
Abstract
Irbesartan, a potent and selective angiotensin II type-1 (AT1) receptor blocker (ARB), is one of the representative medications for the treatment of hypertension. Cytochrome P450 (CYP) 2C9 is primarily involved in the oxidation of irbesartan. CYP2C9 is highly polymorphic, and genetic polymorphism of this enzyme is the leading cause of significant alterations in the pharmacokinetics of irbesartan. This study aimed to establish the physiologically based pharmacokinetic (PBPK) model to predict the pharmacokinetics of irbesartan in different CYP2C9 genotypes. The irbesartan PBPK model was established using the PK-Sim® software. Our previously reported pharmacogenomic data for irbesartan was leveraged in the development of the PBPK model and collected clinical pharmacokinetic data for irbesartan was used for the validation of the model. Physicochemical and ADME properties of irbesartan were obtained from previously reported data, predicted by the modeling software, or optimized to fit the observed plasma concentration–time profiles. Model evaluation was performed by comparing the predicted plasma concentration–time profiles and pharmacokinetic parameters to the observed results. Predicted plasma concentration–time profiles were visually similar to observed profiles. Predicted AUCinf in CYP2C9*1/*3 and CYP2C9*1/*13 genotypes were increased by 1.54- and 1.62-fold compared to CYP2C9*1/*1 genotype, respectively. All fold error values for AUC and Cmax in non-genotyped and CYP2C9 genotyped models were within the two-fold error criterion. We properly established the PBPK model of irbesartan in different CYP2C9 genotypes. It can be used to predict the pharmacokinetics of irbesartan for personalized pharmacotherapy in individuals of various races, ages, and CYP2C9 genotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertension is one of the major risk factors for a variety of cardiovascular diseases (CVDs) including heart failure, acute coronary syndrome, and atrial fibrillation, and cerebral stroke (Kjeldsen 2018). Hypertension is a globally prevalent disease, affecting approximately 1.28 billion people aged 30–79 years in 2019 (NCD Risk Factor Collaboration (NCD-RisC) 2021) and causing 12.8% of the total annual deaths worldwide (World Health Organization 2021). Nevertheless, blood pressure control using antihypertensive medications was adequately achieved in less than 50 percent of patients with hypertension (Cooper-DeHoff and Johnson 2016; Mann and Flack 2023). The inter-individual variability of the drug responses may be mainly attributed to genetic factors associated with the disposition of drugs, such as polymorphisms of drug-transporters (Shin et al. 2020; Magadmi et al. 2023; Jeong et al. 2023) and drug-metabolizing enzymes (Lee et al. 2018; Byeon et al. 2019, 2023; Bae et al. 2020; Kim et al. 2022; Cho et al. 2023a, b; Kang et al. 2023), and non-genetic factors including age, body weight, gender, race, co-administered drugs, etc. (Weinshilboum 2003; Lee et al. 2019; Jung et al. 2020). Individualized therapy considering various factors that influence the antihypertensive drug response is needed to reduce adverse sequelae and achieve proper therapeutic effects.
Irbesartan, a potent and selective angiotensin II type-1 (AT1) receptor blocker (ARB), is one of the representative medications for the treatment of hypertension (Marino and Vachharajani 2001). Glucuronide conjugation (Perrier et al. 1994) and oxidation are the main metabolic pathways of irbesartan. Cytochrome P450 (CYP) 2C9 is primarily involved in the oxidation route, while CYP3A4 has a negligible contribution (Bourrié et al. 1999). Previous in vitro studies reported that the active hepatic uptake of irbesartan is mediated by organic anion-transporting polypeptide (OATP) 1B1 and 1B3 (Chapy et al. 2015; Izumi et al. 2018; McFeely et al. 2019). Irbesartan and its metabolites are excreted by both biliary and renal routes (Brunner 1997), with less than 2% of the dose as unchanged form in the urine (European Medicines Agency 2023).
CYP2C9 is highly polymorphic, and genetic polymorphism of this enzyme leads to significant alterations in the pharmacokinetics and pharmacodynamics of clinically used CYP2C9 substrate drugs (Perini et al. 2005; Bae et al. 2011a, 2012; Choi et al. 2011; Lee et al. 2016; Kim et al. 2017, 2022). To date, more than 85 different CYP2C9 allelic variants and subvariants (CYP2C9*1B to *85) have been identified (https://www.pharmvar.org/gene/CYP2C9). Of these alleles, CYP2C9*2 (rs1799853, c.430C > T, p.Arg144Cys) and CYP2C9*3 (rs1057910, c.1075A > C, p.Ile359Leu), the two most common alleles (Daly et al. 2017), exhibit impaired catalytic activity compared to the normal allele both in vitro and in vivo (Tang et al. 2001; Lee et al. 2003; Perini et al. 2005). CYP2C9*13 (rs72558187, c.269 T > C, p.Leu90Pro), an allele only observed in East Asians with an extremely low frequency of 0.2–0.7% (Bae et al. 2011b; Daly et al. 2017), also shows reduced enzyme activity (Guo et al. 2005; Wang et al. 2015). Thus, dose adjustment of irbesartan according to genetic variations of individuals could be recommended for optimal antihypertensive therapy.
The physiologically based pharmacokinetic (PBPK) model, a mechanistic approach to describe the pharmacokinetics of xenobiotics, is widely used to estimate drug exposures in special populations such as pediatrics, pregnant women, obesity, and those with organ impairments (Kuepfer et al. 2016; Zhuang and Lu 2016; Marsousi et al. 2017). The PBPK model also aids in scrutinizing the pharmacokinetic alterations according to drug-drug interactions (Min and Bae 2017) and the effects of genetic polymorphisms (Cho et al. 2021b; Lee et al. 2022; Rüdesheim et al. 2022; Yang et al. 2022). Previously, PBPK models of irbesartan focused on oral absorption (Kaur et al. 2020) and hepatic uptake (Chapy et al. 2015) have been reported. However, no study has examined the effect of irbesartan according to CYP2C9 genetic polymorphism. In this study, we aimed to establish the PBPK model of irbesartan in different CYP2C9 genotypes.
Methods
Software
PK-Sim® version 10.0 (Bayer AG, Leverkusen, Germany) was used for the building of the irbesartan PBPK model, sensitivity analysis, and parameter optimization. Plasma concentration–time profiles in previous publications were digitized using Engauge Digitizer® version 12.1 (https://markummitchell.github.io/engauge-digitizer/). Pharmacokinetic parameters that were not extracted from previous publications were estimated via non-compartmental analysis (NCA) using the BA Calc 2007 analysis program (MFDS, Cheongju, Republic of Korea) based on the obtained plasma concentration–time profiles.
Clinical pharmacokinetic data
Our previously reported pharmacogenomic data in which 150 mg single oral dose of irbesartan was administered in healthy Korean subjects with three different genotype groups (Choi et al. 2012) was used for the development (to determine input parameters) of the PBPK model. Clinical pharmacokinetic studies were extensively investigated and the pharmacokinetic data with single and multiple oral dose administrations of irbesartan in healthy adults under fasting state were collected and used for the validation (to verify the developed model) of the PBPK model.
Model building
The “middle-out” strategy was used for the PBPK model building. Previously reported literature or drug databases were extensively investigated to obtain the physicochemical and absorption, distribution, metabolism, and excretion (ADME) characteristics of irbesartan, thereafter incorporating them into the PBPK model. Of those, Log P and pKa were adjusted within the previously reported ranges. Gastric emptying time was assigned as 40 min based on Klingensmith et al. (2010). The specific intestinal and organ permeabilities were optimized and calculated by the quantitative structure–activity relationship (QSAR) method built in the PK-Sim® software, respectively.
CYP2C9 and UDP-glucuronosyltransferase (UGT) 1A3 enzymes were incorporated to reflect the metabolism of irbesartan. Michaelis–Menten constant (Km) values for CYP2C9 and UGT1A3 were obtained from Bourrié et al. (1999) and Chapy et al. (2015) respectively, and the value for CYP2C9 was minorly modified. Turnover number (kcat) values of CYP2C9 and UGT1A3 for the non-genotyped model were simultaneously optimized to recover the mean observed plasma concentration–time profile obtained from our pharmacogenomic study (Choi et al. 2012). Thereafter, the developed non-genotyped model was scaled to the PBPK model for different CYP2C9 genotypes, where the kcat values in different CYP2C9 genotypes were optimized to describe the plasma concentration–time profiles for each genotype in Choi et al. (2012). The OATP1B1 and 1B3 were incorporated to describe the transport of irbesartan. The kcat and Km values for OATP1B1 and 1B3 were optimized and obtained from Chapy et al. (2015), respectively. The reference concentration of CYP2C9 was 3.84 μmol/L (Rodrigues 1999) and UGT1A3, OATP1B1, and 1B3 were 1.00 μmol/L, the default value of PK-Sim®. Relative expression values for CYP2C9 and UGT1A3 were obtained from the reverse transcription-polymerase chain reaction (RT-PCR) data (Nishimura et al. 2003; Nishimura and Naito 2005, 2006) and values for OATPs were obtained from ArrayExpress data (http://www.ebi.ac.uk/microarray-as/ae/).
Dissolution characteristics were described using the Lint80 function and the dissolution time was extracted from the dissolution profile of the commercial product (Khullar et al. 2015). Model input parameters were refined by iteratively performing sensitivity analysis and parameter identification based on the observed data at each step and the Levenberg–Marquardt algorithm was adopted for parameter optimization. Estimation methods for the partition coefficients and cellular permeabilities were Poulin and Theil (Poulin and Theil 2000; Poulin et al. 2001; Poulin and Theil 2002a, b) and Charge dependent Schmitt normalized to PK-Sim® (Hindmarsh et al. 2023), respectively.
Sensitivity analysis
A sensitivity analysis was performed to confirm which input parameters had an impact on the area of the plasma concentration–time curve from 0 to infinity (AUCinf) and maximum plasma concentration (Cmax) of irbesartan. Input parameters that were optimized, related to optimized parameters, or might have a marked influence on calculation methods used in the model, were included in the analysis. The sensitivity was calculated as follows (Eq. 1):
where \(S\) is the sensitivity, \(PK\) is the initial value of the pharmacokinetic parameter, \(\Delta PK\) is the change of the pharmacokinetic parameter from initial value, \(p\) is the initial value of the examined parameter, and \(\Delta p\) is the change of the examined parameter from initial value. A sensitivity of + 1.0 indicates that a + 10% change in an examined input parameter causes a + 10% change in the predicted pharmacokinetic parameter.
Model evaluation
Both graphical and numerical evaluation methods were used to evaluate the PBPK model. Predicted plasma concentration–time profiles plotting the arithmetic mean and 90% prediction interval (i.e. 5th and 95th percentile range) for the virtual population (n = 100) with close demographic characteristics to those of the subject populations in clinical studies were visually compared to the observed profiles. The demographic data not reported in previous studies were generated based on the implemented algorithm in the PK-Sim® software. A two-fold error criterion for AUC and Cmax, commonly used in previous studies (Abduljalil et al. 2015; Sager et al. 2015), was used as the numerical evaluation criterion. The fold error value was calculated as follows (Eq. 2):
To quantitatively measure the model’s predictive performance, the mean relative deviation (MRD) of all predicted plasma concentrations and the geometric mean fold error (GMFE) for predicted AUC and Cmax were calculated according to Eqs. 3 and 4, respectively:
where \({\mathrm{C}}_{\mathrm{pred},\mathrm{i}}\) is the i-th predicted plasma concentration, \({\mathrm{C}}_{\mathrm{obs},\mathrm{i}}\) is the corresponding observed plasma concentration, and N is the number of observations.
where \({\mathrm{PK}}_{\mathrm{pred},\mathrm{i}}\) is the i-th predicted AUC or Cmax value, \({\mathrm{PK}}_{\mathrm{obs},\mathrm{i}}\) is the corresponding observed value, and n is the number of the collected pharmacokinetic parameter data.
Results
Model building and evaluation
A total of nineteen clinical pharmacokinetic data, consisting of four data for development (our pharmacogenomic data) and fifteen data for validation (collected data from previous publications), were used to build the PBPK model. Of those, sixteen data were used in the non-genotyped model (Table 1) and three data were used in the genotyped model (Table 2). The demographic, genotype, and dose administration information of clinical studies used to build the irbesartan model are presented in Tables 1 and 2. Input parameters for the irbesartan PBPK model are summarized in Table 3.
The comparison between the predicted and observed plasma concentration–time profiles after single or multiple oral doses of irbesartan at 150–900 mg dose range is visualized in Fig. 1. All fold error values for AUC and Cmax in the non-genotyped model were included within the two-fold range criterion, with respective ranges being 0.52–1.95 and 0.83–1.94 (Fig. 2A and B). GMFE for AUC was 1.30 and 62.5% (10/16) and 75.0% (12/16) of fold error values for AUC were within the 1.25-fold and 1.5-fold range, respectively. For Cmax, 62.5% (10/16) and 68.8% (11/16) of the predicted values were included in the 1.25-fold and 1.5-fold range of the corresponding observed values, respectively, and GMFE was 1.40. As illustrated in the goodness-of-fit plot in Fig. 2C, the percentage of the predicted plasma concentrations within the 1.25- and two-fold range of the observed plasma concentrations were 41.5% and 86.9%, respectively, with an overall MRD of 1.57. The detailed results are presented in Table 4.

Predicted and observed plasma concentration–time profiles of irbesartan after A–J single (A development) and K–P multiple oral doses in non-genotyped populations. Solid lines and shaded areas indicate arithmetic mean and 90% prediction interval (5th and 95th percentile range) of predicted plasma concentrations, respectively. Circles indicate the mean of observed plasma concentrations. Profiles are shown as linear and semi-logarithmic scale. SD single dose, daily once daily dose

Goodness-of-fit plots comparing the predicted versus observed A AUC B Cmax and C plasma concentration of irbesartan in non-genotyped populations. Closed circles indicate single dose administrations and open circles indicate multiple dose administrations. Solid lines indicate the line of unity, dashed lines indicate the two-fold range and dotted lines indicate the 1.25-fold range
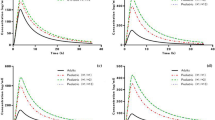
The comparison between the predicted and observed plasma concentration–time profiles in different CYP2C9 genotypes is shown in Fig. 3. Predicted profiles for three different genotypes were visually similar to the observed profiles. Predicted AUCinf in CYP2C9*1/*3 and CYP2C9*1/*13 genotypes were increased by 1.54- and 1.62-fold compared to CYP2C9*1/*1 genotype, respectively. Predicted Cmax for the population with the CYP2C9*3 and *13 carriers were 1.27- and 1.28-fold higher than the wild-type carrier, respectively. Goodness-of-fit plots for AUCinf (Fig. 4A) and Cmax (Fig. 4B) in the irbesartan model related to CYP2C9 genetic polymorphism exhibited that the predicted values were almost consistent with their corresponding observed values with all fold error values included in the 1.25-fold range (AUCinf range 0.98–1.09, Cmax range 0.93–1.14). GMFE for AUCinf was 1.04 and for Cmax was 1.08. As visualized in Fig. 4C, 58.3% and 94.4% of fold error values for plasma concentrations were included in the 1.25- and two-fold range, respectively, and overall MRD was 1.37. The detailed results are presented in Table 5.

Predicted and observed plasma concentration–time profiles of irbesartan after 150 mg single oral dose in A CYP2C9*1/*1, B CYP2C9*1/*3, and C CYP2C9*1/*13 genotypes. Solid lines and shaded areas indicate arithmetic mean and 90% prediction interval (5th and 95th percentile range) of predicted plasma concentrations, respectively. Symbols (inverted triangle; CYP2C9*1/*1, triangle; *1/*3, and rectangle; *1/*13) and error bars indicate the mean and standard deviation of observed plasma concentrations, respectively. Profiles are shown as linear and semi-logarithmic scale. SD single dose

Goodness-of-fit plots comparing the predicted versus observed A AUCinf B Cmax and C plasma concentration of irbesartan in different CYP2C9 genotypes. Inverted triangle, triangle, and rectangle symbols indicate CYP2C9*1/*1, *1/*3, and *1/*13 genotypes, respectively. Solid lines indicate the line of unity, dashed lines indicate the two-fold range and dotted lines indicate the 1.25-fold range
Sensitivity analysis
The results of the sensitivity analysis are illustrated in Fig. 5. Input parameters with sensitivity values calculated as greater than 0.5 were considered sensitive. Sensitive input parameters for AUCinf and Cmax of irbesartan were as follows, in order of higher to lower impact; lipophilicity, acidic pKa, administered dose, and fraction unbound were sensitive input parameters to AUCinf of irbesartan. Lipophilicity and dose were sensitive input parameters to the Cmax of irbesartan.

Results of sensitivity analysis to single parameters, measured as the change of predicted A AUCinf and B Cmax following the administration of 150 mg single oral dose of irbesartan
Discussion
Genetic polymorphism of drug-metabolizing enzymes and transporters is one of the principal issues in achieving adequate control of blood pressure using antihypertensive agents (Oliveira-Paula et al. 2019). Concurrently, several factors including age, gender, and obesity are the potential indicators that could contribute to the inter-individual variations of antihypertensive response (Chapman et al. 2002; Hiltunen et al. 2007). PBPK modeling enables the prediction of the pharmacokinetics of drugs simultaneously considering all of the characteristics mentioned above, and therefore, it may be a desirable approach for the implementation of individualized pharmacotherapy in each patient (Kim et al. 2018, 2021; Cho et al. 2021a, b, 2022; Whang et al. 2022). To date, there have been several attempts to establish the PBPK models in populations with different genotypes for antihypertensive agents such as candesartan (Jung et al. 2021), losartan (Tanveer et al. 2022), and metoprolol (Rüdesheim et al. 2020).
In the present study, a whole-body PBPK model of irbesartan according to CYP2C9 genetic polymorphism was appropriately established. All predicted AUC and Cmax values in the non-genotyped populations and different CYP2C9 genotypes were within the two-fold range of the observed values and calculated GMFE and MRD values showed a good predictive performance. The developed model properly predicted the irbesartan pharmacokinetics in different demographic characteristics and oral dose administrations and it also captured the previous finding in which AUCinf and Cmax of the CYP2C9*1/*3 and CYP2C9*1/*13 genotypes were significantly higher than the CYP2C9*1/*1 genotype (Choi et al. 2012). This suggests the present model could be applied to predict the pharmacokinetics of irbesartan after single and multiple dose administrations with a dose range of 150–900 mg/d in healthy subjects with different CYP2C9 genotypes.
Extensive prior knowledge of ADME and drug-dependent properties of irbesartan were consolidated in this PBPK model. In the metabolism of irbesartan, CYP2C9 is the only allocated enzyme for the oxidation pathway due to the negligible effects of CYP3A4 (Bourrié et al. 1999). Likewise, the glucuronidation pathway was assumed to be mediated by UGT1A3 based on the fact that UGT1A3 is highly selective toward N2-glucuronidation of tetrazoles (Alonen et al. 2008) and irbesartan is metabolized to tetrazole-N2-glucuronide conjugate (Perrier et al. 1994; Chando et al. 1998). Metabolites of irbesartan are pharmacologically inactive (Gillis and Markham 1997), thereby the PBPK model for those was not established in this study. Both OATP1B1 and 1B3 transporters are known to be responsible for the hepatic uptake of irbesartan. Uptake contributions of OATP1B1 and 1B3 in our model are consistent with the previous in vitro studies in which the relative contribution of OATP1B1 is much higher than OATP1B3 (Chapy et al. 2015; Izumi et al. 2018).
A slight increase or stagnation of irbesartan plasma concentration was noted at 10–12 h after administration in our pharmacogenomic data (Choi et al. 2012), but the present model did not capture this phenomenon. Previous studies proposed that the phenomenon could be triggered by the enterohepatic circulation of irbesartan and its glucuronide metabolite (Davi et al. 2000; Chapy et al. 2015). Meanwhile, Karatza and Karalis (2020) suggested that there is a possibility that absorption complexities, representatively irregular gastric emptying time, may contribute to the double-peak of irbesartan and properly captured it using a population pharmacokinetic approach. To the best of our knowledge, no irbesartan PBPK model has elucidated this phenomenon to date, including our study. PBPK models focusing on enterohepatic circulation and/or complex absorption kinetics could allow a more accurate capture of the plasma concentration–time profiles of irbesartan.
Five clinical studies have assessed the impact of CYP2C9 genetic polymorphism on the pharmacokinetics and/or pharmacodynamics of irbesartan (Hallberg et al. 2002; Wen et al. 2003; Hong et al. 2005; Chen et al. 2006; Choi et al. 2012). Chen et al. (2006) and Hong et al. (2005) reported that plasma irbesartan concentration at 6 h after dosing in Chinese hypertensive patients carrying the CYP2C9*3 allele variant was significantly higher than those carrying the wild-type allele. Choi et al. (2012) showed that CYP2C9*3 and *13 alleles markedly altered the AUC, Cmax, half-life (t1/2), and apparent clearance (CL/F) of irbesartan in healthy Korean subjects. Chen et al. (2006) and Hallberg et al. (2002) reported a notable reduction of diastolic blood pressure (DBP) in Chinese hypertensive patients with CYP2C9*1/*3 genotype and Swedish patients with CYP2C9*1/*2 genotype, respectively. On the other hand, Wen et al. (2003) and Hong et al. (2005) reported that the impacts of the CYP2C9*3 variant on the therapeutic efficacy of irbesartan were not significant. Although controversial results in the aspects of pharmacodynamics have been shown, it seems that the genetic polymorphism of CYP2C9 may be one of the predictive indicators for the antihypertensive effects of irbesartan treatment. Among these studies, Choi et al. (2012), our previous pharmacogenomic study, was the only study that included the information to develop the PBPK model such as plasma concentration–time profiles and pharmacokinetic parameters including AUC and Cmax according to CYP2C9 genetic polymorphism. Accordingly, we developed the PBPK model for CYP2C9*3 and *13 allele variants using these data (Choi et al. 2012), and the model for CYP2C9*2 allele was not established because there was no available data in previous studies. Since the CYP2C9*2 allele is the most frequently observed variant globally (Daly et al. 2017), further studies on this variant should be performed.
Albeit the importance of the CYP2C9 genetic polymorphism on the pharmacokinetics or therapeutic efficacy of irbesartan, considerations for the genetic polymorphisms of UGT and SLCO genes may also be desirable. UGT gene is highly polymorphic with more than 200 allele variants identified and these variants are known to influence the expression levels and/or enzymatic activity of UGT (Stingl et al. 2014). The SLCO gene, encoding the OATP transporter, is also polymorphic, by which the efficacy and safety of OATP substrates are affected (Nakanishi and Tamai 2012). Previous studies have shown that the UGT and/or SLCO genetic polymorphisms could have a potential role in the inter-individual variations of the drug responses for some ARB class drugs (Suwannakul et al. 2008; Hirvensalo et al. 2020; Song et al. 2021). However, to our knowledge, the effects of genetic polymorphisms of UGT and SLCO genes on the plasma concentrations or antihypertensive responses of irbesartan have been not identified. Thus, we established the PBPK model of irbesartan associated with the CYP2C9 genetic polymorphism in the present study. Future pharmacogenomic and PBPK modeling studies related to these genes may be needed.
In summary, we established the PBPK model of irbesartan, through which the pharmacokinetic alterations according to CYP2C9 genetic polymorphism were properly described. The present model could contribute to personalized antihypertensive pharmacotherapy of irbesartan via pharmacokinetic predictions considering together the various causes related to the inter-individual variability of drug response.
References
Abduljalil K, Cain T, Humphries H, Rostami-Hodjegan (2015) Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos 42(9):1478–1484. https://doi.org/10.1124/dmd.114.058099
Alonen A, Finel M, Kostiainen R (2008) The human UDP-glucuronosyltransferase UGT1A3 is highly selective towards N2 in the tetrazole ring of losartan, candesartan, and zolarsartan. Biochem Pharmacol 76(6):763–772. https://doi.org/10.1016/j.bcp.2008.07.006
Bae SK, Kim MJ, Shim EJ, Cho DY, Shon JH, Liu KH, Kim EY, Shin JG (2009) HPLC determination of irbesartan in human plasma: its application to pharmacokinetic studies. Biomed Chromatogr 23(6):568–572. https://doi.org/10.1002/bmc.1154
Bae JW, Choi CI, Jang CG, Lee SY (2011a) Effects of CYP2C9*1/*13 on the pharmacokinetics and pharmacodynamics of meloxicam. Br J Clin Pharmacol 71(4):550–555. https://doi.org/10.1111/j.1365-2125.2010.03853.x
Bae JW, Choi CI, Kim MJ, Oh DH, Keum SK, Park JI, Kim BH, Bang HK, Oh SG, Kang BS, Park HJ, Kim HD, Ha JH, Shin HJ, Kim YH, Na HS, Chung MW, Jang CG, Lee SY (2011b) Frequency of CYP2C9 alleles in Koreans and their effects on losartan pharmacokinetics. Acta Pharmacol Sin 32(10):1303–1308. https://doi.org/10.1038/aps.2011.100
Bae JW, Choi CI, Lee HI, Lee YJ, Jang CG, Lee SY (2012) Effects of CYP2C9*1/*3 and *1/*13 on the pharmacokinetics of losartan and its active metabolite E-3174. Int J Clin Pharmacol Ther 50(9):683–689. https://doi.org/10.5414/cp201467
Bae JW, Oh KY, Yoon SJ, Shin HB, Jung EH, Cho CK, Lim CW, Kang P, Choi CI, Jang CG, Lee SY, Lee YJ (2020) Effects of CYP2D6 genetic polymorphism on the pharmacokinetics of metoclopramide. Arch Pharm Res 43(11):1207–1213. https://doi.org/10.1007/s12272-020-01293-4
Bourrié M, Meunier V, Berger Y, Fabre G (1999) Role of cytochrome P-4502C9 in irbesartan oxidation by human liver microsomes. Drug Metab Dispos 27(2):288–296
Brunner HR (1997) The new angiotensin II receptor antagonist, irbesartan: pharmacokinetic and pharmacodynamic considerations. Am J Hypertens 10(S9):311S-317S. https://doi.org/10.1016/S0895-7061(97)00391-9
Byeon JY, Lee CM, Lee YJ, Kim YH, Kim SH, Jung EH, Chae WK, Lee YJ, Jang CG, Lee SY (2019) Influence of CYP2D6 genetic polymorphism on pharmacokinetics of active moiety of tolterodine. Arch Pharm Res 42(2):182–190. https://doi.org/10.1007/s12272-018-1099-y
Byeon JY, Cho CK, Kang P, Kim SH, Jang CG, Lee SY, Lee YJ (2023) Effects of CYP2D6 and CYP2C19 genetic polymorphisms and cigarette smoking on the pharmacokinetics of tolperisone. Arch Pharm Res 46(8):713–721. https://doi.org/10.1007/s12272-023-01462-1
Cagigal E, Gonzalez L, Alonso RM, Jiménez RM (2001) pKa determination of angiotensin II receptor antagonists (ARA II) by spectrofluorimetry. J Pharm Biomed Anal 26(3):477–486. https://doi.org/10.1016/s0731-7085(01)00413-7
Chando TJ, Everett DW, Kahle AD, Starrett AM, Vachharajani N, Shyu WC, Kripalani KJ, Barbhaiya RH (1998) Biotransformation of irbesartan in man. Drug Metab Dispos 26(5):408–417
Chapman AB, Schwartz GL, Boerwinkle E, Turner ST (2002) Predictors of antihypertensive response to a standard dose of hydrochlorothiazide for essential hypertension. Kidney Int 61(3):1047–1055. https://doi.org/10.1046/j.1523-1755.2002.00200.x
Chapy H, Klieber S, Brun P, Gerbal-Chaloin S, Boulenc X, Nicolas O (2015) PBPK modeling of irbesartan: incorporation of hepatic uptake. Biopharm Drug Dispos 36(8):491–506. https://doi.org/10.1002/bdd.1961
Chen G, Jiang S, Mao G, Zhang S, Hong X, Tang G, Li Z, Liu X, Zhang Y, Xing H, Wang B, Yu Y, Xu X (2006) CYP2C9 Ile359Leu polymorphism, plasma irbesartan concentration and acute blood pressure reductions in response to irbesartan treatment in Chinese hypertensive patients. Methods Find Exp Clin Pharmacol 28(1):19–24. https://doi.org/10.1358/mf.2006.28.1.962773
Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2021a) Physiologically based pharmacokinetic (PBPK) modelling of tamsulosin related to CYP2D6*10 allele. Arch Pharm Res 44(11):1037–1049. https://doi.org/10.1007/s12272-021-01357-z
Cho CK, Park HJ, Kang P, Moon S, Lee YJ, Bae JW, Jang CG, Lee SY (2021b) Physiologically based pharmacokinetic (PBPK) modeling of meloxicam in different CYP2C9 genotypes. Arch Pharm Res 44(12):1076–1090. https://doi.org/10.1007/s12272-021-01361-3
Cho CK, Kang P, Park HJ, Ko E, Mu CY, Lee YJ, Choi CI, Kim HS, Jang CG, Bae JW, Lee SY (2022) Physiologically based pharmacokinetic (PBPK) modeling of piroxicam with regard to CYP2C9 genetic polymorphism. Arch Pharm Res 45(5):352–366. https://doi.org/10.1007/s12272-022-01388-0
Cho CK, Byeon JY, Kang P, Park JI, Jang CG, Lee SY, Choi CI, Bae JW, Lee YJ (2023a) Effects of CYP2D6*10 allele on the pharmacokinetics of tolperisone. Arch Pharm Res 46(1):59–64. https://doi.org/10.1007/s12272-022-01422-1
Cho CK, Byeon JY, Kang P, Park HJ, Ko E, Mu CY, Jang CG, Lee SY, Lee YJ (2023b) Effects of CYP2C19 genetic polymorphism on the pharmacokinetics of tolperisone in healthy subjects. Arch Pharm Res 46(2):111–116. https://doi.org/10.1007/s12272-022-01423-0
Choi CI, Kim MJ, Jang CG, Park YS, Bae JW, Lee SY (2011) Effects of the CYP2C9*1/*13 genotype on the pharmacokinetics of lornoxicam. Basic Clin Pharmacol Toxicol 109(6):476–480. https://doi.org/10.1111/j.1742-7843.2011.00751.x
Choi CI, Kim MJ, Chung EK, Lee HI, Jang CG, Bae JW, Lee SY (2012) CYP2C9 *3 and *13 alleles significantly affect the pharmacokinetics of irbesartan in healthy Korean subjects. Eur J Clin Pharmacol 68(2):149–154. https://doi.org/10.1007/s00228-011-1098-0
Choi HY, Lim HS, Kim YH, Jeon HS, Kim MJ, Lee SH, Jung JH, Lee YK, Kim HJ, Bae KS (2015) Evaluation of the pharmacokinetics of the DPP-4 inhibitor gemigliptin when coadministered with rosuvastatin or irbesartan to healthy subjects. Curr Med Res Opin 31(2):229–241. https://doi.org/10.1185/03007995.2014.980886
Cooper-DeHoff RM, Johnson JA (2016) Hypertension pharmacogenomics: in search of personalized treatment approaches. Nat Rev Nephrol 12(2):110–122. https://doi.org/10.1038/nrneph.2015.176
Daly AK, Rettie AE, Fowler DM, Miners JO (2017) Pharmacogenomics of CYP2C9: functional and clinical considerations. J Pers Med 8(1):1. https://doi.org/10.3390/jpm8010001
Davi H, Tronquet C, Miscoria G, Perrier L, DuPont P, Caix J, Simiand J, Berger Y (2000) Disposition of irbesartan, an angiotensin II AT1-receptor antagonist, in mice, rats, rabbits, and macaques. Drug Metab Dispos 28(1):79–88
El-Desoky HS, Ghoneim MM, Habazy A (2011) Voltammetry of irbesartan drug in pharmaceutical formulations and human blood: quantification and pharmacokinetic studies. J Braz Chem Soc 22(2):239–247. https://doi.org/10.1590/S0103-50532011000200008
European Medicines Agency (2023) Aprovel: EPAR - Product Information. https://www.ema.europa.eu/en/documents/product-information/aprovel-epar-product-information_en.pdf. Accessed 06 Nov 2023
Gillis JC, Markham A (1997) Irbesartan. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in the management of hypertension. Drugs 54(6):885–902. https://doi.org/10.2165/00003495-199754060-00007
Guo Y, Zhang Y, Wang Y, Chen X, Si D, Zhong D, Fawcett JP, Zhou H (2005) Role of CYP2C9 and its variants (CYP2C9*3 and CYP2C9*13) in the metabolism of lornoxicam in humans. Drug Metab Dispos 33(6):749–753. https://doi.org/10.1124/dmd.105.003616
Hallberg P, Karlsson J, Kurland L, Lind L, Kahan T, Malmqvist K, Öhman KP, Nyström F, Melhus H (2002) The CYP2C9 genotype predicts the blood pressure response to irbesartan: results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation vs Atenolol (SILVHIA) trial. J Hypertens 20(10):2089–2093. https://doi.org/10.1097/00004872-200210000-00030
Hiltunen TP, Suonsyrjä T, Hannila-Handelberg T, Paavonen KJ, Miettinen HE, Strandberg T, Tikkanen I, Tilvis R, Pentikäinen PJ, Virolainen J, Kontula K (2007) Predictors of antihypertensive drug responses: initial data from a placebo-controlled, randomized, cross-over study with four antihypertensive drugs (The GENRES Study). Am J Hypertens 20(3):311–318. https://doi.org/10.1016/j.amjhyper.2006.09.006
Hindmarsh AC, Reynolds DR, Serban R, Woodward CS, Gardner, DJ, Cohen SD, Taylor A, Peles S, Banks L, Shumaker D (2023) Open Systems Pharmacology Suite Manual version 11 update 1. https://docs.open-systems-pharmacology.org/copyright. Accessed 06 Nov 2023
Hirvensalo P, Tornio A, Launiainen T, Paile-Hyvärinen M, Tapaninen T, Neuvonen M, Backman JT, Niemi M (2020) UGT1A3 and sex are major determinants of telmisartan pharmacokinetics—a comprehensive pharmacogenomic study. Clin Pharmacol Ther 108(4):885–895. https://doi.org/10.1002/cpt.1928
Hong X, Zhang S, Mao G, Jiang S, Zhang Y, Yu Y, Tang G, Xing H, Xu X (2005) CYP2C9* 3 allelic variant is associated with metabolism of irbesartan in Chinese population. Eur J Clin Phamracol 61(9):627–634. https://doi.org/10.1007/s00228-005-0976-8
Huang XH, Li J, Qiu FR, Xie HT, Huang JH, Li JC, Zheng QS (2006) PK-PD modeling of irbesartan in healthy Chinese adult volunteers under non-steady-state conditions. Eur J Drug Metab Pharmacokinet 31(4):259–264. https://doi.org/10.1007/BF03190465
Izumi S, Nozaki Y, Kusuhara H, Hotta K, Mochizuki T, Komori T, Maeda K, Sugiyama Y (2018) Relative activity factor (RAF)-based scaling of uptake clearance mediated by organic anion transporting polypeptide (OATP) 1B1 and OATP1B3 in human hepatocytes. Mol Pharm 15(6):2277–2288. https://doi.org/10.1021/acs.molpharmaceut.8b00138
Jeong SH, Jang JH, Lee YB (2023) P-glycoprotein mechanical functional analysis using in silico molecular modeling: Pharmacokinetic variability according to ABCB1 c.2677G > T/A genetic polymorphisms. Int J Biol Macromol 249:126777. https://doi.org/10.1016/j.ijbiomac.2023.126777
Jung EH, Lee YJ, Kim DH, Kang P, Lim CW, Cho CK, Jang CG, Lee SY, Bae JW (2020) Effects of paroxetine on the pharmacokinetics of atomoxetine and its metabolites in different CYP2D6 genotypes. Arch Pharm Res 43(12):1356–1363. https://doi.org/10.1007/s12272-020-01300-8
Jung EH, Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Choi CI, Jang CG, Lee SY (2021) Physiologically based pharmacokinetic modeling of candesartan related to CYP2C9 genetic polymorphism in adult and pediatric patients. Arch Pharm Res 44(12):1109–1119. https://doi.org/10.1007/s12272-021-01363-1
Kang P, Cho CK, Jang CG, Lee SY, Lee YJ, Choi CI, Bae JW (2023) Effects of CYP2C9 and CYP2C19 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of gliclazide in healthy subjects. Arch Pharm Res 46(5):438–447. https://doi.org/10.1007/s12272-023-01448-z
Karatza E, Karalis V (2020) Delay differential equations for the description of Irbesartan pharmacokinetics: A population approach to model absorption complexities leading to dual peaks. Eur J Pharm Sci 153:105498. https://doi.org/10.1016/j.ejps.2020.105498
Kaur N, Thakur PS, Shete G, Gangwal R, Sangamwar AT, Bansal AK (2020) Understanding the oral absorption of irbesartan using biorelevant dissolution testing and PBPK modeling. AAPS PharmSciTech 21(3):1–13. https://doi.org/10.1208/s12249-020-01643-x
Khullar P, Kolhe V, Kulkarni A, Patel S, Phadke Y, Saravanan D, Shingte M (2015) Solid pharmaceutical fixed dose compositions comprising irbesartan and amlodipine, their preparation and their therapeutic application. United States Patent No. US009173848B2. https://patentimages.storage.googleapis.com/90/e0/b6/0df075f6d20b3a/US9173848.pdf. Accessed 06 Nov 2023
Kim SH, Kim DH, Byeon JY, Kim YH, Kim DH, Lim HJ, Lee CM, Whang SS, Choi CI, Bae JW, Lee YJ, Jang CG, Lee SY (2017) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of celecoxib and its carboxylic acid metabolite. Arch Pharm Res 40(3):382–390. https://doi.org/10.1007/s12272-016-0861-2
Kim SH, Byeon JY, Kim YH, Lee CM, Lee YJ, Jang CG, Lee SY (2018) Physiologically based pharmacokinetic modelling of atomoxetine with regard to CYP2D6 genotypes. Sci Rep 8(1):12405. https://doi.org/10.1038/s41598-018-30841-8
Kim YH, Kang P, Cho CK, Jung EH, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2021) Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch Pharm Res 44(7):713–724. https://doi.org/10.1007/s12272-021-01346-2
Kim NT, Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2022) Effects of CYP2C9*3 and *13 alleles on the pharmacokinetics and pharmacodynamics of glipizide in healthy Korean subjects. Arch Pharm Res 45(2):114–121. https://doi.org/10.1007/s12272-021-01366-y
Kjeldsen SE (2018) Hypertension and cardiovascular risk: general aspects. Pharmacol Res 29:95–99. https://doi.org/10.1016/j.phrs.2017.11.003
Klingensmith WC, Rhea KL, Wainwright EA, Hopper OW (2010) The gastric emptying study with oatmeal: reference range and reproducibility as a function of age and sex. J Nucl Med Technol 38(4):186–190. https://doi.org/10.2967/jnmt.110.077065
Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, Block M, Eissing T, Teutonico D (2016) Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacomet Syst Pharmacol 5(10):516–531. https://doi.org/10.1002/psp4.12134
Lee CR, Pieper JA, Hinderliter AL, Blaisdell JA, Goldstein JA (2003) Losartan and E3174 pharmacokinetics in cytochrome P450 2C9*1/*1, *1/*2, and *1/*3 individuals. Pharmacotherapy 23(6):720–725. https://doi.org/10.1592/phco.23.6.720.32187
Lee HJ, Kim YH, Kim SH, Lee CM, Yang AY, Jang CG, Lee SY, Bae JW, Choi CI (2016) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of zafirlukast. Arch Pharm Res 39(7):1013–1019. https://doi.org/10.1007/s12272-016-0785-x
Lee HI, Byeon JY, Kim YH, Lee CM, Choi CI, Jang CG, Bae JW, Lee YJ, Lee SY (2018) Effects of CYP2C19 and CYP3A5 genetic polymorphisms on the pharmacokinetics of cilostazol and its active metabolites. Eur J Clin Pharmacol 74(11):1417–1426. https://doi.org/10.1007/s00228-018-2522-5
Lee CM, Jung EH, Byeon JY, Kim SH, Jang CG, Lee YJ, Lee SY (2019) Effects of steady-state clarithromycin on the pharmacokinetics of zolpidem in healthy subjects. Arch Pharm Res 42(12):1101–1106. https://doi.org/10.1007/s12272-019-01201-5
Lee CM, Kang P, Cho CK, Park HJ, Lee YJ, Bae JW, Choi CI, Kim HS, Jang CG, Lee SY (2022) Physiologically based pharmacokinetic modelling to predict the pharmacokinetics of metoprolol in different CYP2D6 genotypes. Arch Pharm Res. https://doi.org/10.1007/s12272-022-01394-2
Magadmi R, Alyoubi R, Moshrif T, Bakhshwin D, Suliman BA, Kamel F, Jamal M, Burzangi AS, Basit S (2023) Polymorphisms in the drug transporter gene ABCB1 are associated with drug response in Saudi epileptic pediatric patients. Biomedicines 11(9):2505. https://doi.org/10.3390/biomedicines11092505
Mann JFE, Flack JM (2023) Choice of drug therapy in primary (essential) hypertension. https://www.uptodate.com/contents/choice-of-drug-therapy-in-primary-essential-hypertension#!. Accessed 06 Nov 2023
Marino MR, Vachharajani NN (2001) Drug interactions with irbesartan. Clin Pharmacokinet 40(8):605–614. https://doi.org/10.2165/00003088-200140080-00004
Marino MR, Langenbacher K, Ford NF, Uderman HD (1998a) Pharmacokinetics and pharmacodynamics of irbesartan in healthy subjects. J Clin Pharmacol 38(3):246–255. https://doi.org/10.1002/j.1552-4604.1998.tb04422.x
Marino MR, Langenbacher KM, Raymond RH, Ford NF, Lasseter KC (1998b) Pharmacokinetics and pharmacodynamics of irbesartan in patients with hepatic cirrhosis. J Clin Pharmacol 38(4):347–356. https://doi.org/10.1002/j.1552-4604.1998.tb04434.x
Marsousi N, Desmeules JA, Rudaz S, Daali Y (2017) Usefulness of PBPK modeling in incorporation of clinical conditions in personalized medicine. J Pharm Sci 106(9):2380–2391. https://doi.org/10.1016/j.xphs.2017.04.035
McFeely SJ, Ritchie TK, Yu J, Nordmark A, Levy RH, Ragueneau-Majlessi I (2019) Identification and evaluation of clinical substrates of organic anion transporting polypeptides 1B1 and 1B3. Clin Transl Sci 12(4):379–387. https://doi.org/10.1111/cts.12623
Min JS, Bae SK (2017) Prediction of drug-drug interaction potential using physiologically based pharmacokinetic modeling. Arch Pharm Res 40(12):1356–1379. https://doi.org/10.1007/s12272-017-0976-0
Nakanishi T, Tamai I (2012) Genetic polymorphisms of OATP transporters and their impact on intestinal absorption and hepatic disposition of drugs. Drug Metab Pharmacokinet 27(1):106–121. https://doi.org/10.2133/dmpk.dmpk-11-rv-099
NCD Risk Factor Collaboration (NCD-RisC) (2021) Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. The Lancet 398(10304):957–980. https://doi.org/10.1016/S0140-6736(21)01330-1
Nishimura M, Naito S (2005) Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet 20(6):452–477. https://doi.org/10.2133/dmpk.20.452
Nishimura M, Naito S (2006) Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet 21(5):357–374. https://doi.org/10.2133/dmpk.21.357
Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T (2003) Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessedby high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi 123(5):369–375. https://doi.org/10.1248/yakushi.123.369
Oliveira-Paula GH, Pereira SC, Tanus-Santos JE, Lacchini R (2019) Pharmacogenomics and hypertension: current insights. Pharmgenomics Pers Med 12:341–359. https://doi.org/10.2147/PGPM.S230201
Perini JA, Vianna-Jorge R, Brogliato AR, Suarez-Kurtz G (2005) Influence of CYP2C9 genotypes on the pharmacokinetics and pharmacodynamics of piroxicam. Clin Pharmacol Ther 78(4):362–369. https://doi.org/10.1016/j.clpt.2005.06.014
Perrier L, Bourrié M, Marti E, Tronquet C, Massé D, Berger Y, Magdalou J, Fabre G (1994) In vitro N-glucuronidation of SB 47436 (BMS 186295), a new AT1 nonpeptide angiotensin II receptor antagonist, by rat, monkey and human hepatic microsomal fractions. J Pharmacol Exp Ther 271(1):91–99
Poulin P, Schoenlein K, Theil FP (2001) Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J Pharm Sci 90(4):436–447. https://doi.org/10.1002/1520-6017(200104)90:4<436::aid-jps1002>3.0.co;2-p
Poulin P, Theil FP (2000) A priori prediction of tissue: plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci 89(1):16–35. https://doi.org/10.1002/(SICI)1520-6017(200001)89:1%3c16::AID-JPS3%3e3.0.CO;2-E
Poulin P, Theil FP (2002a) Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91(1):129–156. https://doi.org/10.1002/jps.10005
Poulin P, Theil FP (2002b) Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 91(5):1358–1370. https://doi.org/10.1002/jps.10128
Rodrigues AD (1999) Integrated cytochrome P450 reaction phenotyping: attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem Pharmacol 57(5):465–480. https://doi.org/10.1016/s0006-2952(98)00268-8
Rüdesheim S, Wojtyniak JG, Selzer D, Hanke N, Mahfoud F, Schwab M, Lehr T (2020) Physiologically based pharmacokinetic modeling of metoprolol enantiomers and α-hydroxymetoprolol to describe CYP2D6 drug-gene interactions. Pharmaceutics 12(12):1200. https://doi.org/10.3390/pharmaceutics12121200
Rüdesheim S, Selzer D, Fuhr U, Schwab M, Lehr T (2022) Physiologically-based pharmacokinetic modeling of dextromethorphan to investigate interindividual variability within CYP2D6 activity score groups. CPT Pharmacomet Syst Pharmacol 11(4):494–511. https://doi.org/10.1002/psp4.12776
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N (2015) Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos 43(11):1823–1837. https://doi.org/10.1124/dmd.115.065920
Shin HB, Jung EH, Kang P, Lim CW, Oh KY, Cho CK, Lee YJ, Choi CI, Jang CG, Lee SY, Bae JW (2020) ABCB1 c.2677G>T/c.3435C>T diplotype increases the early-phase oral absorption of losartan. Arch Pharm Res 43(11):1187–1196. https://doi.org/10.1007/s12272-020-01294-3
Song G, Chung JE, Yee J, Lee KE, Park K, Gwak HS (2021) Effects of SLCO1B1 and SLCO1B3 genetic polymorphisms on valsartan pharmacokinetics in healthy Korean volunteers. J Pers Med 11(9):862. https://doi.org/10.3390/jpm11090862
Stingl JC, Bartels H, Viviani R, Lehmann M, Brockmöller J (2014) Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: a quantitative systematic review. Pharmacol Ther 141(1):92–116. https://doi.org/10.1016/j.pharmthera.2013.09.002
Suwannakul S, Ieiri I, Kimura M, Kawabata K, Kusuhara H, Hirota T, Irie S, Sugiyama Y, Higuchi S (2008) Pharmacokinetic interaction between pravastatin and olmesartan in relation to SLCO1B1 polymorphism. J Hum Genet 53(10):899–904. https://doi.org/10.1007/s10038-008-0324-9
Tang C, Shou M, Rushmore TH, Mei Q, Sandhu P, Woolf EJ, Rose MJ, Gelmann A, Greenberg HE, De Lepeleire I, Hecken AV, De Schepper PJ, Ebel DL, Schwartz JI, Rodrigues AD (2001) In-vitro metabolism of celecoxib, a cyclooxygenase-2 inhibitor, by allelic variant forms of human liver microsomal cytochrome P450 2C9: correlation with CYP2C9 genotype and in-vivo pharmacokinetics. Pharmacogenetics 11(3):223–235. https://doi.org/10.1097/00008571-200104000-00006
Tanveer A, Hussain K, Tasneem H, Arif I, Rashid M, Abbas N, Shamim R, Shah PA, Bukhari NI (2022) Prediction of CYP-mediated silybin A-losartan pharmacokinetic interactions using physiological based pharmacokinetic modeling. J Pharmacokinet Pharmacodyn 49(3):311–323. https://doi.org/10.1007/s10928-022-09804-0
Wang L, Bao SH, Pan PP, Xia MM, Chen MC, Liang BQ, Dai DP, Cai JP, Hu GX (2015) Effect of CYP2C9 genetic polymorphism on the metabolism of flurbiprofen in vitro. Drug Dev Ind Pharm 41(8):1363–1367. https://doi.org/10.3109/03639045.2014.950274
Weinshilboum R (2003) Inheritance and drug response. N Engl J Med 348(6):529–537. https://doi.org/10.1056/NEJMra020021
Wen SY, Wang H, Sun OJ, Wang SQ (2003) Rapid detection of the known SNPs of CYP2C9 using oligonucleotide microarray. World J Gastroenterol: World J Gastroenterol 9(6):1342–1346. https://doi.org/10.3748/wjg.v9.i6.1342
Whang SS, Cho CK, Jung EH, Kang P, Park HJ, Lee YJ, Choi CI, Bae JW, Kim HS, Jang CG, Lee SY (2022) Physiologically based pharmacokinetic (PBPK) modeling of flurbiprofen in different CYP2C9 genotypes. Arch Pharm Res 45(8):584–595. https://doi.org/10.1007/s12272-022-01403-4
World Health Organization (2021) The global health observatory: indicator metadata registry list: blood pressure/hypertension. https://www.who.int/data/gho/indicator-metadata-registry/imr-details/3155. Accessed 06 Nov 2023
Yang H, Yang L, Zhong X, Jiang X, Zheng L, Wang L (2022) Physiologically based pharmacokinetic modeling of brivaracetam and its interactions with rifampin based on CYP2C19 phenotypes. Eur J Pharm Sci 177:106258. https://doi.org/10.1016/j.ejps.2022.106258
Zhuang X, Lu C (2016) PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 6(5):430–440. https://doi.org/10.1016/j.apsb.2016.04.004
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (Grant No. NRF-2019R1A2C1004582).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cho, CK., Kang, P., Jang, CG. et al. Physiologically based pharmacokinetic (PBPK) modeling to predict the pharmacokinetics of irbesartan in different CYP2C9 genotypes. Arch. Pharm. Res. 46, 939–953 (2023). https://doi.org/10.1007/s12272-023-01472-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-023-01472-z