
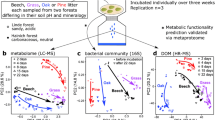
Abstract
Approaches for the cultivation-independent analysis of microbial communities are summarized as meta’omics, which predominantly includes metagenomic, -transcriptomic, -proteomic and -metabolomic studies. These have shown that endophytic, root-associated and soil fungal communities are strongly shaped by associated plant species. The impact of plant identity on the composition of its litter-associated fungal community remains to be disentangled from the impact of litter chemistry. The composition of the plant community also shapes the fungal community. Most strikingly, adjacent plant species may share mycorrhizal symbionts even if the plants usually have different types of mycorrhizal fungi associated with them (ectomycorrhizal, ericoid and arbuscular mycorrhizal fungi). Environmental parameters weakly explain fungal community composition globally, and their effect is inconsistent at local and regional scales. Decrease in similarity among communities with increasing distance (i.e. distance decay) has been reported from local to global scales. This pattern is only exceptionally caused by spatial dispersal limitation of fungal propagules, but mostly due to the inability of the fungi to establish at the particular locality (i.e. environmental filtering or competitive exclusion). Fungal communities usually undergo pronounced seasonal changes and also differ between consecutive years. This indicates that development of the communities is usually not solely cyclic. Meta’omic studies challenge the classical view of plant litter decomposition. They show that mycorrhizal and (previously) endophytic fungi may be involved in plant litter decomposition and only partly support the idea of a succession from an Ascomycota to a Basidiomycota-dominated community. Furthermore, vertical separation of saprotrophic and mycorrhizal species in soil and sequential degradation from easily accessible to ‘recalcitrant’ plant compounds, such as lignin, can probably not be generalized. The current models of litter decomposition may therefore have to be eventually refined for certain ecosystems and environmental conditions. To gain deeper insights into fungal ecology, a meta’omic study design is outlined which focuses on environmental processes, because fungal communities are usually taxonomically diverse, but functionally redundant. This approach would initially identify dynamics of chemical shifts in the host and/or substrate by metametabolomics. Detected shifts would be subsequently linked to microbial activity by correlation with metatranscriptomic and/or metaproteomic data. A holistic trait-based approach might finally identify factors shaping taxonomic composition in communities against the dynamics of the environmental process(es) they are involved in.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
All living plants are associated with microorganisms. These associations may be so intimate that they may help the host plants to adapt to changing environmental conditions (Redman et al. 2011). The importance of these symbiont-mediated adaptations means that plants should not be considered alone, but the ‘holobiont plant’, as the basic unit in ecology and evolution (Hawksworth 1991). This holobiont comprises a host macro-organism and all of its associated micro-organisms (Rosenberg et al. 2010; Feldhaar 2011). Plant-associated fungi are usually categorized according to their functional roles. Due to their importance in plant nutrition, mycorrhizal fungi represent the best-studied group among the mutualistic fungal symbionts, (Smith and Read 2008). Non-mycorrhizal root-associated fungi, however, may also facilitate plant nutrition (Kernaghan 2013) and access additional nutrient sources (Behie et al. 2012). Endophytic fungi represent another taxonomically highly diverse group of plant-associated microorganisms, the functional roles of which, however, are highly diverse and hitherto remain widely unidentified (Hyde and Soytong 2008; Porras-Alfaro and Bayman 2011). The symbiosis between grasses and their systemic endophytes is rather well understood and known to affect their hosts in many ways (Tanaka et al. 2012). Even though prominent taxonomic groups of leaf-endophytes of forest trees, such as the Xylariales, are known to produce a broad spectrum of antibiotically active metabolites (Bills et al. 2012), reports of positive effects on host plant fitness are scarce (Saikkonen et al. 2010). An involvement in litter decomposition has been suggested for other leaf-endophytes (Purahong and Hyde 2011). In this key process to nutrient cycling of plant biomass (Berg and McClaugherty 2014), also mycorrhizal fungi may be directly involved (Ekblad et al. 2013). The decomposer community may therefore be regarded as being plant-associated in a more general sense, in particular since saprotrophic fungi may also prefer ecological niches confined to higher plants (Zhou and Hyde 2001). As a consequence, non-mycorrhizal soil fungi, involved in root-litter decomposition (Solly et al. 2014) and nutrient mobilization from minerals (Gadd 2010) should be considered in the context of plant-associated fungal communities. Wood decay is also an important part of nutrient cycling and strongly dependent on fungal communities (Bradford et al. 2014; Valentín et al. 2014; Van der Wal et al. 2014), but the wood decay communities are different from those associated with living plants (see Stokland et al. 2012).
Assessing the entire species richness in environmental samples is still challenging (Tedersoo et al. 2014a), but hundreds to thousands of fungal species have been detected (Hawksworth 2012). This richness makes complete sampling of communities by cultivation nearly unachievable. Even construction and subsequent screening of reasonably exhaustive clone-libraries from environmental DNA is rather laborious, in particular if functionality is considered in addition to taxonomic diversity (Daniel 2005). The development of massively parallel sequencing technologies (‘Next-Generation-Sequencing’, NGS) provides new opportunities, allowing for simultaneous sequencing of billions of molecules in a nucleic acid extract (Buermans and Dunnen 2014). This advancement also boosted the field of proteomics, because assignment of MS- and NMR-based peptide spectra depend on in silico-generated reference data for identification, and NGS rapidly increased the amount of reference sequences and facilitated the parallel assessment of protein-encoding transcripts (Seifert et al. 2013). At the same time, MS and NMR technologies were further developed (Lankadurai et al. 2013) and now allows the assessment of metabolite profiles of whole holobionts (Hernández et al. 2009; Walker et al. 2014a) as well as of the composition of chemical compounds in environmental samples (Wallenstein et al. 2013; Jones et al. 2014). These technological advances together represent a major breakthrough for microbial ecology, because they allow for insights into structure and function of even the most complex microbial communities in their natural environment.
Cultivation-independent studies on plant-associated fungal communities are reviewed herein, to elucidate the factors shaping community composition and function. The focus is on the role of host plant identity and community in structuring the associated fungal communities. The impact of environmental factors on the spatiotemporal structures and dynamics is discussed and linked to the functional role of the respective communities. Against this background, current challenges and knowledge gaps are highlighted and topics of major interest for future research identified. Focusing on plant holobionts with respect to nutrient cycling, the directly plant-associated endophytic and root-associated fungal communities are addressed, as well as those linked to plant nutrition, i.e. litter decomposing and soil fungi. Single species associations, such as plant pathogens, are not addressed in this context, and the specialized wood decay communities are excluded, as well as the flower microbiomes, which are mostly relevant to reproduction biology (Aleklett et al. 2014). Discussion of fungal groups in the habitats living leaf, litter, roots and soil, is preceded by a brief circumscription of the term “meta’omics” and an overview on the current methodological challenges.
A brief history of the term “meta’omics”
Terminology in ‘omics’ disciplines dates back to 1920, when Hans Winkler introduced the term genome (Winkler 1920). Etymology of the term is disputable, but most likely Hans Winkler combined the ‘gene’ with the suffix ‘-ome’ to describe the entirety of genes in an organism, in line with by then already introduced terms such as ‘rhizome’, which defines the entirety of roots (Lederberg and McCray 2001). Because he could not have been aware of the non-coding DNA elements, the current understanding of the meaning of ‘genome’ slightly differs, referring to “the complete genetic makeup of an organism” (Yadav 2007). The term ‘genomics’ was coined in 1986, as title for a journal launched in 1987 (Kuska 1998). In the following, the suffix ‘omics’ has been taken over by several disciplines, such as transcriptomics, proteomics and metabolomics (e.g., Abbot 1999; Joyce and Palsson 2006). Handelsman et al. (1998) introduced the term ‘metagenome’ of soil for the “collective genomes of soil microflora” and later defined ‘metagenomics’ as “the culture-independent genomic analysis of microbial communities” (Schloss and Handelsman 2003). This was again followed by emergence of the terms ‘metatranscriptomics’, ‘metaproteomics’ and ‘metametabolomics’ (Schneider and Riedel 2010; Zengler and Palsson 2012; Jones et al. 2014). While each of these terms is supposed to depict culture-independent analyses of microbial communities, in analogy to ‘metagenomics’, some variants have been proposed: In distinction of ‘metaproteomics’, ‘community proteomics’ was suggested to be used for the analysis of less complex communities while ‘environmental proteomics’ was suggested as an umbrella term (VerBerkmoes et al. 2009; Schneider and Riedel 2010). Differentiation of approaches based on the complexity of the analyzed communities is, however, vague. In particular the rate of proteins assignable to taxa, which was suggested as distinctive feature, heavily depends on the ever increasing volume of reference data and is therefore certainly not stable through time. Jones et al. (2014) suggested using ‘community metabolomics’ for the analyses of metabolites from an entire community in a given sample. This term was presumably only introduced to avoid the rather clumsy term ‘metametabolomics’, which would, in analogy to ‘metagenomics’, perfectly fit their circumscription. A debatable exception may be approaches not primarily targeting the organisms, but their substrates, i.e. decaying organic material. In biological and ecological contexts, such studies usually analyze the effect of decomposer community activities on shifts in the chemical composition of the substrates (e.g., de Marco et al. 2012). The analyzed samples include organisms inseparable from the substrate, and thus their metametabolome. The fact that chemical composition of the substrate is revealed by the analyses in addition does not make a specific term for delimitation of such approaches indispensable. After all, decaying organic matter may, in the widest sense of nutrient cycling, be regarded as kind of metabolome at the ecosystem scale. Refining the terminology for culture-independent analyses of organismic communities is therefore not necessarily needed and I recommend to avoid confusion and preserve interdisciplinary compatibility by using ‘metagenomics’, ‘metatranscriptomics’, ‘metaproteomics’ and ‘metametabolomics’ in their original, straightforward circumscription. As an umbrella term for these approaches, the term ‘metaomics’ (e.g., Worden and Allen 2010), also spelled as ‘meta-omics’ (e.g., Fritz et al. 2013) and ‘meta’omics’ (e.g., Segata et al. 2013), recently emerged. It is supposed to designate the study of organismic assemblages in the sense of Handelsman’s ‘metagenomics’ by any ‘omics’ discipline. Without knowledge of the historical background, ‘metaomics’ and also ‘meta-omics’ imply that multiple ‘omic’ approaches were applied to study the organism(s) of interest. Addressing studies of organismic assemblages by any ‘omic’ approach collectively as ‘meta’omics’ avoids such confusion.
Methodological challenges
A common challenge for most meta’omic approaches is extracting target molecules in high purity and quality from environmental samples, which is discussed in the first part of this section. Subsequently, a brief overview on the widely differing analytical workflows is given and the section concludes with addressing quantification, which is again an issue shared among all meta’omic disciplines.
Extraction of target molecules
A prerequisite for reliable results in meta’omics is certainly the reproducible extraction of the molecules of interest from environmental samples. Considerable progress has been made in the purification of nucleic acids and proteins in the past years (Tan and Yiap 2009) and several options are available for the analysis of fungal consortia in living hosts. Suitability of the plenty of protocols and commercial kits available for nucleic acid (e.g., Drábková et al. 2002; Fredricks et al. 2005; Kennedy et al. 2014; Mertens et al. 2014) and protein (e.g., Wright et al. 2014; Wu et al. 2014; Zheng et al. 2007) extraction differs among sample types. Studies of decaying tissues and soil have to cope with absorptive molecules and minerals (e.g., Keiblinger et al. 2012; Sagova-Mareckova et al. 2008). This is especially challenging for the extraction of nucleic acids. The currently available commercial kits only yield reproducible results for one or few certain soil types (Dineen et al. 2010). The variability in composition and concentration of humic substances among different soils and organic layers is a major challenge for comparative studies (Peršoh et al. 2008). Considering that humic substances may irreversibly bind to nucleic acids (Crecchio and Stotzky 1998), it is essential to remove or inactivate these prior to cell disruption. Posterior purification also removes the bound nucleic acids, and it is currently unknown whether or not the reaction between humic substances and nucleic acids is random. Among the tested pretreatments, flocculation of humic substances with Al2(SO4)3 currently yields the highest purity of extracted nucleic acids (Peršoh et al. 2008). Another, less laborious option is the addition of competitor DNA (Paulin et al. 2013). Because the added DNA will at least partially remain in the extract, this procedure is most suitable if selective primers are used for amplification, or if DNA is subsequently digested for RNA analyzes. A major advantage of the approach is that competitor DNA occupies all potential absorption sites for the targeted DNA or RNA, i.e. those of humic substances and minerals (Paulin et al. 2013). This allows for removing humic substances subsequent to cell disruption, when time is no longer critical, because all fractions and chemicals, including RNAse inhibitors, are homogeneously mixed.
Compared to nucleic acids a universal protocol for the quantitative extraction of proteins from soil samples seems still further away (Taylor and Williams 2010). Spiking experiments currently only yield satisfactory recovery rates from relatively simple matrices, such as pure sand. About half of the reference material is recovered from soils with higher organic and clay contents (Keiblinger et al. 2012, and references therein). From marine biofilms, three different extraction protocols yielded largely different protein compositions (Leary et al. 2013). Metaproteomics is therefore still in urgent need of protocols yielding reproducible results within and especially among varying matrices (VerBerkmoes et al. 2009; Schneider and Riedel 2010).
Sample preparation for metabolite extraction requires immediate freezing and lyophilization to avoid post-sampling modification of metabolites (Sardans et al. 2011). Individual extraction protocols have to be developed for each sample type and metabolite composition and different solvents are required to obtain polar and non-polar metabolites, respectively (Lankadurai et al. 2013). While detailed extraction protocols are available for plant tissues (Kaiser et al. 2009; Kim and Verpoorte 2010), metabolites from soil and litter have so far only been extracted using a straightforward standard protocol (Jones et al. 2014). In contrast to solution NMR, powderized samples may be directly applied to solid state NMR (Preston et al. 1990; Preston 2014). This approach is therefore not prone to extraction biases.
Sample analyzes
Downstream analyses of the different extracts from environmental samples differ significantly. The broadest spectrum of biotechnological manipulations is available for the analyses of nucleic acid extracts. Analytical workflows targeting a limited number of genes usually commence with amplification of DNA or reverse transcribed RNA (cDNA) by Polymerase Chain Reactions (PCRs), i.e. with environmental PCR. The obtained amplicons may then be analyzed as a whole (‘community fingerprint’), separated by subcloning, or directly sequenced in parallel. A priori anonymous metagenomic fingerprints group all organisms which correspond in the profiling criterion, such as sequence length in ARISA signatures (Popa et al. 2009). Fingerprinting approaches provide limited insights into the structure of the community structure, but they may straightforwardly reveal compositional differences in microbial consortia for monitoring or prescreening purposes (Weig et al. 2013). Subcloning of genes or amplicons allows for their proliferation and separation (e.g., Taylor et al. 2007). The resulting clone libraries are, after sequence analysis, still available for additional analyses, such as expression in transformant model organisms (e.g., Gatte-Picchi et al. 2014). In metagenomic biodiversity studies, clone libraries are mostly constructed to separate amplicons of barcoding genes among different organisms (e.g., Timling et al. 2014). Fingerprint methods, such as RFLP, are then usually applied to differentiate between genotypes, before representatives are sequenced for taxonomic assignment (Oros-Sichler et al. 2007). Parallel sequencing of multiple sequences by Next Generation Sequencing (NGS) technologies makes previous separation of genotypes dispensable, because thousands to billions of individual sequences may be obtained from single or multiple, differentially labeled, nucleic acid extracts (Logares et al. 2012; Kemler et al. 2013; Schmidt et al. 2013; Soon et al. 2013). The high sequencing depths achievable due to the ever increasing capacity of NGS devices significantly improves explanatory power in ecological contexts (Smith and Peay 2014). The possibility of multiplexing hundreds of different samples in a single sequencing run (Hamady et al. 2008) even renders NGS technologies more cost-efficient than PhyloChip microarrays (Cao et al. 2013).
A major challenge for all sequence-based meta’omic studies (including metaproteomics, see below) is linking biological information to sequence data via their taxonomic assignment. Using publicly available reference databases and software pipelines such as CloVR-ITS (White et al. 2013), QIIME (Caporaso et al. 2010) or SEED (Větrovský and Baldrian 2013), the obtained sequences may be assigned to taxa, thus linking a plethora of taxon-related biological and ecological information (Kõljalg et al. 2013). Reliability of the taxonomic assignment and thus the related information may be judged from scores on sequence similarities and alignment coverage by quality criteria (Peršoh et al. 2010) or phylogenetic analyses (Begerow et al. 2010). A mostly considerable proportion of the sequences will remain unidentified or only identifiable to higher taxonomic levels (e.g. ‘Ascomycota sp.’). Since biological and ecological traits may already differ significantly among congeneric taxa (e.g., Crous 2009; Bensch et al. 2012), contributions of such assignments to understanding the principles underlying community structure are often limited (Bálint et al. 2014). The impact of host plant identity on endophytic community composition was clearly higher when analyzed on the basis of operational taxonomic units (OTUs), than when analyzed on the basis of taxa, to which the OTUs were assigned (Peršoh et al. 2010). The authors concluded that sequence-similarity-based OTUs were more likely to represent meaningful biological (i.e. reproductive) units, than the groups emerging from taxonomic assignment. Statistical analyses based on exclusively sequence-similarity based OTUs therefore minimize the risk of biases due to artificial groupings. Species which are artificially split to different OTUs due to too stringent similarity thresholds do usually not compromise the results, because they are easily recognized as very similar sequences with similar ecological preferences and/or distribution patterns (Peršoh 2013). Reproducibility of taxon-name-based statistics is, on the other hand, susceptible to impairment by (i) a rapidly growing, but still limited taxon coverage in the reference databases, (ii) taxonomic misassignments of reference sequences (Bridge et al. 2003; Vilgalys 2003; Nilsson et al. 2006), and (iii) limitations of resolving all taxa by a single barcoding gene (Schoch et al. 2012). The latter issue is mostly caused by an insufficient barcoding gap (i.e., intra- vs. interspecific sequence variability) in certain taxa. It has also to be considered if biological and ecological information is directly deduced from contextual data or metadata accompanying environmental sequence data (Peršoh and Rambold 2012), because representatives of different species with identical barcoding sequences, but different ecology, may conceal existing patterns.
Functional microarrays provide a valuable tool to assess functional diversity in complex environmental samples, by targeting specific genes or transcripts (Roh et al. 2010; He et al. 2012; Nikolaki and Tsiamis 2013). They are particularly suitable for monitoring projects, because the approach targets known genes and may not provide de novo sequence information, even though genes from unknown organisms may be detected by targeting conserved regions (Peršoh et al. 2012). Sequencing of genomes and transcriptomes in environmental samples becomes more and more feasible due to consistently decreasing costs and increasing capacities of NGS technologies (Segata et al. 2013; Buermans and Dunnen 2014). Because such non-targeted approaches are realizable without extensive amplification, they are also less prone to (mostly quantitative) biases (Eisen 2007; Simon and Daniel 2011; Lewin et al. 2013). A major challenge is the assembly of the numerous sequence reads from complex communities (Wooley et al. 2010). This error-prone step may be circumvented by directly mapping the (trimmed) raw reads of metagenomic and metatranscriptomic sequences against reference databases (Davenport and Tümmler 2013).
Advances in metaproteomics are largely coupled to those in sequencing technologies and mass spectroscopy (MS, VerBerkmoes et al. 2009), because the analysis of complex protein extracts is not feasible by de novo assessment of amino acid sequences using Edman Degradation (Edman and Begg 1967) and MS-based de novo sequencing (Seidler et al. 2010) has not yet reached maturity (Schneider and Riedel 2010; Seifert et al. 2013). Instead, reference spectra are generated by in silico transcription of gene sequences and subsequent in silico simulation of digestion patterns of the putative proteins (VerBerkmoes et al. 2009). Analyses of extracts usually begin with separation of the purified proteins according to their isoelectric points and sizes by 2-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (2-D SDS-PAGE), and is followed by proteolytic degradation of the proteins. Alternatively, degradation may be accomplished after 1D SDS-PAGE and the peptides separated by one to multi-dimensional liquid chromatography (LC). Finally, the peptides are ionized and analyzed by MS. Resulting spectra are compared to the references generated in silico (Graham et al. 2011; Becher et al. 2013). These developments enable high-throughput analyses of metaproteomes from even complex environmental samples (e.g., Schulze et al. 2005; Wilmes et al. 2008; Wu et al. 2011, Kolmeder et al. 2012). However, data evaluation strongly depends on the availability of reference genomes/transcriptomes and while the reference databases are rapidly growing, huge amounts of data push the computationally demanding analyses to their limits. This development was suggested to be encountered by compiling environment-specific reference datasets or complementing metaproteomic assessments by metagenomic sequencing (Becher et al. 2013).
Metabolomics also greatly benefitted from the improvement of MS technologies, in particular with regard to the detection of novel rare metabolites (Pan and Raftery 2007; Ban et al. 2012). Mass spectroscopy usually requires time-consuming pretreatments, such as capillary electrophoresis (CE), gas chromatography (GC) or LC (Robertson 2005), which are suitable for single holobionts (Hernández et al. 2009; Walker et al. 2014a), but not particularly compatible with high-throughput analyses of environmental samples. Nuclear magnetic resonance (NMR) technologies require less laborious preparation of samples than MS approaches (Lankadurai et al. 2013) and are therefore preferably used in metametabolomic studies (Jones et al. 2014). Among the various NMR systems available for analyzing solutes, high-end devices may even compensate for the generally lower sensitivity as compared to MS, why affordability of such high-end instrumentation is certainly the major issue currently limiting the number of metametabolomic environmental studies (Lankadurai et al. 2013). Solid-state 13C NMR reveals types of carbon bonds in dried and fine ground samples, which provides detailed insights into the chemical composition of environmental samples (Preston 2014). It is particularly suitable to track chemical shifts in organic material during biological degradation (Preston et al. 2009; Ono et al. 2011; de Marco et al. 2012; Wallenstein et al. 2013; Osono et al. 2014). The method is already well established, with the minor shortcoming that it does not identify chemical compounds, but carbon bonds which partly co-occur in different molecules, thus complicating quantification of specific molecules (Berg and McClaugherty 2014).
Quantification
Quantification of meta’omics data primarily depends on the quantifiable and reproducible extraction of the respective molecules from the environmental samples, as discussed above. The next big challenge is quantifying the molecules in the extracts. This applies in particular for metagenome analyses based on amplified barcoding markers, due to the extensive manipulations applied during preparation of the template (library) for sequencing (Lindahl et al. 2013). Only direct, non-targeted shotgun sequencing of environmental metagenomes circumvents at least the PCR-associated biases (Eisen 2007; Simon and Daniel 2011; Lewin et al. 2013). Metagenomics reveal the genetic capacity of a community, while transcription levels and post-transcriptional modifications, are usually not predictable from the obtained sequences (Moran 2009; Simon and Daniel 2011). If a gene is present, the community has the potential to express this gene, but quantities of the product may not directly dependent on gene abundance. The presence of a certain gene therefore seems more important than its actual abundance and quantification is of minor relevance. Compositional comparisons of microbial communities in environmental samples are, however, usually based on taxon abundances inferred from quantities of barcoding genes (Lindahl et al. 2013), even though it has been shown that the detected abundances of amplicons do not necessarily reflect biological abundances (Amend et al. 2010; Baldrian et al. 2013). Amplification rates during PCR-reactions are particularly prone to quantitative biases caused by primer selectivity and binding kinetics, as well as by differences in secondary structure, length, composition and nucleotide sequence of the amplicons (Polz and Cavanaugh 1998; Bellemain et al. 2010; Engelbrektson et al. 2010; Toju et al. 2012; Werner et al. 2012; Huang et al. 2014; U’Ren et al. 2014). Amplicon abundance, therefore, does not necessarily reflect the corresponding genome abundance. The ratio may be calibrated against internal standards, i.e. quantified pure DNA from the taxa of interest, may allow inference of genome abundances in metagenomic extracts. Inference of biological abundance (e.g., biomass) from amplicon abundance, again, requires laborious experiments including the corresponding reference strains (Raidl et al. 2005; Tellenbach et al. 2010). Despite these limitations of quantifying absolute abundances within a single sample, comparative analyses of relative abundances among metagenomic extracts are largely reliable if similar samples, including similar taxonomic diversity and similar concentration and composition of co-extracted contaminants, are compared (Amend et al. 2010; Davey et al. 2013a). This was supported by the finding that randomization of OTU-abundances within samples had only negligible impact on the results on community level when equally applied across similar samples (Peršoh 2013).
Metatranscriptomic studies are usually interested in the actual activity of microbial communities (Baldrian and López-Mondéjar 2014). This makes quantitative data very important and the short-lived nature of mRNA requires special care during sample preparation. In addition to differences in properties among samples, as discussed above, processing time may be a crucial factor. Half-life times range from less than 10 to more than 60 min for different mRNA species (Geisberg et al. 2014). The mRNA profile has therefore to be stabilized immediately after sampling. Delays and inconsistencies in processing time are likely to bias the results, but their impact on expression profiles in environmental communities has not been studied in detail so far. Quantitative biases introduced during downstream analyses by enzymatic reactions are circumvented by microarray experiments using enzyme-free preparation of labelled templates from RNA extracts (Peršoh et al. 2012). Accounting for technical variance by co-spotted references (Liang et al. 2010) or multiple probe replications (Pozhitkov et al. 2014) even allows for absolute quantification of the signals if the probes are calibrated. This is already challenging for a limited number of known genomes (Pozhitkov et al. 2014), why thorough probe calibration appears currently not achievable for microarray experiments including extracts from environmental samples with unknown composition. Normalization based on overall signal intensities is not advisable for microarrays targeting a subset of the transcripts in environmental samples. A reasonable measure to compare data from similar samples is the ratio of signals from functional probes to those from universal RNA probes, as it reflects expression of a certain gene against the whole community present in the sample (Peršoh et al. 2012). Such an approach is also feasible for de novo generated sequence data by NGS (Wang et al. 2009), in particular if RNA fractions are not, moderately, or at least reproducibly enriched during library preparation (Urich et al. 2008; Simon and Daniel 2011). It is, however, highly recommended to use internal standards for quantitative metatranscriptomics (Satinsky et al. 2013).
Metaproteomics may straightforwardly quantify proteins or peptides by analyzing spot intensities on gels (Seifert et al. 2013). For gel-free LC approaches, tagging the proteins has been suggested to quantitatively compare samples (Thompson et al. 2003; Ross et al. 2004), but also signal intensities or counts of spectra may be used as measures for relative abundances (Nahnsen et al. 2013; Pan and Banfield 2014). The normalized spectral abundance factor (NSAF) is most commonly applied to compare label-free experiments, accounting for protein lengths during normalization (Paoletti et al. 2006). In a study on decomposition processes, these NSAF of cellulases and xylanases, however, did not correlate to the actual independently measured enzyme activities (Schneider et al. 2012). Complementary analyses by independent approaches therefore appear currently advisable to infer ecological processes from metaproteomic data. Metabolic activity of organisms may also directly be assessed by quantifying the incorporation of stable isotopes from labelled substrates into the respective proteins (von Bergen et al. 2013).
Quantification in non-targeted metametabolomic analyses exclusively depends on the extraction method, because MS techniques are largely, and solvent NMR techniques fully quantitative, using an internal standard (Lin et al. 2006; Lankadurai et al. 2013). For solid state NMR, quantification of carbon bonds in complex environmental samples is an issue, which may, however, be compensated by multiple measurements applying different settings (Knicker 2011; Preston 2014).
Factors shaping plant-associated fungal communities
This section discusses the insights meta’omic studies have provided into functional diversity of plant-associated fungal communities. Predictability of fungal community composition from host plant identity and composition of the plant community is one of the focal points. The impact of distance decay, i.e. increasing dissimilarity between fungal communities with spatial distance, is also addressed. The potential drivers of community composition are discussed against the background of the functional role of the fungi in their respective habitats and the temporal dynamics in the communities. Following the communities associated with living leaves, those in decaying leaves (i.e. the soil-covering organic layers) are addressed. Subsequently, root-associated communities are discussed and finally saprotrophic soil fungi.
Phyllosphere communities
Cultivation-based studies revealed an immense diversity of fungi colonizing the plant phyllosphere (for reviews see e.g., Hawksworth 2001; Arnold 2007; Sieber 2007; Rodriguez et al. 2009; Porras-Alfaro and Bayman 2011; Suryanarayanan 2011; Unterseher 2011), but already the first application of next-generation sequencing (NGS) indicated that these estimates may be even too low (Jumpponen and Jones 2009; 2010). Variability of fungal communities in the phyllosphere of a given host actually exceeded variation among host individuals (Cordier et al. 2012a). The high diversity was confirmed by all studies focusing either on the phyllosphere communities as a whole, or only on the endophytic part (see below), while epiphyllous fungi have only been addressed specifically in studies of sooty mould symptoms. Composition of those conspicuous epiphytic communities seems to differ fundamentally between temperate and warmer climates (Chomnunti et al. 2014): culture-derived sequences indicate them to be composed of few taxa in Germany (Flessa et al. 2012), while an unexpectedly high diversity was revealed by NGS in New Zealand (Dhami et al. 2013). Composition of sooty mould communities is generally independent of the host taxon identity. In warm climates, it obviously depends on the identity of the sap-sucking insects excreting honeydew, the nutritional source of the sooty moulds (Dhami et al. 2013). The counterparts from temperate regions are obviously rather uniformly composed (Chomnunti et al. 2014). Exceptions are those living directly on plant exudates, such as the Capnocheirides rhododendri-dominated community of Rhododendron ferrugineum. Flessa and Rambold (2013) showed that growth of the epiphyte C. rhododendri was indeed restricted to the leaf surface, but they also found many epiphyllous fungi co-occurring within the leaves. This is consistent with the two most common taxa of European sooty mould communities, i.e. Aureobasidium and Cladosporium (Flessa et al. 2012), often also dominating endophytic fungal communities (e.g., Jumpponen and Jones 2010; Wearn et al. 2012). Because different genotypes, at least of Cladosporium, showed different preferences for the leaf interior and exterior (Flessa and Rambold 2013), conspecificity of the respective genotypes would have to be established to confirm that they are actually predominantly epiphytic fungi invading the plant tissues only occasionally. Linking the dynamics of epi- and endophytic communities by concerted assessment of both communities in future studies may actually provide a key to understanding distribution patterns of the fungal community in the phyllosphere.
Factors shaping endophytic community structure were only recently addressed by metagenomic studies. Host plant identity clearly separated the endophytic communities in mistletoes from those of their pine hosts (Peršoh 2013). Comparing angiosperm and gymnosperm host trees based on ARISA-profiling, the impact of the host was even more striking (Weig et al. 2013): Host plant identity explained mostly more than 80 % of the differences between host species of the two groups at the mixed forest site. While the angiosperm species also hosted different endophytic fungal communities, the two analyzed pinacean species were indistinguishable in this regard. The communities in Rhododendron were also similar among host species (Raizen 2013) and eleven tropical grass species hosted similar endophyte spectra (Higgins et al. 2014). Endophytic communities in four moss species in southern Norway largely overlapped in composition, but were still clearly distinct (Davey et al. 2013b). Below species level, different genotypes of Fagus sylvatica, Populus angustifolia and P. balsamifera were shown to host significantly different fungal communities (Cordier et al. 2012a; Bálint et al. 2013; Lamit et al. 2014). The impact of phylogenetic and genotypic affiliation of the host on composition of endophytic communities is therefore not consistent among host taxa, i.e. endophytic communities may differ among genotypes of a certain host species, but may be indistinguishable within other host genera or even families.
Fungal spectra isolated from different above-ground organs and tissues of the same host species usually clearly differed (see Peršoh et al. 2010; Moricca et al. 2012; Sun et al. 2012; Tateno et al. 2014, and references therein), but cultivation-independent studies addressing within host distribution of endophytes are scarce. A single pyrosequencing approach partly confirmed the cultivation-based findings, by showing that stems and leaves of pine trees hosted different fungal communities (Peršoh 2013). Those in the corresponding organs of mistletoes, however, were indistinguishable. The discrepancy was hypothesized to be due to the presence of stomata on leaves and stems of mistletoes, which allows for invasion of both organs by the same infection pathway. The cultivatable endophyllous fungal community has also been shown to depend on leaf exposure (Unterseher et al. 2007), but cultivation-independent evidence on exposure-effects is restricted to bark-colonizing communities (Beck et al. 2014). The authors addressed a mixture of rather complex communities, including potentially epi-, endophloedic and lichen-associated fungi of a corticolous lichen community. They revealed that the seasonal development of this assemblage differed according to exposure, but the heterogeneity of the sampled community complicated a final discussion. To study the effect of stressors on endophytic communities, however, exposure-differences are in principal promising, because restriction to a single host individual standardizes the impacts of host genotype, ecotype and microbiome.
A relocation experiment of P. balsamifera from their natural southern to their northern distribution limit revealed significantly different endophyte communities between locations (Bálint et al. 2014). The difference was primarily explained by the escape from pathogenic fungi (i.e. enemy release) due to the abrupt relocation, and hypothesized to be less pronounced in slow naturally shifting host populations. Fungal phyllosphere communities were highly similar in beech trees (Fagus sylvatica) in the Alps and the Vosges, but differed largely from trees sampled in the Pyrenees (Coince et al. 2014). Geographical distance therefore at least not inevitably causes differences in the phyllosphere communities of naturally grown trees, but a conclusive discussion of the contradictory results was hampered by the fact that a methodological bias could not be excluded. Conspecific hosts (i.e. Pinus sylvestris and Viscum album) from geographically distinct, but ecologically similar sites hosted quite similar endophyte communities (Peršoh 2013). Such a low effect of spatial distance at regional scales is pre-conditional for studies focusing on regional and local factors varying between geographically distinct sites (e.g., Jumpponen and Jones 2009; 2010; Zimmerman and Vitousek 2012). The studies by Jumpponen and Jones (2009; 2010) revealed a significant difference of phyllosphere communities between urban and nonurban environments. While autocorrelation of many factors due to land use effects hampered ascertainment of the cause, the authors suggested differences in stand size, fertilization, litter removal, and nutrient and pollutant concentrations, as possible agents. Environmental conditions also shaped endophytic fungal communities across a Hawaiian landscape, with rainfall and temperature having major impacts (Zimmerman and Vitousek 2012). The latter was caused by altitudinal differences, which have also been shown to influence fungal endophyte and phyllosphere communities of mosses and beech, respectively (Davey et al. 2013a; Cordier et al. 2012b; Coince et al. 2014). The authors concertedly argued that altitudinal differences are not necessarily caused by differences in temperature alone, but studies separately targeting correlated factors, such as radiation, diurnal temperature variations and functional plant traits, have not been published so far (see Abbate and Antonovics 2014).
Community composition of phyllosphere fungi changed significantly throughout the years course in Quercus macrocarpa (Jumpponen and Jones 2010), Quercus ilex (Peñuelas et al. 2012) and Fagus crenata (Tateno et al. 2014), as well as in two out of three analyzed moss species (Davey et al. 2013b). Temporal shifts became already apparent within timeframes of one month and two weeks, respectively (Cordier et al. 2012b; Peršoh 2013). Turnover of fungal taxa and not their accumulation caused the seasonal changes in bryophyte associated fungal communities (Davey et al. 2012). The instability of the endophytic community structure demonstrated by these studies implies that the influence of a certain factor on endophytic fungal communities may only be conclusively discussed against the background of seasonal dynamics. Jumpponen and Jones (2010) recovered most of the fungal taxa from the previous year by regular sampling throughout the two growing seasons, while differences between subsequent years were rather pronounced in the beech phyllosphere (Cordier et al. 2012b). Significant interannual variation of cultivatable fungi was also reported in tropical grasses (Higgins et al. 2014), providing additional evidence that endophytic fungal communities may not follow solely cyclic developmental courses across years.
The vertically transmitted Clavicipitaceae endophytes of grasses complete their whole life cycle within host plants (see Tanaka et al. 2012). The vast majority of the horizontally transmitted endophytes are known from other substrates, indicating that the phyllosphere is only one habitat in their life cycle (Peršoh 2013). Growing evidence suggests that some endophytic fungi become saprotrophic decomposers after leaf fall, and inhabit living leaves as dormant saprobes (see Suryanarayanan 2013 and references therein). Others may be beneficial for the host plant due to their antibiotic (Kumar and Kaushik 2012) or antioxidant activity (Hamilton et al. 2012) or by increasing stress tolerance or host fitness in general (White et al. 2014). Some endophytes other than Clavicipitaceae may also be transmitted vertically (Hodgson et al. 2014; Tello et al. 2014) and therefore presumably intimately interact with their hosts, but the nature of these interactions remains speculative. Functional meta’omics approaches have so far not been applied to the phyllosphere microbiome, with the exception of epiphyllous bacteria (Delmotte et al. 2009). A major methodological challenge is certainly the dominance of plant material in leaf extracts, which complicates non-targeted environmental sequencing approaches. Functional microarrays target specific genes and may thus may provide an alternative to gain deeper insights into the ecological roles of phyllosphere fungi in the future.
Litter decomposers
Organic layers of decaying plant material cover the actual mineral soil in almost all ecosystems. The fungal community in decaying beech leaf litter was shown to be similar to that in living leaves when assessed by RFLP-analysis (Peršoh et al. 2013), indicating that the composition of the initial fungal decomposer community may, as the endophytic community, depend on the plant community. Next Generation Sequencing data revealed a rapid turnover in the dominant taxa after the abscission of oak leaves, but half of the dominant phyllosphere taxa persisted in the litterbags for considerable time (Voříšková and Baldrian 2013). Basidiomyceteous yeasts, which were also present in the different compositional stages of the beech litter, were still abundant in the oak litter after eight months of incubation. Cultivation-based studies indicate that phyllosphere yeasts show low host preference (Fonseca and Inácio 2006). Their co-occurrence in living and decaying leaves is therefore unlikely to cause major differences between litter types. Distribution patterns of these yeasts should, however, be interpreted with caution, because species identification is rather challenging in these groups and the most ‘ubiquitous species’ have indeed been shown to be phylogenetically rather heterogeneous (Fonseca and Inácio 2006). To conclusively discuss the role of previously endophytic fungi in the formation of potential host plant- or community-specific decomposer communities, their full life cycle has to be considered, by analyzing phyllosphere and pedosphere communities of different hosts concertedly (Peršoh 2013).
Significantly different fungal communities were found in decomposing leaves of different host species, when litterbags were incubated beneath pure stands of the respective host tree (Kubartová et al. 2009). Because the litter was previously sterilized (Kubartová, pers. comm.), the finding supports the idea that the soil-borne decomposer communities are adapted to degrading the litter they mostly encounter, i.e. to that from the tree species above (Strickland et al. 2009). Due to this ‘home field advantage’ (Gholz et al. 2000; Prescott and Grayston 2013), litter is decomposed on average 8 % faster by native than by exotic decomposer communities (Ayres et al. 2009). Distinct decomposer communities developed in bags with litter of Populus tremuloides and Picea mariana, respectively, at a mixed forest stand (Treseder et al. 2014). The communities in the two litter types only differed from the second year on, which indicates that the differences may be ascribed to an early, but not to the initial community of soil-inhabiting decomposers. Consistent with leaf chemistry, lignocellulolytic fungi preferred spruce, while those capable of using tannin-protein complexes were preferably found in the aspen litter. Feinstein and Blackwood (2013) found the fungal communities in litter collected from mixed deciduous forests to differ only at four out of six sites between American Beech (Fagus grandifolia) and Sugar Maple (Acer saccharum), and only marginally. The saprotrophic basidiomycete community was not structured by the vegetation type in the litter of three different forest ecosystems in North America (Edwards and Zak 2010). Host species identity, again, significantly shaped the fungal community in forest litter in the Czech Republic (Urbanová et al. 2015). More than one third of the fungi in the organic layers significantly preferred litter of one of the of seven analyzed tree species, which could not be explained by litter chemistry. Bray et al. (2012) used common garden experiments to exclude the impact of an adapted local microbial community. Their study found a higher impact of the chemical composition of the litter (i.e. litter quality) than of host identity on the decomposer community. The efficiency of non-adapted artificial communities in litter decomposition thereby seems to depend more on their phylogenetic distinctiveness than on species richness (Kivlin and Treseder 2014). Nutrient content and C:N:P ratios of the litter shaped the colonizing fungal community in European Beech along a natural gradients (Schneider et al. 2012) and across forest management practices (Purahong et al. 2015). Fertilization experiments in tropical forests also revealed that nutrient availability significantly affects community structure of litter decomposers (Kerekes et al. 2013). These studies show that litter chemistry strongly shapes fungal decomposer communities, at least in the litter of the same plant species. Composition of the vegetation cover has a major impact as well, but it may not be conclusively discussed to what degree plant identity directly influences the decomposer community, and which part is ascribable to differences in litter quality between plant species. The decomposer communities appear to be structured at small spatial scales, in addition, the underlying mechanisms of which remain unclear (Feinstein and Blackwood 2013). Forestry significantly affected composition and succession of fungal communities in litter, but causal connections could not be detailed due to the multitude of usually concertedly applied measures (Purahong et al. 2015). Disentangling the effects of the various factors shaping natural decomposer communities appears particularly important, because the same litter is degraded differently by different communities (Wallenstein et al. 2013). Composition of the decomposer community may therefore affect the chemical composition of organic matter entering the soil, which again is of outermost importance for ecosystem functioning (Wickings et al. 2012).
Assessing temporal shifts in litter decomposer communities is challenging, because seasonal and successional shifts both occur along the time scale. Successional shifts in decaying pine needles were recently confirmed by coinciding results from cultivation-based and cultivation-independent approaches (Haňáčková et al. 2015). Enzyme assays and measurements of leaf chemistry in litterbags in an oak forest were mostly in accordance with the widely accepted three-stage model of litter decomposition (see below), but composition of the communities changed not only between, but also during the three stages (Voříšková and Baldrian 2013). This indicates that that the sequential change in substrate quality was not the only factor shaping the community. The authors reported seasonal patterns of several enzyme activities earlier, some of which were strikingly similar between different communities in different litter decomposition stages, i.e. in the litter and organic layers (Voříšková et al. 2013). This is in accordance with Andreetta et al. (2012) reporting seasonal differences in urease and phosphatase activity in the organic layer of a Mediterranean oak forest. Fungal community composition differed more between autumn and spring within the Oh layer, than between the Oh layer and the underlying mineralic E horizon in autumn. Seasonality may therefore overlay succession in litter decomposing fungal community dynamics.
Taxonomic diversity patterns in the temperate oak litter revealed by cellobiohydrolase genes (chbI) were neither accordable to the patterns arising from the ITS barcoding gene, nor to the cellobiohydrolase activities measured using enzyme assays (Voříšková and Baldrian 2013). The authors considered the inconsistencies between the whole and the chbI-possessing fungal community as indication for a proportional shift of cellulolytic fungi during decomposition. Differences in relative chbI abundance and enzyme activities were supposed to result from specific fungi having specific requirements to become active. However, discrepancies between targeted (gene amplification) and non-targeted (enzyme assays) approach may also result from the activity of organisms which were not targeted, such as bacteria (see Štursová et al. 2012), and therefore only detected by one approach. In a (non-targeted) metaproteomic study on litter decomposition, Schneider et al. (2012) found all decomposition-related proteins to derive from fungi, and none from bacteria. Enzyme abundance and fungal community composition remained relatively constant between February and May at the four sampling sites, but the cellulases were produced by clearly different taxa at the two sampling events. This indicates that while being present throughout the study period, the prerequisites for activity of most taxa were only fulfilled in one of the two months. Differences between DNA and RNA barcoding gene profiles indicated that only part of the present fungal community was active in a Scottish moorland, which supports the hypothesis of unequal activity of the present taxa (Curlevski et al. 2011). Present (DNA-derived) and active (RNA-derived) communities in a spruce mountain forest were even more striking, with abundance of 18 % of the active fungal OTUs falling below the detection limit in the DNA pool (Baldrian et al. 2012). Of the chbI genes, 15 and 27 % were only detected as RNA and DNA, respectively. These data clearly demonstrate that only a part of the present fungal community is actively involved in decomposition at a given time, while even taxa representing a negligible proportion of the present community may significantly contribute to the decomposition process.
Metagenomic approaches consistently confirmed an increase in the relative abundance of Basidiomycota at the expense of Ascomycota with progressing decomposition. This succession is already detectable at time scales of weeks to months (Kuramae et al. 2013a; Schneider et al. 2012; Voříšková and Baldrian 2013), but still observable after years along vertical soil profiles (Lindahl et al. 2007). The taxonomic succession is usually accompanied by a functional shift from saprobic to mycorrhizal taxa. This amplifies the trend in habitats dominated by ectomycorrhizal symbioses (predominantly formed by Basidiomycota), but lessens it in environments of ericoid mycorrhizal symbioses, formed by Ascomycota (Clemmensen et al. 2013). Succession within the saprobic community is thought to be coupled to changes in the chemical composition of the plant litter, with the respective most easily degradable substances available being decomposed in three sequential stages: the loss of extractables is followed by degradation of hemicelluloses and cellulose, and finally the most ‘recalcitrant’ compounds, such as lignin are attacked (Berg and Staaf 1980, Moorhead and Sinsabaugh 2006; Šnajdr et al. 2011). Since the Basidiomycota are generally better equipped for degradation of lignin (e.g., Baldrian 2008; Lundell et al. 2010), succession within the saprobic community is directed from Ascomycota to Basidiomycota. However, major concern has been expressed against the categorization of certain litter compounds as ‘recalcitrant’ (Kleber 2010) and the concept of successive decomposition of organic matter compounds is challenged by analyses on residence times of organic matter compounds in soil (Schmidt et al. 2011; Gougoulias et al. 2014). These were shorter for compounds such as lignin than, e.g., for proteins and saccharides, which were so far supposed to be rapidly degraded. Application of NMR techniques in litter decomposition studies may also eventually change the concept of ‘recalcitrant’ lignin (Berg 2014). While the three-phase model of decomposition seems still appropriate in the light of the new data (de Marco et al. 2012), it was suggested to be refined and less focused on lignin (Preston et al. 2009). Actually, activities of all measured organic matter degrading enzymes decreased from the organic layer to the topsoil across North American soils, except for an increase in peroxidase by 9 % (Talbot et al. 2014). Activities of ligninolytic enzymes were even highest in the uppermost litter layer and decreased with proceeding composition (Voříšková et al. 2013). Successive degradation of the most easily degradable compounds, however, was found using litterbag experiments at the same location, with lignolytic activity becoming characteristic of the decomposition process only in the second year (Šnajdr et al. 2011; Voříšková and Baldrian 2013). It remains to be clarified if this discrepancy may be ascribed to interannual variability or a methodological bias associated with either the experimental or the descriptive approach. Transcripts related to lignin degradation were not detected during early decomposition of maize leaves (Kuramae et al. 2013a), but the experimental study was conducted under laboratory conditions and a comparison to natural conditions would be desirable to draw final conclusions, in particular because cellulolytic activity was also not detected. With the meta’omic studies partly supporting an early degradation of lignin and partly not, it seems essential to exclude methodological biases (e.g., Hatfield and Fukushima 2005) to ascertain if leaf litter decomposition generally follows the same course, or if fundamental differences exist.
The idea of an early attack of lignin seems not to fit to the well-established view of a succession from Ascomycota to Basidiomycota (see Frankland 1998), which is seems, at first view, to be also supported by the meta’omic studies discussed above. The reported successional shifts in community composition are, however, based on relative abundances. Considering fungal biomass per soil dry weight, density of Basidiomycota in oak forest soil was highest in the litter layer, i.e. in the uppermost organic layer (Voříšková et al. 2013). Absolute abundance of both Ascomycota and Basidiomycota decreased with progressing decomposition. The observed high density of potentially ligninolytic saprotrophic Basidiomycota in the uppermost layer is well accordable with the corresponding enzyme activities and an early attack of lignin. Biomass of ectomycorrhizal fungi also decreased with depth at the same site (Voříšková et al. 2013). A significant decline of ectomycorrhizal mycelium with depth, i.e. progressing decomposition, is already apparent from small-scale distribution patterns (Anderson et al. 2014). These findings indicate that a considerable fraction of the nutrients EcM fungi provide to the host plant may come from only recently fallen and not yet highly decomposed or even mineralized leaves of the host. While conclusive evidence for EcM fungi utilizing carbon from litter as energy source is still missing, their contribution to decomposition by releasing exoenzymes to exploit nitrogen sources is now undisputed (Ekblad et al. 2013). Genomic analyses revealed that EcM Bsidiomycota are reasonably well equipped to degrade structural plant compounds and lignin in particular (Kohler et al. 2015). Host plants provide energy to EcM fungi and lignin degradation generates little energy, at most (Schimel and Weintraub 2003). Occurrence of EcM fungi in freshly fallen leaves is therefore well accordable to an early attack of lignin. Peroxidase activity, which contributes to lignin degradation, was indeed significantly higher in litterbags enriched in EcM fungi by sand barriers as compared to communities of mostly saprotrophic fungi (Phillips et al. 2014). Significant correlations of proteases and peroxidases with abundance of EcM fungi in Californian pine forests further supports an involvement of EcM fungi in lignin degradation (Talbot et al. 2013) and peroxidase activity was also positively correlated to the ratio of EcM to saprotrophic fungi in pine forests across North America (Talbot et al. 2014). By linking cause (gene expression) and effect (chemical changes), meta’omic studies have the potential to elucidate the complex decomposition processes in detail. Enzyme efficiencies under environmental conditions, however, will have to be accounted for in future, because outside the cells, the released exoenzymes interact with the complex soil/litter matrix (Trumbore 2009; Wang et al. 2012). Assessing the impact of the exact chemical structure of heterogeneous macromolecules, such as lignin, on decomposition dynamics will be another challenge for future studies (Talbot et al. 2012).
Root-associated communities
Meta’omic studies and anonymous fingerprinting approaches usually assess the whole fungal community in root samples, including arbuscular mycorrhizal (AM) ectomycorrhizal (EcM), ericoid mycorrhizal (ErM), pathogenic and ‘endophytic’ fungi, as well as not further classifiable associated fungi. Following sequence-based taxonomic identification, the groups may be separated a posteriori. Specific primers only allow for selective amplification of AM fungi, which all belong to the phylogenetically distinct Glomeromycota (Krüger et al. 2009, Öpik et al. 2009). The patterns discussed in the following therefore apply for the heterogeneous assemblage of all root-associated fungi in general, if not specified otherwise.
Similarity among EcM communities was well explained, when phylogenetic relationships of the hosts were considered (Tedersoo et al. 2013). The phylogenetic relationships among herbaceous Asteraceae host plants in grasslands were also reflected by the root-colonizing fungal community (Wehner et al. 2014) and AM communities clearly differed between the distantly related hosts Lolium perenne and Trifolium repens across Ireland (Hazard et al. 2013). Root-associated fungi, including ErM fungi, differed between Ericacean host species in boreal forests (Bougoure et al. 2007; Ishida and Nordin 2010) and between Ericacean and grass hosts planted into the same soil in single-species mesocosm experiments (Kreyling et al. 2012). Host plant identity also had a strong effect on the fungal community associated with ectomycorrhizal roots in wet sclerophyll forests (Tedersoo et al. 2009). All these studies reveal a strong impact of host identity and/or phylogenetic affiliation on the root-associated fungal community structure, independent of the fungal community consisting of AM, EcM, or ErM fungi, or if all root-associated fungi were considered. Only three species of arctic Ericaceae shared highly similar fungal communities (Walker et al. 2011). This exception may be due to the specific circumstances, i.e. due to the plant community in the Arctic tundra being rather old and uniform, consisting of phylogenetically and spatially closely connected plants and being adapted to the extreme environment, as discussed in more detail below. The majority of root-associated fungi in temperate and subtropical forests have recently been shown to be shared among different host taxa in the same habitat (Toju et al. 2013a, 2014). Distance decay patterns, i.e. the decrease in similarity between communities with spatial distance, indicated that plant species share more fungi the closer they grow together (Toju et al. 2014). Fungal community composition in the roots of Calluna vulgaris changed along a vegetation gradient, with the Ericacean shrubs sharing the EcM symbionts of the trees at forest sites (Bougoure et al. 2007). In Ericaceae-dominated environments, again, ErM fungi regularly associate with non-Ericacean hosts (Chambers et al. 2008). These findings indicate that less abundant plant species predominantly associate with compatible fungi among the preferred associates of the dominant host plants, as suggested by the data of Toju et al. (2013b, 2014). Correlations with plant community have already been shown for EcM and AM communities (Edwards and Zak 2010; Kivlin et al. 2011; Wu et al. 2013). The root-associated fungal community in a certain plant seems to be partly structured by the host plant identity and partly by the whole plant community according to these results.
Öpik et al. (2009) showed that the AM community associated with generalist plants (occurring in various ecosystems) represented a subset of that associated with forest-specialized understory plants in the studied spruce forest. Non-native plants were supposed to recruit their EcM symbionts among compatible associates of native plants in an urban environment (Lothamer et al. 2014). These findings suggest that distinctiveness of the root-associated fungal community may depend on the adaption of the host to the local habitat; however, the underlying mechanisms may differ. Native fungal associates may not be available for introduced exotic plants due to dispersal limitation, i.e. the plant was transplanted outside the distribution areas of the usually associated fungi. The abovementioned understory plants of both specialization categories are native to the local forest habitat and therefore not spatially separated from their associated fungi. That generalist plants are more flexible in recruiting root-associated fungi than habitat-adapted specialists would be a hypothesis in line with the observation. Dispersal limitation has predominantly a significant impact on root-associated fungal communities during early succession (Dickie et al. 2013), i.e. at disturbed sites (Walker and Jones 2013) and recently established (Lekberg et al. 2007) or currently establishing plant communities (Williams et al. 2013). Ecosystems which are rich in plant species represent a fragmented habitat for host-selective root-associated fungi due to low host density. This causes a positive correlation of distance decay with host diversity, i.e. the spatial decrease in similarity between root-associated fungal communities correlates with plant diversity (Bahram et al. 2013). It is unlikely that dispersal limitation causes distance decay patterns in species-rich ecosystems. Abiotic factors (i.e. environmental filtering) and biotic factors (i.e. competitive exclusion) most likely prevent universal establishment of most fungi in these ecosystems (see Kraft et al. 2014), but the underlying mechanism will have to be studied in more detail. When distance decay is caused by dispersal limitation, the established communities reflect the different dispersal strategies of the fungi (Walker and Jones 2013). Distance decay may affect AM communities for a prolonged period of time during succession, due to their lack of efficient mechanisms for long-distance dispersal (Dickie et al. 2013). This may explain why geographical distance had still a major effect on AM community composition in agricultural fields, converted from native vegetation 10–15 years ago (Lekberg et al. 2007), while AM communities were not spatially structured in native plants across Ireland (Hazard et al. 2013).
Cultivation-independent analyses of presumably mostly EcM fungi in soil of a northern hardwood forest confirmed rather short turnover times for the fungal communities (Burke et al. 2011). These were already indicated by EcM-morphotype-based analyses to occur in the range of one month (Courty et al. 2008). The fungal community also changed clearly throughout the growing season of 2007 In roots of the EcM host genus Quercus (Jumpponen et al. 2010). A sampling campaign extended to additional sites in 2011, saw a less pronounced seasonality (Lothamer et al. 2014). These successional studies therefore indicate differences in community dynamics across years. In studies with a sufficient sampling period to address interannual changes, EcM community composition consistently differed significantly between years (Parrent and Vilgalys 2007; Courty et al. 2008). The AM community in a northern hardwood forest underwent no seasonal shifts throughout the growing season (Burke et al. 2011) and it was stable through time in a mixed forest in southeastern Estonia (Davison et al. 2012). The AM community in grasslands changed only negligibly in the warmer months, but was clearly distinct between summer and winter communities (Dumbrell et al. 2011). Metagenomic studies therefore indicate different dynamics for different types of mycorrhizal communities, the underlying mechanisms for which remain to be clarified.
The root-associated fungal community has been found to respond to soil acidity in almost all cultivation-independent studies so far. Soil texture (Wubet et al. 2012), soil type (Hazard et al. 2013), electric conductivity (Gryndler et al. 2010), content of humified organic matter (Gryndler et al. 2010), organic carbon (Corg) content (Wubet et al. 2012), nitrogen (N) content (Walker et al. 2014b, but see also Jumpponen et al. 2010), and C:N ratio (Wubet et al. 2012) also had an effect. All measured soil parameters mostly had a certain impact on the root-associated fungal communities, indicating that their composition is strongly shaped by abiotic factors. Notably, the EcM communities in Chinese subtropical forest neither responded to C:N ratio, pH, nor Corg (Wu et al. 2013). Temperatures (annual mean and range) were correlated with EcM and AM community composition across Chinese forests, while C:N ratios and mean annual precipitation only affected the EcM, but not the AM communities (Shi et al. 2014). Composition of the root-associated community correlated with mean annual temperature and pH along elevation gradients in France, but both factors were correlated with additional soil parameters, why a direct effect could not be ascertained (Coince et al. 2014). The AM community associated with potato roots in the Andes was quite conserved along elevational and environmental gradients (Senés-Guerrero and Schüßler 2015). Soil moisture and temperature were correlated to AM community composition on the global scale, but explained their composition only to a small degree (Kivlin et al. 2011). Relevance of the abiotic factors changed with season in a beech-maple forest (Burke et al. 2009): while soil pH and moisture were most important in June, phosphorous content had the major impact in September. The impact of abiotic factors is therefore best analyzed against seasonal community dynamics. Selections of soil parameters have been assessed concertedly with root-associated fungal community structure, and only as co-variables in descriptive studies. Experimental studies on the community scale as well as descriptive studies focusing on abiotic factors along consistent plant communities and hosts in comparable habitats would be beneficial to ascertain their actual impact. The new meta’omics approaches strongly facilitate assessments of microbial communities, which might be a chance to place more emphasis on assessing or controlling the numerous environmental parameters in future studies.
Soil communities
The mineral soil horizons are usually easily distinguishable from the covering organic layers in the field. Separating bulk soil from the rhizosphere in soil samples is much more challenging, because even if roots are separated, hyphae emerging from root-associated fungi remain in the sample. Coince et al. (2013) showed that the community in soil samples freed of roots still reflects the mycorrhizal community. Taxonomic assignment via barcoding sequences may allow to identify certain Operational Taxonomic Units (OTUs) as mycorrhizal fungi. However, a considerable proportion of sequences are not identifiable to a taxonomic level required for ecological categorization. Many root-associated but non-mycorrhizal fungi remain unidentified as such due to their immense diversity. Studies focusing on soil fungi therefore usually detect the entire soil fungal community and assign a subset as known mycorrhizal taxa. The proportion of OTUs categorized as mycorrhizal fungi in mineral soil often ranges from 40–50 % (e.g., Wubet et al. 2012; Coince et al. 2013; Wu et al. 2013; Shi et al. 2014), but lower (e.g., Lindahl et al. 2007) and higher (e.g., Voříšková et al. 2013) proportions have been reported. Mycorrhizal fungi therefore comprise a significant part of the fungi in soil samples , which may not be separated from saprotrophic soil fungi in anonymous fingerprinting approaches. An unknown proportion of non-mycorrhizal root-associated fungi may be also expected in soil samples, which demands caution in discussing results against the assumption that soil samples exclusively include saprotrophic fungi.
Distinct fungal communities developed in bulk soil of microcosms planted with different grass species (Mouhamadou et al. 2013). Fungi in agricultural soils also differed between crop types (Rice and Gowda 2011). Replacing native Australian eucalypt forests with pine plantations entailed significant shifts in the forest soil fungal community (Bastias et al. 2007) and the communities differed among plots planted with six different tree species at a site in France (Buée et al. 2009). These experimental setups provide strong evidence for dependency of the soil fungal community on the dominant plant species. Analyses in natural single and mixed tree communities revealed that plant identity is more important than plant species richness for the structure of microbial soil communities (Scheibe et al. 2015). Plant and soil fungal community structure were also correlated in tropical (Peay et al. 2013), subtropical (Wu et al. 2013) and temperate forests (Weig et al. 2013), Mediterranean ecosystems (Orgiazzi et al. 2013) and alpine tundra (Lentendu et al. 2011). Soil beneath populations of a single plant species was colonized by different fungi than soil in adjacent areas (Hovatter et al. 2011; Roy et al. 2013). The non-mycorrhizal community beneath Pinus muricata was structured by distance from the plant (Branco et al. 2013). Distribution patterns of primarily saprobic ascomycetous yeasts (Saccharomycetales) did not depend on plant community composition in Amazonian rainforests, in contrast to those of predominantly biotrophic lineages (Peay et al. 2013). Similar analyses of specific soil fungal taxa with relatively uniform biology might help to ascertain the impact of plant cover on their distribution. Disentangling direct and indirect effects of factors affecting plant and fungal communities is certainly another major challenge for future studies (Coince et al. 2013; Scheibe et al. 2015).
Green et al. (2004) conducted a comprehensive study in Australia revealing that similarity between soil fungal communities decreases with spatial distance (i.e., distance decay). While the community remained anonymous due to their applied fingerprinting approach, a pronounced distance-decay pattern was reported along a transect in China for soil fungi in general and non-mycorrhizal fungi in particular (Shi et al. 2014). Differences between Mediterranean soil fungal communities were also only conclusively explained when distance-decay was considered (Orgiazzi et al. 2013). Compositional differences between soil fungal communities in the proximity of Lobelia siphilitica plants were not explained by geographical distance along a 508 km transect in eastern North America, while similarity between bacterial communities decreased with distance (Hovatter et al. 2011). Across the whole continent of North America, however, spatial distance explained much (R 2 = 0.34) of the fungal community structure in pine forest soils (Talbot et al. 2014). The mechanisms underlying distance decay patterns in soil fungal communities have recently been addressed in two studies. In the first, fungal inoculum was transferred between experimental sites to exclude dispersal limitation (Glinka and Hawkes 2014). This had no effect on the communities. The second study compared the fungal communities in soil and air across an area of 40,000 km2 in California and also revealed no evidence for dispersal limitation causing distance decay patterns (Kivlin et al. 2014). Distance decay patterns in both studies were therefore caused by abiotic (i.e. environmental filtering) or biotic (i.e. competitive exclusion) factors preventing establishment of the fungi. However, Kivlin et al. (2014) showed that abundance of hypogeous fungi differed locally. Dispersal limitation may play a role for these fungi, due to selective below-ground distribution of fungal spores by the soil fauna (Werner et al. 2012). Distance decay patterns at small scales, as found along a 270 m transect in a meadow in Germany (Schmidt et al. 2013), could therefore be caused by dispersal limitation.
Seasonal variation of fungal communities was less pronounced in mineral soil than in the organic layers (Andreetta et al. 2012; Voříšková et al. 2013). Soil communities were, however, more efficient in degrading complex plant compounds in winter, while glucose was more efficiently metabolized in summer (Koranda et al. 2013). These results indicate a seasonal variation in functional properties, while the soil fungal community composition remains comparatively stable. Shifts in the active part of the fungal community might explain such effects, but evidence for the expectable differences in RNA and DNA derived profiles is currently only availabble from experimental setups (Kuramae et al. 2013b) and organic soils (Curlevski et al. 2011). Long-term studies under natural conditions are required to establish if soil fungal communities are stable in composition through time, but respond to seasonal changes with changes in activity of specific taxa. The microbial soil community is also structured by acyclic disturbances of the ecosystem, such as wildfire or tree dieback, but studies on their impact on composition and function of the soil fungal community are rare (Štursová et al. 2014, Buscardo et al. 2015).
In beech forests, the entire soil fungal community was affected by the same soil parameters (i.e. Corg, pH, C:N ratio) as the root-associated community (Wubet et al. 2012). In subtropical forest soil, however, it was shown that of Corg, pH and C:N ratio, only Corg had an impact, and only on the whole community (Wu et al. 2013). The soil fungal community was dependent on the sand fraction in both studies. The communities correlated with none of the measured soil parameters (pH, C content, exchangeable calcium) along a transect from temperate to tropical forests across China, but responded to mean annual temperature, temperature range and precipitation (Shi et al. 2014). Coince et al. (2013) showed that the soil fungi in a French beech forest varied with P content, in contrast to EcM fungi, while both groups were sensitive to C:N ratios, but not to acidity. Similarly, a much stronger effect of C:N ratios, than acidity on the community in agricultural soils was reported from Japan (Bao et al. 2012). In contrast, the fungi in a pasture soil in New Zealand did not respond to C:N ratios (or nitrate content), but to acidity (Stevenson et al. 2014). In other studies, the C:N ratios and acidity both shaped the soil fungal community (Dennis et al. 2012; Wubet et al. 2012), or had no effect (Hovatter et al. 2011; Wu et al. 2013; Shi et al. 2014). In arable soils in an experimental field plot in Rothamstead, England, with a pH gradient from 4.0–8.3, but otherwise relatively constant soil parameters, the soil fungal community responded only weakly to pH (Rousk et al. 2010). At a continental scale, environmental variables including soil chemistry accounted only for a small variation in fungal communities (Talbot et al. 2014). Fungal community composition and soil pH were correlated at global scale, but acidity explained only 2.1 % of the saprobic and 1.5 % of the total community structure (Tedersoo et al. 2014b). The best predictor of community composition was the potential evapotranspiration, explaining 3.8 % of the saprobic and 2.4 % of the total community variation. Because of the overall low explanatory values of the environmental parameters, it was concluded that they only have a minor influence on soil fungal community composition at the global scale. This is in agreement with the regional studies discussed above, which indicate that impact of environmental factors on fungal communities differs between regions.
In contrast to taxonomic composition, the major ecosystem functions fulfilled by the fungal communities were not spatially structured in pine forests across North America, but well explained (R 2 = 0.48) by soil chemistry (Talbot et al. 2014). Activity of hydrolases, which degrade a multitude of molecules, such as cellulose and proteins, was best explained by substrate chemistry. Phenol oxidases and peroxidases, which are involved in lignin degradation, correlated with climate, soil moisture, and pH. These correlations were consistent throughout the organic layers and mineral soil horizons. While gene expression profiles of the microbial communities were quite similar in the organic layer and the mineral soil in a French spruce forest, the relative abundance of certain functional groups differed significantly (Uroz et al. 2013). It was concluded that the community in the organic layer is better adapted to degrade easily accessible carbon substrates, while those in the underlying (top) soil can better utilize leaching by-products from organic matter decomposition, such as amino acid derivatives and proteins. Most of the microbial transcripts in the top soil were related to protein and amino acid metabolism after snow melt in Germany, followed by those involved in carbohydrate metabolism (Urich et al. 2008). Lipid metabolism dominated over carbohydrate and amino acid metabolism in sandy pine forest soil on the French coast (Bailly et al. 2007). In a forest soil in Japan most fungal transcript were related to carbohydrate metabolism, followed by amino acid and lipid metabolism in October (Takasaki et al. 2013). These isolated studies reveal no conclusive picture on the functional role of the saprotrophic soil community. They were conducted in different regions and soils and the varying environmental factors have certainly a major impact on functionality of the soil communities. Seasonal shifts in functionality (Koranda et al. 2013) will have to be accounted for in future studies. To establish causal connections between environmental variables and fungal response, it may be helpful to analyze proteins involved in the internal cellular metabolism separately from secreted proteins mobilizing compounds from the environment. Approaches assessing treatment effects on functionality of the soil fungal community are rare. Damon et al. (2012) compared the eukaryotic metatranscriptomes between spruce and beech forest soils in summer in central France. They found a significant higher abundance of membrane transporters, lignin-oxidative enzymes and carbohydrate-active enzymes in the spruce forest soil, while no group was significantly enriched in the beech forest soil. Protein spectra in the soil organic matter also differed between coniferous and broad-leaf forest soils (Schulze et al. 2005). Unfortunately, the corresponding proteins were not further specified; probably due to the poorly populated reference databases of the time.
Meta’omic studies have begun to identify differences in the functionality of soil microbial communities between ecosystems. Further details on the dependency of soil microbial functional diversity or redundancy on ecosystem structure and functioning are, however, required. A key element in linking functionality of the soil microbial community to ecosystem functioning are certainly plant roots (Silver and Miya 2001; Bardgett et al. 2014; Solly et al. 2014). The amount of root litter is similar to leaf litter, but formed within the soil (Berg and McClaugherty 2014). Root litter therefore represents a major energy and nutrient source for soil microbes, which has been widely neglected in meta’omic studies so far.
Conclusion
Meta’omic studies showed that identity of plants strongly shapes the fungal communities in their phyllosphere, rhizosphere, and, possibly to a lesser extent, in the surrounding pedosphere. This justifies the holobiont concept for ecological studies, but delimitation of holobiont systems in natural ecosystems is challenging, because composition of the microbial communities associated with each host depends on the entire plant community structure. Structure and function of microbial communities change with time. A holobiont concept in the wider ecological sense has therefore to be dynamic, not static. Despite these challenges, the close host-fungus interdependencies suggest that taking the holobiont plant as fundamental unit in ecological studies would certainly facilitate our understanding of ecosystem functioning.
The vast majority of meta’omic studies focused on spatial patterns so far, revealing a plethora of environmental parameters to correlate with taxonomic community composition. Explanatory power of these parameters, however, remained mostly poor. DNA-based assessments probably complicate the discovery of existing correlations, because they detect all present taxa. A considerable proportion of taxa was shown to be at least temporarily inactive and resting stages may persist in the environment for years (Nguyen et al. 2012). Presence of these taxa is not necessarily explained by parameters measured within a restricted study period and they may blur actually existing correlations. Assessments of the active fungal communities based on RNA reflect responses to influencing factors more directly. Temporal shifts in community structures, and also in the shaping factors, indicate that such correlations may only emerge within a reasonable study period. The pronounced seasonal dynamics in almost all plant-associated communities did not result in recurring community structures in subsequent years. This indicates that the development in taxonomic composition may not be cyclic, which complicates achieving reproducible results even at the local scale. Predictability of microbial community composition is therefore still limited, but may improve by focusing on active taxa and by accounting for temporal variability.
To improve predictability in microbial ecology, a shift in focus from taxonomic diversity to functional traits has recently been suggested (Crowther et al. 2014). Trait-based approaches also facilitate testing causal connections predicted by modelling approaches or hypothesized on the basis of correlations (Powell et al. 2013). A comprehensive database including fungal traits is currently available for lichenized fungi and Erysiphales pathogens (i.e. LIAS; see Rambold et al. 2001 onwards, 2014), and traits of ectomycorrhizal fungi have been compiled in DEEMY (Agerer and Rambold 2004–2015; Rambold and Agerer 1997). Crucial traits to be included in a future database were recently suggested for root-infecting fungi (Aguilar-Trigueros et al. 2014). The patchy coverage of traits of plant-associated fungi in reference databases makes a trait-based approach currently laborious, and only feasible for assessing the impact of one to few specific factors on fungal communities (Crowther et al. 2014). The factors controlling community composition are, however, mostly unknown in ecological studies, may be manifold and vary with time.
A meta’omics approach complementing a trait-based approach by a process-focused study design is outlined below to cope with such complexity. Due to the functional redundancy of fungal communities (Talbot et al. 2014), focussing on environmental processes reduces complexity, at least in the initial phase. The outlined approach seeks to explain temporal shifts in chemical composition or properties in the habitat by the activity of the microbial communities. Climatic and local environmental parameters and finally the evolutionary background would be considered as controlling factors. Integrating hypothesis-driven and data-driven research approaches to a systems biological perspective (Castell and Ernst 2012; O’Malley and Soyer 2012) may contribute to a detailed understanding of the functional role of fungal communities. Covering the plethora of possibly affective environmental parameters in such a context is a major challenge for isolated research projects, why embedment in international research initiatives (e.g., futureearth.org; igfagcr.org) might be considered for mutual benefits. Local networking or connection to data repositories (e.g., datadryad.org, dataone.org, pangaea.de) may also maximize the availability of parameters for later analyses.
A meta’omic study on the functional structure of a plant-associated fungal community could commence with identifying chemical variation and transformation in the environment by metametabolomic analyses of host and/or substrate (soil, litter). Correlation with metatranscriptomic and/or metaproteomic assessments could identify the processes probably causing the observed chemical shifts. Time series would allow differentiating continuous, seasonally varying and occasionally occurring processes. Modelling the former two against the environmental parameters would predict factors controlling the fundamental processes. Predictions could be tested in reduced experimental setups using model organisms possessing the respective functional traits (Powell et al. 2013). Processes may also be affected by seemingly unrelated trait complexes such as those involved in fungus-plant-interactions affecting litter degradation processes by root-associated or previously endophytic fungi. Explanatory value of the data from such a study would therefore greatly benefit from parallel analyses of fungal habitats (phyllosphere, pedosphere) and substrates (litter, soil); i.e. from focusing on holobiont functioning in the ecosystem. Only identification of the corresponding taxa will, however, eventually yield a comprehensive picture on ecosystem functioning; in particular if evolutionary scenarios determine functional traits (e.g., Wolfe et al. 2012, Delaye et al. 2013). Linking traits via species identification may even be indispensable to explain ecosystem functioning in cases where cause (e.g., selectivity of host plant for endophytic fungi) and effect (e.g., decomposition activity of previously endophytic fungi) are disconnected in time. Taxonomic identification of key players may be achieved via correlation of functional gene expression with abundance of rRNA-derived barcoding markers. Direct links to transcripts may be achieved by whole genome sequencing of isolated strains, which may be preselected by screening strains for the genes of interest. Shifts in taxonomic composition of communities responsible for a certain process may then be explained by taxon-specific trait complexes, e.g., responsible for competitiveness and stress tolerance (Crowther et al. 2014).
References
Abbate JL, Antonovics J (2014) Elevational disease distribution in a natural plant-pathogen system: insights from changes across host populations and climate. Oikos 123:1126–1136. doi:10.1111/oik.01001
Abbot A (1999) Proteomics, transcriptomics: what’s in a name? Nature 402:715. doi:10.1038/45354
Agerer R, Rambold G (2004–2015) DEEMY – an information system for characterization and determination of ectomycorrhizae. www.deemy.de – München, Germany
Aguilar-Trigueros CA, Powell JR, Anderson IC, Antonovics J, Rillig MC (2014) Ecological understanding of root-infecting fungi using trait-based approaches. Trends Plant Sci 19:432–438. doi:10.1016/j.tplants.2014.02.006
Aleklett K, Hart M, Shade A (2014) The microbial ecology of flowers: an emerging frontier in phyllosphere research 1. Botany 92:253–266. doi:10.1139/cjb-2013-0166
Amend AS, Seifert KA, Bruns TD (2010) Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol Ecol 19:5555–5565. doi:10.1111/j.1365-294X.2010.04898.x
Anderson IC, Genney DR, Alexander IJ (2014) Fine-scale diversity and distribution of ectomycorrhizal fungal mycelium in a Scots pine forest. New Phytol 201:1423–1430. doi:10.1111/nph.12637
Andreetta A, Macci C, Ceccherini MT, Cecchini G, Masciandaro G, Pietramellara G, Carnicelli S (2012) Microbial dynamics in Mediterranean Moder humus. Biol Fertil Soils 48:259–270. doi:10.1007/s00374-011-0622-9
Arnold AE (2007) Understanding the diversity of foliar endophytic fungi: progress, challenges, and frontiers. Fungal Biol Rev 21:51–66. doi:10.1016/j.fbr.2007.05.003
Ayres E, Steltzer H, Simmons BL, Simpson RT, Steinweg JM, Wallenstein MD, Mellor N, Parton WJ, Moore JC, Wall DH (2009) Home-field advantage accelerates leaf litter decomposition in forests. Soil Biol Biochem 41:606–610. doi:10.1016/j.soilbio.2008.12.022
Bahram M, Kõljalg U, Courty P, Diédhiou AG, Kjøller R, Põlme S, Ryberg M, Veldre V, Tedersoo L, Wurzburger N (2013) The distance decay of similarity in communities of ectomycorrhizal fungi in different ecosystems and scales. J Ecol 101:1335–1344. doi:10.1111/1365-2745.12120
Bailly J, Fraissinet-Tachet L, Verner M, Debaud J, Lemaire M, Wésolowski-Louvel M, Marmeisse R (2007) Soil eukaryotic functional diversity, a metatranscriptomic approach. ISME J 1:632–642. doi:10.1038/ismej.2007.68
Baldrian P (2008) Enzymes of saprotrophic basidiomycetes. In: Boddy L, Frankland JC, van West P (eds) British Mycological Society Symposia Series: Ecology of Saprotrophic Basidiomycetes, Volume 28. Academic Press, pp 19–41
Baldrian P, López-Mondéjar R (2014) Microbial genomics, transcriptomics and proteomics: new discoveries in decomposition research using complementary methods. Appl Microbiol Biotechnol 98:1531–1537. doi:10.1007/s00253-013-5457-x
Baldrian P, Kolařík M, Štursová M, Kopecký J, Valášková V, Větrovský T, Zifčáková L, Šnajdr J, Rídl J, Vlček C, Voříšková J (2012) Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J 6:248–258. doi:10.1038/ismej.2011.95
Baldrian P, Větrovský T, Cajthaml T, Dobiášová P, Petránková M, Šnajdr J, Eichlerová I (2013) Estimation of fungal biomass in forest litter and soil. Fungal Ecol 6:1–11. doi:10.1016/j.funeco.2012.10.002
Bálint M, Tiffin P, Hallström B, O’Hara RB, Olson MS, Fankhauser JD, Piepenbring M, Schmitt I (2013) Host genotype shapes the foliar fungal microbiome of balsam poplar (Populus balsamifera). PLoS ONE 8, e53987. doi:10.1371/journal.pone.0053987
Bálint M, Bartha L, O’Hara RB, Olson MS, Otte J, Pfenninger M, Robertson AL, Tiffin P, Schmitt I (2014) Relocation, high-latitude warming and host genetic identity shape the foliar fungal microbiome of poplars. Mol Ecol. doi:10.1111/mec.13018
Ban E, Park SH, Kang M, Lee H, Song EJ, Yoo YS (2012) Growing trend of CE at the omics level: the frontier of systems biology – an update. Electrophoresis 33:2–13. doi:10.1002/elps.201100344
Bao Z, Ikunaga Y, Matsushita Y, Morimoto S, Takada-Hoshino Y, Okada H, Oba H, Takemoto S, Niwa S, Ohigashi K, Suzuki C, Nagaoka K, Takenaka M, Urashima Y, Sekiguchi H, Kushida A, Toyota K, Saito M, Tsushima S (2012) Combined analyses of bacterial, fungal and nematode communities in andosolic agricultural soils in Japan. Microbes Environ 27:72–79. doi:10.1264/jsme2.ME11281
Bardgett RD, Mommer L, Vries D, Franciska T (2014) Going underground: root traits as drivers of ecosystem processes. Trends Ecol Evol 29:692–699. doi:10.1016/j.tree.2014.10.006
Bastias BA, Anderson IC, Xu Z, Cairney JW (2007) RNA- and DNA-based profiling of soil fungal communities in a native Australian eucalypt forest and adjacent Pinus elliotti plantation. Soil Biol Biochem 39:3108–3114. doi:10.1016/j.soilbio.2007.06.022
Becher D, Bernhardt J, Fuchs S, Riedel K (2013) Metaproteomics to unravel major microbial players in leaf litter and soil environments: challenges and perspectives. Proteomics 13:2895–2909. doi:10.1002/pmic.201300095
Beck A, Peršoh D, Rambold G (2014) First evidence for seasonal fluctuations in lichen- and bark-colonising fungal communities. Folia Microbiol 59:155–157. doi:10.1007/s12223-013-0278-y
Begerow D, Nilsson H, Unterseher M, Maier W (2010) Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl Microbiol Biotechnol 87:99–108. doi:10.1007/s00253-010-2585-4
Behie SW, Zelisko PM, Bidochka MJ (2012) Endophytic insect-parasitic fungi translocate nitrogen directly from insects to plants. Science 336:1576–1577. doi:10.1126/science.1222289
Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H (2010) ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol 10:189. doi:10.1186/1471-2180-10-189
Bensch K, Braun U, Groenewald JZ, Crous PW (2012) The genus Cladosporium. Stud Mycol 72:1–401. doi:10.3114/sim0003
Berg B (2014) Foliar litter decomposition: a conceptual model with focus on Pine (Pinus) litter—a genus with global distribution. ISRN Forestry 2014:1–22. doi:10.1155/2014/838169
Berg B, McClaugherty C (2014) Plant Litter. Decomposition, humus formation, carbon sequestration, 3rd edn. Springer, Berlin
Berg B, Staaf H (1980) Decomposition rate and chemical changes of Scots pine needle litter. II. Influence of chemical composition. Ecol Bullet 32:373–390
Bills GF, González-Menéndez V, Martín J, Platas G, Fournier J, Peršoh D, Stadler M (2012) Hypoxylon pulicicidum sp. nov. (Ascomycota, Xylariales), a pantropical insecticide-producing endophyte. PLoS ONE 7, e46687. doi:10.1371/journal.pone.0046687
Bougoure DS, Parkin PI, Cairney JWG, Alexander IJ, Anderson IC (2007) Diversity of fungi in hair roots of Ericaceae varies along a vegetation gradient. Mol Ecol 16:4624–4636. doi:10.1111/j.1365-294X.2007.03540.x
Bradford MA, Warren RJ II, Baldrian P, Crowther TW, Maynard DS, Oldfield EE, Wieder WR, Wood SA, King JR (2014) Climate fails to predict wood decomposition at regional scales. Nat Clim Chang 4:625–630. doi:10.1038/NCLIMATE2251
Branco S, Bruns TD, Singleton I (2013) Fungi at a small scale: spatial zonation of fungal assemblages around single trees. PLoS ONE 8, e78295. doi:10.1371/journal.pone.0078295
Bray SR, Kitajima K, Mack MC (2012) Temporal dynamics of microbial communities on decomposing leaf litter of 10 plant species in relation to decomposition rate. Soil Biol Biochem 49:30–37. doi:10.1016/j.soilbio.2012.02.009
Bridge PD, Roberts PJ, Spooner BM, Panchal G (2003) On the unreliability of published DNA sequences. New Phytol 160:43–48. doi:10.1046/j.1469-8137.2003.00861.x
Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F (2009) 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184:449–456. doi:10.1111/j.1469-8137.2009.03003.x
Buermans HPJ, den Dunnen JT (2014) Next generation sequencing technology: advances and applications. Biochim Biophys Acta 1842:1932–1941. doi:10.1016/j.bbadis.2014.06.015
Burke DJ, Lopez-Gutierrez JC, Smemo KA, Chan CR (2009) Vegetation and soil environment influence the spatial distribution of root-associated fungi in a mature beech-maple forest. Appl Environ Microbiol 75:7639–7648. doi:10.1128/AEM.01648-09
Burke DJ, Weintraub MN, Hewins CR, Kalisz S (2011) Relationship between soil enzyme activities, nutrient cycling and soil fungal communities in a northern hardwood forest. Soil Biol Biochem 43:795–803. doi:10.1016/j.soilbio.2010.12.014
Buscardo E, Rodríguez-Echeverría S, Freitas H, de Angelis P, Pereira J, Muller LH (2015) Contrasting soil fungal communities in Mediterranean pine forests subjected to different wildfire frequencies. Fungal Divers 70:85–99. doi:10.1007/s13225-014-0294-5
Cao Y, Van De Werfhorst LC, Dubinsky EA, Badgley BD, Sadowsky MJ, Andersen GL, Griffith JF, Holden PA (2013) Evaluation of molecular community analysis methods for discerning fecal sources and human waste. Water Res 47:6862–6872. doi:10.1016/j.watres.2013.02.061
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi:10.1038/NMETH.F.303
Castell WZ, Ernst D (2012) Experimental ‘omics’ data in tree research: facing complexity. Trees 26:1723–1735. doi:10.1007/s00468-012-0777-5
Chambers SM, Curlevski NJA, Cairney JWG (2008) Ericoid mycorrhizal fungi are common root inhabitants of non-Ericaceae plants in a south-eastern Australian sclerophyll forest. FEMS Microbiol Ecol 65:263–270. doi:10.1111/j.1574-6941.2008.00481.x
Chomnunti P, Hongsanan S, Aguirre-Hudson B, Tian Q, Peršoh D, Dhami MK, Alias AS, Xu J, Liu X, Stadler M, Hyde KD (2014) The sooty moulds. Fungal Divers 66:1–36. doi:10.1007/s13225-014-0278-5
Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H, Stenlid J, Finlay RD, Wardle DA, Lindahl BD (2013) Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 339:1615–1618. doi:10.1126/science.1231923
Coince A, Caël O, Bach C, Lengellé J, Cruaud C, Gavory F, Morin E, Murat C, Marçais B, Buée M (2013) Below-ground fine-scale distribution and soil versus fine root detection of fungal and soil oomycete communities in a French beech forest. Fungal Ecol 6:223–235. doi:10.1016/j.funeco.2013.01.002
Coince A, Cordier T, Lengellé J, Defossez E, Vacher C, Robin C, Buée M, Marçais B (2014) Leaf and root-associated fungal assemblages do not follow similar elevational diversity patterns. PLoS ONE 9, e100668. doi:10.1371/journal.pone.0100668
Cordier T, Robin C, Capdevielle X, Desprez-Loustau M, Vacher C (2012a) Spatial variability of phyllosphere fungal assemblages: genetic distance predominates over geographic distance in a European beech stand (Fagus sylvatica). Fungal Ecol 5:509–520. doi:10.1016/j.funeco.2011.12.004
Cordier T, Robin C, Capdevielle X, Fabreguettes O, Desprez-Loustau M, Vacher C (2012b) The composition of phyllosphere fungal assemblages of European beech (Fagus sylvatica) varies significantly along an elevation gradient. New Phytol 196:510–519. doi:10.1111/j.1469-8137.2012.04284.x
Courty P, Franc A, Pierrat J, Garbaye J (2008) Temporal changes in the ectomycorrhizal community in two soil horizons of a temperate oak forest. Appl Environ Microbiol 74:5792–5801. doi:10.1128/AEM.01592-08
Crecchio C, Stotzky G (1998) Binding of DNA on humic acids: effect on transformation of Bacillus subtilis and resistance to DNase. Soil Biol Biochem 30:1061–1067
Crous PW (2009) Taxonomy and phylogeny of the genus mycosphaerella and its anamorphs. Fungal Divers 38:1–24
Crowther TW, Maynard DS, Crowther TR, Peccia J, Smith JR, Bradford MA (2014) Untangling the fungal niche: the trait-based approach. Front Microbiol 5:579. doi:10.3389/fmicb.2014.00579
Curlevski NJA, Artz RRE, Anderson IC, Cairney JWG (2011) Response of soil microbial communities to management strategies for enhancing Scots pine (Pinus sylvestris) establishment on heather (Calluna vulgaris) moorland. Plant Soil 339:413–424. doi:10.1007/s11104-010-0593-x
Damon C, Lehembre F, Oger-Desfeux C, Luis P, Ranger J, Fraissinet-Tachet L, Marmeisse R (2012) Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. PLoS ONE 7. doi:10.1371/journal.pone.0028967
Daniel R (2005) The metagenomics of soil. Nat Rev Microbiol 3:470–478. doi:10.1038/nrmicro1160
Davenport CF, Tümmler B (2013) Advances in computational analysis of metagenome sequences. Environ Microbiol 15:1–5. doi:10.1111/j.1462-2920.2012.02843.x
Davey ML, Heegaard E, Halvorsen R, Kauserud H, Ohlson M (2012) Seasonal trends in the biomass and structure of bryophyte-associated fungal communities explored by 454 pyrosequencing. New Phytol 195:844–856. doi:10.1111/j.1469-8137.2012.04215.x
Davey ML, Heegaard E, Halvorsen R, Kauserud H, Ohlson M (2013a) Amplicon-pyrosequencing-based detection of compositional shifts in bryophyte-associated fungal communities along an elevation gradient. Mol Ecol 22:368–383. doi:10.1111/mec.12122
Davey ML, Heimdal R, Ohlson M, Kauserud H (2013b) Host- and tissue-specificity of moss-associated Galerina and Mycena determined from amplicon pyrosequencing data. Fungal Ecol 6:179–186. doi:10.1016/j.funeco.2013.02.003
Davison J, Öpik M, Zobel M, Vasar M, Metsis M, Moora M (2012) Communities of arbuscular mycorrhizal fungi detected in forest soil are spatially heterogeneous but do not vary throughout the growing season. PLoS ONE 7, e41938. doi:10.1371/journal.pone.0041938
de Marco A, Spaccini R, Vittozzi P, Esposito F, Berg B, Virzo De Santo A (2012) Decomposition of black locust and black pine leaf litter in two coeval forest stands on Mount Vesuvius and dynamics of organic components assessed through proximate analysis and NMR spectroscopy. Soil Biol Biochem 51:1–15. doi:10.1016/j.soilbio.2012.03.025
Delaye L, García-Guzmán G, Heil M (2013) Endophytes versus biotrophic and necrotrophic pathogens—are fungal lifestyles evolutionarily stable traits? Fungal Divers 60:125–135. doi:10.1007/s13225-013-0240-y
Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, Schlapbach R, von Mering C, Vorholt JA (2009) Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc Natl Acad Sci U S A 106:16428–16433. doi:10.1073/pnas.0905240106
Dennis PG, Rushton SP, Newsham KK, Lauducina VA, Ord VJ, Daniell TJ, O’Donnell AG, Hopkins DW (2012) Soil fungal community composition does not alter along a latitudinal gradient through the maritime and sub-Antarctic. Fungal Ecol 5:403–408. doi:10.1016/j.funeco.2011.12.002
Dhami MK, Weir BS, Taylor MW, Beggs JR (2013) Diverse honeydew-consuming fungal communities associated with scale insects. PLoS ONE 8, e70316. doi:10.1371/journal.pone.0070316
Dickie IA, Martínez-García LB, Koele N, Grelet G, Tylianakis JM, Peltzer DA, Richardson SJ (2013) Mycorrhizas and mycorrhizal fungal communities throughout ecosystem development. Plant Soil 367:11–39. doi:10.1007/s11104-013-1609-0
Dineen SM, Aranda R, Dietz ME, Anders DL, Robertson JM (2010) Evaluation of commercial RNA extraction kits for the isolation of viral MS2 RNA from soil. J Virol Methods 168:44–50. doi:10.1016/j.jviromet.2010.04.014
Drábková L, Kirschner J, Vlĉek Ĉ (2002) Comparison of seven DNA extraction and amplification protocols in historical herbarium specimens of juncaceae. Plant Mol Biol Rep 20:161–175. doi:10.1007/BF02799431
Dumbrell AJ, Ashton PD, Aziz N, Feng G, Nelson M, Dytham C, Fitter AH, Helgason T (2011) Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol 190:794–804. doi:10.1111/j.1469-8137.2010.03636.x
Edman P, Begg G (1967) A protein sequenator. Eur J Biochem 1:80–91. doi:10.1111/j.1432-1033.1967.tb00047.x
Edwards IP, Zak DR (2010) Phylogenetic similarity and structure of Agaricomycotina communities across a forested landscape. Mol Ecol 19:1469–1482. doi:10.1111/j.1365-294X.2010.04566.x
Eisen JA (2007) Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes. PLoS Biol 5, e82. doi:10.1371/journal.pbio.0050082
Ekblad A, Wallander H, Godbold DL, Cruz C, Johnson D, Baldrian P, Björk RG, Epron D, Kieliszewska-Rokicka B, Kjøller R, Kraigher H, Matzner E, Neumann J, Plassard C (2013) The production and turnover of extramatrical mycelium of ectomycorrhizal fungi in forest soils: role in carbon cycling. Plant Soil 366:1–27. doi:10.1007/s11104-013-1630-3
Engelbrektson A, Kunin V, Wrighton KC, Zvenigorodsky N, Chen F, Ochman H, Hugenholtz P (2010) Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J 4:642–647. doi:10.1038/ismej.2009.153
Feinstein LM, Blackwood CB (2013) The spatial scaling of saprotrophic fungal beta diversity in decomposing leaves. Mol Ecol 22:1171–1184. doi:10.1111/mec.12160
Feldhaar H (2011) Bacterial symbionts as mediators of ecologically important traits of insect hosts. Ecol Entomol 36:533–543. doi:10.1111/j.1365-2311.2011.01318.x
Flessa F, Rambold G (2013) Diversity of the Capnocheirides rhododendri-dominated fungal community in the phyllosphere of Rhododendron ferrugineum L. Nova Hedwigia 97:19–53
Flessa F, Peršoh D, Rambold G (2012) Annuality of Central European deciduous tree leaves delimits community development of epifoliar pigmented fungi. Fungal Ecol 5:554–561. doi:10.1016/j.funeco.2011.12.005
Fonseca Á, Inácio J (2006) Phylloplane yeasts. In: Péter G, Rosa C (eds) Biodiversity and ecophysiology of yeasts. Springer, Berlin, pp 263–301
Frankland JC (1998) Fungal succession – unravelling the unpredictable. Mycol Res 102:1–15
Fredricks DN, Smith C, Meier A (2005) Comparison of six DNA extraction methods for recovery of fungal DNA as assessed by quantitative PCR. J Clin Microbiol 43:5122–5128. doi:10.1128/JCM.43.10.5122-5128.2005
Fritz JV, Desai MS, Shah P, Schneider JG, Wilmes P (2013) From meta-omics to causality: experimental models for human microbiome research. Microbiome 1:14. doi:10.1186/2049-2618-1-14
Gadd GM (2010) Metals, minerals and microbes: geomicrobiology and bioremediation. Microbiology (Reading, England) 156:609–643. doi:10.1099/mic.0.037143-0
Gatte-Picchi D, Weiz A, Ishida K, Hertweck C, Dittmann E (2014) Functional analysis of environmental DNA-derived microviridins provides new insights into the diversity of the tricyclic peptide family. Appl Environ Microbiol 80:1380–1387. doi:10.1128/AEM.03502-13
Geisberg JV, Moqtaderi Z, Fan X, Ozsolak F, Struhl K (2014) Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell 156:812–824. doi:10.1016/j.cell.2013.12.026
Gholz HL, Wedin DA, Smitherman SM, Harmon ME, Parton WJ (2000) Long-term dynamics of pine and hardwood litter in contrasting environments: toward a global model of decomposition. Glob Chang Biol 6:751–765
Glinka C, Hawkes CV (2014) Environmental controls on fungal community composition and abundance over 3 years in native and degraded shrublands. Microb Ecol 68:807–817. doi:10.1007/s00248-014-0443-0
Gougoulias C, Clark JM, Shaw LJ (2014) The role of soil microbes in the global carbon cycle: tracking the below-ground microbial processing of plant-derived carbon for manipulating carbon dynamics in agricultural systems. J Sci Food Agric 94:2362–2371. doi:10.1002/jsfa.6577
Graham C, McMullan G, Graham RLJ (2011) Proteomics in the microbial sciences. Bioeng Bugs 2:17–30. doi:10.4161/bbug.2.1.14413
Green JL, Holmes AJ, Westoby M, Oliver I, Briscoe D, Dangerfield M, Gillings M, Beattie AJ (2004) Spatial scaling of microbial eukaryote diversity. Nature 432:747–750. doi:10.1038/nature03034
Gryndler M, Soukupová L, Gryndlerová H, Baldrian P, Hršelová H (2010) Local distribution of ectomycorrhizae-associated basidiomycetes in forest soil correlates with the degree of soil organic matter humification and available electrolytes. Folia Microbiol 55:454–460. doi:10.1007/s12223-010-0076-8
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R (2008) Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5:235–237. doi:10.1038/NMETH.1184
Hamilton CE, Gundel PE, Helander M, Saikkonen K (2012) Endophytic mediation of reactive oxygen species and antioxidant activity in plants: a review. Fungal Divers 54:1–10. doi:10.1007/s13225-012-0158-9
Haňáčková Z, Koukol O, Štursová M, Kolařík M, Baldrian P (2015) Fungal succession in the needle litter of a montane Picea abies forest investigated through strain isolation and molecular fingerprinting. Fungal Ecol 13:157–166. doi:10.1016/j.funeco.2014.09.007
Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5:R245. doi:10.1016/S1074-5521(98)90108-9
Hatfield R, Fukushima RS (2005) Can lignin be accurately measured? Crop Sci 45:832. doi:10.2135/cropsci2004.0238
Hawksworth DL (1991) The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol Res 95:641–655
Hawksworth DL (2001) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105:1422–1432. doi:10.1017/S0953756201004725
Hawksworth DL (2012) Global species numbers of fungi: are tropical studies and molecular approaches contributing to a more robust estimate? Biodivers Conserv 21:2425–2433. doi:10.1007/s10531-012-0335-x
Hazard C, Gosling P, van der Gast CJ, Mitchell DT, Doohan FM, Bending GD (2013) The role of local environment and geographical distance in determining community composition of arbuscular mycorrhizal fungi at the landscape scale. ISME J 7:498–508. doi:10.1038/ismej.2012.127
He Z, Deng Y, Zhou J (2012) Development of functional gene microarrays for microbial community analysis. Curr Opin Biotechnol 23:49–55. doi:10.1016/j.copbio.2011.11.001
Hernández G, Valdés-López O, Ramírez M, Goffard N, Weiller G, Aparicio-Fabre R, Fuentes SI, Erban A, Kopka J, Udvardi MK, Vance CP (2009) Global changes in the transcript and metabolic profiles during symbiotic nitrogen fixation in phosphorus-stressed common bean plants. Plant Physiol 151:1221–1238. doi:10.1104/pp. 109.143842
Higgins KL, Arnold AE, Coley PD, Kursar TA (2014) Communities of fungal endophytes in tropical forest grasses: highly diverse host- and habitat generalists characterized by strong spatial structure. Fungal Ecol 8:1–11. doi:10.1016/j.funeco.2013.12.005
Hodgson S, de Cates C, Hodgson J, Morley NJ, Sutton BC, Gange AC (2014) Vertical transmission of fungal endophytes is widespread in forbs. Ecol Evol 4:1199–1208. doi:10.1002/ece3.953
Hovatter SR, Dejelo C, Case AL, Blackwood CB (2011) Metacommunity organization of soil microorganisms depends on habitat defined by presence of Lobelia siphilitica plants. Ecology 92:57–65
Huang C, Jian F, Huang H, Chang W, Wu W, Hwang C, Lee R, Chiang T (2014) Deciphering mycorrhizal fungi in cultivated Phalaenopsis microbiome with next-generation sequencing of multiple barcodes. Fungal Divers 66:77–88. doi:10.1007/s13225-014-0281-x
Hyde KD, Soytong K (2008) The fungal endophyte dilemma. Fungal Divers 33:163–173
Ishida TA, Nordin A (2010) No evidence that nitrogen enrichment affect fungal communities of Vaccinium roots in two contrasting boreal forest types. Soil Biol Biochem 42:234–243. doi:10.1016/j.soilbio.2009.10.021
Jones OAH, Sdepanian S, Lofts S, Svendsen C, Spurgeon DJ, Maguire ML, Griffin JL (2014) Metabolomic analysis of soil communities can be used for pollution assessment. Environ Toxicol Chem 33:61–64. doi:10.1002/etc.2418
Joyce AR, Palsson BØ (2006) The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol 7:198–210. doi:10.1038/nrm1857
Jumpponen A, Jones KL (2009) Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere. New Phytol 184:438–448. doi:10.1111/j.1469-8137.2009.02990.x
Jumpponen A, Jones KL (2010) Seasonally dynamic fungal communities in the Quercus macrocarpa phyllosphere differ between urban and nonurban environments. New Phytol 186:496–513. doi:10.1111/j.1469-8137.2010.03197.x
Jumpponen A, Jones KL, David Mattox J, Yaege C (2010) Massively parallel 454-sequencing of fungal communities in Quercus spp. ectomycorrhizas indicates seasonal dynamics in urban and rural sites. Mol Ecol 19(Suppl 1):41–53. doi:10.1111/j.1365-294X.2009.04483.x
Kaiser KA, Barding GA, Larive CK (2009) A comparison of metabolite extraction strategies for 1H-NMR-based metabolic profiling using mature leaf tissue from the model plant Arabidopsis thaliana. Magn Reson Chem 47(Suppl 1):S147–S156. doi:10.1002/mrc.2457
Keiblinger KM, Wilhartitz IC, Schneider T, Roschitzki B, Schmid E, Eberl L, Riedel K, Zechmeister-Boltenstern S (2012) Soil metaproteomics - comparative evaluation of protein extraction protocols. Soil Biol Biochem 54:14–24. doi:10.1016/j.soilbio.2012.05.014
Kemler M, Garnas J, Wingfield MJ, Gryzenhout M, Pillay K, Slippers B (2013) Ion Torrent PGM as tool for fungal community analysis: a case study of endophytes in Eucalyptus grandis reveals high taxonomic diversity. PLoS ONE 8, e81718. doi:10.1371/journal.pone.0081718
Kennedy NA, Walker AW, Berry SH, Duncan SH, Farquarson FM, Louis P, Thomson JM, Satsangi J, Flint HJ, Parkhill J, Lees CW, Hold GL (2014) The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS ONE 9, e88982. doi:10.1371/journal.pone.0088982
Kerekes J, Kaspari M, Stevenson B, Nilsson RH, Hartmann M, Amend A, Bruns TD (2013) Nutrient enrichment increased species richness of leaf litter fungal assemblages in a tropical forest. Mol Ecol 22:2827–2838. doi:10.1111/mec.12259
Kernaghan G (2013) Functional diversity and resource partitioning in fungi associated with the fine feeder roots of forest trees. Symbiosis 61:113–123. doi:10.1007/s13199-013-0265-8
Kim HK, Verpoorte R (2010) Sample preparation for plant metabolomics. Phytochem Anal 21:4–13. doi:10.1002/pca.1188
Kivlin SN, Treseder KK (2014) Initial phylogenetic relatedness of saprotrophic fungal communities affects subsequent litter decomposition rates. Microb Ecol. doi:10.1007/s00248-014-0509-z
Kivlin SN, Hawkes CV, Treseder KK (2011) Global diversity and distribution of arbuscular mycorrhizal fungi. Soil Biol Biochem 43:2294–2303. doi:10.1016/j.soilbio.2011.07.012
Kivlin SN, Winston GC, Goulden ML, Treseder KK (2014) Environmental filtering affects soil fungal community composition more than dispersal limitation at regional scales. Fungal Ecol 12:14–25. doi:10.1016/j.funeco.2014.04.004
Kleber M (2010) What is recalcitrant soil organic matter? Environ Chem 7:320–322. doi:10.1071/EN10006
Knicker H (2011) Solid state CPMAS 13C and 15N NMR spectroscopy in organic geochemistry and how spin dynamics can either aggravate or improve spectra interpretation. Org Geochem 42:867–890. doi:10.1016/j.orggeochem.2011.06.019
Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäck B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Costa MD, Doré J, Floudas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Högberg N, Johansson T, Khouja H, LaButti K, Lahrmann U, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, Tunlid A, Grigoriev IV, Hibbett DS, Martin F (2015) Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet. doi:10.1038/ng.3223
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Brandon PM, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüßler A, Scott JA, Senés C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson K (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277. doi:10.1111/mec.12481
Kolmeder CA, de Been M, Nikkilä J, Ritamo I, Mättö J, Valmu L, Salojärvi J, Palva A, Salonen A, de Vos WM (2012) Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE 7, e29913. doi:10.1371/journal.pone.0029913
Koranda M, Kaiser C, Fuchslueger L, Kitzler B, Sessitsch A, Zechmeister-Boltenstern S, Richter A (2013) Seasonal variation in functional properties of microbial communities in beech forest soil. Soil Biol Biochem 60:95–104. doi:10.1016/j.soilbio.2013.01.025
Kraft NJB, Adler PB, Godoy O, James EC, Fuller S, Levine JM, Fox J (2014) Community assembly, coexistence and the environmental filtering metaphor. Funct Ecol. doi:10.1111/1365-2435.12345
Kreyling J, Peršoh D, Werner S, Benzenberg M, Wöllecke J (2012) Short-term impacts of soil freeze-thaw cycles on roots and root-associated fungi of Holcus lanatus and Calluna vulgaris. Plant Soil 353:19–31. doi:10.1007/s11104-011-0970-0
Krüger M, Stockinger H, Krüger C, Schüssler A (2009) DNA-based species level detection of Glomeromycota: one PCR primer set for all arbuscular mycorrhizal fungi. New Phytol 183:212–223. doi:10.1111/j.1469-8137.2009.02835.x
Kubartová A, Ranger J, Berthelin J, Beguiristain T (2009) Diversity and decomposing ability of saprophytic fungi from temperate forest litter. Microb Ecol 58:98–107. doi:10.1007/s00248-008-9458-8
Kumar S, Kaushik N (2012) Metabolites of endophytic fungi as novel source of biofungicide: a review. Phytochem Rev 11:507–522. doi:10.1007/s11101-013-9271-y
Kuramae EE, Hillekens RHE, de Hollander M, van der Heijden MGA, van den Berg M, van Straalen NM, Kreyling J (2013a) Structural and functional variation in soil fungal communities associated with litter bags containing maize leaf. FEMS Microbiol Ecol 84:519–531. doi:10.1111/1574-6941.12080
Kuramae EE, Verbruggen E, Hillekens R, de Hollander M, Röling WFM, van der Heijden MGA, Kowalchuk GA (2013b) Tracking fungal community responses to maize plants by DNA- and RNA-based pyrosequencing. PLoS ONE 8, e69973. doi:10.1371/journal.pone.0069973
Kuska B (1998) Beer, Bethesda, and biology: how “genomics” came into being [news]. J Natl Cancer Inst 90:93
Lamit LJ, Lau MK, Sthultz CM, Wooley SC, Whitham TG, Gehring CA (2014) Tree genotype and genetically based growth traits structure twig endophyte communities. Am J Bot 101:467–478. doi:10.3732/ajb.1400034
Lankadurai BP, Nagato EG, Simpson MJ (2013) Environmental metabolomics: an emerging approach to study organism responses to environmental stressors. Environ Rev 21:180–205. doi:10.1139/er-2013-0011
Leary DH, Hervey WJ, Deschamps JR, Kusterbeck AW, Vora GJ (2013) Which metaproteome? The impact of protein extraction bias on metaproteomic analyses. Mol Cell Probes 27:193–199. doi:10.1016/j.mcp.2013.06.003
Lederberg J, McCray AT (2001) ’Ome Sweet ’Omics—A geneological Treasureof words. Scientist 15:8
Lekberg Y, Koide RT, Rohr JR, Aldrich-Wolfe L, Morton JB (2007) Role of niche restrictions and dispersal in the composition of arbuscular mycorrhizal fungal communities. J Ecol 95:95–105. doi:10.1111/j.1365-2745.2006.01193.x
Lentendu G, Zinger L, Manel S, Coissac E, Choler P, Geremia RA, Melodelima C (2011) Assessment of soil fungal diversity in different alpine tundra habitats by means of pyrosequencing. Fungal Divers 49:113–123. doi:10.1007/s13225-011-0101-5
Lewin A, Wentzel A, Valla S (2013) Metagenomics of microbial life in extreme temperature environments. Curr Opin Biotechnol 24:516–525. doi:10.1016/j.copbio.2012.10.012
Liang Y, He Z, Wu L, Deng Y, Li G, Zhou J (2010) Development of a common oligonucleotide reference standard for microarray data normalization and comparison across different microbial communities. Appl Environ Microbiol 76:1088–1094. doi:10.1128/AEM.02749-09
Lin CY, Viant MR, Tjeerdema RS (2006) Metabolomics: methodologies and applications in the environmental sciences. J Pestic Sci 31:245–251
Lindahl BD, Ihrmark K, Boberg J, Trumbore SE, Högberg P, Stenlid J, Finlay RD (2007) Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol 173:611–620. doi:10.1111/j.1469-8137.2006.01936.x
Lindahl BD, Nilsson RH, Tedersoo L, Abarenkov K, Carlsen T, Kjøller R, Kõljalg U, Pennanen T, Rosendahl S, Stenlid J, Kauserud H (2013) Fungal community analysis by high-throughput sequencing of amplified markers – a user’s guide. New Phytol 199:288–299. doi:10.1111/nph.12243
Logares R, Haverkamp THA, Kumar S, Lanzén A, Nederbragt AJ, Quince C, Kauserud H (2012) Environmental microbiology through the lens of high-throughput DNA sequencing: synopsis of current platforms and bioinformatics approaches. J Microbiol Methods 91:106–113. doi:10.1016/j.mimet.2012.07.017
Lothamer K, Brown SP, Mattox JD, Jumpponen A (2014) Comparison of root-associated communities of native and non-native ectomycorrhizal hosts in an urban landscape. Mycorrhiza 24:267–280. doi:10.1007/s00572-013-0539-2
Lundell TK, Mäkelä MR, Hildén K (2010) Lignin-modifying enzymes in filamentous basidiomycetes – ecological, functional and phylogenetic review. J Basic Microbiol 50:5–20. doi:10.1002/jobm.200900338
Mertens K, Freund L, Schmoock G, Hänsel C, Melzer F, Elschner MC (2014) Comparative evaluation of eleven commercial DNA extraction kits for real-time PCR detection of Bacillus anthracis spores in spiked dairy samples. Int J Food Microbiol 170:29–37. doi:10.1016/j.ijfoodmicro.2013.10.022
Moorhead DL, Sinsabaugh RL (2006) A theoretical model of litter decay and microbial interaction. Ecol Monogr 76:151–174
Moran MA (2009) Metatranscriptomics: eavesdropping on complex microbial communities. Microbe 4:329–335
Moricca S, Ginetti B, Ragazzi A (2012) Species- and organ-specificity in endophytes colonizing healthy and declining Mediterranean oaks. Phytopathol Mediterr 51:587–598
Mouhamadou B, Puissant J, Personeni E, Desclos-Theveniau M, Kastl EM, Schloter M, Zinger L, Roy J, Geremia RA, Lavorel S (2013) Effects of two grass species on the composition of soil fungal communities. Biol Fertil Soils 49:1131–1139. doi:10.1007/s00374-013-0810-x
Nahnsen S, Bielow C, Reinert K, Kohlbacher O (2013) Tools for label-free peptide quantification. Mol Cell Proteomics 12:549–556. doi:10.1074/mcp.R112.025163
Nguyen NH, Hynson NA, Bruns TD (2012) Stayin’ alive: survival of mycorrhizal fungal propagules from 6-yr-old forest soil. Fungal Ecol 5:741–746. doi:10.1016/j.funeco.2012.05.006
Nikolaki S, Tsiamis G (2013) Microbial diversity in the era of omic technologies. Biomed Res Int 2013:958719. doi:10.1155/2013/958719
Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson K, Kõljalg U (2006) Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS ONE 1, e59. doi:10.1371/journal.pone.0000059
O’Malley MA, Soyer OS (2012) The roles of integration in molecular systems biology. Stud Hist Philos Biol Biomed Sci 43:58–68. doi:10.1016/j.shpsc.2011.10.006
Ono K, Hiradate S, Morita S, Ohse K, Hirai K (2011) Humification processes of needle litters on forest floors in Japanese cedar (Cryptomeria japonica) and Hinoki cypress (Chamaecyparis obtusa) plantations in Japan. Plant Soil 338:171–181. doi:10.1007/s11104-010-0397-z
Öpik M, Metsis M, Daniell TJ, Zobel M, Moora M (2009) Large-scale parallel 454 sequencing reveals host ecological group specificity of arbuscular mycorrhizal fungi in a boreonemoral forest. New Phytol 184:424–437. doi:10.1111/j.1469-8137.2009.02920.x
Orgiazzi A, Bianciotto V, Bonfante P, Daghino S, Ghignone S, Lazzari A, Lumini E, Mello A, Napoli C, Perotto S, Vizzini A, Bagella S, Murat C, Girlanda M (2013) 454 pyrosequencing analysis of fungal assemblages from geographically distant, disparate soils reveals spatial patterning and a core mycobiome. Diversity 5:73–98. doi:10.3390/d5010073
Oros-Sichler M, Costa R, Heuer H, Smalla K (2007) Molecular fingerprinting techniques to analyze soil microbial communities. In: van Elsas JD, Jansson JK, Trevors JT (eds) Modern soil microbiology. CRC Press, Oxon, pp 355–386
Osono T, Azuma J, Hirose D (2014) Plant species effect on the decomposition and chemical changes of leaf litter in grassland and pine and oak forest soils. Plant Soil 376:411–421. doi:10.1007/s11104-013-1993-5
Pan C, Banfield JF (2014) Quantitative metaproteomics: functional insights into microbial communities. Methods Mol Biol 1096:231–240. doi:10.1007/978-1-62703-712-9_18
Pan Z, Raftery D (2007) Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal Bioanal Chem 387:525–527. doi:10.1007/s00216-006-0687-8
Paoletti AC, Parmely TJ, Tomomori-Sato C, Sato S, Zhu D, Conaway RC, Conaway JW, Florens L, Washburn MP (2006) Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc Natl Acad Sci U S A 103:18928–18933. doi:10.1073/pnas.0606379103
Parrent JL, Vilgalys R (2007) Biomass and compositional responses of ectomycorrhizal fungal hyphae to elevated CO2 and nitrogen fertilization. New Phytol 176:164–174. doi:10.1111/j.1469-8137.2007.02155.x
Paulin MM, Nicolaisen MH, Jacobsen CS, Gimsing AL, Sørensen J, Bælum J (2013) Improving Griffith’s protocol for co-extraction of microbial DNA and RNA in adsorptive soils. Soil Biol Biochem 63:37–49. doi:10.1016/j.soilbio.2013.02.007
Peay KG, Baraloto C, Fine PVA (2013) Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J 7:1852–1861. doi:10.1038/ismej.2013.66
Peñuelas J, Rico L, Ogaya R, Jump AS, Terradas J (2012) Summer season and long-term drought increase the richness of bacteria and fungi in the foliar phyllosphere of Quercus ilex in a mixed Mediterranean forest. Plant Biol 14:565–575. doi:10.1111/j.1438-8677.2011.00532.x
Peršoh D (2013) Factors shaping community structure of endophytic fungi–evidence from the Pinus-Viscum-system. Fungal Divers 60:55–69. doi:10.1007/s13225-013-0225-x
Peršoh D, Rambold G (2012) Lichen-associated fungi of the Letharietum vulpinae. Mycol Progress 11:753–760. doi:10.1007/s11557-011-0786-6
Peršoh D, Theuerl S, Buscot F, Rambold G (2008) Towards a universally adaptable method for quantitative extraction of high-purity nucleic acids from soil. J Microbiol Methods 75:19–24. doi:10.1016/j.mimet.2008.04.009
Peršoh D, Melcher M, Flessa F, Rambold G (2010) First fungal community analyses of endophytic ascomycetes associated with Viscum album ssp. austriacum and its host Pinus sylvestris. Fungal Biol 114:585–596. doi:10.1016/j.funbio.2010.04.009
Peršoh D, Weig AR, Rambold G (2012) A Transcriptome—targeting EcoChip for assessing functional mycodiversity. Microarrays 1:25–41. doi:10.3390/microarrays1010025
Peršoh D, Segert J, Zigan A, Rambold G (2013) Fungal community composition shifts along a leaf degradation gradient in a European beech forest. Plant Soil 362:175–186. doi:10.1007/s11104-012-1271-y
Phillips LA, Ward V, Jones MD (2014) Ectomycorrhizal fungi contribute to soil organic matter cycling in sub-boreal forests. ISME J 8:699–713. doi:10.1038/ismej.2013.195
Polz MF, Cavanaugh CM (1998) Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol 64:3724–3730
Popa R, Popa R, Mashall MJ, Nguyen H, Tebo BM, Brauer S (2009) Limitations and benefits of ARISA intra-genomic diversity fingerprinting. J Microbiol Methods 78:111–118. doi:10.1016/j.mimet.2009.06.005
Porras-Alfaro A, Bayman P (2011) Hidden fungi, emergent properties: endophytes and microbiomes. Annu Rev Phytopathol 49:291–315. doi:10.1146/annurev-phyto-080508-081831
Powell JR, Anderson IC, Rillig MC (2013) A new tool of the trade: plant-trait based approaches in microbial ecology. Plant Soil 365:35–40. doi:10.1007/s11104-012-1581-0
Pozhitkov AE, Noble PA, Bryk J, Tautz D (2014) A revised design for microarray experiments to account for experimental noise and uncertainty of probe response. PLoS One 9, e91295. doi:10.1371/journal.pone.0091295
Prescott CE, Grayston SJ (2013) Tree species influence on microbial communities in litter and soil: Current knowledge and research needs. For Ecol Manag 309:19–27. doi:10.1016/j.foreco.2013.02.034
Preston CM (2014) Environmental NMR: solid-state methods. eMagRes 3:29–42. doi:10.1002/9780470034590.emrstm1338
Preston CM, Sollins P, Sayer BG (1990) Changes in organic components for fallen logs in old-growth Douglas-fir forests monitored by 13C nuclear magnetic resonance spectroscopy. Can J For Res 20:1382–1391. doi:10.1139/x90-183
Preston CM, Nault JR, Trofymow JA (2009) Chemical changes during 6 years of decomposition of 11 litters in some Canadian forest sites. Part 2. 13C abundance, solid-state 13C NMR spectroscopy and the meaning of “Lignin”. Ecosystems 12:1078–1102. doi:10.1007/s10021-009-9267-z
Purahong W, Hyde KD (2011) Effects of fungal endophytes on grass and non-grass litter decomposition rates. Fungal Divers 47:1–7. doi:10.1007/s13225-010-0083-8
Purahong W, Kapturska D, Pecyna MJ, Jariyavidyanont K, Kaunzner J, Juncheed K, Uengwetwanit T, Rudloff R, Schulz E, Hofrichter M, Schloter M, Krüger D, Buscot F (2015) Effects of forest management practices in temperate beech forests on bacterial and fungal communities involved in leaf litter degradation. Microb Ecol. doi:10.1007/s00248-015-0585-8
Raidl S, Bonfigli R, Agerer R (2005) Calibration of quantitative real-time TaqMan PCR by correlation with hyphal biomass and ITS copies in mycelia of Piloderma croceum. Plant Biol 7:713–717. doi:10.1055/s-2005-873003
Raizen N (2013) Fungal endophyte diversity in foliage of native and cultivated rhododendron species determined by culturing, ITS sequencing, and pyrosequencing. Masters Thesis, Oregon State University, Corvallis
Rambold G, Agerer R (1997) DEEMY – the concept of a characterization and determination system for ectomycorrhizae. Mycorrhiza 7:113–116. doi:10.1007/s005720050171
Rambold G, Davydov E, Elix JA, Nash III TH, Scheidegger C, Zedda L (2001 onwards) LIAS light – A Database for Rapid Identification of Lichens. liaslight.lias.net
Rambold G, Elix JA, Heindl-Tenhunen B, Köhler T, Nash TH III, Neubacher D, Reichert W, Zedda L, Triebel D (2014) LIAS light – towards the ten thousand species milestone. MC 8:11–16
Redman RS, Kim YO, Woodward CJDA, Greer C, Espino L, Doty SL, Rodriguez RJ (2011) Increased fitness of rice plants to abiotic stress via habitat adapted symbiosis: a strategy for mitigating impacts of climate change. PLoS ONE 6, e14823. doi:10.1371/journal.pone.0014823
Rice WC, Gowda PH (2011) Influence of geographical location, crop type and crop residue cover on bacterial and fungal community structures. Geoderma 160:271–280. doi:10.1016/j.geoderma.2010.09.003
Robertson DG (2005) Metabonomics in toxicology: a review. Toxicol Sci 85:809–822. doi:10.1093/toxsci/kfi102
Rodriguez RJ, White JF, Arnold AE, Redman RS (2009) Fungal endophytes: diversity and functional roles. New Phytol 182:314–330. doi:10.1111/j.1469-8137.2009.02773.x
Roh SW, Abell GCJ, Kim K, Nam Y, Bae J (2010) Comparing microarrays and next-generation sequencing technologies for microbial ecology research. Trends Biotechnol 28:291–299. doi:10.1016/j.tibtech.2010.03.001
Rosenberg E, Sharon G, Atad I, Zilber-Rosenberg I (2010) The evolution of animals and plants via symbiosis with microorganisms. Environ Microbiol Rep 2:500–506. doi:10.1111/j.1758-2229.2010.00177.x
Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3:1154–1169. doi:10.1074/mcp.M400129-MCP200
Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351. doi:10.1038/ismej.2010.58
Roy J, Albert CH, Ibanez S, Saccone P, Zinger L, Choler P, Clément J, Lavergne S, Geremia RA (2013) Microbes on the cliff: alpine cushion plants structure bacterial and fungal communities. Front Microbiol 4:64. doi:10.3389/fmicb.2013.00064
Sagova-Mareckova M, Cermak L, Novotna J, Plhackova K, Forstova J, Kopecky J (2008) Innovative methods for soil DNA purification tested in soils with widely differing characteristics. Appl Environ Microbiol 74:2902–2907. doi:10.1128/AEM.02161-07
Saikkonen K, Saari S, Helander M (2010) Defensive mutualism between plants and endophytic fungi? Fungal Divers 41:101–113. doi:10.1007/s13225-010-0023-7
Sardans J, Peñuelas J, Rivas-Ubach A (2011) Ecological metabolomics: overview of current developments and future challenges. Chemoecol 21:191–225. doi:10.1007/s00049-011-0083-5
Satinsky BM, Gifford SM, Crump BC, Moran MA (2013) Use of internal standards for quantitative metatranscriptome and metagenome analysis. Meth Enzymol 531:237–250. doi:10.1016/B978-0-12-407863-5.00012-5
Scheibe A, Steffens C, Seven J, Jacob A, Hertel D, Leuschner C, Gleixner G (2015) Effects of tree identity dominate over tree diversity on the soil microbial community structure. Soil Biol Biochem 81:219–227. doi:10.1016/j.soilbio.2014.11.020
Schimel JP, Weintraub MN (2003) The implications of exoenzyme activity on microbial carbon and nitrogen limitation in soil: a theoretical model. Soil Biol Biochem 35:549–563. doi:10.1016/S0038-0717(03)00015-4
Schloss PD, Handelsman J (2003) Biotechnological prospects from metagenomics. Curr Opin Biotechnol 14:303–310. doi:10.1016/S0958-1669(03)00067-3
Schmidt MWI, Torn MS, Abiven S, Dittmar T, Guggenberger G, Janssens IA, Kleber M, Kögel-Knabner I, Lehmann J, Manning DAC, Nannipieri P, Rasse DP, Weiner S, Trumbore SE (2011) Persistence of soil organic matter as an ecosystem property. Nature 478:49–56. doi:10.1038/nature10386
Schmidt P, Bálint M, Greshake B, Bandow C, Römbke J, Schmitt I (2013) Illumina metabarcoding of a soil fungal community. Soil Biol Biochem 65:128–132. doi:10.1016/j.soilbio.2013.05.014
Schneider T, Riedel K (2010) Environmental proteomics: analysis of structure and function of microbial communities. Proteomics 10:785–798. doi:10.1002/pmic.200900450
Schneider T, Keiblinger KM, Schmid E, Sterflinger-Gleixner K, Ellersdorfer G, Roschitzki B, Richter A, Eberl L, Zechmeister-Boltenstern S, Riedel K (2012) Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J 6:1749–1762. doi:10.1038/ismej.2012.11
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A 109:6241–6246. doi:10.1073/pnas.1117018109
Schulze WX, Gleixner G, Kaiser K, Guggenberger G, Mann M, Schulze E (2005) A proteomic fingerprint of dissolved organic carbon and of soil particles. Oecologia 142:335–343. doi:10.1007/s00442-004-1698-9
Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C (2013) Computational meta’omics for microbial community studies. Mol Syst Biol 9:666. doi:10.1038/msb.2013.22
Seidler J, Zinn N, Boehm ME, Lehmann WD (2010) De novo sequencing of peptides by MS/MS. Proteomics 10:634–649. doi:10.1002/pmic.200900459
Seifert J, Herbst F, Halkjaer Nielsen P, Planes FJ, Jehmlich N, Ferrer M, von Bergen M (2013) Bioinformatic progress and applications in metaproteogenomics for bridging the gap between genomic sequences and metabolic functions in microbial communities. Proteomics 13:2786–2804. doi:10.1002/pmic.201200566
Senés-Guerrero C, Schüßler A (2015) A conserved arbuscular mycorrhizal fungal core-species community colonizes potato roots in the Andes. Fungal Divers. doi:10.1007/s13225-015-0328-7
Shi L, Mortimer PE, Ferry Slik JW, Zou X, Xu J, Feng W, Qiao L (2014) Variation in forest soil fungal diversity along a latitudinal gradient. Fungal Divers 64:305–315. doi:10.1007/s13225-013-0270-5
Sieber TN (2007) Endophytic fungi in forest trees: are they mutualists? Fungal Biol Rev 21:75–89. doi:10.1016/j.fbr.2007.05.004
Silver WL, Miya RK (2001) Global patterns in root decomposition: comparisons of climate and litter quality effects. Oecologia 129:407–419. doi:10.1007/s004420100740
Simon C, Daniel R (2011) Metagenomic analyses: past and future trends. Appl Environ Microbiol 77:1153–1161. doi:10.1128/AEM.02345-10
Smith DP, Peay KG (2014) Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS ONE 9, e90234. doi:10.1371/journal.pone.0090234
Smith SE, Read D (eds) (2008) Mycorrhizal symbiosis, 3rd edn. London, Academic Press
Šnajdr J, Cajthaml T, Valášková V, Merhautová V, Petránková M, Spetz P, Leppänen K, Baldrian P (2011) Transformation of Quercus petraea litter: successive changes in litter chemistry are reflected in differential enzyme activity and changes in the microbial community composition. FEMS Microbiol Ecol 75:291–303. doi:10.1111/j.1574-6941.2010.00999.x
Solly EF, Schöning I, Boch S, Kandeler E, Marhan S, Michalzik B, Müller J, Zscheischler J, Trumbore SE, Schrumpf M (2014) Factors controlling decomposition rates of fine root litter in temperate forests and grasslands. Plant Soil 382:203–218. doi:10.1007/s11104-014-2151-4
Soon WW, Hariharan M, Snyder MP (2013) High-throughput sequencing for biology and medicine. Mol Syst Biol 9:640. doi:10.1038/msb.2012.61
Stevenson BA, Hunter DW, Rhodes PL (2014) Temporal and seasonal change in microbial community structure of an undisturbed, disturbed, and carbon-amended pasture soil. Soil Biol Biochem 75:175–185. doi:10.1016/j.soilbio.2014.04.010
Stokland JN, Siitonen J, Jonsson BG (2012) Biodiversity in dead wood. Ecology, biodiversity, and conservation. Cambridge Univ. Press, Cambridge
Strickland MS, Lauber C, Fierer N, Bradford MA (2009) Testing the functional significance of microbial community composition. Ecology 90:441–451
Štursová M, Zifčáková L, Leigh MB, Burgess R, Baldrian P (2012) Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol Ecol 80:735–746. doi:10.1111/j.1574-6941.2012.01343.x
Štursová M, Šnajdr J, Cajthaml T, Bárta J, Šantrůčková H, Baldrian P (2014) When the forest dies: the response of forest soil fungi to a bark beetle-induced tree dieback. ISME J 8:1920–1931. doi:10.1038/ismej.2014.37
Sun X, Ding Q, Hyde KD, Guo LD (2012) Community structure and preference of endophytic fungi of three woody plants in a mixed forest. Fungal Ecol 5:624–632. doi:10.1016/j.funeco.2012.04.001
Suryanarayanan T (2011) Diversity of fungal endophytes in tropical trees. In: Pirttilä AM, Frank AC (eds) Endophytes of forest trees, vol 80. Springer, Netherlands, pp 67–80
Suryanarayanan TS (2013) Endophyte research: going beyond isolation and metabolite documentation. Fungal Ecol 6:561–568. doi:10.1016/j.funeco.2013.09.007
Takasaki K, Miura T, Kanno M, Tamaki H, Hanada S, Kamagata Y, Kimura N (2013) Discovery of glycoside hydrolase enzymes in an aviceladapted forest soil fungal community by a metatranscriptomic approach. PLoS ONE 8. doi:10.1371/journal.pone.0055485
Talbot JM, Yelle DJ, Nowick J, Treseder KK (2012) Litter decay rates are determined by lignin chemistry. Biogeochem 108:279–295. doi:10.1007/s10533-011-9599-6
Talbot JM, Bruns TD, Smith DP, Branco S, Glassman SI, Erlandson S, Vilgalys R, Peay KG (2013) Independent roles of ectomycorrhizal and saprotrophic communities in soil organic matter decomposition. Soil Biol Biochem 57:282–291. doi:10.1016/j.soilbio.2012.10.004
Talbot JM, Bruns TD, Taylor JW, Smith DP, Branco S, Glassman SI, Erlandson S, Vilgalys R, Liao H, Smith ME, Peay KG (2014) Endemism and functional convergence across the North American soil mycobiome. Proc Natl Acad Sci U S A 111:6341–6346. doi:10.1073/pnas.1402584111
Tan SC, Yiap BC (2009) DNA, RNA, and protein extraction: the past and the present. J Biomed Biotechnol 2009:574398. doi:10.1155/2009/574398
Tanaka A, Takemoto D, Chujo T, Scott B (2012) Fungal endophytes of grasses. Curr Opin Plant Biol 15:462–468. doi:10.1016/j.pbi.2012.03.007
Tateno O, Hirose D, Osono T, Takeda H (2014) Beech cupules share endophytic fungi with leaves and twigs. Mycosci. doi:10.1016/j.myc.2014.07.005
Taylor EB, Williams MA (2010) Microbial protein in soil: influence of extraction method and C amendment on extraction and recovery. Microb Ecol 59:390–399. doi:10.1007/s00248-009-9593-x
Taylor DL, Herriott IC, Long J, O’Neill K (2007) TOPO TA is A-OK: a test of phylogenetic bias in fungal environmental clone library construction. Environ Microbiol 9:1329–1334. doi:10.1111/j.1462-2920.2007.01253.x
Tedersoo L, Pärtel K, Jairus T, Gates G, Põldmaa K, Tamm H (2009) Ascomycetes associated with ectomycorrhizas: molecular diversity and ecology with particular reference to the Helotiales. Environ Microbiol 11:3166–3178. doi:10.1111/j.1462-2920.2009.02020.x
Tedersoo L, Mett M, Ishida TA, Bahram M (2013) Phylogenetic relationships among host plants explain differences in fungal species richness and community composition in ectomycorrhizal symbiosis. New Phytol 199:822–831. doi:10.1111/nph.12328
Tedersoo L, Bahram M, Dickie IA (2014a) Does host plant richness explain diversity of ectomycorrhizal fungi? Re-evaluation of Gao et al. (2013) data sets reveals sampling effects. Mol Ecol 23:992–995. doi:10.1111/mec.12660
Tedersoo L, Bahram M, Polme S, Koljalg U, Yorou NS, Wijesundera R, Villarreal Ruiz L, Vasco-Palacios AM, Thu PQ, Suija A, Smith ME, Sharp C, Saluveer E, Saitta A, Rosas M, Riit T, Ratkowsky D, Pritsch K, Poldmaa K, Piepenbring M, Phosri C, Peterson M, Parts K, Partel K, Otsing E, Nouhra E, Njouonkou AL, Nilsson RH, Morgado LN, Mayor J, May TW, Majuakim L, Lodge DJ, Lee SS, Larsson K, Kohout P, Hosaka K, Hiiesalu I, Henkel TW, Harend H, Guo L, Greslebin A, Grelet G, Geml J, Gates G, Dunstan W, Dunk C, Drenkhan R, Dearnaley J, de Kesel A, Dang T, Chen X, Buegger F, Brearley FQ, Bonito G, Anslan S, Abell S, Abarenkov K (2014b) Fungal biogeography. Global diversity and geography of soil fungi. Science (New York, NY) 346:1256688. doi:10.1126/science.1256688
Tellenbach C, Grünig CR, Sieber TN (2010) Suitability of quantitative real-time PCR to estimate the biomass of fungal root endophytes. Appl Environ Microbiol 76:5764–5772. doi:10.1128/AEM.00907-10
Tello SA, Silva-Flores P, Agerer R, Halbwachs H, Beck A, Peršoh D (2014) Hygrocybe virginea is a systemic endophyte of Plantago lanceolata. Mycol Progress 13:471–475. doi:10.1007/s11557-013-0928-0
Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75:1895–1904. doi:10.1021/ac0262560
Timling I, Walker DA, Nusbaum C, Lennon NJ, Taylor DL (2014) Rich and cold: diversity, distribution and drivers of fungal communities in patterned-ground ecosystems of the North American Arctic. Mol Ecol 23:3258–3272. doi:10.1111/mec.12743
Toju H, Tanabe AS, Yamamoto S, Sato H (2012) High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 7, e40863. doi:10.1371/journal.pone.0040863
Toju H, Sato H, Yamamoto S, Kadowaki K, Tanabe AS, Yazawa S, Nishimura O, Agata K (2013a) How are plant and fungal communities linked to each other in belowground ecosystems? A massively parallel pyrosequencing analysis of the association specificity of root-associated fungi and their host plants. Ecol Evol 3:3112–3124. doi:10.1002/ece3.706
Toju H, Yamamoto S, Sato H, Tanabe AS, Gilbert GS, Kadowaki K (2013b) Community composition of root-associated fungi in a Quercus-dominated temperate forest: “codominance” of mycorrhizal and root-endophytic fungi. Ecol Evol 3:1281–1293. doi:10.1002/ece3.546
Toju H, Sato H, Tanabe AS (2014) Diversity and spatial structure of belowground plant-fungal symbiosis in a mixed subtropical forest of ectomycorrhizal and arbuscular mycorrhizal plants. PLoS ONE 9, e86566. doi:10.1371/journal.pone.0086566
Treseder KK, Bent E, Borneman J, McGuire KL (2014) Shifts in fungal communities during decomposition of boreal forest litter. Fungal Ecol 10:58–69. doi:10.1016/j.funeco.2013.02.002
Trumbore S (2009) Radiocarbon and Soil Carbon Dynamics. Annu Rev Earth Planet Sci 37:47–66. doi:10.1146/annurev.earth.36.031207.124300
U’Ren JM, Riddle JM, Monacell JT, Carbone I, Miadlikowska J, Arnold AE (2014) Tissue storage and primer selection influence pyrosequencing-based inferences of diversity and community composition of endolichenic and endophytic fungi. Mol Ecol Resour 14:1032–1048. doi:10.1111/1755-0998.12252
Unterseher M (2011) Diversity of fungal endophytes in temperate forest trees. In: Pirttilä AM, Frank AC (eds) Endophytes of forest trees, 80. Springer, Netherlands, pp 31–46
Unterseher M, Reiher A, Finstermeier K, Otto P, Morawetz W (2007) Species richness and distribution patterns of leaf-inhabiting endophytic fungi in a temperate forest canopy. Mycol Progress 6:201–212. doi:10.1007/s11557-007-0541-1
Urbanová M, Šnajdr J, Baldrian P (2015) Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol Biochem 84:53–64. doi:10.1016/j.soilbio.2015.02.011
Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC (2008) Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3, e2527. doi:10.1371/journal.pone.0002527
Uroz S, Ioannidis P, Lengelle J, Cébron A, Morin E, Buée M, Martin F (2013) Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a Norway spruce plantation. PLoS ONE 8, e55929. doi:10.1371/journal.pone.0055929
Valentín L, Rajala T, Peltoniemi M, Heinonsalo J, Pennanen T, Mäkipää R (2014) Loss of diversity in wood-inhabiting fungal communities affects decomposition activity in Norway spruce wood. Front Microbiol 5:230. doi:10.3389/fmicb.2014.00230
van der Wal A, Ottosson E, de Boer W (2014) Neglected role of fungal community composition in explaining variation in wood decay rates. Ecology. doi:10.1890/14-0242.1
VerBerkmoes NC, Denef VJ, Hettich RL, Banfield JF (2009) Systems biology: Functional analysis of natural microbial consortia using community proteomics. Nat Rev Microbiol 7:196–205. doi:10.1038/nrmicro2080
Větrovský T, Baldrian P (2013) Analysis of soil fungal communities by amplicon pyrosequencing: current approaches to data analysis and the introduction of the pipeline SEED. Biol Fertil Soils 49:1027–1037. doi:10.1007/s00374-013-0801-y
Vilgalys R (2003) Taxonomic misidentification in public DNA databases. New Phytol 160:4–5. doi:10.1046/j.1469-8137.2003.00894.x
von Bergen M, Jehmlich N, Taubert M, Vogt C, Bastida F, Herbst F, Schmidt F, Richnow H, Seifert J (2013) Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology. ISME J 7:1877–1885. doi:10.1038/ismej.2013.78
Voříšková J, Baldrian P (2013) Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J 7:477–486. doi:10.1038/ismej.2012.116
Voříšková J, Brabcová V, Cajthaml T, Baldrian P (2013) Seasonal dynamics of fungal communities in a temperate oak forest soil. New Phytol 201:269–278. doi:10.1111/nph.12481
Walker JKM, Jones MD (2013) Little evidence for niche partitioning among ectomycorrhizal fungi on spruce seedlings planted in decayed wood versus mineral soil microsites. Oecologia 173:1499–1511. doi:10.1007/s00442-013-2713-9
Walker JF, Aldrich-Wolfe L, Riffel A, Barbare H, Simpson NB, Trowbridge J, Jumpponen A (2011) Diverse Helotiales associated with the roots of three species of Arctic Ericaceae provide no evidence for host specificity. New Phytol 191:515–527. doi:10.1111/j.1469-8137.2011.03703.x
Walker A, Pfitzner B, Neschen S, Kahle M, Harir M, Lucio M, Moritz F, Tziotis D, Witting M, Rothballer M, Engel M, Schmid M, Endesfelder D, Klingenspor M, Rattei T, Castell WZ, de Angelis MH, Hartmann A, Schmitt-Kopplin P (2014a) Distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J 8:2380–2396. doi:10.1038/ismej.2014.79
Walker JKM, Phillips LA, Jones MD (2014b) Ectomycorrhizal fungal hyphae communities vary more along a pH and nitrogen gradient than between decayed wood and mineral soil microsites. Botany 92:453–463. doi:10.1139/cjb-2013-0239
Wallenstein MD, Haddix ML, Ayres E, Steltzer H, Magrini-Bair KA, Paul EA (2013) Litter chemistry changes more rapidly when decomposed at home but converges during decomposition–transformation. Soil Biol Biochem 57:311–319. doi:10.1016/j.soilbio.2012.09.027
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi:10.1038/nrg2484
Wang G, Post WM, Mayes MA, Frerichs JT, Sindhu J (2012) Parameter estimation for models of ligninolytic and cellulolytic enzyme kinetics. Soil Biol Biochem 48:28–38. doi:10.1016/j.soilbio.2012.01.011
Wearn JA, Sutton BC, Morley NJ, Gange AC (2012) Species and organ specificity of fungal endophytes in herbaceous grassland plants. J Ecol 100:1085–1092. doi:10.1111/j.1365-2745.2012.01997.x
Wehner J, Powell JR, Muller LAH, Caruso T, Veresoglou SD, Hempel S, Rillig MC, van der Heijden M (2014) Determinants of root-associated fungal communities within Asteraceae in a semi-arid grassland. J Ecol 102:425–436. doi:10.1111/1365-2745.12197
Weig AR, Peršoh D, Werner S, Betzlbacher A, Rambold G (2013) Diagnostic assessment of mycodiversity in environmental samples by fungal ITS1 rDNA length polymorphism. Mycol Prog 12:719–725. doi:10.1007/s11557-012-0883-1
Werner S, Peršoh D, Rambold G (2012) Basidiobolus haptosporus is frequently associated with the gamasid mite Leptogamasus obesus. Fungal Biol 116:90–97. doi:10.1016/j.funbio.2011.10.004
White JR, Maddox C, White O, Angiuoli SV, Fricke WF (2013) CloVR-ITS: Automated internal transcribed spacer amplicon sequence analysis pipeline for the characterization of fungal microbiota. Microbiome 1:6. doi:10.1186/2049-2618-1-6
White JF Jr, Torres MS, Johnson H, Irizarry I, Tadych M (2014) A functional view of plant microbiomes: endosymbiotic systems that enhance plant growth and survival. In: Verma VC, Gange AC (eds) Advances in endophytic research. Springer, India, pp 425–439
Wickings K, Grandy AS, Reed SC, Cleveland CC (2012) The origin of litter chemical complexity during decomposition. Ecol Lett 15:1180–1188. doi:10.1111/j.1461-0248.2012.01837.x
Williams RJ, Hallgren SW, Wilson GW, Palmer MW (2013) Juniperus virginiana encroachment into upland oak forests alters arbuscular mycorrhizal abundance and litter chemistry. Appl Soil Ecol 65:23–30. doi:10.1016/j.apsoil.2012.12.020
Wilmes P, Wexler M, Bond PL (2008) Metaproteomics provides functional insight into activated sludge wastewater treatment. PLoS ONE 3, e1778. doi:10.1371/journal.pone.0001778
Winkler H (1920) Verbreitung und Ursache der Parthenogenesis im Pflanzen- und Tierreiche. Fischer, Jena
Wolfe BE, Tulloss RE, Pringle A (2012) The irreversible loss of a decomposition pathway marks the single origin of an ectomycorrhizal symbiosis. PLoS ONE 7, e39597. doi:10.1371/journal.pone.0039597
Wooley JC, Godzik A, Friedberg I (2010) A primer on metagenomics. PLoS Comput Biol 6, e1000667. doi:10.1371/journal.pcbi.1000667
Worden AZ, Allen AE (2010) The voyage of the microbial eukaryote. Curr Opin Microbiol 13:652–660. doi:10.1016/j.mib.2010.08.001
Wright EP, Partridge MA, Padula MP, Gauci VJ, Malladi CS, Coorssen JR (2014) Top-down proteomics: enhancing 2D gel electrophoresis from tissue processing to high-sensitivity protein detection. Proteomics 14:872–889. doi:10.1002/pmic.201300424
Wu L, Wang H, Zhang Z, Lin R, Zhang Z, Lin W (2011) Comparative metaproteomic analysis on consecutively Rehmannia glutinosa-monocultured rhizosphere soil. PLoS ONE 6, e20611. doi:10.1371/journal.pone.0020611
Wu YT, Wubet T, Trogisch S, Both S, Scholten T, Bruelheide H, Buscot F (2013) Forest Age and Plant Species Composition Determine the Soil Fungal Community Composition in a Chinese Subtropical Forest. PLoS ONE 8, e66829. doi:10.1371/journal.pone.0066829
Wu X, Gong F, Wang W (2014) Protein extraction from plant tissues for 2DE and its application in proteomic analysis. Proteomics 14:645–658. doi:10.1002/pmic.201300239
Wubet T, Christ S, Schöning I, Boch S, Gawlich M, Schnabel B, Fischer M, Buscot F (2012) Differences in soil fungal communities between European beech (Fagus sylvatica L.) dominated forests are related to soil and understory vegetation. PLoS ONE 7, e47500. doi:10.1371/journal.pone.0047500
Yadav SP (2007) The Wholeness in Suffix -omics, −omes, and the Word Om. J Biomol Tech 18:277
Zengler K, Palsson BO (2012) A road map for the development of community systems (CoSy) biology. Nat Rev Microbiol 10:366–372. doi:10.1038/nrmicro2763
Zheng Q, Song J, Doncaster K, Rowland E, Byers DM (2007) Qualitative and quantitative evaluation of protein extraction protocols for apple and strawberry fruit suitable for two-dimensional electrophoresis and mass spectrometry analysis. J Agric Food Chem 55:1663–1673. doi:10.1021/jf062850p
Zhou DQ, Hyde KD (2001) Host-specificity, host-exclusivity, and host-recurrence in saprobic fungi. Mycol Res 105:1449–1457
Zimmerman NB, Vitousek PM (2012) Fungal endophyte communities reflect environmental structuring across a Hawaiian landscape. Proc Natl Acad Sci U S A 109:13022–13027. doi:10.1073/pnas.1209872109
Acknowledgments
I thank Gerhard Rambold (Bayreuth) for his valuable comments on the initial manuscript. Constructive criticism by Kevin D. Hyde (Chiang Rai) and an anonymous reviewer helped to improve the manuscript significantly. My position was funded by the Deutsche Forschungsgemeinschaft (DFG, project PE 1673/4-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Peršoh, D. Plant-associated fungal communities in the light of meta’omics. Fungal Diversity 75, 1–25 (2015). https://doi.org/10.1007/s13225-015-0334-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-015-0334-9
Keywords
- Meta’omics
- Metaomics
- Fungal community
- Metagenomics
- Metaproteomics
- Metatranscriptomics
- Metametabolomics
- Phyllosphere fungi
- Endophytic fungi
- Litter decomposition
- Decomposer fungi
- Root-associated fungi
- Arbuscular mycorrhiza
- Ectomycorrhiza
- Ericoid mycorrhiza
- Soil fungi
- Functional diversity
- Environmental processes
- Distance decay
- Environmental filtering
- Saisonality
- Temporal shift
- Fungal traits