
Abstract
The aim of this study was to establish the physiologically based pharmacokinetic (PBPK) model of flurbiprofen related to CYP2C9 genetic polymorphism and describe the pharmacokinetics of flurbiprofen in different CYP2C9 genotypes. PK-Sim® software was used for the model development and validation. A total of 16 clinical pharmacokinetic data for flurbiprofen in different CYP2C9 genotypes, dose regimens, and age groups were used for the PBPK modeling. Turnover number (kcat) of CYP2C9 values were optimized to capture the observed profiles in different CYP2C9 genotypes. In the simulation, predicted fraction metabolized by CYP2C9, fraction excreted to urine, bioavailability, and volume of distribution were similar to previously reported values. Predicted plasma concentration-time profiles in different CYP2C9 genotypes were visually similar to the observed profiles. Predicted AUCinf in CYP2C9*1/*2, CYP2C9*1/*3, and CYP2C9*3/*3 genotypes were 1.44-, 2.05-, and 3.67-fold higher than the CYP2C9*1/*1 genotype. The ranges of fold errors for AUCinf, Cmax, and t1/2 were 0.84–1.00, 0.61–1.22, and 0.74–0.94 in development and 0.59–0.98, 0.52–0.97, and 0.61–1.52 in validation, respectively, which were within the acceptance criterion. Thus, the PBPK model was successfully established and described the pharmacokinetics of flurbiprofen in different CYP2C9 genotypes, dose regimens, and age groups. The present model could guide the decision-making of tailored drug administration strategy by predicting the pharmacokinetics of flurbiprofen in various clinical scenarios.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flurbiprofen, a nonsteroidal anti-inflammatory drug (NSAID), is indicated for symptomatic alleviation of osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis (Buchanan and Kassam 1986). Most frequently reported adverse events of flurbiprofen are gastrointestinal related, including abdominal pain, dyspepsia, nausea, diarrhea and constipation (Pfizer 2016). Flurbiprofen is generally marketed as racemates and it has more ulcerogenic potential than its (S)-enantiomer (Wechter et al. 1993). Flurbiprofen is rapidly absorbed with maximum plasma concentration observed between 0.5 and 3 h after oral administration and highly bound (> 99%) to plasma albumin (Davies 1995). Flurbiprofen is extensively metabolized via hydroxylation by cytochrome P450 (CYP) 2C9 (Tracy et al. 1996) and glucuronidation by UDP-glucuronosyltransferase (UGT) 2B7 and UGT1A9 (Wang et al. 2011). The metabolites are primarily eliminated in the kidney and an excreted ratio of unchanged flurbiprofen in urine is less than 3% (Pfizer 2016).
CYP2C9, accounting for up to 20% of P450 contents in the total human liver (Shimada et al. 1994), is responsible for the metabolism of approximately 15–20% of drugs that undergo Phase I metabolism (Evans and Relling 1999; Rettie and Jones 2005). CYP2C9 gene is highly polymorphic with at least 85 allele variants or sub-variants (CYP2C9*1B to CYP2C9*85) reported (https://www.pharmvar.org/gene/CYP2C9). Of these alleles, CYP2C9*2 (rs1799853, c.430 C > T, p.Cys144Arg) and CYP2C9*3 (rs1057910, c.1075 A > C, p.Ile359Leu), the most frequently observed allele variants worldwide (Daly et al. 2017), exhibit the reduction of enzyme activity for the substrate of CYP2C9 in vitro and in vivo (Yasar et al. 2001; Perini et al. 2005; Bae et al. 2011b; Lee et al. 2014). Wang et al. (2015) showed the catalytic activities of CYP2C9*2 and *3 variants for flurbiprofen in vitro were decreased by 61.4 and 24.3% compared to the wild-type allele, respectively. Several studies reported that genetic polymorphism of CYP2C9 significantly affected the pharmacokinetics of flurbiprofen in humans (Lee et al. 2003, 2015; Kumar et al. 2008). Clinical Pharmacogenetic Implementation Consortium (CPIC) proposed 50–75% dose reduction for NSAIDs with a short half-life such as celecoxib, flurbiprofen, ibuprofen and lornoxicam in CYP2C9 poor metabolizers (CYP2C9PMs) (Theken et al. 2020).
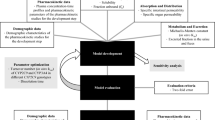
Physiologically based pharmacokinetic (PBPK) modeling is a mechanistic approach to predict the dispositions of xenobiotics in humans and other animal species (Kuepfer et al. 2016; Zhuang and Lu 2016). It is a valuable tool for tailoring drug administration strategies according to the alteration of physiological characteristics (Abduljalil and Badhan 2020; Verscheijden et al. 2020; Heimbach et al. 2021), drug–drug interactions (Abouir et al. 2021; Ferreira et al. 2021), cigarette consumption (Plowchalk and Rowland Yeo 2012), and genetic polymorphism (Cho et al. 2021a, 2021b; Jung et al. 2021; Kim et al. 2021; Rüdesheim et al. 2022). In the present study, we aimed to develop and validate the PBPK model for flurbiprofen related to the genetic polymorphism of CYP2C9.
Materials and methods
Software and data source
PBPK model was developed and validated using PK-Sim® version 10.0 (Bayer AG, Leverkusen, Germany). Plasma concentration-time profiles for model development were digitized with Engauge Digitizer® version 12.1 (https://markummitchell.github.io/engauge-digitizer/) according to the proposed digitization algorithm in Wojtyniak et al. (2020). Pharmacokinetic parameters which were not obtained from the publications were estimated via non-compartmental analysis (NCA) with the BA calc 2007 analysis program (MFDS, Cheongju, Republic of Korea).
A total of 16 clinical pharmacokinetic data for flurbiprofen were collected and used to develop and validate the PBPK model. The data for fifteen subjects with CYP2C9*1/*1 (n = 5), CYP2C9*1/*2 (n = 5), and CYP2C9*1/*3 (n = 5) genotypes from the report of Lee et al. (2003) and for two subjects with CYP2C9*3/*3 genotype from the report of Kumar et al. (2008) were used to develop the model. Other 12 data were used to validate the developed model. Clinical data from studies without information on the CYP2C9 genotype were assumed that the all subjects were carrying the CYP2C9*1/*1 genotype.
Model building
“Middle-out” strategy was used for the model building. Physicochemical properties of the drug were collected from previous publications or drug databases. The specific intestinal and organ permeabilities were calculated in the software (Thelen et al. 2011, 2012). CYP2C9, UGT1A9, and UGT2B7 enzymes were incorporated to describe the metabolism of flurbiprofen. Michaelis–Menten constant (Km) values for CYP2C9 (Wang et al. 2015) and UGTs (Wang et al. 2011) were obtained from previous studies. CYP2C9 turnover number (kcat) values in different CYP2C9 genotypes were optimized to capture the observed profiles. The kcat values for UGT1A9 and UGT2B7 were optimized based on clinical pharmacokinetic data for CYP2C9*3/*3 genotype in Kumar et al. (2008) in which the effects of UGT1A9 or UGT2B7 on the total clearance of flurbiprofen were expected to be sensitive. The reference concentration of CYP2C9 was 3.84 µmol/L (Rodrigues 1999) and UGTs was 1.00 µmol/L, the default value of PK-Sim®. Relative expression values for CYP2C9 and UGTs in each organ were obtained from the reverse transcription-polymerase chain reaction (RT-PCR) data (Nishimura et al. 2003; Nishimura and Naito 2005, 2006). Hepatic plasma clearance was incorporated with considerations of minor enzymatic pathways, those not mediated by CYP2C9, UGT1A9, and UGT2B7. Renal plasma clearance was adjusted to recover the excreted fraction in urine as unchanged form (< 3% of dose, Pfizer 2016). Dissolution time (80% dissolved) was adjusted from the dissolution profiles for marketed tablet of Daravath et al. (2018) and lag time was optimized to capture the observed profiles more accurately. Parameter optimization was performed via the Levenberg-Marquardt algorithm in the PK-Sim® software. Schmitt (2008) and PK-Sim® standard method (Hindmarsh et al. 2021) was used to estimate partition coefficients and cellular permeabilities, respectively.
Sensitivity analysis
Sensitivity analysis was performed in the PK-Sim® software. In the analysis, a total of 241 input parameters were evaluated for the area under the plasma concentration-time curve from time zero to infinity (AUCinf) and peak plasma concentration (Cmax). The sensitivity was calculated as follows
where \(S\) is the sensitivity, \(PK\) is the initial values of the pharmacokinetic parameter, \({\Delta PK}\) is the change of the pharmacokinetic parameters from initial values, \(p\) is the initial values of the evaluated input parameter, and \({\Delta p}\) is the change of evaluated input parameters from initial values, respectively. A sensitivity of + 1.0 indicates that + 10% change of an evaluated input parameter causes + 10% change of the predicted pharmacokinetic parameters.
Model evaluation
The PBPK model was evaluated using visual and numerical methods. Observed plasma concentration-time profiles were visually compared with the predicted profiles by plotting the geometric mean and 5th–95th percentiles for a virtual population (n = 100). Demographic data for virtual populations were designated to be similar to those of the observed populations. The demographic data which could not be obtained from previous studies were generated using the implemented algorithm in the PK-Sim® software. Standard deviations for the reference concentration of CYP2C9 and UGTs were assigned as 1.15 and 0.30 µmol/L, respectively, to reflect moderate variability (30% of the mean). The PBPK model were numerically evaluated by comparing the observed and predicted AUCinf, Cmax, and half-life (t1/2) values. A two-fold error range for the pharmacokinetic parameters was used as the evaluation criterion. The PBPK model was acceptable if the fold error (predicted value divided by observed value) is within the 0.5–2 range.
Results
The summary of input parameters for the PBPK model is presented in Table 1. In the simulation shown in Fig. 1, estimated values for the fraction metabolized by CYP2C9 (fm,CYP2C9) and fraction excreted to urine of 71.8 and 2.91%, respectively, were very close to the reported values of 71 (Patel et al. 2003; Loisios-Konstantinidis et al. 2020) and < 3%, respectively (Pfizer 2016). Bioavailability and volume of distribution (Vd/F) were estimated as 0.94 and 0.11 L/kg, similar to the reported values of 0.96 and 0.12 L/kg, respectively (Pfizer 2016).

Predicted and observed profiles of flurbiprofen after 50 mg single oral administration. The black solid, red dashed, and blue dotted lines indicate plasma concentration, fraction excreted to urine, and fraction metabolized by CYP2C9, respectively. Black open circles and error bars indicate the mean and standard deviation of observed plasma concentration, respectively. Red and blue open circles indicate the observed fraction excreted to urine and fraction metabolized by CYP2C9, respectively
Predicted plasma concentration-time profiles for CYP2C9 allele variants were visually similar to the observed profiles (Fig. 2). Predicted AUCinf in CYP2C9*1/*2, CYP2C9*1/*3, and CYP2C9*3/*3 genotypes were 1.44-, 2.05-, and 3.67-fold higher than CYP2C9*1/*1 genotype, respectively. Significant differences for predicted Cmax in different CYP2C9 genotypes were not identified (5.1–5.8 µg/mL). The ranges of fold errors for AUCinf, Cmax, and t1/2 in development were 0.84–1.00, 0.61–1.22, and 0.74–0.94, respectively, which were within the acceptance criterion (Table 2).

Predicted and observed plasma concentration–time profiles of flurbiprofen in different CYP2C9 genotypes. Solid and dashed lines indicate geometric mean and 5th–95th percentile, respectively. Open circles indicate the mean of observed plasma concentrations. Profiles are expressed using linear and semi-logarithmic scales
Sensitivity analysis is shown in Fig. 3. Dose had the equally highest impact on both AUCinf and Cmax. Several physicochemical parameters including lipophilicity and fraction unbound were sensitive to AUCinf and Cmax. Parameters related to CYP2C9 and UGT2B7 enzymatic pathways such as Km, kcat, and reference concentration were identified as sensitive but UGT1A9 was not. Renal plasma clearance and pKa had slight influence on AUCinf and Cmax, respectively.

Results of sensitivity analysis toward AUCinf (A) and Cmax (B). x-axis and y-axis indicate sensitivity values and lists of sensitive parameters, respectively
A total of 12 clinical data for the validation included 40–150 mg single dose regimen in various age groups (from 6 to 83 years). In validation, the ranges of fold errors for AUCinf, Cmax, and t1/2 were 0.59–0.98, 0.52–0.97, and 0.61–1.52, respectively, which is within the acceptance criterion (Table 2).
Discussion
The activity of the drug metabolizing enzymes and transporters is closely related to the disposition of the drug in the body. Genetic polymorphism of drug metabolizing enzymes and transporters causes the inter-individual variability in drug responses. Various studies to investigate the influences of genetic variants of drug metabolizing enzymes (Choi et al. 2012; Byeon et al. 2015; Bae et al. 2020; Jung et al. 2020a, b; Kim et al. 2022) and transporters (Sai et al. 2010; Choi et al. 2013; Shin et al. 2020) on the pharmacokinetics or pharmacodynamics of clinically used drugs have been reported. As the utilization of the PBPK modeling approach has been rapidly increasing in the last couple of decades (El-Khateeb et al. 2021), access to a tailored drug administration strategy, considering physiological characteristics, genotypes, diseases, and drug interactions of individuals, has been attempted using the PBPK model (Rüdesheim et al. 2020, 2022; Cho et al. 2021a, b; Jung et al. 2021; Kim et al. 2021; Marok et al. 2021).
Majority of NSAIDs including celecoxib, lornoxicam, meloxicam, naproxen, and piroxicam are metabolized by CYP2C9 and the influences of CYP2C9 allele variants on the pharmacokinetics and pharmacodynamics of NSAIDs have been studied (Perini et al. 2005; Bae et al. 2009, 2011a, b; Choi et al. 2011; Kim et al. 2017). Likewise, significant influences on the pharmacokinetics of flurbiprofen according to CYP2C9 genetic polymorphism have been identified (Lee et al. 2003, 2015; Kumar et al. 2008). CPIC and drug label recommended dose reduction of flurbiprofen in the CYP2C9PM phenotype (Pfizer 2016; Theken et al. 2020). They suggested that the genetic polymorphism of CYP2C9 is one of the important factors causing inter-individual variability of the responses of flurbiprofen.
The present PBPK model was developed by leveraging a number of information related to physicochemical and pharmacokinetic (absorption, distribution, metabolism, and excretion [ADME]) characteristics of flurbiprofen. An in vitro study showed the major oxidative pathway in flurbiprofen metabolism was conversion to a 4′-hydroxy metabolite mediated by CYP2C9 (Tracy et al. 1996). Also, UGT2B7 and UGT1A9 predominantly and minorly contributed in the glucuronidation of flurbiprofen, respectively (Mano et al. 2007; Wang et al. 2011). The results of sensitivity analysis demonstrated that the present model sufficiently reflected the contributions of the metabolizing enzymes (Fig. 3). Mano et al. (2007) also reported UGT1A1, UGT1A3, and UGT2B4 exhibit the glucuronidation activity of flurbiprofen. Hepatic plasma clearance was additionally incorporated to consider the contributions of these enzymes. In the initial model, in vitro metabolism data (Wang et al. 2015) was incorporated without any modification and AUC was highly overestimated. Lee et al. (2021) reported that in vitro test tends to underpredict the in vivo clearance and recommended the optimization for metabolism parameters based on in vivo data. Thus, CYP2C9 kcat value was optimized to capture the observed profiles. The estimated values including fm,CYP2C9, urine excretion, bioavailability, and volume of distribution, were almost similar to previously reported values and this suggests that our model can predict the pharmacokinetics of flurbiprofen.
Our model properly captured the pharmacokinetics of flurbiprofen not only for the CYP2C9*1/*1 genotype but also for genotypes with CYP2C9 allele variants. The predicted profiles were visually similar to the observed profiles in different CYP2C9 genotypes (Fig. 2). Also, the range of fold error values for AUCinf, Cmax, and t1/2 in development and validation satisfied the acceptance criterion (Table 2). This suggests that the PBPK model could capture the disposition of flurbiprofen in different dose regimens, demographic characteristics, and genotypes. Although observed Cmax of CYP2C9*3/*3 genotype slightly deviated from the predicted range visually, the qualified model can provide reasonable information for the pharmacokinetics of flurbiprofen in different CYP2C9 genotypes with reduction in the risk of adverse events by administration of the drug.
The present model properly described the pharmacokinetics of flurbiprofen in special populations including the pediatric and geriatric populations. PK-Sim® provides the quantitative data for age-dependent physiological alterations in pediatric and geriatric populations (Edginton et al. 2006; Schlender et al. 2016). It easily enables the prediction of the pharmacokinetics of drugs in various age groups. Thus, we validated the applicability of the model using the clinical data on children (Scaroni et al. 1984) or elderly subjects (Kean et al. 1992) and identified that the PBPK model could be applied to these populations.
Modeling studies to describe the pharmacokinetics of flurbiprofen in humans have been previously identified. Kumpulainen et al. (2010) and Zhang et al. (2018) reported population pharmacokinetic models in healthy children and Chinese patients with postoperative pain, respectively. As these models only applied mathematical methods based on the clinical data to predict the pharmacokinetics of the drug, limitations exist in that physicochemical and physiological characteristics could not be incorporated. Verscheijden et al. (2019) developed a pediatric brain PBPK model to predict the cerebrospinal fluid drug concentrations and flurbiprofen was used as a drug for model validation. However, their model was focused on the pediatric population and the pharmacokinetics of flurbiprofen in the adult population was not presented. Loisios-Konstantinidis et al. (2020) developed the flurbiprofen PBPK model using Simcyp® software to predict the effects of various factors including CYP2C9 genetic polymorphism, co-administration, and formulation. Although their model can properly describe the pharmacokinetic alterations of flurbiprofen according to CYP2C9 genetic polymorphism, we developed the flurbiprofen model using PK-Sim® software because there are differences in the description of ADME characteristics between platforms and it can cause discrepant simulation consequences from the equal input parameters. Furthermore, they successfully established the flurbiprofen model in the adult populations. Whether their model could adequately be applied in the pediatric or geriatric populations is uncertain. In the present study, we developed the flurbiprofen model and validated it using the clinical data for a wide range of ages groups including the pediatric and geriatric populations.
The established PBPK model for flurbiprofen has several limitations. Validation was not performed in CYP2C9*1/*2 and CYP2C9*3/*3 genotypes due to the lack of pharmacogenetic studies for these genotypes. To our knowledge, only one clinical pharmacokinetic data for each of the CYP2C9*1/*2 (Lee et al. 2003) and CYP2C9*3/*3 genotypes (Kumar et al. 2008) were identified to date, and these were used in the development. Furthermore, we did not consider the physiological alterations in the patients with arthritis. In our study, we developed the PBPK model using the pharmacogenetic studies for healthy subjects and validated the model by using equal parameters in healthy subjects and patients with arthritis. Further research for the physiological differences between healthy and patient populations may improve the model. It would be better to apply the present model under the consideration of these potential limitations.
In conclusion, the flurbiprofen PBPK model in different CYP2C9 genotypes was successfully established. The present model could guide the decision-making of tailored drug administration strategy by predicting the pharmacokinetics of flurbiprofen in various clinical scenarios.
References
Abduljalil K, Badhan RKS (2020) Drug dosing during pregnancy-opportunities for physiologically based pharmacokinetic models. J Pharmacokinet Pharmacodyn 47(4):319–340. https://doi.org/10.1007/s10928-020-09698-w
Abouir K, Samer CF, Gloor Y, Desmeules JA, Daali Y (2021) Reviewing data integrated for PBPK model development to predict metabolic drug–drug interactions: shifting perspectives and emerging trends. Front Pharmacol 12:708299. https://doi.org/10.3389/fphar.2021.708299
Bae JW, Kim JH, Choi CI, Kim MJ, Kim HJ, Byun SA, Chang YS, Jang CG, Park YS, Lee SY (2009) Effect of CYP2C9*3 allele on the pharmacokinetics of naproxen in Korean subjects. Arch Pharm Res 32(2):269–273. https://doi.org/10.1007/s12272-009-1232-z
Bae JW, Choi CI, Jang CG, Lee SY (2011a) Effects of CYP2C9*1/*13 on the pharmacokinetics and pharmacodynamics of meloxicam. Br J Clin Pharmacol 71(4):550–555. https://doi.org/10.1111/j.1365-2125.2010.03853.x
Bae JW, Choi CI, Kim MJ, Oh DH, Keum SK, Park JI, Kim BH, Bang HK, Oh SG, Kang BS, Park HJ, Kim HD, Ha JH, Shin HJ, Kim YH, Na HS, Chung MW, Jang CG, Lee SY (2011b) Frequency of CYP2C9 alleles in Koreans and their effects on losartan pharmacokinetics. Acta Pharmacol Sin 32(10):1303–1308. https://doi.org/10.1038/aps.2011.100
Bae JW, Oh KY, Yoon SJ, Shin HB, Jung EH, Cho CK, Lim CW, Kang P, Choi CI, Jang CG, Lee SY, Lee YJ (2020) Effects of CYP2D6 genetic polymorphism on the pharmacokinetics of metoclopramide. Arch Pharm Res 43(11):1207–1213. https://doi.org/10.1007/s12272-020-01293-4
Buchanan WW, Kassam YB (1986) European experience with flurbiprofen: a new analgesic/anti-inflammatory agent. Am J Med 80(3):145–152. https://doi.org/10.1016/0002-9343(86)90134-8
Byeon JY, Kim YH, Na HS, Jang JH, Kim SH, Lee YJ, Bae JW, Kim IS, Jang CG, Chung MW, Lee SY (2015) Effects of the CYP2D6*10 allele on the pharmacokinetics of atomoxetine and its metabolites. Arch Pharm Res 38(11):2083–2091. https://doi.org/10.1007/s12272-015-0646-z
Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2021a) Physiologically based pharmacokinetic (PBPK) modelling of tamsulosin related to CYP2D6*10 allele. Arch Pharm Res 44(11):1037–1049. https://doi.org/10.1007/s12272-021-01357-z
Cho CK, Park HJ, Kang P, Moon S, Lee YJ, Bae JW, Jang CG, Lee SY (2021b) Physiologically based pharmacokinetic (PBPK) modeling of meloxicam in different CYP2C9 genotypes. Arch Pharm Res 44(12):1076–1090. https://doi.org/10.1007/s12272-021-01361-3
Choi CI, Kim MJ, Jang CG, Park YS, Bae JW, Lee SY (2011) Effects of the CYP2C9*1/*13 genotype on the pharmacokinetics of lornoxicam. Basic Clin Pharmacol Toxicol 109(6):476–480. https://doi.org/10.1111/j.1742-7843.2011.00751.x
Choi CI, Kim MJ, Chung EK, Lee HI, Jang CG, Bae JW, Lee SY (2012) CYP2C9*3 and *13 alleles significantly affect the pharmacokinetics of irbesartan in healthy Korean subjects. Eur J Clin Pharmacol 68(2):149–154. https://doi.org/10.1007/s00228-011-1098-0
Choi CI, Bae JW, Keum SK, Lee YJ, Lee HI, Jang CG, Lee SY (2013) Effects of OCT2 c.602 C > T genetic variant on the pharmacokinetics of lamivudine. Xenobiotica 43(7):636–640. https://doi.org/10.3109/00498254.2012.747710
Daly AK, Rettie AE, Fowler DM, Miners JO (2017) Pharmacogenomics of CYP2C9: functional and clinical considerations. J Pers Med 8(1):1. https://doi.org/10.3390/jpm8010001
Daravath B, Naveen C, Vemula SK, Tadikonda RR (2018) Solubility and dissolution enhancement of flurbiprofen by solid dispersion using hydrophilic carriers. Braz J Pharm Sci 53(4):e00010. https://doi.org/10.1590/s2175-97902017000400010
Davies NM (1995) Clinical pharmacokinetics of flurbiprofen and its enantiomers. Clin Pharmacokinet 28(2):100–114. https://doi.org/10.2165/00003088-199528020-00002
Edginton AN, Schmitt W, Willmann S (2006) Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet 45(10):1013–1034. https://doi.org/10.2165/00003088-200645100-00005
El-Khateeb E, Burkhill S, Murby S, Amirat H, Rostami-Hodjegan A, Ahmad A (2021) Physiological-based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20-years; in-depth analysis of applications, organizations, and platforms. Biopharm Drug Dispos 42(4):107–117. https://doi.org/10.1002/bdd.2257
Evans WE, Relling MV (1999) Pharmacogenomics: translating functional genomics into rational therapeutics. Science 286(5439):487–491. https://doi.org/10.1126/science.286.5439.487
Ferreira A, Lapa R, Vale N (2021) PBPK modeling and simulation and therapeutic drug monitoring: possible ways for antibiotic dose adjustment. Processes 9(11):2087. https://doi.org/10.3390/pr9112087
Hanley MJ, Masse G, Harmatz JS, Court MH, Greenblatt DJ (2012) Pomegranate juice and pomegranate extract do not impair oral clearance of flurbiprofen in human volunteers: divergence from in vitro results. Clin Pharmacol Ther 92(5):651–657. https://doi.org/10.1038/clpt.2012.170
Heimbach T, Chen Y, Chen J, Dixit V, Parrott N, Peters SA, Poggesi I, Sharma P, Snoeys J, Shebley M, Tai G, Tse S, Upreti VV, Wang YH, Tsai A, Xia B, Zheng M, Zhu AZX, Hall S (2021) Physiologically-based pharmacokinetic modeling in renal and hepatic impairment populations: a pharmaceutical industry perspective. Clin Pharmacol Ther 110(2):297–310. https://doi.org/10.1002/cpt.2125
Hindmarsh AC, Reynolds DR, Serban R, Woodward CS, Gardner DJ, Cohen SD, Taylor A, Peles S, Banks L, Shumaker D (2021) Open systems pharmacology suite manual version 10. Available from https://docs.open-systems-pharmacology.org/. Accessed 23 May 2022
Jung EH, Lee CM, Byeon JY, Shin HB, Oh KY, Cho CK, Lim CW, Jang CG, Lee SY, Lee YJ (2020a) Relationship between plasma exposure of zolpidem and CYP2D6 genotype in healthy Korean subjects. Arch Pharm Res 43(9):976–981. https://doi.org/10.1007/s12272-020-01250-1
Jung EH, Lee YJ, Kim DH, Kang P, Lim CW, Cho CK, Jang CG, Lee SY, Bae JW (2020b) Effects of paroxetine on the pharmacokinetics of atomoxetine and its metabolites in different CYP2D6 genotypes. Arch Pharm Res 43(12):1356–1363. https://doi.org/10.1007/s12272-020-01300-8
Jung EH, Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Choi CI, Jang CG, Lee SY (2021) Physiologically based pharmacokinetic modeling of candesartan related to CYP2C9 genetic polymorphism in adult and pediatric patients. Arch Pharm Res 44(12):1109–1119. https://doi.org/10.1007/s12272-021-01363-1
Kean WF, Antal EJ, Grace EM, Cauvier H, Rischke J, Buchanan WW (1992) The pharmacokinetics of flurbiprofen in younger and elderly patients with rheumatoid arthritis. J Clin Pharmacol 32(1):41–48. https://doi.org/10.1002/j.1552-4604.1992.tb03786.x
Kim SH, Kim DH, Byeon JY, Kim YH, Kim DH, Lim HJ, Lee CM, Whang SS, Choi CI, Bae JW, Lee YJ, Jang CG, Lee SY (2017) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of celecoxib and its carboxylic acid metabolite. Arch Pharm Res 40(3):382–390. https://doi.org/10.1007/s12272-016-0861-2
Kim YH, Kang P, Cho CK, Jung EH, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2021) Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch Pharm Res 44(7):713–724. https://doi.org/10.1007/s12272-021-01346-2
Kim NT, Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2022) Effects of CYP2C9*3 and *13 alleles on the pharmacokinetics and pharmacodynamics of glipizide in healthy Korean subjects. Arch Pharm Res 45(2):114–121. https://doi.org/10.1007/s12272-021-01366-y
Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, Block M, Eissing T, Teutonico D (2016) Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol 5(10):516–531. https://doi.org/10.1002/psp4.12134
Kumar V, Brundage RC, Oetting WS, Leppik IE, Tracy TS (2008) Differential genotype dependent inhibition of CYP2C9 in humans. Drug Metab Dispos 36(7):1242–1248. https://doi.org/10.1124/dmd.108.020396
Kumpulainen E, Välitalo P, Kokki M, Lehtonen M, Hooker A, Ranta VP, Kokki H (2010) Plasma and cerebrospinal fluid pharmacokinetics of flurbiprofen in children. Br J Clin Pharmacol 70(4):557–566. https://doi.org/10.1111/j.1365-2125.2010.03720.x
Lee CR, Pieper JA, Frye RF, Hinderliter AL, Blaisdell JA, Goldstein JA (2003) Differences in flurbiprofen pharmacokinetics between CYP2C9*1/*1, *1/*2, and *1/*3 genotypes. Eur J Clin Pharmacol 58(12):791–794. https://doi.org/10.1007/s00228-003-0574-6
Lee HI, Bae JW, Choi CI, Lee YJ, Byeon JY, Jang CG, Lee SY (2014) Strongly increased exposure of meloxicam in CYP2C9*3/*3 individuals. Pharmacogenet Genomics 24(2):113–117. https://doi.org/10.1097/fpc.0000000000000025
Lee YJ, Byeon JY, Kim YH, Kim SH, Choi CI, Bae JW, Sohn UD, Jang CG, Lee J, Lee SY (2015) Effects of CYP2C9*1/*3 genotype on the pharmacokinetics of flurbiprofen in Korean subjects. Arch Pharm Res 38(6):1232–1237. https://doi.org/10.1007/s12272-015-0580-0
Lee J, Yang Y, Zhang X, Fan J, Grimstein M, Zhu H, Wang Y (2021) Usage of in vitro metabolism data for drug–drug interaction in physiologically based pharmacokinetic analysis submissions to the US Food and Drug Administration. J Clin Pharmacol 61(6):782–788. https://doi.org/10.1002/jcph.1819
Liu YM, Liu GY, Liu Y, Li SJ, Jia JY, Zhang MQ, Lu C, Zhang YM, Li XN, Yu C (2009) Pharmacokinetic and bioequivalence comparison between orally disintegrating and conventional tablet formulations of flurbiprofen: a single-dose, randomized-sequence, open-label, two-period crossover study in healthy chinese male volunteers. Clin Ther 31(8):1787–1795. https://doi.org/10.1016/j.clinthera.2009.08.008
Loisios-Konstantinidis I, Cristofoletti R, Jamei M, Turner D, Dressman J (2020) Physiologically based pharmacokinetic/pharmacodynamic modeling to predict the impact of CYP2C9 genetic polymorphisms, co-medication and formulation on the pharmacokinetics and pharmacodynamics of flurbiprofen. Pharmaceutics 12(11):1049. https://doi.org/10.3390/pharmaceutics12111049
Mano Y, Usui T, Kamimura H (2007) Predominant contribution of UDP-glucuronosyltransferase 2B7 in the glucuronidation of racemic flurbiprofen in the human liver. Drug Metab Dispos 35(7):1182–1187. https://doi.org/10.1124/dmd.107.015347
Marok FZ, Fuhr LM, Hanke N, Selzer D, Lehr T (2021) Physiologically based pharmacokinetic modeling of bupropion and its metabolites in a CYP2B6 drug–drug–gene interaction network. Pharmaceutics. https://doi.org/10.3390/pharmaceutics13030331
Nishimura M, Naito S (2005) Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet 20(6):452–477. https://doi.org/10.2133/dmpk.20.452
Nishimura M, Naito S (2006) Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet 21(5):357–374. https://doi.org/10.2133/dmpk.21.357
Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T (2003) Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi 123(5):369–375. https://doi.org/10.1248/yakushi.123.369
Patel B, Jackson S, Swift C, Hutt A (2003) Disposition of flurbiprofen in man: influence of stereochemistry and age. Xenobiotica 33(10):1043–1057. https://doi.org/10.1080/00498250310001602739
Perini JA, Vianna-Jorge R, Brogliato AR, Suarez‐Kurtz G (2005) Influence of CYP2C9 genotypes on the pharmacokinetics and pharmacodynamics of piroxicam. Clin Pharmacol Ther 78(4):362–369. https://doi.org/10.1016/j.clpt.2005.06.014
Pfizer (2016) ANSAID® (flurbiprofen) prescribing information. Available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/018766s020lbl.pdf. Accessed 23 May 2022
Plowchalk DR, Rowland Yeo K (2012) Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Clin Pharmacol 68(6):951–960. https://doi.org/10.1007/s00228-011-1189-y
Qayyum A, Najmi MH, Abbas M (2013) Comparative bioavailability and pharmacokinetics of two oral formulations of flurbiprofen: a single-dose, randomized, open-label, two-period, crossover study in Pakistani subjects. Pak J Pharm Sci 26(6):1221–1227
Rettie AE, Jones JP (2005) Clinical and toxicological relevance of CYP2C9: drug–drug interactions and pharmacogenetics. Annu Rev Pharmacol Toxicol 45:477–494. https://doi.org/10.1146/annurev.pharmtox.45.120403.095821
Rodrigues AD (1999) Integrated cytochrome P450 reaction phenotyping—attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem Pharmacol 57(5):465–480. https://doi.org/10.1016/s0006-2952(98)00268-8
Rüdesheim S, Wojtyniak JG, Selzer D, Hanke N, Mahfoud F, Schwab M, Lehr T (2020) Physiologically based pharmacokinetic modeling of metoprolol enantiomers and α-hydroxymetoprolol to describe CYP2D6 drug–gene interactions. Pharmaceutics 12(12):1200. https://doi.org/10.3390/pharmaceutics12121200
Rüdesheim S, Selzer D, Fuhr U, Schwab M, Lehr T (2022) Physiologically-based pharmacokinetic modeling of dextromethorphan to investigate interindividual variability within CYP2D6 activity score groups. CPT Pharmacometrics Syst Pharmacol 11(4):494–511. https://doi.org/10.1002/psp4.12776
Sai K, Saito Y, Maekawa K, Kim SR, Kaniwa N, Nishimaki-Mogami T, Sawada J, Shirao K, Hamaguchi T, Yamamoto N, Kunitoh H, Ohe Y, Yamada Y, Tamura T, Yoshida T, Matsumura Y, Ohtsu A, Saijo N, Minami H (2010) Additive effects of drug transporter genetic polymorphisms on irinotecan pharmacokinetics/pharmacodynamics in Japanese cancer patients. Cancer Chemother Pharmacol 66(1):95–105. https://doi.org/10.1007/s00280-009-1138-y
Scaroni C, Mazzoni P, D’Amico E, Benvenuti C, Hind I (1984) Pharmacokinetics of oral and rectal flurbiprofen in children. Eur J Clin Pharmacol 27(3):367–369. https://doi.org/10.1007/BF00542178
Schlender JF, Meyer M, Thelen K, Krauss M, Willmann S, Eissing T, Jaehde U (2016) Development of a whole-body physiologically based pharmacokinetic approach to assess the pharmacokinetics of drugs in elderly individuals. Clin Pharmacokinet 55(12):1573–1589. https://doi.org/10.1007/s40262-016-0422-3
Schmitt W (2008) General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro 22(2):457–467. https://doi.org/10.1016/j.tiv.2007.09.010
Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270(1):414–423
Shin HB, Jung EH, Kang P, Lim CW, Oh KY, Cho CK, Lee YJ, Choi CI, Jang CG, Lee SY, Bae JW (2020) ABCB1 c.2677G > T/c.3435 C > T diplotype increases the early-phase oral absorption of losartan. Arch Pharm Res 43(11):1187–1196. https://doi.org/10.1007/s12272-020-01294-3
Theken KN, Lee CR, Gong L, Caudle KE, Formea CM, Gaedigk A, Klein TE, Agúndez JAG, Grosser T (2020) Clinical Pharmacogenetics Implementation Consortium guideline (CPIC) for CYP2C9 and nonsteroidal anti-inflammatory drugs. Clin Pharmacol Ther 108(2):191–200. https://doi.org/10.1002/cpt.1830
Thelen K, Coboeken K, Willmann S, Burghaus R, Dressman JB, Lippert J (2011) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: oral solutions. J Pharm Sci 100(12):5324–5345. https://doi.org/10.1002/jps.22726
Thelen K, Coboeken K, Willmann S, Dressman JB, Lippert J (2012) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: extension to describe performance of solid dosage forms. J Pharm Sci 101(3):1267–1280. https://doi.org/10.1002/jps.22825
Tracy TS, Marra C, Wrighton SA, Gonzalez FJ, Korzekwa KR (1996) Studies of flurbiprofen 4′-hydroxylation: additional evidence suggesting the sole involvement of cytochrome P450 2C9. Biochem Pharmacol 52(8):1305–1309. https://doi.org/10.1016/0006-2952(96)00501-1
Verscheijden LF, Koenderink JB, de Wildt SN, Russel FG (2019) Development of a physiologically-based pharmacokinetic pediatric brain model for prediction of cerebrospinal fluid drug concentrations and the influence of meningitis. PLoS Comput Biol 15(6):e1007117. https://doi.org/10.1371/journal.pcbi.1007117
Verscheijden LFM, Koenderink JB, Johnson TN, de Wildt SN, Russel FGM (2020) Physiologically-based pharmacokinetic models for children: starting to reach maturation? Pharmacol Ther 211:107541. https://doi.org/10.1016/j.pharmthera.2020.107541
Volak LP, Hanley MJ, Masse G, Hazarika S, Harmatz JS, Badmaev V, Majeed M, Greenblatt DJ, Court MH (2013) Effect of a herbal extract containing curcumin and piperine on midazolam, flurbiprofen and paracetamol (acetaminophen) pharmacokinetics in healthy volunteers. Br J Clin Pharmacol 75(2):450–462. https://doi.org/10.1111/j.1365-2125.2012.04364.x
Wang H, Yuan L, Zeng S (2011) Characterizing the effect of UDP-glucuronosyltransferase (UGT) 2B7 and UGT1A9 genetic polymorphisms on enantioselective glucuronidation of flurbiprofen. Biochem Pharmacol 82(11):1757–1763. https://doi.org/10.1016/j.bcp.2011.08.004
Wang L, Bao SH, Pan PP, Xia MM, Chen MC, Liang BQ, Dai DP, Cai JP, Hu GX (2015) Effect of CYP2C9 genetic polymorphism on the metabolism of flurbiprofen in vitro. Drug Dev Ind Pharm 41(8):1363–1367. https://doi.org/10.3109/03639045.2014.950274
Wechter WJ, Bigornia AE, Murray ED Jr, Levine BH, Young JW (1993) Rac-flurbiprofen is more ulcerogenic than its (S)‐enantiomer. Chirality 5(7):492–494. https://doi.org/10.1002/chir.530050703
Wojtyniak JG, Britz H, Selzer D, Schwab M, Lehr T (2020) Data digitizing: accurate and precise data extraction for quantitative systems pharmacology and physiologically-based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol 9(6):322–331. https://doi.org/10.1002/psp4.12511
Yasar Ü, Tybring G, Hidestrand M, Oscarson M, Ingelman-Sundberg M, Dahl ML, Eliasson E (2001) Role of CYP2C9 polymorphism in losartan oxidation. Drug Metab Dispos 29(7):1051–1056
Yaşin DS, Yilmaz Ş, Teksin Z (2018) Evaluation of biorelevant media to investigate the dissolution properties on flurbiprofen and to assess cytotoxicity effects on Caco-2 cell line. J Pharm Istanbul Univ 48(3):82–88. https://doi.org/10.26650/IstanbulJPharm.2018.180013
Zhang J, Zhang H, Zhao L, Gu J, Feng Y, An H (2018) Population pharmacokinetic modeling of flurbiprofen, the active metabolite of flurbiprofen axetil, in Chinese patients with postoperative pain. J Pain Res 11:3061–3070. https://doi.org/10.2147/JPR.S176475
Zhuang X, Lu C (2016) PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 6(5):430–440. https://doi.org/10.1016/j.apsb.2016.04.004
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (Grant No. NRF-2019R1A2C1004582).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interest for this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Whang, SS., Cho, C., Jung, E.H. et al. Physiologically based pharmacokinetic (PBPK) modeling of flurbiprofen in different CYP2C9 genotypes. Arch. Pharm. Res. 45, 584–595 (2022). https://doi.org/10.1007/s12272-022-01403-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01403-4