest—I. Hosts. Wash Agric Exp Stat Bull 765:1–121
Shimizu T, Ito T, Kanematsu S (2012) Transient and multivariate system for transformation of a fungal plant pathogen, Rosellinia necatrix, using autonomously replicating vectors. Curr Genet 58:129–138
Simmons EG (1992) Alternaria taxonomy: current status, viewpoint, challenge. Alternaria biology, plant diseases and metabolites, pp 1–35
Silvestro D, Michalak I (2012) raxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12:335–337
Singtripop C, Hongsanan S, Li J, De Silva NI, Phillips AJ, Jones GE, Bahkali AH, Hyde KD (2016) Chaetothyrina mangiferae sp. nov., a new species of Chaetothyrina. Phytotaxa 255:21–33
Sivanesan A (1984) The bitunicate ascomycetes and their anamorphs. J. Cramer, Vaduz, Liechtenstein
Sivanesan A, Holliday P (1972) Rosellinia necatrix. CMI Descr Pathog Fungi Bact 352:1–2
Slippers B, Crous PW, Denman S, Countinho TA, Wingfield BD, Wingfield MJ (2004a) Combined multiple gene genealogies and phenotypic characters differentiate several species previously identified as Botryosphaeria dothidea. Mycologia 96:83–101
Slippers B, Fourie G, Crous PW, Countinho TA, Wingfield BD, Carnegie AJ, Wingfield MJ (2004b) Speciation and distribution of Botryosphaeria spp. on native and introduced Eucalyptus trees in Australia and South Africa. Stud Mycol 50:343–358
Slippers B, Fourie G, Crous PW, Coutinho TA, Wingfield BD, Wingfield MJ (2004c) Multiple gene sequences delimit Botryosphaeria australis sp. nov. from B. lutea. Mycologia 96:1030–1041
Slippers B, Boissin E, Phillips AJL, Groenewald JZ, Lombard L, Wingfield MJ, Postma A, Burgess T, Crous PW (2013) Phylogenetic lineages in the Botryosphaeriales: a systematic and evolutionary framework. Stud Mycol 76:31–49
Slippers B, Roux J, Wingfield MJ, Van der Walt FJJ, Jami F, Mehl JWM, Marais GJ (2014) Confronting the constraints of morphological taxonomy in the Botryosphaeriales. Persoonia 33:155–168
Solarte F, Muñoz CG, Maharachchikumbura SS, Álvarez E (2018) Diversity of Neopestalotiopsis and Pestalotiopsis spp., causal agents of guava scab in Colombia. Plant Dis 102:49–59
Stark C (1961) Das Auftreten der Verticillium-Tracheomykosen in Hamburger Gartenbaukulturen: Ein Beitrag zur Kenntnis ihrer Erreger. Die Gartenbauwissenschaft 26:493–528
Steenkamp ET, Wingfield BD, Desjardins AE, Marasas WFO, Wingfield MJ (2002) Cryptic speciation in Fusarium subglutinans. Mycologia 94:1032–1043
Steyaert RL (1949) Contributions etude monographique de Pestalotia de Not. et Monochaetia Sacc. (Truncatella gen. nov. et Pestalotiopsis gen. nov.). Bull JardinBotanique de l’EtatBruxelles 19:285–354
Su H, Li QR, Kang JC, Wen TC, Hyde KD (2016) Rosellinia convexa sp. nov. (Xylariales, Pezizomycotina) from China. Mycoscience 3:164–170
Sun FF, Sun SL, Zhu ZD (2016) Occurrence of Verticillium wilt caused by Verticillium dahliae on mung bean in Northern China. Plant Dis 100:1792–1792
Sutton BC (1977) Coelomycetes VI. Nomenclature of generic names proposed for Coelomycetes. Mycol Pap 141:1–253
Sutton BC, Marasas WFO (1976) Observations on Neottiosporina and Tiarosporella. Trans Br Mycol Soc 67:69–76
Swofford D (2002) PAUP*: phylogenetic analysis using parsimony (*and other methods) Version 4.0b10. Sinauer, Sunderland
Sydow H, Petrak F (1922) EinBeitragzurKenntnisder PilzfloraNordamerikas, insbesondere der nordwestlichenStaaten. Ann Myco-logici 20:178–218
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Tennakoon DS, Phillips AJL, Phookamsak R, Ariyawansa HA, Bahkali AH, Hyde KD (2016) Sexual morph of Lasiodiplodia pseudotheobromae (Botryosphaeriaceae, Botryosphaeriales, Dothideomycetes) from China. Mycosphere 7:990–1000
ten Hoopen GM, Krauss U (2006) Biology and control of Rosellinia bunodes, Rosellinia necatrix and Rosellinia pepo: a review. Crop Prot 25:89–107
Tibpromma S, Hyde KD, Jeewon R, Maharachchikumbura SS, Liu JK, Bhat DJ, Jones EBG, McKenzie E, Camporesi E, Bulgakov TS, Doilom M, Santiago AM, Das K, Manimohan P, Gibertoni TB, Lim YW, Ekanayaka AH, Thongbai B, Lee HB, Yang J, Kirk PM, Sysouphanthong P, Singh SK, Boonmee S, Dong W, Raj KN, Latha KP, Phookamsak R, Phukhamsakda C, Konta S, Jayasiri SC, Norphanphoun C, Tennakoon D, Li J, Dayarathne MC, Perera RH, Xiao Y, Wanasinghe DN, Senanayake IC, Goonasekara ID, Silva NI, Mapook A, Jayawardena RS, Dissanayake AJ, Manawasinghe IS, Chethana KW, Luo Z, Hapuarachchi KK, Baghela A, Soares AM, Vizzini A, Meiras-Ottoni A, Mešić A, Dutta AK, Souza CA, Richter C, Lin C, Chakrabarty D, Daranagama DA, Lima DX, Chakraborty D, Ercole E, Wu F, Simonini G, Vasquez G, Silva GA, Plautz HL, Ariyawansa HA, Lee HS, Kušan I, Song J, Sun J, Karmakar J, Hu K, Semwal KC, Thambugala KM, Voigt K, Acharya K, Rajeshkumar KC, Ryvarden L, Jadan M, Hosen MI, Mikšík M, Samarakoon MA, Wijayawardene NN, Kim NK, Matočec N, Singh PN, Tian Q, Bhatt RP, Oliveira RJ, Tulloss RE, Aamir S, Kaewchai S, Marathe SD, Khan S, Hongsanan S, Adhikari S, Mehmood T, Bandyopadhyay TK, Svetasheva TY, Nguyen TT, Antonín V, Li W, Wang Y, Indoliya Y, Tkalčec Z, Elgorban AM, Bahkali AH, Tang A, Su H, Zhang H, Promputtha I, Luangsa-Ard J, Xu J, Yan J, Kang JC, Stadler M, Mortimer PE, Chomnunti P, Zhao Q, Phillips AJ, Nontachaiyapoom S, Wen T, Karunarathna SC (2017) Fungal diversity notes 491–602: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 83:1–261
Thambugala KM, Daranagama DA, Camporesi E, Singtripop C, Liu Z-Y, Hyde KD (2014) Multi-locus phylogeny reveals the sexual state of Tiarosporella in Botryosphaeriaceae. Cryptogam Mycol 35:359–367
Thaxter R (1922) Note on two remarkable Ascomycetes. Proc Am Acad Arts Sci 57:425–436
Theissen FSJ (1913) Die Gattung Asterina. Abh Zool Botanischen Ges Wien 7:1–130
Thines M, Kamoun S (2010) Oomycete-plant coevolution: recent advances and future prospects. Curr Opin Plant Biol 13:1–7
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 25:4876–4882
Thynne E, McDonald MC, Evans M, Wallwork H, Neate S, Solomon PS (2015) Reclassification of the causal agent of white grain disorder on wheat as three separate species of Eutiarosporella. Australas Plant Pathol 44:527–539
Trakunyingcharoen T, Lombard L, Groenewald JZ, Cheewangkoon R, To-Anun C, Crous PW (2015) Caulicolous Botryosphaeriales from Thailand. Persoonia 34:87–99
Tsai I, Maharachchikumbura SS, Hyde KD, Ariyawansa HA (2018) Molecular phylogeny, morphology and pathogenicity of Pseudopestalotiopsis species on Ixora in Taiwan. Mycol Prog 17:941–952
Vaghefi N, Pethybridge SJ, Ford R, Nicolas ME, Crous PW, Taylor PWJ (2012) Stagonosporopsis spp. associated with ray blight disease of Asteraceae. Australas Plant Pathol 41:675–686
Vaghefi N, Hay FS, Pethybridge SJ, Ford R, Taylor PW (2016) Development of a multiplex PCR diagnostic assay for the detection of Stagonosporopsis species associated with ray blight of Asteraceae. Eur J Plant Pathol 146:581–595
Van der Meer JHH (1925) Verticillium wilt of herbaceous and woody plants. Meded Landbouwhogesch Wagening 28:1–82
Varela CP, Casal OA, Padin MC, Martinez VF, Oses MS, Scauflaire J, Munaut F, Castro MB, Vázquez JM (2013) First report of Fusarium temperatum causing seedling blight and stalk rot on maize in Spain. Plant Dis 97:1252–1252
Vitale A, Aiello D, Guarnaccia V, Perrone G, Stea G, Polizzi G (2012) First outbreak of root rot caused by Neonectria (Ilyonectria) macrodidyma on avocado (Persea americana). Eur J Phytopathol 160:156–159
Voglmayr H, Constantinescu O (2008) Revision and reclassification of three Plasmopara species based on morphological and molecular phylogenetic data. Mycol Res 112:487–501
Voglmayr H, Riethmüller A, Göker M, Weiß M, Oberwinkler F (2004) Phylogenetic relationships of Plasmopara, Bremia and other genera of downy mildews with pyriform haustoria based on Bayesian analysis of partial LSU rDNA sequence data. Mycol Res 108:1011–1024
Walz A, de Hoog GS (1987) A new species of Cyphellophora. Anton Leeuw Int J G 53:143–146
Wang Q, Coleman JJ (2019) CRISPR/Cas9-mediated endogenous gene tagging in Fusarium oxysporum. Fungal Genet Biol 126:17–24
Wang Z, Johnston PR, Takamatsu S, Spatafora JW, Hibbett DS (2006) Toward a phylogenetic classification of the Leotiomycetes based on rDNA data. Mycologia 98:1065–1075
Wang H, Chen D, Li C, Tian N, Zhang J, Xu JR, Wang C (2019) Stage-specific functional relationships between Tub1 and Tub2 beta-tubulins in the wheat scab fungus Fusarium graminearum. Fungal Genet Biol 132:103251
Ward TJ, Bielawski JP, Kistler HC, Sullivan E, O’Donnell K (2002) Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc Natl Acad Sci 99:9278–9283
Wehmeyer LE (1975) The pyrenomycetous fungi. Mycol Mem 6:1–250
Wendt L, Sir EB, Kuhnert E, Heitkämper S, Lambert C, Hladki AI, Romero AI, Luangsa-ard JJ, Srikitikulchai P, Peršoh D, Stadler M (2018) Resurrection and emendation of the Hypoxylaceae, recognised from a multigene phylogeny of the Xylariales. Mycol Prog 17:115–154
Wenneker M, de Jong PF, Joosten NN, Goedhart PW, Thomma BP (2017) Development of a method for detection of latent European fruit tree canker (Neonectria ditissima) infections in apple and pear nurseries. Eur J Plant Pathol 148:631–635
Wijayawardene NN, Crous PW, Kirk PM, Hawksworth DL, Boonmee S, Braun U, Dai DQ, D’souza MJ, Diederich P, Dissanayake AJ, Doilom M, Hongsanan S, Jones EBG, Groenewald JZ, Jayawardena R, Lawrey JD, Liu JK, Lücking R, Madrid H, Manamgoda DS, Muggia L, Nelsen MP, Phookamsak R, Suetrong S, Tanaka K, Thambugala KM, Wanasinghe DN, Wikee S, Zhang Y, Aproot A, Ariyawansa HA, Bahkali AH, Bhat DJ, Gueidan C, Chomnunti P, De Hoog GS, Knudsen K, Li WJ, McKenzie EHC, Miller AN, Phillips AJL, Piątek M, Raja HA, Shivas RS, Slippers B, Taylor JE, Tian Q, Wang Y, Woudenberg JHC, Cai L, Jaklitsch WM, Hyde KD (2014) Naming and outline of Dothideomycetes—2014 including proposals for the protection or suppression of generic names. Fungal Divers 69:1–55
Wijayawardene NN, Hyde KD, Rajeshkumar KC, Hawksworth DL, Madrid H, Kirk PM, Braun U, Singh RV, Crous PW, Kukwa M, Lucking R, Kurtzman CP, Yurkov A, Haelewaters D, Aptroot A, Lumbsch HT, Timdal E, Ertz D, Etayo J, Phillips AJL, Groenewald JZ, Papizadeh M, Selbmann L, Dayarathne MC, Weerakoon G, Jones EBG, Suetrong S, Tian Q, Castaneda-Ruiz RF, Bahkali AH, Pang K-L, Tanaka K, Dai DQ, Sakayaroj J, Hujslova M, Lombard L, Shenoy BD, Suija A, Maharachchikumbura SSN, Thambugala KM, Wanasinghe DN, Sharma BO, Gaikwad S, Pandit G, Zucconi L, Onofri S, Egidi E, Raja HA, Kodsueb R, Cáceres MES, Pérez-Ortega S, Fiuza PO, Monteiro JS, Vasilyeva LN, Shivas RG, Prieto M, Wedin M, Olariaga I, Lateef AA, Agrawal Y, Fazeli SAS, Amoozegar MA, Zhao GZ, Pfliegler WP, Sharma G, Oset M, Abdel-Wahab MA, Takamatsu S, Bensch K, de Silva NI, De Kese A, Karunarathna A, Boonmee S, Pfister DH, Lu Y-Z, Luo Z-L, Boonyuen N, Daranagama DA, Senanayake IC, Jayasiri SC, Samarakoon MC, Zeng X-Y, Doilom M, Quijada L, Rampadarath S, Heredia G, Dissanayake AJ, Jayawardana RS, Perera RH, Tang LZ, Phukhamsakda C, Hernández-Restrepo M, Ma X, Tibpromma S, Gusmao LFP, Weerahewa D, Karunarathna SC (2017) Notes for genera-ascomycota. Fungal Divers 86:1–594
Wijayawardene NN, Hyde KD, Lumbsch HT, Liu JK, Maharachchikumbura SSN, Ekanayaka AH, Tian Q, Phookamsak R (2018) Outline of ascomycota: 2017. Fungal Divers 88:167–263
Wilcox WF, Gubler WD, Uyemoto JK (eds) (2015) Compendium of grape diseases, disorders, and pests 39–45. APS Press, The American Phytopathological Society, Paul, MN
Williamson SM, Sutton TB (2000) Sooty blotch and flyspeck of apple: etiology, biology, and control. Plant Dis 84:714–724
Wilson GW (1907) An historical review of the proposed genera of Phycomycetes. J Mycol 13:205–209
Wollenweber HW (1917) Fusaria autographice delineata. Ann Mycol 15:1–56
Woudenberg JHC, Groenewald JZ, Binder M, Crous PW (2013) Alternaria redefined. Stud Mycol 75:171–212
Xu J, Ebada SS, Proksch P (2010) Pestalotiopsis a highly creative genus: chemistry and bioactivity of secondary metabolites. Fungal Divers 44:15–31
Xu J, Yang X, Lin Q (2014) Chemistry and biology of Pestalotiopsis -derived natural products. Fungal Divers 66:37–68
Yang T, Groenewald JZ, Cheewangkoon R, Jami F, Abdollahzadeh J, Lombard L, Crous PW (2017) Families, genera, and species of Botryosphaeriales. Fungal Biol 21:322–346
Yang XB, Navi SS (2005) First report of charcoal rot epidemics caused by Macrophomina phaseolina in soybean in Iowa. Plant dis 89:526–526
Yang H, Hyde KD, Karunarathna SC, Deng C, Gu CH, Yang SA, Zhang ZC (2018) New species of Camptophora and Cyphellophora from China, and first report of sexual morphs for these genera. Phytotaxa 343:149–159
Yang Q, Fan ZL, Crous PW, Liang YM, Tian CM (2015) Cytospora from Ulmus pumila in Northern China. Mycol Progress 14:74
Zare R, Gams W (2001a) A revision of Verticillium section Prostrata. IV. The genera Lecanicillium and Simplicillium gen. nov. Nova Hedwig 73:1–50
Zare R, Gams W (2001b) A revision of Verticillium section Prostrata. VI. The genus Haptocillium. Nova Hedwig 73:271–292
Zare R, Gams W (2008) A revision of the Verticillium fungicola species complex and its affinity with the genus Lecanicillium. Mycol Res 112:811–824
Zare R, Gams W (2016) More white verticillium-like anamorphs with erect conidiophores. Mycol Prog 15:993–1030
Zare R, Gams W, Schroers HJ (2004) The type species of Verticillium is not congeneric with the plant-pathogenic species placed in Verticillium and it is not the anamorph of ‘Nectria’ inventa. Mycol Res 108:576–582
Zare R, Gams W, Starink-Willemse M, Summerbell RC (2007) Gibellulopsis, a suitable genus for Verticillium nigrescens, and Musicillium, a new genus for V. theobromae. Nova Hedwig 85:463–489
Zeng XY, Wu HX, Hongsanan S, Jeewon R, Wen TC, Maharachchikumbura SS, Chomnunti P, Hyde KD (2019) Taxonomy and the evolutionary history of Micropeltidaceae. Fungal Divers 97:393–436
Zhang W, Manawasinghe IS, Zhao W, Xu J, Brooks S, Zhao X, Xu JP, Brooks S, Zhao X, Hyde KD, Chethana KWT, Liu J, Li XH, Yan JY (2017) Multiple gene genealogy reveals high genetic diversity and evidence for multiple origins of Chinese Plasmopara viticola population. Sci Rep 7:17304
Zhao P, Luo J, Zhuang W, Liu X, Wu B (2011) DNA barcoding of the fungal genus Neonectria and the discovery of two new species. Sci China Life Sci 54:664–674
Zhuang JL, Zhu MQ, Zhang R, Yin H, Lei YP, Sun GY, Mark LG (2010) Phialophora sessilis, a species causing flyspeck signs on bamboo in China. Mycotaxon 113:405–413
Acknowledgements
This work was funded by the grants of the project of National Natural Science Foundation of China (No. 31560489), Talent project of Guizhou science and technology cooperation platform ([2017]5788-5) and Guizhou science, technology department international cooperation base project ([2018]5806). Kevin D. Hyde would like to thank “the future of specialist fungi in a changing climate: baseline data for generalist and specialist fungi associated with ants, Rhododendron species and Dracaena species” (Grant No. DBG6080013), Thailand Research Fund (TRF) grant no RSA5980068 entitled Biodiversity, phylogeny and role of fungal endophytes on above parts of Rhizophora apiculata and Nypa fruticans and“Impact of climate change on fungal diversity and biogeography in the Greater Mekong Subregion” (RDG6130001). Rajesh Jeewon would like to thank Mae Fah Luang University and the University of Mauritius for research support. Alan J.L. Phillips acknowledges the support from Biosystems and Integrative Sciences Institute (BioISI, FCT/UID/ Multi/04046/2013).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jayawardena, R.S., Hyde, K.D., McKenzie, E.H.C. et al. One stop shop III: taxonomic update with molecular phylogeny for important phytopathogenic genera: 51–75 (2019). Fungal Diversity 98, 77–160 (2019). https://doi.org/10.1007/s13225-019-00433-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-019-00433-6