
Abstract
In plant pathology, the correct naming of a species is essential for determining the causal agents of disease. Species names not only serve the general purpose of concise communication, but also are critical for effective plant quarantine, preventing the introduction of new pathogens into a territory. Many phytopathogenic genera have multiple species and, in several genera, disagreements between the multiple prevailing species concept definitions result in numerous cryptic species. Some of these species were previously called by various names; forma speciales (specialised forms), subspecies, or pathotypes. However, based on new molecular evidence they are being assigned into new species. The frequent name changes and lack of consistent criteria to delineate cryptic species, species, subspecies, forms, and races create increasing confusion, often making communication among biologists arduous. Furthermore, such ambiguous information can convey misleading evolutionary concepts and species boundaries. The aim of this paper is to review these concepts, clarify their use, and evaluate them by referring to existing examples. We specifically address the question, “Do plant pathogens require a different ranking system?” We conclude that it is necessary to identify phytopathogens to species level based on data from multiple approaches. Furthermore, this identification must go beyond species level to clearly classify hitherto known subspecies, forms and races. In addition, when naming phytopathogenic genera, plant pathologists should provide more information about geographic locations and host ranges as well as host specificities for individual species, cryptic species, forms or races. When describing a new phytopathogen, we suggest that authors provide at least three representative strains together with pathogenicity test results. If Koch’s postulates cannot be fulfilled, it is necessary to provide complementary data such as associated disease severity on the host plant. Moreover, more sequenced collections of species causing diseases should be published in order to stabilise the boundaries of cryptic species, species, subspecies, forms, and races.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungal plant pathogens are an extremely important group of organisms as they impact all aspects of agriculture and food security. Moreover, this is one of the highly published areas of research work (Fig. 1). For any effective disease management, accurate species diagnosis is the foundation. This includes three main steps: (1) isolation of the pathogen, (2) correct species identification and (3) confirmation of pathogenicity by testing and fulfilling Koch’s postulates. Isolation and pathogenicity assays involve several laboratory techniques while correct species identification requires a comprehensive understanding of the taxonomy of organisms closely related to the target species. While all three steps are critical, incorrect species identification due to a taxonomic error can cause the entire disease management process to become unnecessarily challenging.

Web of Science data showing the number of publications appearing per year on plant pathology from 1960 to 2020
Difficulty in identifying and naming pathogens has particularly affected plant pathologists, who are at the frontline of dealing with plant fungal pathogens. As most plant pathologists are not taxonomists, naming and identifying pathogens can often be a challenging task. One contributing factor to the issue is that the majority of fungal pathogens can occur in different forms: sexual morph, asexual morph or a combination of both (Wingfield et al. 2012). This phenomenon, known as pleomorphism, has placed fungal taxonomy in a complex situation since the mid-nineteenth century as many pathologists may not have been aware of these multiple forms. This has landed plant pathologists in the chaotic situation where either one fungus may have several different names or the identification of some species stops at the genus level. Since January 1st, 2013, one fungus can now have only one name, thus, ending the system of permitting separate names for asexual morphs (Wingfield et al. 2012). This means that the legitimate name proposed for the species, regardless of the stage in the life cycle they are typified by, serves as the standard and correct name for that species. This new consistency in naming procedures, along with the introduction and development of evolving molecular techniques, represents a light at the end of the tunnel for the problem of fungal species ambiguity.
Development and enhancement of molecular techniques in fungal taxonomy and systematics in the last two decades has provided a wealth of data (Guarro et al. 1999). Though the identification of fungus by sequencing has limitations (Thines 2019; Lücking et al. 2020), sequencing and molecular phylogeny have emerged as the most convenient and standardised method. In many cases, molecular barcoding with one or a few loci is sufficient for species-level identification in plant pathology. Furthermore, some species have varieties (subspecies, forms, and races) and the same species may exhibit different levels of pathogenicity depending on the plant species. Within a species, however, there can be different groups of strains exhibiting different levels of pathogenicity, which can also vary among host plant species (Shang et al. 2016). Moreover, the genetic variations among strains within pathogenic species could be the basis for the emergence of a new disease or a more virulent strain causing outbreaks (Milgroom 2017).
Looking at recent history, wheat stem rust is one of the best examples to show the necessity of studying beyond the species level in plant pathology. In 2015, an asexual lineage of Puccinia graminis began to spread through Africa and the Middle East resulting in a devastating epidemic (Singh et al. 2015). This epidemic was caused by the Ug99 strain of race TTKSK, which belongs to Puccinia graminis f. sp. tritici (Pgt) (Li et al. 2019). This is a real-time indication of a fungus that nuclear exchange and recombination of asexual fungi can create novel and more virulent strains (Li et al. 2019). Another example is Plasmopara halstedii, the causal agent of sunflower head blight (Gascuel et al. 2015). There are at least 36 pathotypes of P. halstedii in the world. In 2010, one new race was identified as the causal agent capable of causing a significant increase in disease severity (Bán et al. 2014). This newly identified race has a broad spectrum of pathogenicity toward the genetic resources that are used in sunflower hybrids (Bán et al. 2014). Thus, the result of a single fungal strain isolated from a single host plant for pathogenicity testing should not be used solely to conclude the pathogenicity of a particular species. Disease development is a combined effect of interactions between pathogen, host plant and environmental factors (Scholthof 2007).
The naming of a phytopathogenic species is often complicated as many plant pathogenic genera and species defined based on one set of criteria (e.g. morphology) are found to comprise numerous cryptic species based on other criteria (e.g. DNA sequences). This raises the question, “Are the different fungal genetic variants actually different species?” In this paper, we debate the different approaches available to resolve “the species” and ask the question, “Does plant pathology require a different ranking system?” The paper starts by giving a brief introduction as to why it is important to define a species and the necessity of breaking it into finer levels for ranking in plant pathology. Then we discuss how and why genotypes and pathotypes are important in plant pathology. This is followed by discussions on cryptic species, focusing on their ambiguity and thereafter, on the importance of naming phytopathogenic taxa. In conclusion, this paper sets up boundaries for introducing new pathogenic species and how to expand it to lower levels with suggested guidelines for future publications.
Importance of identification of fungal plant pathogens
Knowledge derived from history can be applied to understand and unravel the potential impacts of fungi on future food security (Thirkell et al. 2017). Critically, plant pathogen induced famines are among the most important ways to demonstrate how pathogenic taxa can impact agricultural crops and the human population. Through the history of human civilisation, several famines due to natural causes have been witnessed. Among them, the Irish Potato famine is considered the most infamous. The potato blight disease caused by Phytophthora infestans, during 1845–1849 is considered the most devastating crop-related disease in the recorded history (Geber and Murphy 2012). The pathogenic nature of this single species resulted in the death of one million people through starvation and famine-related diseases, as well as causing two million people to emigrate from Ireland, mainly to the United States of America (Paping et al. 2007; Geber and Murphy 2012). Another example was Hemileia vastatrix, which causes coffee leaf rust (Talhinhas et al. 2017). The disease was first reported in Sri Lanka in 1869 and caused enormous damage to productivity. By 1890, the Sri Lankan and Indonesian coffee industries were almost completely destroyed (McCook and Vandermeer 2015). Sri Lankan coffee producers switched to tea production while Indonesian farmers started to grow rubber. Coffee production moved to Brazil and Central America (McCook 2006; Talhinhas et al. 2017). However, major epidemics of coffee rust appeared in Brazil and Central America in 2012 and 2013 (McCook 2006; Avelino et al. 2015). Such major historical events highlight the impact of plant pathogens on human civilization and suggest that accurately identifying fungal pathogens before a crisis, could be of great benefit in adverting future famines.
In addition to plant pathogens, fungal toxins produced by certain groups of fungi are another important threat to agriculture and human and animal health. Mycotoxins have adverse effects on both humans and livestock (Zain 2011; Omotayo et al. 2019). These toxins can occur in cereals and pasture causing acute poisoning leading to long-term ill-effects such as cancer (Zain 2011; WHO 2021). Overall, phytopathogens are a threat to food security and ecosystem stability. Hence it is important to develop an effective framework for early detection and quarantine, to overcome the biological invasion of phytopathogenic fungi (Luchi et al. 2020).
To develop such frameworks, a key tool is correct species diagnosis or species identification, and this in turn relies on a stable and robust means to apply meaningful names to plant pathogens. Plant disease epidemics are rare (or non-existent) in natural populations of plants within their native range and without invasion of alien pathogens without an invasive event. Major epidemics are caused either by the introduction of alien pathogens into a region where they previously did not exist, or by introduction of a host into a new region where they are exposed to pathogens that they have not encountered in their normal geographic range (Morse 2001). Proper identification of both the disease and the causal organism is vital to prevent pathogens from entering new geographical localities. The international trading of plants and animals is one of the key sources of introduction of non-native plant pathogens (Jones and Baker 2007). Therefore, each country has developed a list of “quarantine pathogens”. Identification and characterisation of fungal quarantine pathogens require the collaboration of plant pathologists and mycologists.
Apart from new introductions, disease emergence can also be due to the generation of a new virulent genotype through mutation of an existing genotype or recombination among native genotypes. These genetic variations among pathogens can result in different levels of responses towards the natural and human-mediated environmental changes. Therefore, it is necessary in plant pathology to identify not only at the species level but also beyond species levels. In the following section, we will discuss why it has become necessary to identify a fungal plant pathogen.
Is it enough to stop at the species level in Plant Pathology?
It is often difficult to identify specific traits that contribute to fungal pathogenesis because most species that are opportunistic pathogens are capable of having saprobic, endophytic and parasitic growth forms (Rogers et al. 2012; Gilbert et al. 2015; Taylor 2015). Within the same species, pathogenicity can vary among strains where both locality and the host play a key role (Manawasinghe et al. 2016). In asexual fungal populations, vegetative reproduction facilitates more stable and uniform gene compositions as compared to cross-fertilization (Nieuwenhuis and James 2016a, b). Therefore, variation in such a population is relatively easy to track. Some of the most common phytopathogenic genera, Alternaria, Bipolaris, Colletotrichum, Curvularia and Diaporthe are predominantly asexual (Hyde et al. 2014). Moreover, species belonging to Botryosphaeriaceae and Didymellaceae are also common disease-causing asexual genera (Hyde et al. 2014). Thus, variation within these genera mainly occur due to mutations.
By definition, all individuals of the same species share the same gene pool. However, for each gene, different strains can have different alleles, especially in sexually reproducing fungi. The combination of alleles at different loci constitutes the genotype of the strains. For this reason, it is the populations that undergo cross-fertilization that can be more challenging to identify due to the high level of allelic variation. The variations that occurred in either in the asexual population or sexual population results in a different number of genotypes within the population. A genotype can be determined based on genetic variations of the entire genome, a certain gene, or a set of genes (Milgroom 2017). These genotypes can be identified using different markers such as random amplification of polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP) analysis, amplified fragment length polymorphism (AFLP) analysis, and single nucleotide polymorphism (SNPs) (McDonald 2004; Milgroom 2017). Genotyping is critical since it can be used to track the origin and spread of pathogens and to infer potential outbreaks.
A disease outbreak could be a result of four major categories: (i) introduction of a novel strain, (ii) changes in host population, and (iii) changes in environmental factors, and iv) introduction of a novel host (Manawasinghe et al. 2018). Since we focus on factors related to fungi, the introduction of a novel strain could be either from a different locality or as a result of the generation of a genetically novel strain (Storfer et al. 2007). The generation of novel genotypes could be either via sexual reproduction (including horizontal gene transfer), a parasexual cycle in asexual reproduction or by mutations (Nieuwenhuis and James 2016a, b). As a result, a single species will contain strains with different genetic compositions (McDonald 2004; Milgroom 2017). They could be classified as “subspecies”, “varieties” and “forms”. These strains will have different genomic composition that can result in different levels of pathogenicity and colonisation patterns. Asexual reproduction is often involved in the rapid spread of infectious disease outbreaks (Ashu and Xu 2015). However, major outbreaks are most frequently linked to the generation of new strains, so genotyping pathogens accurately at the subspecies level is essential for tracking potential diseases.
Creating boundaries: species and beyond species level in Plant Pathology
Defining a “species”
Over the years, biologists have been applied various species concepts such as the biological, ecological, and phylogenetic species concepts to define boundaries (Xu 2020). Additionally, the Genealogical concordance phylogenetic species recognition (GCPSR) approach has also been used and merits consideration (Taylor et al. 2000; Laurence et al. 2014). In each of these concepts, different characters and characteristics are used to recognise a species, resulting in disagreements in classification based on the concepts used. Unfortunately, what is currently missing is the fundamental concept of what defines a species (Dvořák et al. 2015). The application of different approaches can vary with the type of organism. In this section we will consider the merits, and shortfalls of each individual species concept and finally consider whether multiple concepts can be used in combination.
According to the Darwin’s theory of evolution, a species can most reliably be defined as; a: specific segmentation of evolutionary lineage that has evolved independently from its closest ally (De Queiroz 2007). While this definition can be used to effectively delineate species of most types of organisms, there are several reasons why Darwin’s evolution theory as well as Linnaeus’s classification are no longer applicable to fungi. Indeed, the oldest method used to define a species was the morphological species concept, in which fungal species were defined based on macromorphological characters and (or) microscopic features (Xu 2020). However, the use of morphological characters has always been problematic, and further issues have become apparent with present-day studies. The problem that arises is that based on morphological characters, the same organism may be defined as one species by one observer in one environment but a different species by another observer in a different environment (Safran and Nosil 2012). Moreover, with cryptic species (to be discussed in further sections), the problem with the morphological species concept will be further illustrated.
Along with the pitfalls of other approaches to defining a species, one of the main reasons why the biological species concept cannot be applied to asexual fungi is due to the lack of inter-breeding in their natural populations (Taylor et al. 2000). In sexual reproduction, the offspring inherit a mixture of alleles from their parents, with individual offspring being genetically different from each other. However, in the kingdom of fungi, asexual reproduction is favoured because it develops offspring with an identical genetic makeup to that of the parent. However, there are very few ancient asexual fungi, and population genetics has uncovered evidence of cryptic sex in almost all fungi analysed so far, likely due to mutational meltdown or parasexual cycles (Xu 2002, 2004; Nieuwenhuis and James 2016a, b). With the advancement of molecular techniques, the sex of these taxa was resolved and linkage between an asexual morph and the complementary sexual morph was established (Judson and Normark 1996; Nieuwenhuis and James 2016a, b). As a result, agreement over the treatment of sexual and asexual morphs represent an identification challenge that does not exist with most living extents.
With the advancement of molecular techniques, the phylogenetic species concept has become dominant in species delineation in fungal taxonomy. Almost all the species introduced at present are based on phylogenetic approaches that are based on various algorithms and hypotheses (Xu 2016, 2020). In phylogenetic species delineation, speciation is determined by the evolutionary lengths of branches and statistical support for a phylogenetic branch (Wróbel 2008; Kumar et al. 2012). However, the statistical support for a phylogenetic branch is primarily related to the number of phylogenetically informative characters, not on the method of analyses (Bordewich et al. 2018). Thus it is necessary to identify the most suitable and accurate gene regions for phylogenetic analysis with informative characters. With the advancement of the genomics era, however, this method has been questioned due to the fact that using whole-genome sequence data, almost any two strains within the same species can be separated with strong statistical support (Xu 2020).
Genealogical concordance phylogenetic species recognition (GCPSR) is another approach used to resolve species complexes or cryptic species (Taylor et al. 2000; Laurence et al. 2014; Nguyen et al. 2015; Liu et al. 2016). The assumption in GCPSR is that recombination within a lineage can cause conflict and incongruence between individual gene trees (Taylor et al. 2000; Udayanga et al. 2014) and the limits of a species lies at the point of transition from congruence to incongruence. This method has been employed to resolve many cryptic genera such as Colletotrichum (Crouch 2014; Liu et al. 2016; Vieira et al. 2017), Diaporthe (Udayanga et al. 2014, 2015) and Neurospora (Dettman et al. 2003; Gladieux et al. 2020). In addition, recent studies have employed recombination tests (Huson and Bryant 2006). This method provides biological evidence that could be used to identify different lineages (Dykhuizen and Green 1991). Using either sequence or genomic data, if it can be proven that two strains do not have evidence of recombination, the two strains belong to separate lineages (Dykhuizen and Green 1991; Huson and Bryant 2006). The most commonly used recombination analysis is the pairwise homoplasy index (PHI) (Bruen et al. 2006). In this analysis, no recombination will be set as the null hypothesis. However, this method requires a relatively large sample size to be meaningful (Bruen et al. 2006; Xu 2020).
Every method listed above has its own pros and cons, and therefore a combination of several methods is needed to be considered significant evidence for species identification. Thus the application of a consolidated species identification method could be the best approach to overcome the issue. Before settling on developing a consolidated approach, several problems need to be addressed (Xu 2020). Foremost, there is a sizeable knowledge gap in determining the type and number of gene regions that should be included in the analysis. For example, due to differences on selection pressure operating among genes, the choice of coding and non-coding gene regions affects the relative number of informative characters in strain and species discrimination. Similarly, the number of genes and the total length of analysed gene fragments can impact the total number of polymorphic nucleotide sites that can influence the statistical support for clades. Whereas non-coding regions are more likely to accumulate random mutations than the coding regions (Subramanian and Kumar 2003). The question of how many gene regions to be included is unclear. The majority of present-day studies are based on 3–5 gene regions, but the use of mating-type gene(s) is comparatively low. Aside from gene selection, another critical aspect is taxon sampling. How many samples must be included to introduce a new species? In recent times, over majority of species introduced have been based on single strains. With a single strain, it is not possible to determine reproductive barriers between sibling species (Xu 2016, 2020) and it is not possible to determine within species genetic variation. Importantly, with increasing sample size, the chances of detecting recombination among organisms within a population increases (Xu 2020). Based on these important points and with a careful consideration of each method, at the end of this paper several recommendations are given to set a standard approach in introducing a new fungal pathogen.
Irrespective of which approach is used to define a species should always be an evolutionarily independent lineage. This will necessitate that in order to define a new species in plant pathology, evidence must be provided to prove that the current species is an independent lineage that has evolved from an existing species. Based on the above facts, even within the same species, different lineages have their morphological characteristics and virulence potentials. Hence the next question that comes up is, “is it enough to stop at the species-level in plant pathology?” In phytopathology research, it must be attempted always to go beyond the species level. In view of this, we will discuss the importance of the identification of genotypes, pathotypes, etc.
Beyond the species level
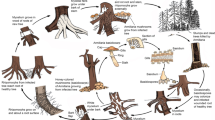
To identify or define beyond species level, a secondary ranking system is created in descending order; subspecies, variety, form, and sub-form by article 4 of the International Code of Nomenclature for Algae, Fungi, and Plants (2012). The term ‘race’ also has been commonly used. Thus, the ranking hierarchy for phytopathogenic genera beyond the species level could be given as follows: (1). Species, (1.2) Subspecies, (1.3) Variety, (1.4) Form, (1.5) Race (based on Shenzhen Code 2018, International Code of Nomenclature for Algae, Fungi, and Plants). In fungal pathogen taxonomy and ranking, “species” identification should be the priority. After the species, the subspecies can be incorporated (Fig. 2). However, naming a subspecies will be mostly based on allopatric speciation. Thus, if there is sufficient data to demonstrate that a certain group is phylogenetically or morphologically different from the type group, it is possible to define it as a subspecies. However, if this variation is basically host dependent, the ranking should come as forms or varieties. Moreover, in a given host if the pathogenicity or virulence between two or more groups of strains are significantly different, then the ranking of ‘race’ should be used. To be able to define beyond the species levels, it requires extended sampling and pathogenicity data. In the following sections, we will define and briefly explain the use of each of these classifications.

Taxonomic ranking system in the kingdom of fungi and beyond species hierarchy for phytopathogenic fungi. Higher level ranking of fungi above the species level. Cryptic species are not included in the hierarchy as it is not common to all genera. Thus, we propose a separate level between genus and species
Subspecies
The use of subspecies is not well established in fungal ranking. The term subspecies has often been confused with variety from the Linnaean period. In early studies, subspecies were defined based on geographical variation (Mayr 1982), as a “group of individuals, those who have different traits as defined by scientists”. Such traits were mainly based on geographic variations or environmental factors. Further, a subspecies can be a distinct natural population, which does not have enough evidence to prove as a separate species. In late 1800, subspecies was proposed as an incipient species (Mayr 1942), as a stage towards complete speciation. Thus, they represent real-time evidence for adaptative responses of specific populations of a species to the given environmental conditions (Mayr 1982; Patten 2015). However, the exact definition for the subspecies is still not established, but often referred to as a “collection of populations occupying a distinct breeding range and diagnosably distinct from other such populations” (Patten 2009) and that “the species addresses reproductive and behavioural criteria, while the subspecies, morphological diagnosability” (Patten 2015). Based on these facts we define a subspecies of fungi as a “group of individuals which are distinct from the type population either morphologically or environmentally, but capable of interbreeding with other populations while sharing similar characteristics”. According to the phylogenetic species concept, subspecies could also be a diagnosable cluster (Oláh et al. 2018). However, there is no threshold level to clarify the boundaries of a subspecies. In the molecular era of fungal taxonomy, meta-barcoding has been employed to identify fungal species and subspecies in mixed samples from a model fungal community (Marcelino et al. 2019).
Variety
Defining varieties started after subspecies and before forms. This lower level is acceptable in botany. However, in mycology the species were earlier classified into varieties. In earlier studies, varieties have been defined for genera such as Alternaria, Cercospora, Colletotrichum, Fusarium, and Plasmopara (www.indexfungorum.org). In many cases the names were given after the host names. Group of populations that share similar inheritable characters, when met with individuals of other populations, it is possible to interbreed within the same geographical entity. These individuals have a broad spectrum of hosts compared to the forms as forms are confined to a specific host group.
Forms
The term forma specialis (f. sp. in plural: f. spp.) classifies individuals of a particular species that infect a specific group of plants, distinct from others of the same species. This intraspecific host specificity occurs even though all members of the species are associated with the same general host range. Forma specialis is usually dependent on several stable genetic determinants in a population. This subspecies grouping implies characteristic adaptation of different genotypes (Edel-Hermann and Lecomte 2019a, b). However, many forma specialis groups have starkly different disease phenotypes depending on the host and the environment. The main problem associated with identifying forma specialis is artificial inoculation on multiple hosts in cross pathogenicity assays. For example, Fusarium oxysporum is pathogenic on Melon (Cucumis melo) causing wilt and root rot. There are two formae speciales of F. oxysporum; F. oxysporum f. sp. melonis responsible for causing fusarium wilt in melon under natural environmental conditions and F. oxysporum f. sp. radicis cucumerinum causing root and stem rot when inoculated artificially (Vakalounakis et al. 2004). The problem associated with artificial inoculation is that the results may depend on inoculation technique and environmental conditions which can cause frequent false-positive results due to factors such as an inappropriately large amount of inoculum and an environmental condition that favoured disease development in an otherwise non-susceptible host. With these complicating factors in mind, it is important to develop boundaries when conducting pathogenicity assays on forma specialis.
Races
Depending on the virulence genes, pathogenic species can be subdivided into different races. Similar to “forma specialis”, the term “race” is not currently considered a formal taxonomic rank as it comes below the species level. A particular race can be identified based on physiological characters, mainly pathogenicity. In a race, all individuals share the same virulence pattern and that virulence pattern is genetically controlled. At present, the ranking or naming of races does not have a standardised method. They are typically named under a particular forma specialis as a number. Naming and characterisation of races mostly depend on the gene-for-gene relationship. In crop breeding, pathologists and plant breeders often confirm with each other which race they are going to develop resistance for, in breeding cultivars. Such a consensus requires knowledge of the genomic diversity of a plant pathogen and the genetic features used to define each race. Here we define a race as the lowest unit in plant pathology, a group of individuals that are confined to a single host, share the same genetic makeup of virulence genes and have a similar level of pathogenicity under the given environmental condition. However, it is a necessity to develop a boundary for practical approaches. Defining a race will be relatively easy for those plant pathogens associated with domestic crops and fruits. However, it can be difficult to define a race in a pathogenic species in natural ecosystems like virgin forests and aquatic environments.
Confusions in ranking beyond the species
Confusions and delineating issues in the lower ranks go back to Darwin’s time. In Darwin’s theory of evolution, he mentioned that there is no difference in varieties and species, and that species could be varieties that diverged more and coexisted without common. However, there is no clear-cut line to define polymorphic variations that occur within a population and how to define those as “races’’ or ‘‘subspecies” (Mallet 2007). Although we can define rankings below species, unlike the higher ranking of the fungal kingdom, there are no defined rules nor an international body controlling these rankings. This has led to different ranks being applied to forms, varieties, and races over time, causing further confusions in the literature. Gulya et al. (1991) established a system to test the virulence of sunflower rust pathogen Plasmopara halstedii. Then they chose pathotype over race, because the classification was based on pathogenicity data or infection bioassays with host plants. Spring and Thines (2010) proposed that the races can be used to define characters without a host association. They proposed that all these clarifications must come together with more biological data associated with pathogenicity. Some studies have defined races by climatic predisposition and the associated differences in plant symptoms. This has resulted in some confusion between forma specialis and its races. Therefore, defining the races should be based on gene-for-gene interaction between the host genotype and pathogen strain genotype or at least on the pathogen “race”, at a clear-cut host cultivar-level specialisation. Such a definition can accommodate the variable virulence patterns within and between races (Parlevliet 1985).
Another confusion is the use of the term “pathotypes”. Pathotypes are classified at the same level as forma specialis. The use of the term pathotype and pathovars are more common in bacteriology than mycology. Therefore, in this study, we prefer to leave them out from the context of lower ranking of pathogenic fungi. Nevertheless, two genera are given as examples, from Index Fungorum. Colletotrichum gloeosporioides consists of five “forms”, eight forma speciales and eight varieties. These classifications were established in the pre-molecular era and most of them were defined based on host species. Similarly, Fusarium avenaceum has four forms, four subspecies, and eight varieties. Even though the epithet “avenaceum” obtained a form, a subspecies and a variety, most strains did not contain three levels of descriptions in their taxonomy. However, most have now been synonymized under Fusarium avenaceum and another four Fusarium species. This synonymization was based on the cryptic species concept after the molecular era. Thus, in the following section, we will discuss why there are cryptic species and how they may have led to confusions in fungal taxonomy.
Fusarium as a case study: importance of formae speciales and problems associated with species delineation
Fusarium taxonomy has been continuously evolving since the first description of a Fusarium fungus by Link in 1809 (Stępień and Chełkowski 2010). Fusarium taxonomy was first based on morphological features and dozens of species were classified into Sections (Burgess et al. 1988). With a growing number of phylogenetic analyses based on genomic data, the original system started to become ambiguous (Watanabe et al. 2011). Complicating matters, recent work has shown that there are 106 formae speciales associated with F. oxysporum (Edel-Hermann and Lecomte 2019a, b). Among these, only 53 are unique to plant species (Supplementary Table S1). Therefore, it is difficult to define the host specificity of F. oxysporum pathogenic strains. It became obvious that a new classification system was needed and DNA sequences subsequently formed the basis for distinguishing Fusarium clades and species complexes. One of the important phytopathogenic genera with a larger number of formae speciales is Fusarium.
Molecular identification of Fusarium species
The availability of fast and affordable DNA sequencing techniques has accelerated the process of accurate identification of the vast number of isolates belonging to multiple species. The universal and relatively evolutionary conserved Internal Transcribed Spacer rDNA (ITS) sequences have become the most popular diagnostic DNA marker region, though its ability to discriminate many closely related Fusarium species has been limited (Stępień et al. 2011). The sequence of the translation elongation factor 1α (EF1) became more useful and allowed an update to uncertain species boundaries in Fusarium (O’Donnell et al. 1998; Geiser et al. 2004). However, other genes and genomic regions (like Inter Genic Sequences—IGS) have also been used to shed additional light on the evolutionary history of the Fusarium genus, including partial sequences of beta–tubulin (tub2), RNA polymerase II gene (rpb2), and secondary metabolite biosynthetic genes, which vary in terms of divergence and resolution (Fan et al. 2013; O’Donnell et al. 2013; Stępień 2014). The global use of sequence-based phylogeny resulted in a proposed revision of the Fusarium taxonomy towards the one fungus—one name rule (Geiser et al. 2013). This approach has also helped to include the controversial and substantially divergent Fusarium solani species complex into the Fusarium genus (Geiser et al. 2020).
Intraspecific variability of Fusarium genotypes
The historical species names of Fusarium were generally very broad and covered most isolates with similar morphological characteristics. As more discriminative methods and descriptions were used, numerous differences were observed within the original species group. Thus, species complexes were proposed, with individual subgroups being first called lineages (like for F. graminearum sensu lato), which later split into many separate species (O’Donnell et al. 2004, 2008). For F. oxysporum, the division into formae speciales was additionally supported by the species-specificity in pathogenic abilities (Edel-Hermann and Lecomte 2019a, b). The high level of intraspecific variability may be reflected in morphological, biochemical, mycotoxigenic and, finally, genetic features. For example, different chemotypes were determined by variable secondary metabolite biosynthetic gene alleles and specific polymorphic sites within evolutionarily conserved genes (Proctor et al. 2009).
Real time observation of Fusarium pathogens
Depending on the species concept used, speciation events have arguably been observed for the Fusarium pathogens, with new variants emerging from previously documented species. For example, F. temperatum emerged from F. subglutinans species (Scauflaire et al. 2011) and appeared to be present in different countries (Czembor et al. 2014; Zhang et al. 2014; Stępień et al. 2019), Moreover, some intraspecific genetic divergence was observed in F. proliferatum showing host-specific sequence variants (Stępień et al. 2015). On the other hand, until now no host specificities were found for most of the main Fusarium species such as F. graminearum, F. avenaceum, F. verticillioides, F. proliferatum or F. poae. The complexities of the Fusarium genus illustrates the key point that the name of a species should reflect the whole variability of the genotypes within the species. The names should be broad enough to encompass both pathogens and non-pathogens, mycotoxin producers and non-producers, while also considering the possibility of as well as non-host-specific isolates.
Cryptic species
What are the cryptic species?
Cryptic species are characterised as at least two groups of organisms that are superficially and morphologically indistinguishable, recently diverged, reproductively isolated and separable only with molecular data (Crespo and Lumbsch 2010; Struck et al. 2018). The use of molecular approaches in species delineation has been the main avenue to highlight the occurrence of cryptic species (Bickford et al. 2007; Struck et al. 2018). The role of cryptic species in evolution, however, is still debatable (Chenuil et al. 2019). Defining a cryptic species is based on its taxonomic nomenclature history, which is found to be unsatisfactory as various taxonomic and biological factors result in erroneous species lumping (Struck et al. 2018). Moreover, it is still unresolved as to how many morphological features should be similar or dissimilar to define a species as cryptic (Struck et al. 2018). Cryptic species are more common in fungi than other organisms, partly due to fungi having a limited number of morphological features that can be used to distinguish closely related taxa, however much divergent they are (Milgroom 2017; Xu 2020).
During the last decade, an increasing number of novel pathogenic taxa have been introduced through phylogenetic approaches. The majority of these new species have been introduced as cryptic species. These studies were based primarily on molecular evidence and employed relatively less morphological support. In addition, analytical methods such as tests for recombination and phylogenetic incompatibility have seldom been employed (Dugan and Everhart 2016; Xu 2016, 2020). Many traditionally defined species that are devastating phytopathogenic genera, such as Colletotrichum (Jayawardena et al. 2020), Diaporthe (Udayanga et al. 2014; Gao et al. 2017; Manawasinghe et al. 2019), Pestalotiopsis (Maharachchikumbura et al. 2014; Solarte et al. 2018) Plasmopara (Rouxel et al. 2013; Zhang et al. 2017) and Fusarium (O’Donnell et al. 2015), are now considered species complexes with each complex containing multiple cryptic candidate species. In the following section, we will discuss these genera, focusing on several controversies involving cryptic species.
The ambiguity of cryptic species
In some cases, the diversity of a fungal species can be deceptive, such that the discovery of what was thought to be one or a small number of fungal species, can later be revealed to be many diverse phylogenetic species. For example, Colletotrichum gloeosporioides is a well-known common causal agent of anthracnose in a wide range of hosts, including both field and plantation crops (Sharma and Kulshrestha 2015). In early studies, C. gloeosporioides was considered to be a single species (Cannon et al. 2012; Weir et al. 2012). However, with molecular data, it was proposed and established that C. gloeosporioides is a species complex that comprises 22 phylogenetic species based on concatenated sequences of five gene fragments and with about two to a dozen strains in each phylogenetic species (Weir et al. 2012). However, based on a larger sample size using sequences from the same gene fragments, Xu et al. (2016) observed significant allele sharing and evidence of recombination between several phylogenetic species including C. fructicola, C. siamense and C. camelliae. Depending on the degree of allele sharing and recombination, it was suggested that all of their 199 isolates belong to two biological species (Xu et al. 2016), different from the six inferred species based on phylogenetic affiliations of type strains as defined earlier using fewer strains (Weir et al. 2012). Indeed, the use of a limited number of strains in most taxonomic studies and the lack of critical evaluation of recombination could lead to the recognition of a large number of phylogenetic species (Xu 2020). Therefore, it is necessary to identify species in a rigorous and consistent manner. The above case study emphasises that caution should be exercised when introducing new species as many closely related genotypes could belong to the same species rather than each genotype belonging to a distinct phylogenetic species.
In contrast to the unexpected diversity of Colletotrichum gloeosporioides, a distinct issue arises when what was originally thought to be a large number of species turns out to be a much less diverse grouping. The Diaporthe eres species complex is another example of such a phytopathogenic cryptic species. In D. eres, sequence heterogeneity of the ITS gene region was revealed to be the reason for over-estimation of species diversity in this complex (Udayanga et al. 2014). Using seven gene regions, Udayanga et al. (2014) resolved the species boundaries within this complex based on the application of GCPSR (Dettman et al. 2003; Taylor 2015). Further, they mentioned factors limiting the cryptic diversity in this complex as biological species recognition, morphology and discordance of genes (Udayanga et al. 2014). Moreover, the authors emphasised the use of additional gene regions and the importance of extended sampling while introducing new species. In addition to these facts, the D. eres species complex is polyphyletic (Udayanga et al. 2014). However, studies published several years later on Diaporthe taxonomy and phylogeny still appeared to ignore this fact. As a result, many species have been introduced as novel taxa in the D. eres complex when only one or two D. eres representative strains were included in the phylogenetic tree. A recent study by Manawasinghe et al. (2019) has shown that in the combined multigene (ITS, tub2, calmodulin (cal), and tef1) phylogenetic tree, D. rosicola and D. mahothocarpus clustered together with D. eres when the number of samples were increased. However, in the original publications of D. rosicola, the authors had included several D. eres strains clustered in an entirely different clade (Wanasinghe et al. 2018). Diaporthe mahothocarpus was introduced using only ITS and tef1 sequence data (Gao et al. 2014), making it difficult to compare with other species.
Botryosphaeria dothidea is common opportunistic fungal pathogen belonging to family Botryosphaeriaceae (Phillips et al. 2013). In Phillips et al. (2013) six Botryosphaeria species were accepted based on morphology and phylogeny. During last few years several new species were introduced to this genus and all were morphologically similar to B. dothidea but phylogenetically distinct (Zhou et al. 2016; Liang et al. 2019). Thus, they were introduced as cryptic species. However, a study conducted by Zhang et al. (2020) revealed that some of these introduced species are likely to be synonyms of B. dothidea as these six species do not show any significant differences from the type sequences of B. dothidea (Zhang et al. 2020). Thus, to define a cryptic species a boundary should be established. Therefore, it is necessary to pay attention on taxon sampling and selecting multiple gene regions for species identification through phylogenetic analysis. Once a novel taxon is identified the next step is to give it a valid name based on the international nomenclatural rules. It is also important to pay serious attention when naming a fungal plant pathogen. In the following section, we will discuss extensively the naming of fungal plant pathogens.
Naming of fungal plant pathogens
Accuracies in naming and species identification are among the most important steps in developing effective biosecurity protocols, trade policies and reducing risks (Hyde et al. 2010). Identification and characterisation of fungal pathogens have been subjected to many changes, and they are still evolving. The species names currently used are binomial, in which the first part is the genus name and the second part a specific epithet (Cantino et al. 1999). The ambiguity in naming is due, in part, because the rules for naming and introducing new species having undergone numerous changes over the years. The international rules governing scientific nomenclature began to be implemented in the nineteenth century. The first International Code for Botanical Nomenclature (ICBN) was published in 1905. This became the International Code of Nomenclature for algae, fungi and plants (ICN) (Hawksworth 2011). In earlier studies, separate nomenclatural status was followed for naming asexual and sexual morphs of the same fungus. This was eliminated by article 59 of the ICBN (McNeill et al. 2012) and this was one of the important milestones, namely the implementation of the “one name-one fungus” rule (McNeill et al. 2012).
Based on DNA sequences and phylogenetic analyses, an increasing number of new species have been reported in the literature. So far, the majority of new phytopathogen names were based on an epithet affiliated with the host (Dayarathne et al. 2016). One problem with this system is that, if we introduce a new species with an epithet linked to the host, it can be misconstrued as a host-specific pathogen. Similarly, another common species epithet used is the locality of the disease where it was first reported (Dayarathne et al. 2016). This might similarly lead to the wrong conclusion that such a pathogen is endemic or even restricted to the place where the disease was first discovered. Above all, the most important factor in practical farming is transferring knowledge about disease agents to farmers, for they are directly involved in disease identification and control for their crop production. Indeed, the use of complex scientific names for pathogens may make them inaccessible to farmers, thus hindering the ability of farmers to control the pathogens when needed. Therefore, giving names to the pathogens should also be compatible with the prior knowledge of farmers. While there is a need for a specific scientific name, there is also an urgency for a pathogenic genus to have a common name as well. One of the frequently faced confusions between farmers and researchers are the diseases caused by the Diaporthe species (Dayarathne et al. 2016). These species were previously known as Phomopsis and many old books on farming still carry the name Phomopsis. Most importantly, the farming knowledge flows through generations and the disease has been established as Phomopsis, while the scientific community calls it Diaporthe. One example is Phomopsis cane blight caused by Diaporthe ampelina. Despite this the disease is still known as Phomopsis cane blight (Guarnaccia et al. 2018). Therefore, the naming of phytopathogenic genera should receive careful attention.
When it comes to the lower ranks there are no firm guidelines to giving names. Each level is identified by an abbreviation given after the species name, in which subspecies are denominated as “subsp.”, varieties as “var.” and forma speciales as “f. sp.”. Then this abbreviation is followed by an epithet based on which they are defined. In here too, most of the time epithet is based on the host or locality. In plant pathology, giving a lower-ranking epithet based on the host is considered informative. When it comes to the races, naming is based on either a number or a code as defined by the person who identified that particular race. For example, the rice blast pathogen, Magnaporthe oryzae has five races namely; IA-1, IA-3, IA-63, IB-3 and IB-59 (Fang et al. 2017). Further, de Labrouhe et al. (2012) proposed a methodology to improve race nomenclature in defining races of sunflower downy mildew pathogen, based on the resistance and susceptibility of sunflower genotypes. Therefore, it is important to follow a specific standard based on the fungal species and the host when defining and naming of races.
As per the foregoing, we believe that when naming a phytopathogenic genus it is important to compare the host specificity and geographical variation of the already known species belonging to the same genus or family. Such knowledge would facilitate establishing the validity of a name for an epithet, based on the host and/or the geography. It will also help in the understanding whether a certain species belongs to a genetically closely related fungal group that causes a specific disease in a host.
Recommendations for introducing new pathogenic species
There are several issues associated with naming and identifying plant fungal pathogens. Every genus and particular group of pathogenic fungi have their questionable areas to be resolved. The common issues include insufficient sampling, lack of data on the plant’s growth conditions, lack of statistical support and lack of taxon sampling. Moreover, there are lesser number of studies to encourage researchers to go beyond species level identification. Therefore, in this study we try to provide some recommendations (i) to avoid wrong species introduction and (ii) to establish the lower levels ranking of phytopathogenic fungi. Below are our general recommendations to introduce novel phytopathogenic fungal species,
Sampling:
-
Collect samples representing diverse host cultivars from different environmental niches and geographical locations.
-
Provide quantitative data on disease intensity, disease severity, and the growth conditions, age or developmental stage of plant hosts.
-
Describe the features of the general ecosystem(s), natural and/or human-made, in which the disease was found.
A well-balanced sampling will facilitate collecting as many genotypes as possible from a particular locality. Even when sampling has been done as above, the majority of the isolated strains will be sorted based on colony morphology before molecular analysis. At this point, the only sorting criteria are colony morphology and if the morphological features of all the cultures are the same, researchers will choose one or two strains for further analyses. Therefore, it is necessary to set up a boundary to choose at least two representative isolates from each sampling category.
Number of strains:
-
Include a minimum of three strains from separate diseased tissues or plants (several single spore isolates/pure strains to conduct further analysis).
-
Use the same strains to assess pathogenicity.
-
The overall sample size for pathogenicity testing needs be increased, when there are multiple cryptic species within the population.
-
Provide sufficient data to understand the distribution and host specificity of allopatric and sympatric strains belonging to closely related taxa.
Assuring sequence quality and data:
-
Sequence length must be similar to the already known species.
-
Provide protein coding sequence data as annotated.
Species delineation:
-
Use more than two species recognition criteria, one of which should be phylogenetic analysis.
-
Taxon sampling in phylogenetic analysis must include type sequences and several representative strains (3–5 based on the availability).
-
To introduce novel species into a well-established genus, authors must provide DNA sequence data for all gene regions commonly used for delineating the existing species.
-
When there is limited bootstrap support for the new species in phylogenetic analyses, it is necessary to provide other evidence (e.g. number of nucleotide differences) to show that the new species is distinct from all existing species and back this up with microscopic or colony morphology features.
-
To show evidence of cryptic species in phytopathogenic genera such as Colletotrichum, Diaporthe and Fusarium, additional analytical methods such as GCPR, Phi index and phylogenetic compatibility are encouraged.
Identification beyond species level (lower-ranking):
-
Lower-level ranking is encouraged to apply on genera that are already defined as containing cryptic species or species complexes.
-
To identify lower ranks, authors should practice randomised sampling, so that samples represent all available host species and ecological niches.
-
The minimum number of samples should be more than ten for each species (for statistical validation).
-
Application of GCPR, Neighbour-joining analysis, haplotype analysis and network analysis are encouraged to identify different grouping patterns rather than phylogenetic trees.
-
Defining each level
-
Subspecies:grouping patterns based on the geography and or climatic variations.
-
Varieties: grouping patterns observed/witnessed in the same geographical range.
-
Forms: can be defined based on host specificity. Availability of cross pathogenicity data will provide better resolution in defining forms. Forms mostly have morphological variations as well. If there is no evidence of subspecies and varieties, naming as forma specialis is accepted after species identification.
-
Races: Characterised by the level of pathogenicity in plant pathology. There are no morphological or geographical difference requirements among races. This highly host-specific groupings, based on gene-by-gene interactions should be classified after forma specialis.
-
Where should we stand in pathogenic species ranking; to the species level and beyond species level
Even though plant pathology has improved tremendously, still one question remains difficult to obtain consensus on: “What is a species?” Here, our objective was to discuss several issues associated with species delineation of phytopathogenic fungi. While it is clear that correct species identification is key to disease control, there remains no specific boundary to clearly define a fungal species. Most studies have attempted to define a species by relying on previously accumulated data for specific groups of organisms. In this study, we have attempted to provide several benchmarks to aid in this critical first step of plant pathology. People working in plant pathology are pathologists, farmers and taxonomists. We need to establish a method to link these three groups together. The knowledge from the field must be shared in the laboratory and similarly the experimental results must be simplified in order to be comprehensible to the farmers. Accordingly, the knowledge of farmers regarding disease occurrence and spread, is one of the key factors in designing a sampling method.
As stated in previous studies, there are over 30 species concepts available to define a species. However, what happens in practice is that species boundaries are made by adopting a species concept(s) based on available data. This has resulted in a tremendous number of novel taxa that cannot be compared using a universal method. While we cannot adopt the same comprehensive method for species delineation, it is necessary to have an all-inclusive method at least for within the same genus. One of the best approaches to overcome this is by adopting a consolidated species concept (Quaedvlieg et al. 2014). We encourage researchers to apply the polyphasic method to define a fungal plant pathogen in which Morphological Species Concept (MSC), Ecological Species Concept (ESC) (van Valen 1976), and the Phylogenetic Species Concept (PSC) (Hennig 1966) can be the baseline. This approach has been adopted by many studies and this combined approach has been named as the “Consolidated Species Concept (CSC)” (Frisvad and Samson 2004; Crous and Groenewald 2005; Leslie and Summerell 2006; Cai et al. 2009; Groenewald et al. 2013).
The use of morphological species and biological species concepts has become less and less efficient for fungi. In contrast, the majority of publications in the last two decades have relied on the phylogenetic species concept with help from DNA sequence data. In the last decade, nearly 2000 new fungal species have been introduced (Hyde et al. 2020) with molecular data by mycological groups worldwide. At this current rate of new species introduction as well as the rate of synonymisations of previously introduced taxa, it will become essential that mycologists in different groups come to a single agreement on how to define a species. It is required to define how much morphological divergence is acceptable? How much sequence divergence is acceptable in a fungal species? These questions are still remaining to be answered.
Defining beyond the species level of plant pathogens has taken place in a few established fungal genera. In addition, some early established forms and varieties have been synonymised into the species. From the viewpoint of fungal taxonomists, this is acceptable, as their role is to define the species. However, from the viewpoint of plant pathologists, it is necessary to define these lower levels, if it has a single asexual strain that has the potential to make a devastating plant disease, separately recognized. Thus, we encourage mycologists who are working on plant pathogens to collect samples to represent all possible variable factors (geography, host, and cultivar) to help define these lower ranks beyond the species level. While being subjective, when defining these lower ranks, it is essential to provide rationales. When defining varieties and forms, cross inoculation experiments and pathogenicity data should become mandatory. As already discussed, ‘race’ should be at a level higher than the ‘strain’, in which several strains could together act as one particular race. While there should be shared features among strains within a race, the genetic differences between races can vary and be difficult to define. These recommendations may not apply to all phytopathogenic genera, and yet maybe valid for majority of genera to avoid introduction of any different genotype as a novel species. Moreover, methods employed in the lower ranking could vary from gene fragment sequencing to whole-genome resequencing. Thus, selection of markers to define a forma specialis and/or a race is always subjective and depends on the time available and cost-effectiveness. It should be noted here that when adding lower ranks, efforts should be made to reduce confusions about names, as the majority of names given are based on host names.
Based on the arguments presented, we propose that plant pathogens need clear and standard identification criteria that go beyond the species level. Even though a particular fungus has received a species name, where possible, lower levels of identifications are encouraged. An established and agreed upon ranking system can help to develop consistent and dependable control measures. With an increasing world population, global warming, drastic environmental changes, and decreasing availability of cultivatable land, agronomists are facing the biggest challenge in feeding current and future world populations. Thus, it is necessary to develop an international framework for controlling disease occurrence and their spread. The knowledge of basic pathological aspects such as identification of causal organisms of particular hosts will help to protect domestic food security as well as to develop quarantine measures to control the introduction of diseases into new localities. Therefore, both fungal taxonomists and plant pathologists have to pay more rigid attention to correct species identification.
References
Ashu EE, Xu J (2015) The roles of sexual and asexual reproduction in the origin and dissemination of strains causing fungal infectious disease outbreaks. Infect Genet Evol 36:199–209
Avelino J, Cristancho M, Georgiou S, Imbach P, Aguilar L, Bornemann G, Läderach P, Anzueto F, Hruska AJ, Morales C (2015) The coffee rust crises in Colombia and Central America (2008–2013): impacts, plausible causes and proposed solutions. Food Secur 7:303–321
Bán R, Kovács A, Körösi K, Perczel M, Turóczi G (2014) First report on the occurrence of a new pathotype, 714, of Plasmopara halstedii (sunflower downy mildew) in Hungary. Plant Dise 98:1580–1580
Bickford D, Lohman DJ, Sodhi NS, Peter KLN, Meier R, Winker K, Krista K, Indraneil Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Bordewich M, Deutschmann IM, Fischer M, Kasbohm E, Semple C, Steel M (2018) On the information content of discrete phylogenetic characters. J Math Biol 77:527–544
Bruen TC, Philippe H, Bryant D (2006) A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681
Burgess LW, Nelson PE, Toussoun TA, Forbes GA (1988) Distribution of Fusarium species in sections Roseum, Arthrosporiella, Gibbosum and Discolor recovered from grassland, pasture and pine nursery soils of Eastern Australia. Mycologia 80:815–824
Cai L, Hyde KD, Taylor PWJ, Weir BS, Waller JM, Abang MM, Zhang JZ, Yang YL, Phoulivong S, Liu ZY, Prihastuti H, Shivas RG, McKenzie EHC, Johnston PR (2009) A polyphasic approach for studying Colletotrichum. Fungal Divers 39:183–204
Cannon PF, Damm U, Johnston PR, Weir BS (2012) Colletotrichum: current status and future directions. Stud Mycol 73:181–213
Cantino DP, Bryant HN, Queiroz KD, Donoghue MJ, Eriksson T, Hillis DM, Michael S, Lee Y (1999) Species names in phylogenetic nomenclature. Syst Biol 48:790–807
Chenuil A, Cahill AE, Délémontey N, Salliant Du, du Luc E, Fanton H (2019) Problems and questions posed by cryptic species. A Framework to Guide Future Studies 24:77–106
Crespo A, Lumbsch HT (2010) Cryptic species in lichen-forming fungi. IMA Fungus 1:167–170
Crouch JA (2014) Colletotrichum caudatum is a species complex. IMA Fungus 5:17–30
Crous PW, Groenewald JZ (2005) Hosts, species and genotypes: opinions versus data. Australas Plant Pathol 34:463–470
Czembor E, Stępień Ł, Waśkiewicz A (2014) Fusarium temperatum as new species causing ear rot on maize in Poland. Plant Dis 98:1001
Dvořák P, Poulíčková A, Hašler P, Belli M, Casamatta DA, Papini A (2015) Species concepts and speciation factors in cyanobacteria, with connection to the problems of diversity and classification. Biodivers Conserv 24:739–757
Edel-Hermann V, Lecomte C (2019a) Current status of Fusarium oxysporum formae speciales and races. Phytopathol 109:512–530
Dayarathne MC, Boonmee S, Braun U, Crous PW, Daranagama DA, Dissanayake AJ, Ekanayaka H, Jayawardena R, Jones EBG, Maharachchikumbura SSN, Perera RH, Phillips AJL, Stadler M, Thambugala KM, Wanasinghe DN, Zhao Q, Hyde KD, Jeewon R (2016) Taxonomic utility of old names in current fungal classification and nomenclature: Conflicts, confusion & clarifications. Mycosphere 7:1622–1648
de Labrouhe DT, Walser P, Jolivot D, Roche S, Serre F, Leguillon M, Delmotte F, Bordat A, Godiard L, Vincourt P, Vear F (2012) Proposal for improvement of sunflower downy mildew race nomenclature. In: Proceedings of the 18th International Sunflower Conference, Mar del Plata pp. 322 –327.
De Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56:879–886
Dettman JR, Jacobson DJ, Taylor JW (2003) a multilocus genealogical approach to phylogenetic species recognition in the model eukaryote neurospora. Evolution (NY) 57:2703
Dugan FM, Everhart S (2016) Cryptic species: a leitmotif of contemporary mycology has challenges and benefits for plant pathologists. Plant Heal Prog 17:250–253
Dykhuizen DE, Green L (1991) Recombination in Escherichia coli and the definition of biological species. J Bacteriol 173:7257–7268
Edel-Hermann V, Lecomte C (2019b) Current status of Fusarium oxysporum Formae Speciales and races. Phytopathology 109:512–530
Fan J, Urban M, Parker JE, Brewer HC, Kelly SL, Hammond-Kosack KE, Fraaije BA, Liu X, Cools HJ (2013) Characterization of the sterol 14α-demethylases of Fusarium graminearum identifies a novel genus-specific CYP51 function. New Phytol 198:821–835
Fang X, Snell P, Barbetti MJ, Lanoiselet V (2017) Races of Magnaporthe oryzae in Australia and genes with resistance to these races revealed through host resistance screening in monogenic lines of Oryza sativa. Eur J Plant Pathol 148:647–656
Frisvad JC, Samson RA (2004) Polyphasic taxonomy of Penicillium subgenus Penicillium : a guide to identification of food and air-borne terverticillate penicillia and their mycotoxins. Stud Mycol 49:1–173
Gao YH, Sun W, Su YY, Cai L (2014) Three new species of Phomopsis in Gutianshan nature reserve in China. Mycol Prog 13:111–121
Gao Y, Liu F, Duan W, Crous PW, Cai L (2017) IMA Fungus 8:153–187
Gascuel Q, Martinez Y, Boniface MC, Vear F, Pichon M, Godiard L (2015) The sunflower downy mildew pathogen Plasmopara halstedii. Mol Plant Pathol 16:109–122
Geber J, Murphy E (2012) Scurvy in the great irish famine: evidence of vitamin c deficiency from a mid-19th century skeletal population. Am J Phys Anthropol 148:512–524
Geiser DM, Aoki T, Bacon CW, Baker SE, Bhattacharyya MK, Brandt ME, Brown DW, Burgess LW, Chulze S, Coleman JJ, Correll JC, Covert SF, Crous PW, Cuomo CA, De Hoog GS, Di Pietro A, Elmer WH, Epstein L, Frandsen RJ, Freeman S, Gagkaeva T, Glenn AE, Gordon TR, Gregory NF, Hammond-Kosack KE, Hanson LE, Jímenez-Gasco Mdel M, Kang S, Kistler HC, Kuldau GA, Leslie JF, Logrieco A, Lu G, Lysøe E, Ma LJ, McCormick SP, Migheli Q, Moretti A, Munaut F, O’Donnell K, Pfenning L, Ploetz RC, Proctor RH, Rehner SA, Robert VA, Rooney AP, Bin Salleh B, Scandiani MM, Scauflaire J, Short DP, Steenkamp E, Suga H, Summerell BA, Sutton DA, Thrane U, Trail F, Van Diepeningen A, Vanetten HD, Viljoen A, Waalwijk C, Ward TJ, Wingfield MJ, Xu JR, Yang XB, Yli-Mattila T, Zhang N (2013) One fungus, one name: defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathol 103:400–408
Geiser DM, Al-Hatmi A, Aoki T, Arie T, Balmas V, Barnes I, Bergstrom GC, Bhattacharyya MKK, Blomquist CL, Bowden R, Brankovics B, Brown DW, Burgess LW, Bushley K, Busman M, Cano-Lira JF, Carrillo JD, Chang HX, Chen CY, Chen W, Chilvers MI, Chulze SN, Coleman JJ, Cuomo CA, de Beer ZW, de Hoog GS, Del Castillo-Múnera J, Del Ponte E, Diéguez-Uribeondo J, Di Pietro A, Edel-Hermann V, Elmer WH, Epstein L, Eskalen A, Esposto MC, Everts KL, Fernández-Pavía SP, da Silva GF, Foroud NA, Fourie G, Frandsen RJN, Freeman S, Freitag M, Frenkel O, Fuller KK, Gagkaeva T, Gardiner DM, Glenn AE, Gold S, Gordon T, Gregory NF, Gryzenhout M, Guarro J, Gugino B, Gutiérrez S, Hammond-Kosack K, Harris LJ, Homa M, Hong CF, Hornok L, Huang JW, Ilkit M, Jacobs A, Jacobs K, Jiang C, Jimenez-Gasco MDM, Kang S, Kasson MT, Kazan K, Kennell JC, Kim H, Kistler HC, Kuldau GA, Kulik T, Kurzai O, Laraba I, Laurence MH, Lee TY, Lee YW, Lee YH, Leslie JF, Liew ECY, Lofton LW, Logrieco A, Sánchez López-Berges M, Luque AG, Lysøe E, Ma LJ, Marra RE, Martin FN, May SR, McCormick S, McGee CT, Meis JF, Migheli Q, Mohamed Nor NMI, Monod M, Moretti A, Mostert D, Mulé G, Munaut F, Munkvold GP, Nicholson P, Nucci M, O'Donnell K, Pasquali M, Pfenning LH, Prigitano A, Proctor R, Ranque S, Rehner S, Rep M, Rodríguez-Alvarado G, Rose LJ, Roth MG, Ruiz-Roldán C, Saleh AA, Salleh B, Sang H, Scandiani M, Scauflaire J, Schmale D 3rd, Short DP, Šišić A, Smith J, Smyth CW, Son H, Spahr E, Stajich JE, Steenkamp E, Steinberg C, Subramaniam R, Suga H, Summerell BA, Susca A, Swett CL, Toomajian C, Torres-Cruz TJ, Tortorano AM, Urban M, Vaillancourt LJ, Vallad GE, van der Lee T, Vanderpool D, van Diepeningen AD, Vaughan M, Venter E, Vermeulen M, Verweij PE, Viljoen A, Waalwijk C, Wallace EC, Walther G, Wang J, Ward T, Wickes B, Wiederhold NP, Wingfield MJ, Wood AKM, Xu JR, Yang XB, Yli-Matilla T, Yun SH, Zakaria L, Zhang H, Zhang N, Zhang S, Zhang X (2020) Phylogenomic analysis of a 55.1 kb 19-gene dataset resolves a monophyletic Fusarium that includes the Fusarium solani Species Complex. Phytopathology doi: https://doi.org/10.1094/PHYTO-08-20-0330-LE.
Geiser DM, Jimenez-Gasco MDM, Kang S, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, O’Donnell K (2004) FUSARIUM-ID v. 1.0: A DNA sequence database for identifying Fusarium. Euro J Plant Pathol 110:473–479
Gilbert AS, Wheeler RT, May RC (2015) Fungal pathogens: survival and replication within macrophages. Cold Spring Harb Perspect Med 5:019661
Gladieux P, De Bellis F, Hann-Soden C, Svedberg J, Johannesson H, Taylor JW (2020) Neurospora from natural populations: Population genomics insights into the Life history of a model microbial Eukaryote. In: Statistical population genomics Humana, New York, NY pp. 313–336.
Groenewald JZ, Nakashima C, Nishikawa J, Shin HD, Park JH, Groenewald M, Braun U, Crous PW (2013) Species concepts in Cercospora: spotting the weeds among the roses. Stud Mycol 75:115–170
Guarnaccia V, Groenewald JZ, Woodhall J, Armengol J, Cinelli T, Eichmeier A (2018) Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 40, 135–153
Guarro J, Gené J, Stchigel AM (1999) Developments in fungal taxonomy. Clin Microbiol Rev 12:454–500
Hawksworth D (2011) A new dawn for the naming of fungi: impacts of decisions made in Melbourne in July 2011 on the future publication and regulation of fungal names. MycoKeys 1:7–20
Hennig W (1966) Phylogenetic systematics. University of Illinois Press, Urbana
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Hyde KD, Chomnunti P, Crous PW, Groenewald JZ, Damm U, Ko TW, Shivas RG, Summerell BA, Tan YP (2010) A case for re-inventory of Australia’s plant pathogens. Persoonia 25:50–60
Hyde KD, Jeewon R, Chen YJ, Bhunjun CS, Calabon MS, Jiang HB, Lin CG, Norphanphoun C, Sysouphanthong P, Pem D, Tibpromma S, Zhang Q, Doilom M, Jayawardena RS, Liu JK, Maharachchikumbura SSN, Phukhamsakda C, Phookamsak R, Al-Sadi AM, Thongklang N, Wang Y, Gafforov Y, Jones EBG, Lumyong S (2020) The numbers of fungi: is the descriptive curve flattening? Fungal Divers 103:219–271
Hyde KD, Nilsson RH, Alias SA, et al. (2014). One stop shop: backbones trees for important phytopathogenic genera: I. Fungal Diversity 67: 21–125.
Jayawardena RS, Hyde KD, Chen YJ, Papp V, Palla B, Papp D, Bhunjun CS, Hurdeal VG, Senwanna C, Manawasinghe IS, Harischandra DL, Gautam AK, Avasthi S, Chuankid B, Goonasekara ID, Hongsanan S, Zeng XY, Liyanage KK, Liu NG, Karunarathna A, Hapuarachchi KK, Luangharn T, Raspé O, Brahmanage R, Doilom M, Lee HB, Mei L, Jeewon R, Huanraluek N, Chaiwan N, Stadler M, Wang Y (2020) One stop shop IV: taxonomic update with molecular phylogeny for important phytopathogenic genera: 76–100. Fungal Divers 103:87–218
Jones DR, Baker RHA (2007) Introductions of non-native plant pathogens into Great Britain. Plant Pathol 56:891–910
Judson OP, Normark BB (1996) Ancient asexual scandals. Trends Ecol Evol 11:41–46
Kumar S, Filipski AJ, Battistuzzi FU, Pond SL, Tamura K (2012) Statistics and truth in phylogenomics. Mol Biol Evol 29:457–472
Laurence MH, Summerell BA, Burgess LW, Liew EC (2014) Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biol 118:374–384
Leslie JF, Summerell BA (2006) The Fusarium laboratory manual. Blackwell Publishing, Ames
Liang LY, Jiang N, Chen WY, Liang YM, Tian CM (2019) Botryosphaeria qinlingensis sp. nov. causing oak frogeye leaf spot in China. Mycotaxon 134:463–473
Li F, Upadhyaya NM, Sperschneider J, Matny O, Nguyen-Phuc H, Mago R, Raley C, Miller ME, Silverstein KA, Henningsen E, Hirsch CD (2019) Emergence of the Ug99 lineage of the wheat stem rust pathogen through somatic hybridisation. Nat Commun 10:1–15
Liu F, Wang M, Damm U, Crous PW, Cai L (2016) Species boundaries in plant pathogenic fungi: a Colletotrichum case study. BMC Evol Biol 16:81. https://doi.org/10.1186/s12862-016-0649-5
Luchi N, Ioos R, Santini A (2020) Fast and reliable molecular methods to detect fungal pathogens in woody plants. Appl microbiol and biotechn 104:2453–2468
Lücking R, Aime MC, Robbertse B, Miller AN, Ariyawansa HA, Aoki T, Cardinali G, Crous PW, Druzhinina IS, Geiser DM, Hawksworth DL, Hyde KD, Irinyi L, Jeewon R, Johnston PR, Kirk PM, Malosso E, May TW, Meyer W, Öpik M, Robert V, Stadler M, Thines M, Vu D, Yurkov AM, Zhang N, Schoch CL (2020) Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 11:14–46
Manawasinghe IS, Zhang W, Li X, Zhao W, Chethana KT, Xu J, Chen Z, Dissanayaka AJ, Mugnai L, Úrbez-Torres JR, Savocchia S (2018) Novel microsatellite markers reveal multiple origins of Botryosphaeria dothidea causing the Chinese grapevine trunk disease. Fungal Ecol 33, 134–142
McNeill J, Barrie FR, Buck R, Demoulin V, Greuter W, Hawksworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, Prud'homme Van Reine WF, Smith GF, Wiersema JH (2012) International Code of Nomenclature for algae, fungi, and plants (Melbourne Code) Regnum Vegetabile 154. ISBN 978-3-87429-425-6 http://www.iapt-taxon.org/nomen/main.php
Maharachchikumbura SSN, Hyde KD, Groenewald JZ, Xu J, Crous PW (2014) Pestalotiopsis revisited. Stud Mycol 79:121–186
Manawasinghe IS, Dissanayake A, Liu M, Wanasinghe D, Xu J, Zhao W, Wei Z, Zhou Y, Hyde KD, Brooks S, Yan J (2019) High genetic diversity and species complexity of Diaporthe associated with grapevine dieback in China. Front Microbiol 10:1936
Manawasinghe IS, Phillips AJL, Hyde KD, Chethana KWT, Zhang W, Zhao WS, Yan JY, Li XH (2016) Mycosphere Essays 14: assessing the aggressiveness of plant pathogenic Botryosphaeriaceae. Mycosphere 7:883–892
Mallet J (2007) Subspecies, semispecies, superspecies. Encyclopedia Biodivers 5:523–526
Marcelino VR, Irinyi L, Eden JS, Meyer W, Holmes EC, Sorrell TC (2019) Metatranscriptomics as a tool to identify fungal species and subspecies in mixed communities–a proof of concept under laboratory conditions. IMA Fungus 10:1–10
McCook S (2006) Global rust belt: Hemileia vastatrix and the ecological integration of world coffee production since 1850. J Glob Hist 1:177–195
McCook S, Vandermeer J (2015) The Big Rust and the red queen: long-term perspectives on coffee rust research. Phytopathology 105:1164–1173
McDonald B (2004) Population genetics of plant pathogens. The Plant Health Instructor. https://doi.org/10.1094/PHI-A-2004-0524-01
Mayr E (1942) Systematics and the origin of species. Columbia University Press, New York
Mayr E (1982) Of what use are subspecies? Auk 99:593–595
Milgroom MG (2017) . Population biology of plant pathogens: genetics, ecology, and evolution. The American Phytopathological Society.
Morse SS (2001) Factors in the emergence of infectious diseases. Plagues Politics 4:8–26
Nieuwenhuis BP, James TY (2016a) The frequency of sex in fungi. Philoso Trans R Soc B 371:20150540
Nguyen HGT, Jančič S, Meijer M, Tanney JB, Zalar P, Gunde-Cimerman N, Seifert KA (2015) Correction: application of the phylogenetic species concept to wallemia sebi from house dust and indoor air revealed by multi-locus genealogical concordance. PLoS ONE 10:e0129752
Nieuwenhuis BPS, James TY (2016b) The frequency of sex in fungi. Philos Trans R Soc B 371:20150540
O’Donnell K, Cigelnik E, Nirenberg HI (1998) Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia 90:465–493
O’Donnell K, Ward TJ, Aberra D, Kistler HC, Aoki T, Orwig N, Kimura M, Bjørnstad S, Klemsdal SS (2008) Multilocus genotyping and molecular phylogenetics resolve a novel head blight pathogen within the Fusarium graminearum species complex from Ethiopia. Fungal Genet Biol 45:1514–1522
O’Donnell K, Ward TJ, Geiser DM, Kistler HC, Aoki T (2004) Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet Biol 41:600–623
O’Donnell K, Rooney AP, Proctor RH, Brown DW, McCormick SP, Ward TJ, Frandsen RJN, Lysøe E, Rehner SA, Aoki T, Robert VARG, Crous PW, Groenewald JZ, Kang S, Geiser DM (2013) RPB1 and RPB2 phylogeny supports an early Cretaceous origin and a strongly supported clade comprising all agriculturally and medically important fusaria. Fungal Genet Biol 52:20–31
O’Donnell K, Ward TJ, Robert VARG, Crous PW, Geiser DM, Kang S (2015) DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43:583–595
Oláh J, Andersen T, Beshkov S, Ciubuc C, Coppa G, Kovács T, Oláh J (2018) Unified phylogenetic species concept: taking subspecies and race out of science: postmodern theory applied to the Potamophylax cingulatus group (Trichoptera, Limnephilidae). Opuscula Zool (budapest) 49:33–70
Omotayo OP, Omotayo AO, Mwanza M, Babalola OO (2019) Prevalence of mycotoxins and their consequences on human health. Toxicol Res 35:1–7
Paping R, Vanhaute E, O Grada C (eds) (2007) When the Potato Failed. Causes and Effects of the Last European Subsistence Crisis, 1845–1850. Brepols Publishers, Turnhout
Patten M (2009) Subspecies’ and “race” should not be used as synonyms. Nature 457:147
Patten MA (2015) Subspecies and the philosophy of science. Auk 132:481–485
Phillips AJL, Alves A, Abdollahzadeh J, Slippers B, Wingfield MJ, JZ (2013) The Botryosphaeriaceae: genera and species known from culture. Stud Mycol 76:51–167
Proctor RH, McCormick SP, Alexander NJ, Desjardins AE (2009) Evidence that a secondary metabolite gene cluster has grown by gene relocation during evolution of the filamentous fungus Fusarium. Mol Microbiol 74:1128–1142
Quaedvlieg W, Binder M, Groenewald JZ, Summerell BA, Carnegie AJ, Burgess TI, Crous PW (2014) Introducing the consolidated species concept to resolve species in the Teratosphaeriaceae. Persoonia Mol Phylogeny Evol Fungi 33:1–40
Rouxel M, Mestre P, Comont G, Lehman BL, Schilder A, Delmotte F (2013) Phylogenetic and experimental evidence for host-specialized cryptic species in a biotrophic oomycete. New Phytol 197:251–263
Safran RJ, Nosil P (2012) Speciation: the origin of new species. Nat Educ Knowl 3:17
Scauflaire J, Gourgue M, Munaut F (2011) Fusarium temperatum sp.nov. from maize, an emergent species closely related to Fusarium subglutinans. Mycologia 103:586–597
Scholthof KBG (2007) The disease triangle: pathogens, the environment and society. Nat Rev Microbiol 5:152–156
Shang Y, Xiao G, Zheng P, Cen K, Zhan S, Wang S (2016) Divergent and convergent evolution of fungal pathogenicity. Genome Biol Evol 8:1374–1387
Sharma M, Kulshrestha S (2015) Colletotrichum gloeosporioides: an anthracnose causing pathogen of fruits and vegetables. Biosci Biotechnol Res Asia 12(2):1233–1246
Singh RP, Hodson DP, Jin Y, Lagudah ES, Ayliffe MA, Bhavani S, Rouse MN, Pretorius ZA, Szabo LJ, Huerta-Espino J, Basnet BR (2015) Emergence and spread of new races of wheat stem rust fungus: continued threat to food security and prospects of genetic control. Phytopathol 105:872–884
Solarte F, Muñoz CG, Maharachchikumbura SSN, Álvarez E (2018) Diversity of Neopestalotiopsis and Pestalotiopsis spp., causal agents of guava scab in Colombia. Plant Dis 102:49–59
Spring O, Thines M (2010) Molecular techniques for classification and diagnosis of plant pathogenic oomycota. In: Molecular identification of fungi. Springer, Berlin pp. 35–50.
Stępień Ł (2014) The use of Fusarium secondary metabolite biosynthetic genes in chemotypic and phylogenetic studies. Crit Rev Microbiol 40:176–185
Stępień Ł, Chełkowski J (2010) Fusarium head blight of wheat: pathogenic species and their mycotoxins. World Mycotox J 3:107–119
Stępień Ł, Gromadzka K, Chełkowski J, Basińska-Barczak A, Lalak-Kańczugowska J (2019) Diversity and mycotoxin production by Fusarium temperatum and Fusarium subglutinans as causal agents of pre-harvest Fusarium maize ear rot in Poland. J Appl Gen 60:113–121
Stępień Ł, Koczyk G, Waśkiewicz A (2011) FUM cluster divergence in fumonisins-producing Fusarium species. Fungal Biol 115:112–123
Stępień Ł, Waśkiewicz A, Wilman K (2015) Host extract modulates metabolism and fumonisin biosynthesis by the plant-pathogenic fungus Fusarium proliferatum. Int J Food Microbiol 193:74–81
Storfer A, Alfaro ME, Ridenhour BJ, Jancovich JK, Mech SG, Parris MJ, Collins JP (2007) Phylogenetic concordance analysis shows an emerging pathogen is novel and endemic. Ecol Lett 10:1075–1083
Struck TH, Feder JL, Bendiksby M, Birkeland S, Cerca J, Gusarov VI, Kistenich S, Larsson KH, Liow LH, Nowak MD, Stedje B, Bachmann L, Dimitrov D (2018) Finding evolutionary processes hidden in cryptic species. Trends Ecol Evol 33:153–163
Subramanian S, Kumar S (2003) Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genom Res 13:838–844
Talhinhas P, Batista D, Diniz I, Vieira A, Silva DN, Loureiro A, Tavares S, Pereira AP, Azinheira HG, Guerra-Guimarães L, Várzea V, do Céu Silva M, (2017) The coffee leaf rust pathogen Hemileia vastatrix : one and a half centuries around the tropics. Mol Plant Pathol 18:1039–1051
Taylor JW (2015) Evolutionary perspectives on human fungal pathogens. Cold Spring Harb Perspect Med 5:e019588
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fishera MC (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Thines M (2019) An evolutionary framework for host shiftsjumping ships for survival. New Phytol 224:605–617
Thirkell TJ, Charters MD, Elliott AJ, Sait SM, Field KJ (2017) Are mycorrhizal fungi our sustainable saviours? Considerations for achieving food security. J Ecol 105:921–929
Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, Hyde KD (2014) Insights into the genus Diaporthe: phylogenetic species delimitation in the D. eres species complex. Fungal Divers 67:203–229
Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, Hyde KD (2015) The Diaporthe sojae species complex: Phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol 119:383–407
Vakalounakis DJ, Wang Z, Fragkiadakis GA, Skaracis GN, Li DB (2004). Characterization of Fusarium oxysporum isolates obtained from cucumber in China by pathogenicity, VCG, and RAPD. Plant Disease, 88:645–649
van Valen L (1976) Ecological species, multispecies and oaks. Taxon 25:233–239
Vieira WA, Lima WG, Nascimento ES, Michereff SJ, Câmara MP, Doyle VP (2017) The impact of phenotypic and molecular data on the inference of Colletotrichum diversity associated with Musa. Mycologia 109:912–934
Wanasinghe DN, Phukhamsakda C, Hyde KD, Jeewon R, Lee HB, Jones EBG, Tibpromma S, Dissanayake AJ, Tennakoon DS, Jayasiri SC, Gafforov Y, Camporesi E, Bulgakov TS, Ekanayake AH, Perera RH, Samarakoon MC, Goonasekara ID, Mapook A, Li WJ, Senanayake IC, Li J, Norphanphoun C, Doilom M, Bahkali AH, Xu J, Mortimer PE, Tibell L, Tibell S, Karunarathna SC, (2018) Fungal diversity notes 709–839: taxonomic and phylogenetic contributions to fungal taxa with an emphasis on fungi on Rosaceae. Fungal Divers 89:1–236
Watanabe M, Yonezawa T, Lee KI, Kumagai S, Sugita-Konishi Y, Goto K, Hara-Kudo Y (2011) Molecular phylogeny of the higher and lower taxonomy of the Fusarium genus and differences in the evolutionary histories of multiple genes. BMC Evol Biol 11:322
Weir BS, Johnston PR, Damm U (2012) The Colletotrichum gloeosporioides species complex. Stud Mycol 73:115–180
WHO (2021) Mycotoxins are naturally occurring toxins,can be found in food & The adverse health effects of as immune deficiency and cancer. Accessed 20 Jan 2021.
Wingfield MJ, Wilhelm De Beer Z, Slippers B, Wingfield BD, Groenewald JZ, Lombard L, Crous PW (2012) One fungus, one name promotes progressive plant pathology. Mol Plant Pathol 13:604–613
Wróbel B (2008) Statistical measures of uncertainty for branches in phylogenetic trees inferred from molecular sequences by using model-based methods. J Appl Genet 49:49–67
Xu J (2002) Estimating the spontaneous mutation rate of loss of sex in the human pathogenic fungus Cryptococcus neoformans. Genetics 162:1157–1167
Xu J (2020) Fungal species concepts in the genomics era. Genome 63:459–468
Xu J (2004) Genotype-environment interactions of spontaneous mutations for vegetative fitness in the human pathogenic fungus Cryptococcus neoformans. Genetics 168:1177–1188
Xu J (2016) Fungal DNA barcoding. Genome 59:913–932
Xu J, Li H, Zhou GY, Liu JA (2016) When do we call genetically distinct strains different species? A cautionary case study of the Colletotrichum gloesporioides species complex. Fungal Genom Biol 06:1–5
Zain ME (2011) Impact of mycotoxins on humans and animals. J Saudi Chem Soc 15:129–144
Zhang H, Luo W, Pan Y, Xu J, Xu JS, Chen WQ, Feng J (2014) First report of Fusarium temperatum causing fusarium ear rot on Maize in Northern China. Plant Dis 98:1273
Zhang W, Groenewald JZ, Lombard L, Schumacher RK, Phillips AJ, Crous PW (2020) Evaluating species in Botryosphaeriales. Persoonia 46:63–115
Zhang W, Manawasinghe IS, Zhao W, Xu J, Brooks S, Zhao X, Hyde KD, Chethana KWT, Liu J, Li XH, Yan JY (2017) Multiple gene genealogy reveals high genetic diversity and evidence for multiple origins of Chinese Plasmopara viticola population. Sci Rep 7:17304
Zhou Y, Dou Z, He W, Zhang X, Zhang Y (2016) Botryosphaeria sinensia sp nov., a new species from China. Phytotaxa 245:43–50
Acknowledgements
We would like to thank the Thailand Research Fund, Grant RDG6130001 entitled “Impact of climate change on fungal diversity and biogeography in the Greater Mekong Subregion”. Kevin D Hyde thanks Chiang Mai University for the award of a Visiting Professor. Ishara S Manawasinghe thank Prof Marco Thines for guiding the development of this paper by providing valuable ideas and comments. Alan JL Phillips acknowledges the support from UIDB/04046/2020 and UIDP/04046/2020 Centre grants from FCT, Portugal (to BioISI).
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
ISM and KDH conceived the basic idea. ISM, AJLP and JX developed the outline. ISM, ŁS, DLH, AK, wrote the manuscript. JW created the graphics in the manuscript. AJLP, JX, AB, MT, ŁS, KDH, JY, RC, ZD, ML, JW revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Handling Editor: Sinang Hongsanan.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Manawasinghe, I.S., Phillips, A.J.L., Xu, J. et al. Defining a species in fungal plant pathology: beyond the species level. Fungal Diversity 109, 267–282 (2021). https://doi.org/10.1007/s13225-021-00481-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-021-00481-x