
Abstract
Piroxicam is a non-steroidal anti-inflammatory drug used to alleviate symptoms of osteoarthritis and rheumatoid arthritis. CYP2C9 genetic polymorphism significantly influences the pharmacokinetics of piroxicam. The objective of this study was to develop and validate the piroxicam physiologically based pharmacokinetic (PBPK) model related to CYP2C9 genetic polymorphism. PK-Sim® version 10.0 was used for the PBPK modeling. The PBPK model was evaluated by predicted and observed plasma concentration–time profiles, fold errors of predicted to observed pharmacokinetic parameters, and a goodness-of-fit plot. The turnover number (kcat) of CYP2C9 was adjusted to capture the pharmacokinetics of piroxicam in different CYP2C9 genotypes. The population PBPK model overall accurately described and predicted the plasma concentration–time profiles in different CYP2C9 genotypes. In our simulations, predicted AUCinf in CYP2C9*1/*2, CYP2C9*1/*3, and CYP2C9*3/*3 genotypes were 1.83-, 2.07-, and 6.43-fold higher than CYP2C9*1/*1 genotype, respectively. All fold error values for AUC, Cmax, and t1/2 were included in the acceptance criterion with the ranges of 0.57–1.59, 0.63–1.39, and 0.65–1.51, respectively. The range of fold error values for predicted versus observed plasma concentrations was 0.11–3.13. 93.9% of fold error values were within the two-fold range. Average fold error, absolute average fold error, and root mean square error were 0.93, 1.27, and 0.72, respectively. Our model accurately captured the pharmacokinetic alterations of piroxicam according to CYP2C9 genetic polymorphism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Piroxicam is a non-steroidal anti-inflammatory drug used to alleviate symptoms of osteoarthritis and rheumatoid arthritis (Weintraub et al. 1977; Dessain et al. 1979). Piroxicam reduces the synthesis of prostaglandins via inhibition of both cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) (Berg et al. 1999; Blanco et al. 1999). The most common adverse events of piroxicam are nausea, constipation, flatulence, abdominal pain, and diarrhea (Pfizer 2016). The risk of serious cardiovascular adverse events, including myocardial infarction and stroke, and serious gastrointestinal adverse events such as bleeding, ulceration, and perforation are increased by piroxicam (Dean 2019). It should be used for the shortest possible duration at the lowest effective dose (Pfizer 2016).
Piroxicam is primarily metabolized to 5’-hydroxypiroxicam by cytochrome P450 2C9 (CYP2C9) (Brogden et al. 1981). CYP2C9 is involved in the metabolism of various clinically used drugs, including glipizide (Kim et al. 2022), losartan (Bae et al. 2011b, 2012), meloxicam (Bae et al. 2011a; Lee et al. 2014), and S-enantiomer of warfarin (Rettie et al. 1992). CYP2C9 is genetically polymorphic and approximately 85 allele variants for CYP2C9 (CYP2C9*1B to CYP2C9*85) have been identified to date (https://www.pharmvar.org/gene/CYP2C9). Among them, CYP2C9*2 (rs1799853, c.430C > T, p.Cys144Arg) and *3 (rs1057910, c.1075A > C, p.Ile359Leu) alleles are the most common variants with overall frequency of 9.14% and 6.37% worldwide, respectively (Daly et al. 2017). These allele variants show impaired enzyme activity toward a number of substrates both in vitro and in vivo (Tang et al. 2001; Kirchheiner et al. 2004; Choi et al. 2012; Lee et al. 2015; Wang et al. 2015; Kim et al. 2017). Tracy et al. (2002) demonstrated that the CYP2C9*3 allele variant reduces enzyme activity for 5’-hydroxylation of piroxicam in vitro. Perini et al. (2005) reported significantly higher exposures and lower clearances for piroxicam in individuals carrying CYP2C9*2 or CYP2C9*3 alleles compared to homozygous CYP2C9*1 allele in vivo. Especially, exposure and half-life (t1/2) in the CYP2C9*3/*3 genotype were increased by 5.31- and 8.75-fold compared to the CYP2C9*1/*1 genotype, respectively (Perini and Suarez-Kurtz 2006). In vivo studies presented notable differences not only in the pharmacokinetics but also pharmacodynamics of piroxicam according to CYP2C9 genetic polymorphism (Perini et al. 2005; Perini and Suarez-Kurtz 2006). The drug label for piroxicam recommends the dose reduction in CYP2C9 poor metabolizers (CYP2C9PM) because they may have abnormally high plasma levels (Pfizer 2016). These studies suggest that responses of piroxicam could be varied according to the genetic polymorphism of CYP2C9.
Physiologically based pharmacokinetic (PBPK) modeling is a mechanistic approach for the prediction of the pharmacokinetics of drugs (Zhuang and Lu 2016; Kim et al. 2018). It is a useful tool to guide dose adjustment in various clinical scenarios such as pediatric populations (Yellepeddi et al. 2019; Verscheijden et al. 2020), pregnancy (Abduljalil and Badhan 2020; Coppola et al. 2021), organ impairments (Suri et al. 2015; Heimbach et al. 2021), and the effects of genetic polymorphisms (Rüdesheim et al. 2020; Cho et al. 2021a; Jung et al. 2021; Xu et al. 2021). Previously, PBPK models for several NSAIDs including celecoxib (Kim et al. 2021), flurbiprofen (Loisios-Konstantinidis et al. 2020), and meloxicam (Cho et al. 2021b) were established in different CYP2C9 genotypes. However, the piroxicam PBPK model related to CYP2C9 genetic polymorphism has not been reported. In this study, we developed and validated the PBPK model for piroxicam in the populations carrying the most two common CYP2C9 allele variants.
Methods
Software and data source
PBPK model of piroxicam was developed and validated using PK-Sim® version 10.0 (Bayer AG, Leverkusen, Germany). Previously published concentration–time profiles were digitized with Engauge Digitizer® version 12.1 (https://markummitchell.github.io/engauge-digitizer/) according to the proposed digitization algorithm in Wojtyniak et al. (2020). Pharmacokinetic parameters which were not obtained from the publications were estimated via non-compartmental analysis (NCA) with the BA Calc 2007 analysis program (MFDS, Cheongju, Republic of Korea).
Previous publications were extensively investigated to obtain information on the absorption, distribution, metabolism, and excretion (ADME) process, physicochemical characteristics, and clinical pharmacokinetic data for piroxicam. In this study, we only gathered the clinical studies in which the administration of piroxicam was an oral route. Among the collected clinical pharmacokinetic data, only two articles contain pharmacogenomic data (Perini et al. 2005; Perini and Suarez-Kurtz 2006). The data of the two articles were used for model development and the others were used for model validation. Information on the collected clinical data for the development and validation of the PBPK model is presented in Table 1.
Model building
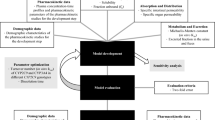
The “middle-out” strategy was used for the PBPK model building. The physicochemical parameters were obtained from drug databases or previous studies. The specific intestinal and organ permeabilities were calculated in the software (Thelen et al. 2011, 2012; Hindmarsh et al. 2021). Fraction metabolized by CYP2C9 (fm, CYP2C9) was estimated as 81.1% using the area under the plasma concentration–time curve from 0 to infinity (AUCinf ) values in CYP2C9*1/*1 and CYP2C9*3/*3 genotypes (Perini and Suarez-Kurtz 2006) based on previously reported methods (Ito et al. 2005; Huang et al. 2017). In the estimation, it was assumed that CYP2C9*1/*1 and CYP2C9*3/*3 genotypes are translated into CYP2C9 extensive metabolizer (CYP2C9EM) and CYP2C9PM phenotypes, respectively. The turnover number (kcat) was optimized to capture the estimated fm, CYP2C9 value and Michaelis–Menten constant (Km) obtained from Tracy et al. (2002) was used. The reference concentration of CYP2C9 was 3.84 μmol/L (Rodrigues 1999). Relative expression values in each organ were obtained from the reverse transcription-polymerase chain reaction (RT-PCR) data (Nishimura et al. 2003; Nishimura and Naito 2005, 2006). Renal clearance value was determined to capture the profile of cumulative excretion as unchanged form in urine within the ranges presented in Ishizaki et al. (1979). Dissolution times (80% dissolved) were adjusted based on dissolution profiles in biorelevant media (Li et al. 2019). Partition coefficients and cellular permeabilities were estimated as Schmitt and PK-Sim® standard methods, respectively (Schmitt 2008; Hindmarsh et al. 2021). kcat values were optimized in different CYP2C9 genotypes based on the previous pharmacogenetic studies (Perini et al. 2005; Perini and Suarez-Kurtz 2006). Parameter optimization was performed as the Levenberg–Marquardt algorithm in the PK-Sim® software. The PBPK model was developed for the different populations and dose regimens in the development dataset and verified using the validation dataset. Clinical studies without information on CYP2C9 genotype were assumed that all subjects of the studies were carrying homozygous CYP2C9*1 allele.
Sensitivity analysis
Sensitivity analysis was performed in the PK-Sim® software. In the analysis, a total of 861 parameters were assessed for AUCinf and maximum plasma concentration (Cmax). The sensitivity was calculated by the following Eq. 1.
where \(S\) is the sensitivity, \(PK\) is the initial values of the pharmacokinetic parameter, \(\Delta PK\) is the change of the pharmacokinetic parameters from initial values, \(p\) is the initial values of the assessed input parameter, and ∆p is the change of assessed input parameters from initial values, respectively. A sensitivity of + 1.0 indicates that + 10% change of an assessed input parameter causes + 10% change of the predicted pharmacokinetic parameters.
Model evaluation
The PBPK model was evaluated using visual and numerical methods. Observed plasma concentration–time profiles were graphically compared with the predicted profiles by plotting the geometric mean and 5th to 95th percentiles for a virtual population (n = 100). Demographic ranges for virtual populations were adjusted to be similar to those of the observed population. If the age and proportion of female (%) range had not been obtained from the clinical studies, it was assumed as 19 to 65 years and 50%, respectively. Others were generated via the implemented algorithm in the PK-Sim® software. The standard deviation for the reference concentration of CYP2C9 was assigned as 1.15 μmol/L to reflect moderate variability (30% of the mean). The PBPK model was numerically evaluated by comparing observed and predicted AUC, Cmax, and t1/2 values. A two-fold error range was used as the acceptance criterion for the model. In other words, the PBPK model could be justified if the fold error (predicted value divided into observed value) is within the 0.5–2 range. Geometric mean values for virtual populations were used as predicted values and reported values in clinical studies or estimated values based on the NCA were used as observed values. Lastly, the overall performance of the PBPK model was evaluated via a goodness-of-fit plot for the predicted versus observed plasma concentrations. Average fold error (AFE), absolute average fold error (AAFE), and root mean square error (RMSE) were used to evaluate the prediction accuracy and precision, respectively. AFE, AAFE, and RMSE were calculated according to Eqs. (2–4).
Results
A total 54 of clinical data were collected. Clinical trial data of various doses, administration period, and age of subjects were included. Most of the clinical studies recruited healthy subjects, but several clinical studies targeted patients with osteoarthritis or rheumatoid arthritis. Data of two pharmacogenomic studies (Perini et al. 2005; Perini and Suarez-Kurtz 2006) were used for development of PBPK model.
The summary of input parameters for the PBPK model is presented in Table 2. The fraction unbound (fu) value was adjusted to be 0.01% lower value than the minimum value of Blocka et al. (1988) to capture the plasma-concentration time profiles more accurately. Simulation for the individual PBPK model after the administration of 20 mg single oral dose properly captured not only the plasma concentration–time profiles but also the metabolized and excreted fractions (Fig. 1). The estimated volume of distribution (Vd) was 0.15 L/kg which is almost consistent with the previously reported value (0.14 L/kg) (Pfizer 2016).

Predicted and observed plasma concentration–time profiles of piroxicam after a single oral dose of piroxicam 20 mg. Solid, dashed, and dotted lines indicate predicted plasma concentration, fraction excreted to urine, and fraction metabolized by CYP2C9, respectively. The open circles and error bars indicated observed mean and standard deviation (or standard error), respectively. Observed plasma concentration and fraction excreted to urine data were obtained from Perini et al. (2005) and Ishizaki et al. (1979), respectively. Fraction metabolized by CYP2C9 data was estimated based on Perini and Suarez-Kurtz (2006)
Predicted plasma concentration–time profiles in different CYP2C9 genotypes were visually similar to the observed profiles (Fig. 2). Predicted AUCinf in CYP2C9*1/*2, CYP2C9*1/*3, and CYP2C9*3/*3 genotypes were 1.83-, 2.07-, and 6.43-fold higher than CYP2C9*1/*1 genotype, respectively, and significant differences for predicted Cmax in different CYP2C9 genotypes were not identified (1.89–1.96 μg/mL) (Table 3) (Fig. 3). In addition, PBPK model overall accurately described and predicted the plasma concentration–time profiles in pediatric, adult, and geriatric populations who received a single or multiple-dose regimens (Fig. 4). All fold error values for AUC, Cmax, and t1/2 were included in the acceptance criterion with the ranges of 0.57–1.59, 0.63–1.39, and 0.65–1.51, respectively (Table 3).

Predicted and observed plasma concentration–time profiles of piroxicam after a single oral dose of piroxicam 20 mg in different CYP2C9 genotypes. Solid and dashed lines indicate geometric mean and 5th to 95th percentiles, respectively. Open circles and error bars indicate observed mean and standard deviation (or standard error), respectively. Observed plasma concentration data except CYP2C9*3/*3 genotype was obtained from Perini et al. (2005) and CYP2C9*3/*3 genotype was obtained from Perini and Suarez-Kurtz (2006). Plasma concentration–time profiles are expressed using linear and semi-logarithmic plots

Results of sensitivity analysis toward AUCinf A and Cmax B. x-axis and y-axis indicate sensitivity and lists of sensitive parameters, respectively

Representative predicted and observed plasma concentration–time profiles of piroxicam for different populations and dose regimens. Solid and dashed lines indicate geometric mean and 5th to 95th percentiles, respectively. Open circles and error bars indicate observed mean and standard deviation (or standard error), respectively. Observed data in Mäkelä et al. (1991) are depicted as individual values
Sensitivity analysis shown in Fig. 3 presented that dose had an equal impact on AUCinf and Cmax (1.00). Lipophilicity which had the highest impact in Cmax (− 1.82) was identified as having a relatively low impact in AUCinf (0.03). The parameters related to the enzymatic pathway of CYP2C9, including kcat, Km, reference concentration, and ontogeny factor, had an impact on the AUCinf and Cmax with higher influences for AUCinf than Cmax. Several organ volumes were identified as sensitive physiological characteristics.
Among the collected articles, except for the two papers used for model establishment, all other papers did not include pharmacogenomic data, so model validation for each genotype using PK data not used for model establishment could not be performed. Therefore, model validation was performed using PK data without pharmacogenomic information. The goodness-of-fit plot for a total 674 of predicted versus observed plasma concentration data is illustrated in Fig. 5. The range of fold error values for plasma concentrations was 0.11–3.13. 93.9% of fold error values were within the two-fold range. AFE, AAFE, and RMSE were 0.93, 1.27, and 0.72, respectively.

Goodness-of-fit plot of predicted versus observed plasma concentrations. Solid and dashed lines indicate line of unity and two-fold range, respectively
Discussion
Genetic polymorphisms of drug metabolizing enzymes and transporters cause the inter-individual variations in drug response to varying degrees (Byeon et al. 2019; Bae et al. 2020; Jung et al. 2020a; Shin et al. 2020; Kim et al. 2022). Also, drug interactions significantly influence the pharmacokinetics of clinically used drugs (Byeon et al. 2018; Lee et al. 2019; Jung et al. 2020b). According to the advance in computational technology, a personalized dose administration strategy considering physiological characteristics of individuals or populations, genetic polymorphisms of drug metabolizing enzymes or transporters, and drug interactions has been proposed via the PBPK modeling approach (Li et al. 2020; Rüdesheim et al. 2020; Cho et al. 2021a, 2021b; Jung et al. 2021; Wojtyniak et al. 2021; Xu et al. 2021).
CYP2C9 is primarily responsible for the metabolism of a number of NSAIDs including ibuprofen, lornoxicam, meloxicam, flurbiprofen, and celecoxib and significant effects of CYP2C9 genetic polymorphism on the pharmacokinetics or pharmacodynamics of these drugs have been reported (Bae et al. 2011a; Choi et al. 2011; Lee et al. 2014, 2015; Ochoa et al. 2015; Kim et al. 2017). Like these NSAIDs, piroxicam is mainly metabolized by CYP2C9 and the influences of CYP2C9 genetic polymorphism on the piroxicam actions are known (Perini et al. 2005; Perini and Suarez-Kurtz 2006; Calvo et al. 2017). CYP2C9 genotype is potentially involved in the adverse events given the relationship between exposure and toxicity of the NSAIDs (Smith et al. 2022). Piroxicam has a longer t1/2 than other NSAIDs metabolized by CYP2C9 such as celecoxib, flurbiprofen, and meloxicam (Theken et al. 2020). This amplifies the potential risks in the patients with reduced CYP2C9 metabolism and hampers dose titration due to lack of data (Theken et al. 2020). Drug label and Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for piroxicam recommend the consideration of dose reduction or the alteration to other NSAIDs which are not metabolized by CYP2C9 or metabolized by CYP2C9 with short half-life in CYP2C9PM group, respectively (Pfizer 2016; Theken et al. 2020).
To overcome the potential risk in patients carrying CYP2C9 allele variants and fulfill the lack of data related to piroxicam and CYP2C9 genotype to some extent, we developed and validated the first PBPK model for piroxicam according to CYP2C9 genetic polymorphism. In the model development, physicochemical and ADME properties for piroxicam were incorporated. In vitro enzyme kinetic data (Tracy et al. 2002) significantly under-estimated in vivo clearance of piroxicam in CYP2C9*1/*1 genotype. Hence, we estimated the fraction metabolized by CYP2C9 based on the previously reported pharmacogenetic study (Perini and Suarez‐Kurtz 2006). CPIC assigned CYP2C9*1/*1 genotype as CYP2C9EM phenotype and CYP2C9*3/*3 genotype as CYP2C9PM phenotype, respectively (Theken et al. 2020). Accordingly, the fraction was calculated based on the ratio of AUCinf in CYP2C9*1/*1 and CYP2C9*3/*3 genotypes (Ito et al. 2005; Perini and Suarez‐Kurtz 2006; Huang et al. 2017). Minor enzymatic pathways such as dealkylation and glucuronidation of piroxicam were identified, but it is not well reported and no candidate genes have been published (Brogden et al. 1981; Milligan et al. 1993). Thus, only the 5’-hydroxylation of piroxicam, the principal metabolic pathway mediated by CYP2C9, was applied in our model.
Our model successfully captured the pharmacokinetic alterations according to CYP2C9 genetic polymorphism. Especially, the pharmacokinetics of piroxicam in the CYP2C9*3/*3 genotype which had been traced for an extremely long period (120 days), were properly captured in this model. Furthermore, the range of fold error values for the pharmacokinetic parameters (AUC 0.57–1.59, Cmax 0.63–1.39, and t1/2 0.65–1.51) was within the acceptance criterion (Table 3) and the goodness-of-fit plot showed that the predicted data were overall in agreement with the observed data (AFE 0.93, AAFE 1.27, and RMSE 0.72). It suggests the present model was properly established for single and multiple doses of 0.4 or 1.0 mg/kg in children and 20–60 mg in adults, including the elderly, in both fasting and eating conditions. Although the sample size used for the modeling of the CYP2C9*3/*3 genotype was very small (n = 1), the present model could provide an insight for grasping the pharmacokinetics of piroxicam, simultaneous considering genetic and non-genetic factors.
Several modeling studies for the pharmacokinetics of piroxicam in humans have been reported (Wang et al. 2000; Tvrdonova et al. 2009; Li et al. 2019). Wang et al. (2000) reported population pharmacokinetic and pharmacodynamic model for piroxicam and piroxicam-β-cyclodextrin to investigate the pharmacokinetic-pharmacodynamic relationship of piroxicam. In their model, the physiological and anatomical characteristics of human were not incorporated because it was conducted as a “top-down” approach based on clinical data. Tvrdonova et al. (2009) showed a physiologically motivated time-delay model. This study accurately captured the multiple peak phenomenon of piroxicam, but in vitro data of piroxicam was not considered. Li et al. (2019) developed the PBPK model to predict the pharmacokinetics of piroxicam in beagle dogs and performed interspecies extrapolation to humans. Albeit a successful model, the application of the PBPK model to pediatric or geriatric populations could not be certain since it was only verified with clinical data on an adult population. In the present study, we developed the PBPK model for piroxicam by incorporating in vitro and in vivo data, robustly validated using a number of clinical studies, and demonstrated the applicability of the PBPK model for almost the entire age populations.
In this study, the PBPK model for only the two most common CYP2C9 allele variants (*2 and *3) was developed because of the lack of available pharmacogenetic studies. Two pharmacogenetic studies used for the PBPK modeling were performed in the Brazilian population (Perini et al. 2005; Perini and Suarez‐Kurtz 2006). CYP2C9*13 (rs72558187, c.269 T > C, p.Leu90Pro) allele variant, found only in East Asians with a frequency of 0.5% (Bae et al. 2011b), significantly influences the plasma exposures for various CYP2C9 substrates (Bae et al. 2011a, 2012; Choi et al. 2011, 2012). Previous studies reported the PBPK models related to CYP2C9*13 allele variants for celecoxib (Kim et al. 2021), meloxicam (Cho et al. 2021b), and candesartan (Jung et al. 2021) based on the clinical trials performed in the healthy Korean population. Further pharmacogenetic and modeling studies would be needed for the proper prescription of piroxicam in the East Asian population carrying the CYP2C9*13 allele.
There were several limitations in the present study. First, physiological differences between healthy volunteers and subjects with arthritis were not considered. Several studies were performed on the subjects with osteoarthritis or rheumatoid arthritis (Darragh et al. 1985; Blocka et al. 1988; Mäkelä et al. 1991; Caldwell 1994), but we developed and validated the PBPK model based on the parameters used in healthy volunteers without any modifications for these diseases. Second, in most clinical studies, all subjects were assumed to be carrying homozygous CYP2C9*1 allele due to the low frequencies of CYP2C9*2 or *3 allele variants (Daly et al. 2017). Nevertheless, a few of the subjects in the clinical data could be carrying allele variants and it could be one of the causes for the discrepancies between the predicted and observed data. Third, digitized plasma concentration–time profiles could be discrepant with the raw data. Wojtyniak et al. (2020) reported the greatest pitfall for the data digitizing comes from the pre-existing errors and they recommended making published data available as raw values. However, raw data for the pharmacokinetics of piroxicam were not available and the resolution of the piroxicam profiles was relatively low since the most of clinical studies used in this study were reported a long time ago. It would be better to interpret the results of the modeling under the consideration of these potential limitations.
In conclusion, the PBPK model for piroxicam related to CYP2C9 genetic polymorphism was properly established and described the pharmacokinetics of piroxicam in different CYP2C9 genotypes. It could be used as a cornerstone to predict the pharmacokinetics of piroxicam in a number of clinical scenarios. We expect the present model could contribute to personalized pharmacotherapy for patients treated with piroxicam.
References
Abduljalil K, Badhan RKS (2020) Drug dosing during pregnancy-opportunities for physiologically based pharmacokinetic models. J Pharmacokinet Pharmacodyn 47(4):319–340. https://doi.org/10.1007/s10928-020-09698-w
Al-Shakargi SDMS (2012) Bioequivalency of two piroxicam products in plasma by high performance liquid chromatography. Tikret Journal of Pharmceutical Sciences 8(2):242–247
Bae JW, Choi CI, Jang CG, Lee SY (2011a) Effects of CYP2C9*1/*13 on the pharmacokinetics and pharmacodynamics of meloxicam. Br J Clin Pharmacol 71(4):550–555. https://doi.org/10.1111/j.1365-2125.2010.03853.x
Bae JW, Choi CI, Kim MJ, Oh DH, Keum SK, Park JI, Kim BH, Bang HK, Oh SG, Kang BS, Park HJ, Kim HD, Ha JH, Shin HJ, Kim YH, Na HS, Chung MW, Jang CG, Lee SY (2011b) Frequency of CYP2C9 alleles in Koreans and their effects on losartan pharmacokinetics. Acta Pharmacol Sin 32(10):1303–1308. https://doi.org/10.1038/aps.2011.100
Bae JW, Choi CI, Lee HI, Lee YJ, Jang CG, Lee SY (2012) Effects of CYP2C9*1/*3 and *1/*13 on the pharmacokinetics of losartan and its active metabolite E-3174. Int J Clin Pharmacol Ther 50(9):683–689. https://doi.org/10.5414/cp201467
Bae JW, Oh KY, Yoon SJ, Shin HB, Jung EH, Cho CK, Lim CW, Kang P, Choi CI, Jang CG, Lee SY, Lee YJ (2020) Effects of CYP2D6 genetic polymorphism on the pharmacokinetics of metoclopramide. Arch Pharm Res 43(11):1207–1213. https://doi.org/10.1007/s12272-020-01293-4
Benveniste C, Striberni R, Dayer P (1990) Indirect assessment of the enterohepatic recirculation of piroxicam and tenoxicam. Eur J Clin Pharmacol 38(6):547–549. https://doi.org/10.1007/BF00278579
Berg J, Fellier H, Christoph T, Grarup J, Stimmeder D (1999) The analgesic NSAID lornoxicam inhibits cyclooxygenase (COX)-1/-2, inducible nitric oxide synthase (iNOS), and the formation of interleukin (IL)-6 in vitro. Inflamm Res 48(7):369–379. https://doi.org/10.1007/s000110050474
Blanco FJ, Guitian R, Moreno J, de Toro FJ, Galdo F (1999) Effect of anti-inflammatory drugs on COX-1 and COX−2 activity in human articular chondrocytes. J Rheumatol 26(6):1366–1373
Blocka KL, Richardson CJ, Wallace SM, Ross SG, Verbeeck RK (1988) The effect of age on piroxicam disposition in rheumatoid arthritis. J Rheumatol 15(5):757–763
Boudinot FD, Ibrahim SS (1988) High-performance liquid chromatographic assay for piroxicam in human plasma. J Chromatogr 430(2):424–428. https://doi.org/10.1016/s0378-4347(00)83181-3
Brogden RN, Heel RC, Speight TM, Avery GS (1981) Piroxicam: a review of its pharmacological properties and therapeutic efficacy. Drugs 22(3):165–187. https://doi.org/10.2165/00003495-198122030-00001
Byeon JY, Lee YJ, Kim YH, Kim SH, Lee CM, Bae JW, Jang CG, Lee SY, Choi CI (2018) Effects of diltiazem, a moderate inhibitor of CYP3A4, on the pharmacokinetics of tamsulosin in different CYP2D6 genotypes. Arch Pharm Res 41(5):564–570. https://doi.org/10.1007/s12272-018-1030-6
Byeon JY, Lee CM, Lee YJ, Kim YH, Kim SH, Jung EH, Chae WK, Lee YJ, Jang CG, Lee SY (2019) Influence of CYP2D6 genetic polymorphism on pharmacokinetics of active moiety of tolterodine. Arch Pharm Res 42(2):182–190. https://doi.org/10.1007/s12272-018-1099-y
Caldwell JR (1994) Comparison of the efficacy, safety, and pharmacokinetic profiles of extended-release ketoprofen and piroxicam in patients with rheumatoid arthritis. Clin Ther 16(2):222–235
Calvo AM, Santos GM, Dionísio TJ, Marques MP, Brozoski DT, Lanchote VL, Fernandes MHR, Faria FAC, Santos CF (2016) Quantification of piroxicam and 5′-hydroxypiroxicam in human plasma and saliva using liquid chromatography–tandem mass spectrometry following oral administration. J Pharm Biomed Anal 120:212–220. https://doi.org/10.1016/j.jpba.2015.12.042
Calvo AM, Zupelari-Gonçalves P, Dionísio TJ, Brozoski DT, Faria FA, Santos CF (2017) Efficacy of piroxicam for postoperative pain after lower third molar surgery associated with CYP2C8*3 and CYP2C9. J Pain Res 10:1581–1589. https://doi.org/10.2147/jpr.S138147
Campbell AJ, Ferry DG, Edwards IR (1985) Pharmacokinetic projections for isoxicam and piroxicam in old and young subjects. Br J Rheumatol 24(2):176–178. https://doi.org/10.1093/rheumatology/24.2.176
Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2021a) Physiologically based pharmacokinetic (PBPK) modelling of tamsulosin related to CYP2D6*10 allele. Arch Pharm Res 44(11):1037–1049. https://doi.org/10.1007/s12272-021-01357-z
Cho CK, Park HJ, Kang P, Moon S, Lee YJ, Bae JW, Jang CG, Lee SY (2021b) Physiologically based pharmacokinetic (PBPK) modeling of meloxicam in different CYP2C9 genotypes. Arch Pharm Res 44(12):1076–1090. https://doi.org/10.1007/s12272-021-01361-3
Choi CI, Kim MJ, Jang CG, Park YS, Bae JW, Lee SY (2011) Effects of the CYP2C9*1/*13 genotype on the pharmacokinetics of lornoxicam. Basic Clin Pharmacol Toxicol 109(6):476–480. https://doi.org/10.1111/j.1742-7843.2011.00751.x
Choi CI, Kim MJ, Chung EK, Lee HI, Jang CG, Bae JW, Lee SY (2012) CYP2C9*3 and *13 alleles significantly affect the pharmacokinetics of irbesartan in healthy Korean subjects. Eur J Clin Pharmacol 68(2):149–154. https://doi.org/10.1007/s00228-011-1098-0
Coppola P, Kerwash E, Cole S (2021) Physiologically based pharmacokinetics model in pregnancy: a regulatory perspective on model evaluation. Front Pediatr 9:687978. https://doi.org/10.3389/fped.2021.687978
Daly AK, Rettie AE, Fowler DM, Miners JO (2017) Pharmacogenomics of CYP2C9: functional and clinical considerations. J Pers Med 8(1):1. https://doi.org/10.3390/jpm8010001
Darragh A, Gordon AJ, O’Byrne H, Hobbs D, Casey E (1985) Single-dose and steady-state pharmacokinetics of piroxicam in elderly vs young adults. Eur J Clin Pharmacol 28(3):305–309. https://doi.org/10.1007/BF00543328
Dean L (2019) Piroxicam Therapy and CYP2C9 Genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kane MS, Kattman BL, Malheiro AJ (ed) Medical Genetics Summaries [Internet]. Bethesda (MD): National Center for Biotechnology Information (US) [Online] Available from https://www.ncbi.nlm.nih.gov/books/NBK537367/ Accessed 18 April 2022
Dessain P, Estabrooks TF, Gordon AJ (1979) Piroxicam in the treatment of osteoarthrosis: A multicentre study in general practice involving 1218 patients. J Int Med Res 7(5):335–343. https://doi.org/10.1177/030006057900700501
Dix P, Prosser DP, Streete P (2004) A pharmacokinetic study of piroxicam in children. Anaesthesia 59(10):984–987. https://doi.org/10.1111/j.1365-2044.2004.03806.x
Dixon JS, Lowe JR, Galloway DB (1984) Rapid method for the determination of either piroxicam or tenoxicam in plasma using high-performance liquid chromatography. J Chromatogr 310(2):455–459. https://doi.org/10.1016/0378-4347(84)80116-4
Dixon JS, Lacey LF, Pickup ME, Langley SJ, Page MC (1990) A lack of pharmacokinetic interaction between ranitidine and piroxicam. Eur J Clin Pharmacol 39(6):583–586. https://doi.org/10.1007/BF00316100
Ferry DG, Gazeley LR, Busby WJ, Beasley DM, Edwards IR, Campbell AJ (1990) Enhanced elimination of piroxicam by administration of activated charcoal or cholestyramine. Eur J Clin Pharmacol 39(6):599–601. https://doi.org/10.1007/BF00316105
Guentert TW, Defoin R, Mosberg H (1988) The influence of cholestyramine on the elimination of tenoxicam and piroxicam. Eur J Clin Pharmacol 34(3):283–289. https://doi.org/10.1007/bf00540957
Hasan MM, Jilani JA, Salem MS, Najib NM, Pillai GK, Ganem E (1997) Bioequivalence of two capsule formulations of piroxicam. Acta Pharm Sci 39(3):93–97
Heimbach T, Chen Y, Chen J, Dixit V, Parrott N, Peters SA, Poggesi I, Sharma P, Snoeys J, Shebley M, Tai G, Tse S, Upreti VV, Wang YH, Tsai A, Xia B, Zheng M, Zhu AZX, Hall S (2021) Physiologically-based pharmacokinetic modeling in renal and hepatic impairment populations: a pharmaceutical industry perspective. Clin Pharmacol Ther 110(2):297–310. https://doi.org/10.1002/cpt.2125
Helmy SA, El-Bedaiwy HM (2014) Piroxicam immediate release formulations: A fasting randomized open-label crossover bioequivalence study in healthy volunteers. Clin Pharmacol Drug Dev 3(6):466–471. https://doi.org/10.1002/cpdd.106
Hindmarsh AC, Reynolds DR, Serban R, Woodward CS, Gardner, DJ, Cohen SD, Taylor A, Peles S, Banks L, Shumaker D (2021) Open Systems Pharmacology Suite Manual version 10. [Online] Available from https://docs.open-systems-pharmacology.org/. Accessed 18 April 2022
Hobbs DC, Twomey TM (1979) Piroxicam pharmacokinetics in man: aspirin and antacid interaction studies. J Clin Pharmacol 19(5–6):270–281. https://doi.org/10.1002/j.1552-4604.1979.tb02480.x
Huang W, Nakano M, Sager J, Ragueneau-Majlessi I, Isoherranen N (2017) Physiologically based pharmacokinetic model of the CYP2D6 probe atomoxetine: extrapolation to special populations and drug-drug interactions. Drug Metab Dispos 45(11):1156–1165. https://doi.org/10.1124/dmd.117.076455
Ishizaki T, Nomura T, Abe T (1979) Pharmacokinetics of piroxicam, a new nonsteroidal anti-inflammatory agent, under fasting and postprandial states in man. J Pharmacokinet Biopharm 7(4):369–381. https://doi.org/10.1007/BF01062535
Ito K, Hallifax D, Obach RS, Houston JB (2005) Impact of parallel pathways of drug elimination and multiple cytochrome P450 involvement on drug-drug interactions: CYP2D6 paradigm. Drug Metab Dispos 33(6):837–844. https://doi.org/10.1124/dmd.104.003715
Jeon SS, Cha HR, Park YJ, Lee BC, Kim ND (1998) Comparison of absorption rate between piroxicam-β-cyclodextrin and piroxicam in Korean healthy subjects after a single dose administration. Korean J Clin Pharm 8(2):95–100
Jung EH, Lee CM, Byeon JY, Shin HB, Oh KY, Cho CK, Lim CW, Jang CG, Lee SY (2020) Lee YJ (2020a) Relationship between plasma exposure of zolpidem and CYP2D6 genotype in healthy Korean subjects. Arch Pharm Res 43(9):976–981. https://doi.org/10.1007/s12272-020-01250-1
Jung EH, Lee YJ, Kim DH, Kang P, Lim CW, Cho CK, Jang CG, Lee SY, Bae JW (2020) Effects of paroxetine on the pharmacokinetics of atomoxetine and its metabolites in different CYP2D6 genotypes. Arch Pharm Res 43(12):1356–1363. https://doi.org/10.1007/s12272-020-01300-8
Jung EH, Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Choi CI, Jang CG, Lee SY (2021) Physiologically based pharmacokinetic modeling of candesartan related to CYP2C9 genetic polymorphism in adult and pediatric patients. Arch Pharm Res 44(12):1109–1119. https://doi.org/10.1007/s12272-021-01363-1
Kim SH, Kim DH, Byeon JY, Kim YH, Kim DH, Lim HJ, Lee CM, Whang SS, Choi CI, Bae JW, Lee YJ, Jang CG, Lee SY (2017) Effects of CYP2C9 genetic polymorphisms on the pharmacokinetics of celecoxib and its carboxylic acid metabolite. Arch Pharm Res 40(3):382–390. https://doi.org/10.1007/s12272-016-0861-2
Kim SH, Byeon JY, Kim YH, Lee CM, Lee YJ, Jang CG, Lee SY (2018) Physiologically based pharmacokinetic modelling of atomoxetine with regard to CYP2D6 genotypes. Sci Rep 8(1):12405. https://doi.org/10.1038/s41598-018-30841-8
Kim YH, Kang P, Cho CK, Jung EH, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY (2021) Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch Pharm Res 44(7):713–724. https://doi.org/10.1007/s12272-021-01346-2
Kim NT, Cho CK, Kang P, Park H-J, Lee YJ, Bae JW, Jang C-G, Lee S-Y (2022) Effects of CYP2C9*3 and *13 alleles on the pharmacokinetics and pharmacodynamics of glipizide in healthy Korean subjects. Arch Pharm Res 45(2):114–121. https://doi.org/10.1007/s12272-021-01366-y
Kirchheiner J, Tsahuridu M, Jabrane W, Roots I, Brockmöller J (2004) The CYP2C9 polymorphism: from enzyme kinetics to clinical dose recommendations. Per Med 1(1):63–84. https://doi.org/10.1517/17410541.1.1.63
Lee HI, Bae JW, Choi CI, Lee YJ, Byeon JY, Jang CG, Lee SY (2014) Strongly increased exposure of meloxicam in CYP2C9*3/*3 individuals. Pharmacogenet Genomics 24(2):113–117. https://doi.org/10.1097/fpc.0000000000000025
Lee YJ, Byeon JY, Kim YH, Kim SH, Choi CI, Bae JW, Sohn UD, Jang CG, Lee J, Lee SY (2015) Effects of CYP2C9*1/*3 genotype on the pharmacokinetics of flurbiprofen in Korean subjects. Arch Pharm Res 38(6):1232–1237. https://doi.org/10.1007/s12272-015-0580-0
Lee CM, Jung EH, Byeon JY, Kim SH, Jang CG, Lee YJ, Lee SY (2019) Effects of steady-state clarithromycin on the pharmacokinetics of zolpidem in healthy subjects. Arch Pharm Res 42(12):1101–1106. https://doi.org/10.1007/s12272-019-01201-5
Li X, Yang Y, Zhang Y, Wu C, Jiang Q, Wang W, Li H, Li J, Luo C, Wu W, Wang Y, Zhang T (2019) Justification of biowaiver and dissolution rate specifications for piroxicam immediate release products based on physiologically based pharmacokinetic modeling: an in-depth analysis. Mol Pharm 16(9):3780–3790. https://doi.org/10.1021/acs.molpharmaceut.9b00350
Li X, Frechen S, Moj D, Lehr T, Taubert M, Hsin C-h, Mikus G, Neuvonen PJ, Olkkola KT, Saari TI, Fuhr U (2020) A physiologically based pharmacokinetic model of voriconazole integrating time-dependent inhibition of CYP3A4, genetic polymorphisms of CYP2C19 and predictions of drug–drug Interactions. Clin Pharmacokinet 59(6):781–808. https://doi.org/10.1007/s40262-019-00856-z
Loisios-Konstantinidis I, Cristofoletti R, Jamei M, Turner D, Dressman J (2020) Physiologically based pharmacokinetic/pharmacodynamic modeling to predict the impact of CYP2C9 genetic polymorphisms, co-medication and formulation on the pharmacokinetics and pharmacodynamics of flurbiprofen. Pharmaceutics 12(11):1049. https://doi.org/10.3390/pharmaceutics12111049
Macek J, Vácha J (1987) Rapid and sensitive method for determination of piroxicam in human plasma by high-performance liquid chromatography. J Chromatogr 420(2):445–449. https://doi.org/10.1016/0378-4347(87)80203-7
Mäkelä AL, Olkkola KT, Mattila MJ (1991) Steady state pharmacokinetics of piroxicam in children with heumatic diseases. Eur J Clin Pharmacol 41(1):79–81. https://doi.org/10.1007/BF00280114
Milligan PA, McGill PE, Howden CW, Kelman AW, Whiting B (1993) The consequences of H2 receptor antagonist—piroxicam coadministration in patients with joint disorders. Eur J Clin Pharmacol 45(6):507–512. https://doi.org/10.1007/BF00315306
Nishimura M, Naito S (2005) Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet 20(6):452–477. https://doi.org/10.2133/dmpk.20.452
Nishimura M, Naito S (2006) Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet 21(5):357–374. https://doi.org/10.2133/dmpk.21.357
Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T (2003) Tissue Distribution of mRNA expression of human cytochrome p450 isoforms assessedby high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi 123(5):369–375. https://doi.org/10.1248/yakushi.123.369
Ochoa D, Prieto-Pérez R, Román M, Talegón M, Rivas A, Galicia I, Abad-Santos F, Cabaleiro T (2015) Effect of gender and CYP2C9 and CYP2C8 polymorphisms on the pharmacokinetics of ibuprofen enantiomers. Pharmacogenomics 16(9):939–948. https://doi.org/10.2217/pgs.15.40
Palma-Aguirre JA, Lopez-Gamboa M, Cariño L, Burke-Fraga V, González-de la Parra M (2010) Relative bioavailability of two oral formulations of piroxicam 20 mg: a single-dose, randomized-sequence, open-label, two-period crossover comparison in healthy Mexican adult volunteers. Clin Ther 32(2):357–364. https://doi.org/10.1016/j.clinthera.2010.02.002
Perini JA, Suarez-Kurtz G (2006) Impact of CYP2C9*3/*3 genotype on the pharmacokinetics and pharmacodynamics of piroxicam. Clin Pharmacol Ther 80(5):549–551. https://doi.org/10.1016/j.clpt.2006.08.003
Perini JA, Vianna-Jorge R, Brogliato AR, Suarez-Kurtz G (2005) Influence of CYP2C9 genotypes on the pharmacokinetics and pharmacodynamics of piroxicam. Clin Pharmacol Ther 78(4):362–369. https://doi.org/10.1016/j.clpt.2005.06.014
Pfizer (2016) FELDENE® (piroxicam) Prescribing information. [Online] Available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/018147s044lbl.pdf. Accessed 18 April 2022
Piscitelli DA, Bigora S, Propst C, Goskonda S, Schwartz P, Lesko LJ, Augsburger L, Young D (1998) The impact of formulation and process changes on in vitro dissolution and the bioequivalence of piroxicam capsules. Pharm Dev Technol 3(4):443–452. https://doi.org/10.3109/10837459809028625
Rahman NU, Ahamd M, Akhtar N (2004) Bioequivalence of a generic piroxicam capsule formulation. J Pure Appl Sci 23(2):52–61
Rasetti-Escargueil C, Grangé V (2005) Pharmacokinetic profiles of two tablet formulations of piroxicam. Int J Pharm 295(1–2):129–134. https://doi.org/10.1016/j.ijpharm.2005.02.006
Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF (1992) Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol 5(1):54–59. https://doi.org/10.1021/tx00025a009
Richardson CJ, Blocka KL, Ross SG, Verbeeck RK (1985) Effects of age and sex on piroxicam disposition. Clin Pharmacol Ther 37(1):13–18. https://doi.org/10.1038/clpt.1985.4
Richardson C, Blocka K, Ross S, Verbeeck R (1987) Piroxicam and 5’-hydroxypiroxicam kinetics following multiple dose administration of piroxicam. Eur J Clin Pharmacol 32(1):89–91. https://doi.org/10.1007/BF00609964
Riedel KD, Laufen H (1983) High-performance thin-layer chromatographic assay for the routine determination of piroxicam in plasma, urine and tissue. J Chromatogr 276:243–248. https://doi.org/10.1016/s0378-4347(00)85090-2
Rodrigues AD (1999) Integrated cytochrome P450 reaction phenotyping: attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biocheml Pharmacol 57(5):465–480. https://doi.org/10.1016/s0006-2952(98)00268-8
Rogers HJ, Spector RG, Morrison PJ, Bradbrook ID (1981) Comparative steady state pharmacokinetic study of piroxicam and flurbiprofen in normal subjects. Eur J Rheumatol Inflamm 4(3):303–308
Rüdesheim S, Wojtyniak JG, Selzer D, Hanke N, Mahfoud F, Schwab M, Lehr T (2020) Physiologically based pharmacokinetic modeling of metoprolol enantiomers and α-Hydroxymetoprolol to Describe CYP2D6 drug-gene interactions. Pharmaceutics 12(12):1200. https://doi.org/10.3390/pharmaceutics12121200
Rudy AC, Figueroa NL, Hall SD, Brater DC (1994) The pharmacokinetics of piroxicam in elderly persons with and without renal impairment. Br J Clin Pharmacol 37(1):1–5. https://doi.org/10.1111/j.1365-2125.1994.tb04230.x
Said SA, Foda AM (1989) Influence of cimetidine on the pharmacokinetics of piroxicam in rat and man. Arzneimittelforschung 39(7):790–792
Schmitt W (2008) General approach for the calculation of tissue to plasma partition coefficients. Toxicol in Vitro 22(2):457–467. https://doi.org/10.1016/j.tiv.2007.09.010
Shahbaz N, Iqbal Z, Nasir F, Khan FU, Hassan AM, Khan SI (2018) Simultaneous determination of piroxicam and 5-hydroxypiroxicam: HPLC/UV method development, validation and application for pharmacokinetic evaluation in Pakistani population. J Chem Soc Pak 40(05):856–865
Shin HB, Jung EH, Kang P, Lim CW, Oh KY, Cho CK, Lee YJ, Choi CI, Jang CG, Lee SY, Bae JW (2020) ABCB1 c.2677G>T/c.3435C>T diplotype increases the early-phase oral absorption of losartan. Arch Pharm Res 43(11):1187–1196. https://doi.org/10.1007/s12272-020-01294-3
Smith DM, Stevenson JM, Ho TT, Formea CM, Gammal RS, Cavallari LH (2022) Pharmacogenetics: A precision medicine approach to combatting the opioid epidemic. J Am Coll Clin Pharm 5(2):239–250. https://doi.org/10.1002/jac5.1582
Song HH, Choi KS, Kim CW, Kwon YE (2009) Pharmacokinetic profiles of two branded formulations of piroxicam 20mg in healthy Korean volunteers by a rapid isocratic HPLC method. J Bioequiv Availab 1(3):74–79
Suri A, Chapel S, Lu C, Venkatakrishnan K (2015) Physiologically based and population PK modeling in optimizing drug development: A predict–learn–confirm analysis. Clin Pharmacol Ther 98(3):336–344. https://doi.org/10.1002/cpt.155
Tang C, Shou M, Rushmore TH, Mei Q, Sandhu P, Woolf EJ, Rose MJ, Gelmann A, Greenberg HE, De Lepeleire I, Van Hecken A, De Schepper PJ, Ebel DL, Schwartz JI, Rodrigues AD (2001) In-vitro metabolism of celecoxib, a cyclooxygenase-2 inhibitor, by allelic variant forms of human liver microsomal cytochrome P450 2C9: correlation with CYP2C9 genotype and in-vivo pharmacokinetics. Pharmacogenet Genomics 11(3):223–235
Theken KN, Lee CR, Gong L, Caudle KE, Formea CM, Gaedigk A, Klein TE, Agúndez JAG, Grosser T (2020) Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2C9 and nonsteroidal anti-inflammatory drugs. Clin Pharmacol Ther 108(2):191–200. https://doi.org/10.1002/cpt.1830
Thelen K, Coboeken K, Willmann S, Burghaus R, Dressman JB, Lippert J (2011) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: oral solutions. J Pharm Sci 100(12):5324–5345. https://doi.org/10.1002/jps.22726
Thelen K, Coboeken K, Willmann S, Dressman JB, Lippert J (2012) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: extension to describe performance of solid dosage forms. J Pharm Sci 101(3):1267–1280. https://doi.org/10.1002/jps.22825
Tilstone WJ, Lawson DH, Omara F, Cunningham F (1981) The steady-state pharmacokinetics of piroxicam: effect of food and iron. Eur J Rheumatol Inflamm 4(3):309–313
Tracy TS, Hutzler JM, Haining RL, Rettie AE, Hummel MA, Dickmann LJ (2002) Polymorphic variants (CYP2C9*3 and CYP2C9*5) and the F114L active site mutation of CYP2C9: effect on atypical kinetic metabolism profiles. Drug Metab Dispos 30(4):385–390. https://doi.org/10.1124/dmd.30.4.385
Trnavská Z, Trnavský K (1984) Plasma protein binding and interaction studies with piroxicam. Naunyn Schmiedeberg’s Arch Pharmacol 327(1):81–85. https://doi.org/10.1007/BF00504996
Tvrdonova M, Dedik L, Mircioiu C, Miklovicova D, Durisova M (2009) Physiologically motivated time-delay model to account for mechanisms underlying enterohepatic circulation of piroxicam in human beings. Basic Clin Pharmacol Toxicol 104(1):35–42. https://doi.org/10.1111/j.1742-7843.2008.00304.x
Verscheijden LFM, Koenderink JB, Johnson TN, de Wildt SN, Russel FGM (2020) Physiologically-based pharmacokinetic models for children: Starting to reach maturation? Pharmacol Ther 211:107541. https://doi.org/10.1016/j.pharmthera.2020.107541
Wang D, Miller R, Zheng J, Hu C (2000) Comparative population pharmacokinetic-pharmacodynamic analysis for piroxicam-beta-cyclodextrin and piroxicam. J Clin Pharmacol 40(11):1257–1266
Wang L, Bao SH, Pan PP, Xia MM, Chen MC, Liang BQ, Dai DP, Cai JP, Hu GX (2015) Effect of CYP2C9 genetic polymorphism on the metabolism of flurbiprofen in vitro. Drug Dev Ind Pharm 41(8):1363–1367. https://doi.org/10.3109/03639045.2014.950274
Wanwimolruk S, Wanwimolruk SZ, Zoest A (1991) A simple and sensitive HPLC assay for piroxicam in plasma and its application to bioavailability study. J Liq Chromatogr 14(12):2373–2381. https://doi.org/10.1080/01483919108049697
Weintraub M, Jacox RF, Angevine CD, Atwater EC (1977) Piroxicam (CP 16171) in rheumatoid arthritis: a controlled clinical trial with novel assessment techniques. J Rheumatol 4(4):393–404
Wojtyniak JG, Britz H, Selzer D, Schwab M, Lehr T (2020) Data digitizing: accurate and precise data extraction for quantitative systems pharmacology and physiologically-based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol 9(6):322–331. https://doi.org/10.1002/psp4.12511
Wojtyniak JG, Selzer D, Schwab M, Lehr T (2021) Physiologically based precision dosing approach for drug-drug-gene interactions: a simvastatin network analysis. Clin Pharmacol Ther 109(1):201–211. https://doi.org/10.1002/cpt.2111
Xu M, Zheng L, Zeng J, Xu W, Jiang X, Wang L (2021) Physiologically based pharmacokinetic modeling of tramadol to inform dose adjustment and drug-drug interactions according to CYP2D6 phenotypes. Pharmacotherapy 41(3):277–290. https://doi.org/10.1002/phar.2494
Yellepeddi V, Rower J, Liu X, Kumar S, Rashid J, Sherwin CMT (2019) State-of-the-art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin Pharmacokinet 58(1):1–13. https://doi.org/10.1007/s40262-018-0677-y
Zhuang X, Lu C (2016) PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 6(5):430–440. https://doi.org/10.1016/j.apsb.2016.04.004
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2019R1A2C1004582).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cho, C., Kang, P., Park, HJ. et al. Physiologically based pharmacokinetic (PBPK) modeling of piroxicam with regard to CYP2C9 genetic polymorphism. Arch. Pharm. Res. 45, 352–366 (2022). https://doi.org/10.1007/s12272-022-01388-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01388-0