
Abstract
Diabetes mellitus is a heterogeneous, multifactorial, chronic disease characterized by hyperglycemia owing to insulin insufficiency and insulin resistance (IR). Recent epidemiological studies showed that the diabetes epidemic affects 382 million people worldwide in 2013, and this figure is expected to be 600 million people by 2035. Diabetes is associated with microvascular and macrovascular complications resulting in accelerated endothelial dysfunction (ED), atherosclerosis, and cardiovascular disease (CVD). Unfortunately, the complex pathophysiology of diabetic cardiovascular damage is not fully understood. Therefore, there is a clear need to better understand the molecular pathophysiology of ED in diabetes, and consequently, better treatment options and novel efficacious therapies could be identified. In the light of recent extensive research, we re-investigate the association between diabetes-associated metabolic disturbances (IR, subclinical inflammation, dyslipidemia, hyperglycemia, dysregulated production of adipokines, defective incretin and gut hormones production/action, and oxidative stress) and ED, focusing on oxidative stress and endothelial progenitor cells (EPCs). In addition, we re-emphasize that oxidative stress is the final common pathway that transduces signals from other conditions—either directly or indirectly—leading to ED and CVD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a heterogeneous, multifactorial, chronic disease characterized mainly by hyperglycemia owing to insulin insufficiency (due to partial or complete β-cell defect) and insulin resistance (IR) (inability of insulin to exert its actions on tissues). Type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM) are the most common forms of diabetes [1]. Recent epidemiological studies showed that the diabetes epidemic affects 382 million people globally in 2013, and this figure is expected to be 600 million people by 2035 [2].
There are many key players which contribute to diabetes pathophysiology: (1) β-cell dysfunction, (2) IR (defects of insulin signaling) in brain, skeletal muscles, and liver, (3) accelerated lipolysis resulting in increased production of non-esterified fatty acids (NEFA), (4) dysregulated production of adipokines, (5) dysregulated action/production of incretin and gut hormones, (6) hyperglucagonemia, (7) subclinical inflammation through production of proinflammatory mediators, (8) increased glucose production and reabsorption from the kidneys, and (9) oxidative stress and excessive production of reactive oxygen species (ROS) [1, 3–5] (Fig. 1).

General pathophysiological pathways contributing to the development of diabetic cardiovascular diseases
Diabetes is associated with microvascular complications (e.g., retinopathy, nephropathy, and neuropathy) and macrovascular complications, culminating in accelerated endothelial dysfunction (ED), atherosclerosis, and cardiovascular disease (CVD) e.g., coronary artery, cerebrovascular, and peripheral vascular diseases [6, 7]. CVD accounts for more than 65 % of all deaths in T2DM [8]. The CVD mortality risk is almost two fold as higher in T2DM cases compared with age-matched control. T2DM increases the risk of coronary and peripheral artery diseases by 2–4 fold and increases the risk of stroke by 10 fold in individuals younger than 55 years old [8]. Unfortunately, the complex pathophysiology of diabetic cardiovascular damage is not completely understood.
The endothelium
The endothelium is a monolayer of endothelial cells (ECs) lining the lumen of all body vasculature. It is now considered a pivotal metabolically active, homeostatic organ which regulates the tone and structure of the blood vessels in response to different stimuli such as shear stress and agonists [e.g., acetylcholine (ACh) and insulin]. Disturbing the tightly controlled balance between the vasodilating [e.g., nitric oxide (NO) and prostacyclin (PGI2)] and vasoconstricting [e.g., endothelin-1 (ET-1) and thromboxane A2 (TXA2)] factors leads to ED. ED is the fundamental trigger of pathophysiological pathways which eventually culminates in atherosclerosis, CVD, and vascular diabetic complications [9–11].
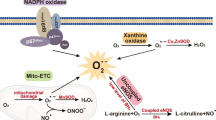
The most important autacoid produced by the ECs is NO. Impaired NO bioavailability is the hallmark of ED. NO is generated from l-arginine by endothelial NO synthase (eNOS), an enzyme which is constitutively expressed in ECs (Fig. 2). NO bioavailability depends on the balance between the rate of its generation by eNOS and its inactivation particularly by ROS [9, 10]. Moreover, NO-mediated signaling pathways could stimulate mobilization of EPCs from bone marrow stem cell niches to the blood circulation [12].

Biosynthesis and physiological role of nitric oxide in endothelial and vascular smooth muscle cells. NO is generated from l-arginine by endothelial NO synthase (eNOS), an enzyme which is constitutively expressed in ECs. O2, reduced nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN) and tetrahydrobiopterin (BH4) are required as cofactors for this reaction. Under normal physiological conditions, stimulation of ECs induces elevation of intracellular Ca2+ concentration which binds to calmodulin, thereby releasing eNOS from the caveolar protein caveolin-1, which inhibits eNOS activity. NO diffuses to the surrounding cells, exerting its cardiovascular protective action. NO activates guanylate cyclase (GC) enzyme which converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). cGMP enhances the activity of a cGMP-dependent protein kinase (PK), leading to decreased intracellular Ca2+ concentration, which eventually inhibits the contractile machinery in the vascular smooth muscle cells (VSMCs) followed by vasodilatation
The exact definition of EPCs is controversial due to the absence of unique marker or combination of markers to identify and characterize progenitor cells that differentiate into ECs in vitro and in vivo [13]. A myriad of cells with complex surface markers and different phenotypes has been referred to as EPCs including colony forming unit-Hill (CFU-Hill) EPCs, circulating angiogenic cells (CACs), and endothelial colony forming cells (ECFCs) [14]. EPCs maintain endothelial integrity through mediating the processes of repair and regeneration for ECs injury [15]. Characterization of EPCs is done by three methods: (1) functional assays that evaluate their adhesion to fibronectin, secretion of growth factors, and ability to correct ischemia in animal models, (2) expression of EPCs-specific surface markers CD34+, KDR+ (Flk-1+ in mice), and CD133, and (3) the ability to form colonies in vitro (stemness) [16]. EPCs contribute to vascular repair by two main mechanisms: (1) EPCs proliferate to regenerate new ECs and (2) EPCs release cytokines and growth factors to stimulate proliferation of resident ECs and other EPCs [17]. The vasculogenic function of EPCs under hypoxic conditions is mediated by hypoxia-inducible factor-1α (HIF-1α) activation of chemokine receptor type-4 (CXCR-4) expression [18]. Moreover, HIF-1α induces expression of stromal cell-derived factor-1 (SDF-1) in ischemic ECs in a hypoxia-dependent manner. The gradient of SDF-1 concentration in ischemic tissue and its interaction with CXCR-4 receptors on EPCs drives migration, homing, and incorporation of EPCs in ischemic tissue for vascular repair [19].
Low levels of circulating EPCs are associated with ED [20]. Decreased EPCs levels in diabetic patients (both T1DM and T2DM) are also implicated in the pathogenesis of vascular complications [21, 22]. Therefore, EPCs could independently predict endothelial function and, consequently, be used for assessing microvascular ED [23]. However, Lombardo et al. demonstrated that T2DM affects the maturation and commitment of EPCs rather than affecting their production and mobilization processes [24].
Oxidative stress and endothelial dysfunction
A common feature of all free radicals is destruction of macromolecules such as proteins and DNA, the latter being of utmost importance because introducing permanent mutations in genetic material stymies normal function of the cell [25].
Oxidative stress in endothelial cells
Oxidative stress activates forkhead transcription factor (FOXO1) that increases the transcription of inducible NOS and disturbs eNOS function [26]. FOXO1 is essential for expression of cell adhesion molecules and activation of inflammatory mediators in ECs. Furthermore, FOXO1 promotes ECs senescence and significantly stymies their proliferation by its effect on pro-apoptotic proteins (Bim) and cell cycle inhibitors such as p53 and p21 [27]. Moreover, H2O2 induces p53 expression in Human umbilical vein endothelial cells (HUVEC), which is known to inhibit cellular proliferation.
The effect of ROS on the activation of certain pathways by transforming growth factor (TGF-β) was demonstrated in two types of cultured ECs. The use of anti-oxidants and their subsequent downstream effects attenuates the activation/phosphorylation of p38 mitogen activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) pathways by TGF-β, thus inhibiting ECs apoptosis [28]. Uncouplers of oxidative phosphorylation decrease ROS production in mitochondria which inhibit inhibitor of kappa B (IκB) phosphorylation, inciting a state of resistance to tumor necrosis factor-α (TNF-α) activation of Nuclear factor κB (NF-κB) signaling pathway and inflammation in ECs [29]. Moreover, H2O2 and peroxynitrite-mediated activation of poly (ADP-ribose) polymerase (PARP-1) depletes cellular ATP and NAD+ that is associated with ECs apoptosis. PARP-1 prevents phosphorylation of Bcl-2-associated death promoter (BAD), thereby inhibiting its pro-apoptotic effects by hindering its association with the multifunctional phosphoserine-binding protein 14-3-3 [30]. Hyperglycemia-induced Reactive nitrogen species (RNS) promote apoptosis, which is mediated by JNK phosphorylation that increases pro-apoptotic Bax and increases cleavage of caspase-3, leading to ECs apoptosis [31]. H2O2 induces dephosphorylation of β-catenin mediated by p66shc, leading to activated downstream Wnt signaling. This leads to ED via decreased NO bioavailability, decreased vasorelaxation induced by ACh, increased TNF-α expression, and monocyte adhesion to ECs [32].
Oxidative stress in endothelial progenitor cells
Despite the discrepancy regarding the anti-oxidant capacity of EPCs among researchers, there is a consensus on the pivotal damaging effect of oxidative stress on EPCs functions [33]. Early-stage EPCs do not exhibit increased ROS in diabetes, and this may be due to increased expression of anti-oxidant enzymes such as catalase in diabetic EPCs [34]. H2O2 significantly decreases EPCs viability, increases apoptosis, and impairs tube formation. These effects are mediated by the increased expression of FOXO3a, which has high basal expression in EPCs and the subsequent activation of its downstream targets such as the pro-apoptotic protein Bim [35]. H2O2 induces EPCs apoptosis in a dose-dependent manner, which is inhibited by SIRT1-induced deacetylation of FOXO3a, thereby increasing FOXO3a ubiquitination and proteasomal degradation [35]. Oxidized low density lipoprotein (ox-LDL) induced activation of Akt/p53/p21 signaling pathway is associated with accelerated EPCs senescence in diabetic patients [36].
H2O2 induces oxidation of sulfhydryl groups of multiple anti-oxidant proteins such as glutaredoxin and thioredoxin, leading to activation of apoptosis signal-regulating kinase 1 [37], which induces EPCs apoptosis [38]. Furthermore, H2O2 induces oxidation of pivotal EPCs proteins such as the T-complex protein 1 subunit α, isoform A of prelamin-A/C, cofilin-1, peroxiredoxin-4, and actin [39]. Despite the relevance of these data in elucidation of ROS-induced impairment of EPCs function, no studies have explored their mechanistic roles in diabetes EPCs dysfunction.
Insulin resistance (IR) and endothelial dysfunction
Insulin resistance-induced ROS
Insulin, a hormone with pleiotropic metabolic and vascular effects such as vasodilation through generation of NO by activation of phosphatidylinositol triphosphate kinase/protein kinase B (PI3K/Akt) [40], as well as vasoconstriction via MAPK-induced production of ET-1 [41]. IR is characterized by pathway specific impairment of insulin signaling where PI3K/Akt signaling is impaired with reduced NO formation [42], meanwhile MAPK/extracellular signal-regulated kinase (ERK) signaling is intact [42] and even enhanced due to hyperinsulinemia, which leads to increased ET-1 production [43] and enhanced proliferation of vascular smooth muscle cells (VSMCs) [44].
Insulin-induced eNOS-Ser1177 phosphorylation/activation is diminished in Zucker Obese (ZO) compared to Zucker Lean (ZL) rats, which explains reduced NO synthesis in ZO rats [45]. Therefore, reduced NO generation will hypothetically lessen NO-mediated ROS neutralization. Inhibition of eNOS activity by nitro-l-arginine methyl ester (L-NAME) paradoxically decreases ROS generation, implicating the role of insulin in induction of eNOS uncoupling, which enhances superoxide production and contributes to oxidative stress [46].
Of note here, superoxide is produced by eNOS as a natural intermediate under physiological conditions but is not released and coupled to NADPH consumption [47]. This effect is mediated by BH4, which stabilizes eNOS dimers and prevents the dissociation of ferrous dioxygen complex of the enzyme’s oxygenase domain. Uncoupled eNOS mainly generates superoxide due to the dissociation of ferrous dioxygen complex [48]. Interestingly, ROS produced by uncoupled eNOS oxidize BH4 into dihydrobiopterin (BH2), reducing BH4 bioavailability and competing with it on active sites. This induces eNOS uncoupling and further production of ROS in a vicious cycle [49].
Insulin promotes the phosphorylation and translocation of p47phox subunit of NADPH oxidase from cytosol to membrane, through activation of the PI3K/protein kinase C (PKC) signaling pathway, hence provoking superoxide generation [50]. Moreover, inhibition of Nox2 in macrophages by RNA interference prevents insulin-stimulated superoxide production, while PKC inhibition prevents the insulin-induced superoxide production without affecting the phosphorylation of Akt [50].
Both Nox2 and Nox4 are expressed in ECs [51–53]. Indeed, insulin stimulates NADPH-dependent superoxide production in fibroblasts, smooth muscle cells, and adipocytes [54–56]. Insulin-treated Sprague–Dawley rats have increased superoxide production in vascular ECs of aortic segments as measured by lucigenin-enhanced chemiluminescence method [57].
Insulin effect on EPCs
Murphy et al. investigated the increased coronary artery disease risk in UK South Asians compared to Caucasians. They attributed this increased risk to the fact that South Asians were more insulin resistant, showing ED as well as reduced EPCs numbers and function [58].
The potential for insulin to affect the mobilization and differentiation of EPCs directly is not known. For example, a study of patients with T2DM has demonstrated that insulin stimulates the outgrowth of EPCs in vitro, and this effect was mediated by insulin growth factor (IGF-I) receptor stimulation of MAPKs and ERK1/2 but not insulin receptors [59]. Therefore, insulin presumably induces proliferation and differentiation of EPCs. However, recent study demonstrates that haploinsufficiency of IGF1-receptor enhances their phenotype despite producing fewer yields of EPCs. These angiogenic EPCs showed accelerated endothelial regeneration in vivo and enhanced tube forming and adhesive potential of angiogenic progenitor cells in vitro as well as overall enhanced vascular repair [60]. In a small study of patients with poorly controlled T2DM, insulin-mediated EPCs mobilization was significantly enhanced in subjects with SDF-1 3′-A/G allele, a polymorphism known to be associated with increased EPCs mobilization [61].
Mouse models heterozygous for insulin receptor knockout (IRKO) are non-diabetic but are characterized by ED and reduced EPCs numbers and function. When IRKO mice crossed with transgenic mice with Tie-2-driven human IR expression in ECs (HIRECO), the offspring is considered to have restored insulin signaling in ECs through insulin receptors. HIRECO × IRKO offspring show improved blood pressure, endothelial function, NO bioavailability, and vascular repair in the setting of global IR. This is not related to glucoregulation or EPCs abundance [62].
Insulin resistant, non-diabetic mice hemizygous for IRKO have a defect in EPCs mobilization manifested as lower numbers of circulating EPCs in peripheral blood but not in bone marrow compared with wild-type mice [63]. Moreover, after arterial injury, endothelial regeneration was delayed in IRKO mice and transfusion of mononuclear cells or c-kit + bone marrow cells from wild-type mice restored endothelial regeneration in IRKO mice [63]. EPCs in diabetic patients have reduced clonogenicity and uncoupled eNOS mediated by ROS, which further contribute to more oxidative stress and impaired vascular repair [22].
Inflammation and endothelial dysfunction
Subclinical inflammation is a hallmark sign of metabolic syndrome and T2DM. Systemic inflammation in these conditions is characterized by elevated levels of C-reactive protein (CRP), TNF-α, and many cytokines, most notable of them are interleukin-1 (IL-1), interleukin-6 (IL-6), and interleukin-18 (IL-18) [64, 65]. These factors interact with different receptors that converge in their final mediators that increase oxidative stress and activate NF-κB in ECs, which eventually result in ED.
C-reactive protein (CRP)
CRP levels are significantly elevated in T1DM and T2DM patients [66, 67]. Furthermore, elevated CRP levels in healthy individuals are highly correlated with markers of metabolic syndrome [66] and could be predictors for the risk of development of T2DM in various populations [68, 69]. As well, CRP has been proven to induce ED in cultured ECs [70]. The role of CRP in ED is related to the presence of other agents such as ox-LDL. In addition, CRP interacts with lecithin-like ox-LDL receptor-1 (LOX-1) and complement proteins to induce inflammation in cultured ECs [71].
The effects of CRP are also mediated by the IgG Fc receptor (FcγRIIB). CRP abolishes insulin activation of eNOS by inhibiting Akt phosphorylation at Ser473 [72]. Furthermore, CRP interaction with FcγRIIB receptor reduces protein nitrosylation in ECs and activates NF-κB pathway by reducing the inhibitory p65 S-nitrosylation. Those effects are also dependent on FcγRII receptors [73]. However, other studies have shown that NF-κB activation is mediated by activation of ERK1/2 pathway, but the exact receptors by which CRP mediates this effect is not elucidated. Also, CRP increases ROS generation by a mechanism which is independent on NADPH oxidase [74].
The effect of CRP on EPCs was also investigated by Chen and colleagues. CRP significantly disrupts EPCs migration, adhesiveness and proliferation. Furthermore, CRP abolishes eNOS expression in EPCs and increases EPCs apoptosis. Surprisingly, all these effects are shown to be mediated through receptors for advanced glycation end products (RAGE) [75]. Fujii et al. reported similar results where CRP instigates apoptosis and necrosis in EPCs. Furthermore, the culprit was shown to be the increase in mitochondrial ROS generation that was aggravated by CRP effect in modulating the expression of the anti-oxidant enzymes glutathione peroxidase and Mn superoxide dismutase (MnSOD) [76]. However, recent work by Fasing et al. showed no association between CRP plasma level and circulating EPCs [77].
Interleukins
IL-1, IL-6, and IL-18 are among the most notable interleukins in diabetic inflammation [65]. IL-1β decreases the expression of PI3K, eNOS, paraoxonase-1 (PON-1) (anti-oxidant gene), and other genes involved in adenosine monophosphate-dependent kinase (AMPK) upstream signaling. Also, IL-1β increases expression of adhesion molecules intercellular adhesion molecule-1 (ICAM-1) and monocyte chemotactic protein-1 (MCP-1) in cultured ECs. These effects are attenuated by β-carotene [78]. Vallejo et al. demonstrated that IL-1β induces ED in diabetic rat model by activating NADPH oxidase. This effect was partially prevented by incubation of the isolated blood vessels with Apocynin (NADPH oxidase inhibitor). Also, normal endothelial function was restored by incubation with a NADPH oxidase, cyclooxygenase-2 (COX-2), eNOS inhibitors, and superoxide dismutase [79], proving the role of oxidative stress in IL-1β-induced ED.
Moreover, IL-1β induces murine EPCs viability, proliferation, and migration both in vivo and in vitro, an effect which is associated with ERK1/2 pathway activation [80]. Also, IL-1β increases mRNA and protein levels of vascular endothelial growth factor (VEGF)-A. Intriguingly, these observed effects of IL-1β on EPCs are mediated via the PI3K/Akt signaling pathway [81]. On the contrary, Mao et al. showed that IL-1β significantly reduces the number and proliferation of pig EPCs. Moreover, IL-1β, through p38 MAPK pathway activation, reduces EPCs migration, adhesion, and angiogenic functions [82].
In addition, serum levels of IL-6, IL-10, and IL-17 are significantly elevated in T2DM patients [83]. Furthermore, IL-18 and IL-12 are associated with T2DM risk and ED in T2DM, respectively [84, 85]. IL-18 induces inflammation in ECs and increases the expression of vascular cell adhesion molecule (VCAM-1), a process that is mediated by PKCβ activity [86]. Moreover, endogenous inhibitor of IL-18 (IL-18BP) is downregulated in aortic ECs from diabetic animals by PKCβ heightened activity [86]. Furthermore, IL-18 suppresses Akt phosphorylation and activates IκB kinase (IKK), resulting in activation of NF-κB. [87]. Regarding the effect of IL-18 on EPCs, Kahlenberg et al. showed that IL-18 reduces the ability of EPCs from healthy individuals to differentiate into mature ECs [88].
IL-6 induces IR in ECs by inhibiting phosphorylation of Tyr612 in insulin receptor substrate-1 (IRS-1), thus inhibiting the IRS-1 downstream signals of PI3K/Akt/eNOS [87, 89]. IL-6 inhibits phosphorylation/activation of AMPK at Thr172 in ECs regardless of insulin presence [90]. On the other hand, Andreozzi et al. presented compelling evidence that IL-6 inhibits IRS-1 via inhibitory phosphorylation on Ser312 and Ser616 through activation of JNK/signal transducer and activator of transcription-3 (STAT3) and ERK1/2 pathways using different experimental settings [91]. In addition, IL-6 enhances EPCs migration, proliferation, and differentiation in cell culture. It also activates both JNK/STAT3 pathway and ERK1/2 pathway [92].
Intriguingly, IL-10 exerts beneficial effects on ECs. IL-10 inhibits ox-LDL-induced ECs apoptosis via inhibition of ox-LDL-mediated activation of p38 MAPK and JNK. This effect was mediated by suppressor of cytokine signaling-3 (SOCS3), which is upregulated by IL-10 [93]. Moreover, IL-10 inhibits the insulting effects of TNF-α on ECs and reduces ICAM-1 expression as well as ROS generation through PI3K activation [94]. Furthermore, IL-10 alone has no effect on EPCs migration and differentiation. However, it significantly increases the expression of VEGF and matrix metallopeptidase-9 (MMP-9) and potentiates the detrimental effects of TNF-α on EPCs [95].
Tumor necrosis factor-α (TNF-α)
TNF-α is a cytokine that has a myriad of functions such as mediating apoptosis, regulation of immunity, and hematopoiesis as well as contributing to the pathophysiology of many diseases. TNF-α exerts its effects among various cells via its action on its TNFR1 and TNFR2 receptors. TNFR1 is expressed ubiquitously and is present in different types of cells; on the other hand, TNFR2 is only expressed in few cell types including ECs [96]. TNFR1 activates NF-κB via the canonical pathway which is dependent on the activation of the IKK complex, which phosphorylates IκB and subsequently results in its proteasomal degradation. This is mediated by the downstream effectors of TNFR1: TNF receptor-associated death domain (TRADD) and TNFR-associated factor 2 (TRAF2) [97, 98]. Moreover, TNFR2 activates non-canonical NF-κB activation by preventing proteasomal degradation of NF-κB-inducing kinase (NIK), which activates IKKα, resulting in an activation of certain isoforms of NF-κB [99].
TNF-α serum level is elevated in T1DM and T2DM and is associated with various complications of diabetes [100, 101]. Studies conducted on Lepr diabetic mice and ZO diabetic rats showed that TNF-α mediates ED by increasing ROS generation via NADPH oxidase, an effect that was related to NF-κB activation [102–104]. Moreover, TNF-α increases the expression of genes encoding Nox2 subunits (p22phox and p40phox) in coronary artery ECs [104]. In cerebral microvascular ECs, TNF-α induces ROS generation mainly via Nox4 [105]. However, recent work conducted on cultured human microvascular ECs showed that TNF-α does not alter the expression of Nox2, p22, and p67, while inducing downexpression of Nox4 [106]. TNF-α mediates ROS production mainly via NADPH oxidase and to a lesser extent by the mitochondria [107]. Also, TNF-α stimulates ROS production in ECs by activating p47 [108] and enhancing its localization as well as the associated subunits p67 and Rac1 to the cell membrane [109, 110]. TNF-α activates p47 phosphorylation at Ser379 by PKC which induces a conformational change that allows binding with both p22 and TRAF4, and stimulates its membrane translocation as well as the downstream activation of ERK1/2, p38 MAPK, and JNK [111]. Furthermore, TNF-α antagonist (infliximab) was successful in alleviating ED in metabolic syndrome patients [112]. However, other studies using different TNF-α antagonist (etanercept) failed to show similar results [113].
TNF-α induces expression of IL-18 which has deleterious effects on EPCs differentiation. TNF-α reduces Akt phosphorylation mediated by insulin and increases EPCs apoptosis. These effects are mediated through NF-κB activation [114]. Additionally, TNF-α inhibits EPCs migration and proliferation in a dose and time-dependent manner. TNF-α mediates downexpression of VEGF-receptor 1 and SDF-1 as well as the inducible and endothelial forms of NOS [115]. Interestingly, some studies have shown that TNF-α enhances EPCs migration, adhesion, and tubule formation [95], but these results were not replicated by other researchers.
Nuclear factor kappa B (NF-κB)
NF-κB is a family of transcription factors highly conserved in several species. Cytoplasmic NF-κB molecules have an inhibitor component that hinders their nuclear localization required for their actions. These inhibitor components are termed inhibitor of kappa B (IκB) and include IκBα, β, and ε as well as the products of precursor proteins p105 and p100 termed IκBγ and IκBδ [116]. The canonical pathway is usually activated by pro-inflammatory cytokines and is dependent upon IKK complex. The non-canonical pathway is mediated solely by IKKα [117].
Sustained activation of NF-κB is a characteristic sign observed in diabetic patients and animal models [118]. Endothelial constitutive inhibition of NF-κB in mice models attenuates age-related changes in ECs as well as decreases and increases the expression of cellular adhesion molecules and eNOS, respectively [119]. Pharmacological inhibition of NF-κB by Salsalate significantly inhibits p65 nuclear translocation, p47 NADPH oxidase subunit expression, and nitrotyrosine levels in cultured HUVECs. Furthermore, Salsalate ameliorates ROS-inhibited vasodilation in a manner similar to that mediated by high dose of anti-oxidant (vitamin C) [120]. These experiments suggest that NF-κB mediates its effects via increasing ECs oxidative stress. However, some reports have demonstrated that NF-κB is activated by ROS [121] and protects ECs from TNF-α-induced apoptosis [122]. In experiments using constitutively inhibited NF-κB [123] and overexpression of IκBα [122], TNF-α stimulation of cultured ECs results in more apoptotic cells. Consistently, IκB decreases the activation of the anti-oxidant/anti-apoptotic heme oxygenase-1 (HO-1) enzyme in cerebral ECs [122].
Ephemeral but stable overexpression of NF-κB in murine embryonic EPCs does not impair migration or vasculogenesis. Moreover, NF-κB overexpression enhances EPCs adherence to endothelium through increasing the expression of adhesion molecules E-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) [124]. In insulin resistant EPCs isolated from ZO rats and stimulated by TNF-α, NF-κB induces apoptosis via caspase-3 [114]. Nonetheless, damage induced by some environmental toxins such as Benzo[a]pyrene on EPCs is mediated by activation of NF-κB which increases ROS production and impairs EPCs migration, proliferation, and vasculogenic activities [125].
Dyslipidemia and endothelial dysfunction
Cholesterol and modified cholesterol
Oxidized LDL (Ox-LDL) particles are LDL lipoproteins that have their protein and/or lipid component oxidatively modified [126]. Ox-LDL blood levels are elevated in T2DM patients and obese individuals [127]. Ox-LDL is considered a prerequisite for the development of atherosclerosis and therefore, is a major contributor to CVD [128]. The main culprit in the generation of ox-LDL is the oxidative stress generated by increased induction of iNOS by FOXO1 [129]. Oxidized and glycated oxidized LDL inhibit aortic dilatation induced by ACh, an effect that is induced by increased ROS generation [26]. Ox-LDL phosphorylates/activates p66shc—an adaptor protein that functions as a redox enzyme—via its activation of LOX-1 receptor [130]. Ox-LDL increases ROS generation in human arterial ECs via activation of p66shc by Ser36 phosphorylation and eNOS uncoupling [131]. Downstream effects of p66shc activation involve PKCβ2 phosphorylation at Thr641 and Ser660, JNK phosphorylation, and increased expression of p47phox [130].
Furthermore, recent work showed that ox-LDL mediates vascular dysfunction by allosteric activation of angiotensin I receptors in ECs. This allosteric activation is mediated by ox-LDL binding to LOX-1 receptor, resulting in increased phosphorylation/activation of ERK1/2, which is a known activator of NF-κB [132]. Ox-LDL obtained from human subjects increases ROS generation, TNF-α expression and NF-κB activation in cultured ECs through binding to LOX-1 [133].
Ox-LDL induces dysfunction of cultured EPCs isolated from healthy subjects [134]. Ox-LDL induces EPCs senescence and inhibits VEGF-mediated differentiation via LOX-1 receptors. Furthermore, ox-LDL increases expression of LOX-1 mRNA in EPCs [135]. Ox-LDL reduces the number of viable EPCs in culture and inhibits their migration and adhesion activities [136, 137]. These effects were shown to be either mediated by both NADPH oxidase and NF-κB activation, or activation of LOX-1 and its subsequent inhibition of Akt/eNOS pathway [136, 137].
Oxidized HDL (ox-HDL) particles exhibit dysfunction in their anti-inflammatory and anti-oxidant activities [138]. Blood levels of ox-HDL and myeloperoxidase (MPO) enzyme are elevated in T2DM patients [127]. Interestingly, HDL particles isolated from diabetic patients are dysfunctional in their ability to induce ECs migration, proliferation, and adhesion compared to HDL isolated from normal healthy patients [139]. Ox-HDL particles lose their ability to attenuate TNF-α inflammatory effect and, surprisingly, activate NF-κB in cultured ECs via activation of IKK complex [140]. Ox-HDL increases ROS generation in cultured ECs via its activation of NADPH oxidase. This effect is greatly attenuated by pretreatment of cultured cells with LOX-1 antibody, suggesting a possible interaction between LOX-1 and ox-HDL [141].
Administration of reconstituted HDL to T2DM patients ameliorates the decline in circulating EPCs [142]. In addition, treatment of cultured EPCs with HDL induces their proliferation and protects them from apoptosis. It also increases their migration, adhesion, and tube formation [143]. Furthermore, HDL protects EPCs from the deleterious effects of ox-LDL. However, high concentration of HDL (>400 μgm/ml) induces EPCs senescence and impairs their tube formation capability via activation of Rho kinase that inhibits Akt and p38 MAPK activation [144]. On the other hand, ox-HDL stimulates EPCs apoptosis in a dose-dependent manner, depending on the surface receptor CD36. In addition, ox-HDL interaction with CD36 increases the activity of NADPH oxidase, upregulates Nox2 mRNA (NADPH oxidase subunit), and activates MAPK/NF-κB pathway [145].
Ceramide and sphingolipids
Ceramide is a major sphingolipid that lies in the crossroad of various degradation and synthesis pathways of sphingolipids. Inhibition of ceramide de novo synthesis ameliorates IR in diabetic mice models [146].
Ceramide accumulation in arteries caused ED in high fat diet (HFD) mice, an effect that is mediated by enhancing the association of protein phosphatase 2A (PP2A) with eNOS, resulting in dephosphorylation of eNOS active form. Furthermore, the effect of ceramide on eNOS activity is not associated with AMPK, Akt, or ERK1/2 phosphorylation [147]. Therefore, inhibition of de novo synthesis of ceramide attenuates ED in HFD mice models [148, 149]. Ceramide increases NADPH oxidase activity and mitochondrial ROS generation in ECs [150]. Ceramide also potentiates the ROS-generating effect of palmitate in cultured bovine aortic ECs in a dose-dependent manner [147].
Additionally, palmitate-induced ED was shown to be mediated by ceramide accumulation in EPCs. Ceramide accumulation inhibits EPCs proliferation, migration, and tube formation via its inhibition of Akt/eNOS pathway [151].
Non-esterified fatty acids (NEFA)
NEFA induce apoptosis and decrease proliferation in cultured ECs [152]. Palmitic and linoleic acids inhibit phosphorylation/activation of eNOS both in basal and insulin-stimulated conditions. However, only palmitic acid inhibits PI3K/Akt/eNOS pathway by inducing expression of PTEN [153]. NEFA induce monocyte adhesion to cultured ECs, which is an essential step in endothelial inflammation [154].
Stearic and α-linoleic acids induce ECs apoptosis via downregulating X-ray repair cross-complementing protein 1 (XRCC1), c-myc, and transcription factor E2F-1 [155]. NEFA also induce apoptosis in cultured ECs via pathways that enhance acyl-coA generation and NF-κB activation [156]. Additionally, palmitic acid induces apoptosis via increasing ROS generation in mesenteric artery ECs, while the anti-oxidant N-acetyl cysteine partially mitigates this effect [157]. Palmitate also activates COX-2 and thromboxane prostanoid (TP) receptor, leading to further ROS generation in mice aortic ECs, possibly by activation of NF-κB [158]. In addition, the isoflavonoid tectorigenin (anti-oxidant) attenuates the palmitate-induced oxidative stress in cultured ECs [159].
Stearic, α-linolenic and arachidonic acids induce apoptosis of cultured EPCs via a caspase-8-dependent mechanism [160]. Palmitic and linoleic acids inhibit EPCs adhesion, migration, proliferation, and vasculogenesis capabilities of EPCs in a dose-dependent manner via inhibition of eNOS. However, only palmitic acid is shown to inhibit Akt phosphorylation [161]. Moreover, palmitic acid effect is shown to be mediated via its binding to peroxisome proliferator activated receptor (PPARγ) and subsequent inhibition of STAT5A expression. PPARγ and STAT5A form heterodimers that enhance transcription of cell cycle progression proteins specifically cyclin D1 and reduces the expression of cell cycle progression inhibitor p21. This decrease in STAT5A is responsible for the halted proliferation of EPCs induced by palmitic acid [160].
Hyperglycemia and endothelial dysfunction
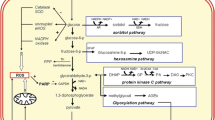
Hyperglycemia is the major causal factor in the development of ED in diabetes. There are several pathways by which hyperglycemia could generate ROS and contribute to oxidative stress, and ultimately developing ED. Here, we briefly highlight these pathways to emphasize the role of glucose toxicity in oxidative stress and ED (Fig. 3).

The central role of mitochondrial ROS in hyperglycemia-induced endothelial dysfunction
Advanced glycation end products (AGEs) pathway
AGEs production is increased in diabetic mice Leprdb compared to non-diabetic controls. Moreover, protein expression of RAGE is higher in diabetic mice than controls [162]. Carboxymethyl lysine (AGEs)-mediated activation of NF-κB was significantly diminished in double negative-RAGE transfected cells, suggesting that AGEs activate NF-κB through binding to RAGE [163]. The use of soluble form of RAGE to inhibit AGEs/RAGE interaction and signaling decreases NADPH oxidase expression (NOX-2, p22phox, and p40phox) and oxidase activity in isolated coronary arterioles, suggesting the role of AGEs/RAGE in Nox activation. The study also suggests that AGEs/RAGE signaling activates TNF-α and induce ED through production of superoxide mediated by NF-κB [162]. AGEs/RAGE stimulates MAPK-mediated NF-κB upregulation and increases COX-2 expression, leading to increased expression of cleaved caspase-3 and cleaved PARP in a dose-dependent manner in pancreatic islet microvascular ECs [164].
Yoon et al. demonstrated that hydroxymethylglutaryl (HMG)-coA reductase inhibitors (statins) reduce intracellular ROS by inhibiting AGEs/RAGE interactions in a dose-dependent manner. Statins are capable of inhibiting all of the stimulatory effects of AGEs on NF-κB, p65, phosphorylated ERK, phosphorylated p38 MAPK, COX-2, and c-Jun [165]. AGEs increase the release of soluble dipeptidyl-peptidase 4 (DPP-4) in HUVECs, where DPP-4 increases RAGE expression and interacts with d-Mannose-6-phosphate/IGF II receptor to dose-dependently increase ROS generation and endothelial damage [166]. Exposure to high glucose AGEs in ECs and EPCs increased cyclin-dependent kinase inhibitors p27 and p57 expression due to down regulation of mir221/mir222 expression. Moreover, AGEs exposure is associated with impaired vessel formation in vivo mouse model [167].
The effect of AGEs on the number and functions of EPCs is profound and has been studied extensively. AGEs reduce NO production and eNOS expression while increasing caspase-3 and Bcl-2 expression as well as EPCs apoptosis in a time-dependent manner. These effects are hampered by MAPK inhibitors [168]. AGEs also significantly decrease anti-oxidant enzymes SOD and glutathione peroxidase expression and confirm the involvement of RAGE in AGEs-mediated ROS production in EPCs [169].
AGEs/RAGE interaction impairs late EPCs function but not proliferation by downregulation of Akt and Cox-2. Furthermore, increasing RAGE expression deteriorates EPCs migration and tube formation in a dose-dependent manner [170]. Even in apparently healthy subjects, serum levels of AGEs correlated with reduced numbers and migratory capacity of EPCs [171], which reflects the possible role of AGEs as independent markers for vascular disease prognosis. In another study, AGEs promoted EPCs apoptosis while decreasing the production of SDF-1, NO, PGI2, and tissue plasminogen activator (tPA) [172]. AGEs modification of basement membrane proteins such as fibronectin in EPCs interfere with signaling through integrin receptor α5β1 by abolishing cell recognition motifs on fibronectin, leading to impaired adhesion of EPCs in diabetic retinopathy [173]. Inflammation is also an important component of RAGE effect on EPCs function as demonstrated by CRP-induced RAGE expression which diminishes eNOS expression and NO production in EPCs [75].
Polyol pathway
The polyol pathway consists of two enzymes: aldose reductase (AR), which reduces glucose to sorbitol using NADPH as a co-factor, and sorbitol dehydrogenase (SDH), which converts sorbitol to fructose with its co-factor NAD+ [174]. Polyol pathway is a major contributor to oxidative stress in states of hyperglycemia [175]. Several mechanisms have been proposed to explain the role of polyol pathway in hyperglycemia-induced oxidative stress. Firstly, the consumption of NADPH in the sorbitol reduction reaction by AR leads to its depletion and inefficient regeneration of glutathione (GSH) by glutathione reductase which uses NADPH as a co-factor [176]. This is supported by the fact that, in hyperglycemia, around 30 % of glucose is shuttled through polyol pathway [176]. Secondly, NADH on the other hand accumulates as a result of the reduction of NAD+ upon oxidizing sorbitol to fructose by SDH [177], creating a state of pseudohypoxia [178]. NADH is the primary substrate for NADH oxidase that produces ROS [179]. Thirdly, the polyol pathway results in oxidation of glucose into fructose. Fructose and its metabolites fructose-3-phosphate and 3-deoxyglucosone are more potent glycation agents than glucose [180]. Therefore, hyperglycemia-induced flux of glucose through polyol pathway increases the production of AGEs and their interaction with RAGE, thereby increasing ROS generation and inducing oxidative stress.
Aldose reductase inhibitors (ARI) improve diabetic complications such as diabetic retinopathy [181] and diabetic nephropathy [182], as well as regenerating GSH [183] in animal models, suggesting the role of AR in ROS production. However, ARI have intrinsic free-radical scavenger activity [184], which may account for restoring GSH levels regardless of ARI activity. Therefore, the use of genetic models to investigate the role of AR in hyperglycemia-induced ROS revealed important insights. Mice normally have low AR in their lenses and are resistant to diabetic cataract. Induction of diabetes in transgenic mice overexpresses AR, specifically in their lenses, leads to cataract with significant decrease in GSH level and significant increase in the level of malondialdehyde (MDA), an oxidative stress marker. Moreover, introducing SDH deficient mutation in transgenic AR mice increases GSH levels and reduces MDA levels, implicating the role of SDH in generation of oxidative stress [175]. On the other hand, induction of diabetes in AR-knockout mice rendered them resistant to diabetic neuropathy with normal nerve conduction velocity and GSH levels [175].
Polyol pathway in hyperglycemic state is characterized by increased cytosolic NADH/NAD+ ratio by enhancing SDH activity and increasing superoxide generation. Free radicals induce single strand breaks that activate PARP, thereby inhibiting the activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and increasing concentrations of triose phosphate [185]. Fragmentation of triose phosphate compounds leads to increased formation of methylglyoxal [186], which forms AGEs with many cellular proteins including HIF-1α (methylgloxal modification of Arg17 and Arg23 in the bHLH domain) and P300 coactivator [187]. This modification impairs both HIF-1α/aryl hydrocarbon receptor nuclear translocator (ARNT) heterodimer formation as well as inhibiting the complex HIF-1α/ARNT/P300 from binding to consensus hypoxia response elements of target genes (such as SDF-1, VEGF, CXCR4 and eNOS) in EPCs required for neovascularization [187, 188]. This reflects the elaborate interplay between hyperglycemia-induced pathways in disrupting normal ECs and EPCs functions.
Protein kinase C (PKC) pathway
Hyperglycemia increases PKC activity in the membranous pool, most probably by translocation of PKC from cytoplasm to cell membrane [189]. In retinal ECs, high glucose concentration inhibits GADPH which leads to accumulation of glycolytic intermediates, dihydroxyacetone phosphate, and glycerol 3-phosphate that undergo acetylation to lysophosphatidic and phosphatidic acids and increase de novo synthesis of diacylglycerol (DAG) [190].
Hyperglycemia increases DAG levels in cultured ECs in a time-dependent manner, as well as increasing PKC-β2 isoform in aorta and heart of diabetic rats [191]. Additionally, hyperglycemia-mediated PKC activation has been shown to impair intercellular communications through gap junction transmembrane channels [192], as well as perturbing endothelial barrier and, thereby, increasing transendothelial albumin permeability [193]. For example, VEGF-induced endothelial permeability requires PKC-dependent phosphorylation of occludin (transmembrane tight junction protein) [194]. Hyperglycemia-induced PKC activation promotes eNOS gene expression and triggers eNOS uncoupling, thus increasing superoxide production and decreasing NO bioavailability. PKC activation in hyperglycemia also increases expression of the NADPH oxidase subunits gp91phox and p22phox [195]. Intermittent high glucose is characterized by increased PKC. Moreover, PKC inhibitors reduce oxidative stress markers and HUVECs apoptosis [196]. Also, PKC activation increases peroxynitrite formation and tyrosine nitration of PGI2 synthetase, thereby reducing PGI2 release [195]. PKC-δ activation, secondary to hyperglycemia, leads to p38 MAPK phosphorylation that increases the expression of Src-homology-2 domain-containing phosphatase-1 (SHP-1), which dephosphorylates platelet-derived growth factor (PDGF) receptor-β and impairs its downstream signaling, resulting in vascular cell apoptosis [197]. PKC-β2 activation by high glucose upregulates p66Shc in human aortic ECs, leading to mitochondrial ROS generation and ED [198].
Hyperglycemia-induced PKCβ activation decreases IL-18 binding protein (IL-18BP) expression (IL-18 inhibitor) which leads to increased IL-18 and VCAM-1, resulting in monocyte adhesion to ECs and ED [86]. High glucose concentration increases NF-κB activation through PKC pathway in ECs [199].
PKC is an essential mediator in EPCs signaling. CXCR2-mediated activation of PLC forms DAG, which activates PKC that triggers abnormal Ca2+ release. Calcium signaling is critical for mobilization and recruitment of EPCs at sites of vascular remodeling and repair [200].
Hexosamine biosynthetic pathway (HSP)
Hexosamine synthetic pathway (HSP) diverges from glycolysis and serves as a cellular nutrient sensor. HSP involves converting fructose-6-phosphate and glutamine into glucosamine-6-phosphate catalyzed by the rate-limiting enzyme glutamine/fructose-6-phosphate amidotransferase (GFAT) and ends with UDP-N-acetylglucosamine (UDP-GlcNAc). O-GlcNAc transferases use UDP-GlcNAc in post-translational modification of proteins by O-GlcNAcylation as well as glycolipids, proteoglycans, and gangliosides [201].
HSP is involved in many diabetic vascular complications, inflammation and oxidative stress. Hyperglycemia-induced ROS inhibits GADPH, thereby inhibiting flux of glyceraldehyde-3-phosphate and upstream metabolites (e.g., fructose-6-phosphate) from flow through glycolysis and inducing their shuttling through HSP. Hyperglycemia-induced HSP stimulates TGFβ1 expression by increasing the expression of upstream stimulatory factor proteins and their binding to glucose-response element in TGFβ1 promoter [202]. Increased HSP activity leads to increased O-GlcNAcylation and activation of transcription factor specificity protein-1 and enhances its binding to multiple genes involved in diabetic complications, such as increasing expression of TGFβ1 and plasminogen activator inhibitor 1 (PAI-1) in ECs [203].
HSP activation induces oxidative stress by inhibiting pentose phosphate shunt, thereby diminishing NADPH and the anti-oxidant GSH [204]. Reciprocal relation between phosphorylation and O-GlcNAcylation on the same or contiguous amino acid residues plays a pivotal role in the regulation of protein activity. HSP-induced O-GlcNAcylation of eNOS at ser1177 decreases phosphorylation at this site and prevents eNOS activation in ECs [205].
Mitochondrial ROS
Hyperglycemia increases the formation of electron donors such as NADH and FADH2 from glycolysis and TCA cycle. In ECs, increased flux of NADH and FADH2 through electron transport chain increases voltage gradient across inner mitochondrial membrane to a critical threshold, thereby, hindering electron transport in complex III and reverting electrons back to coenzyme Q, which catalyze superoxide generation by reducing molecular oxygen [206, 207]. It has been established that hyperglycemia-induced mitochondrial ROS generation is the unifying mechanism, which underlies all previously discussed pathways in hyperglycemia [208]. Supporting this idea, dissipating voltage gradient across mitochondrial membrane by uncoupling protein 1 (UCP-1) or neutralizing superoxide by MnSOD prevents hyperglycemia-induced ROS or even activation of other pathways [209]. Moreover, hyperglycemia fails to promote superoxide generation in rho zero ECs that have been depleted of their mitochondrial DNA thus having non-functional mitochondrial electron transport chain [208].
Mitochondrial ROS acts as initiating event in hyperglycemia-induced cellular damage, since these free radicals promote DNA single-strand breaks, which activate PARP. Activated PARP then adds poly-ADP polymers to nuclear proteins and to GADPH (which shuttles in and out of the nucleus to participate in DNA repair), thereby inhibiting GADPH [185]. Inhibition of GADPH blocks glycolysis and leads to accumulation of glycolytic intermediates upstream of glyceraldehyde-3-phosphate (GA-3P), hence activating all the previously discussed pathways and amplifying the signal of ROS production initiated in mitochondria. Accumulated GA-3P non-enzymatically forms methylglyoxal that act as a potent AGEs and activates RAGE expression. GA-3P and DHAP also fragment into DAG, which activates PKC pathway [210].
Upstream of GA-3P, accumulation of fructose-6-phosphate activate HSP and induce formation of UDP–GlcNAc which modifies many cellular proteins post-translationally. Ultimately, inactivation of GAPDH leads to accumulation of glucose, which activates polyol pathway through AR, consuming NADPH and generating NADH, thereby compromising inherent anti-oxidant capacity such as GSH reductase. Besides mitochondrial ROS, the activity of xanthine oxidase increases in plasma of diabetic animals due to increased release of the enzyme from hepatocytes. Xanthine oxidase binds to vascular ECs by sulfated glycosaminoglycan [211]. Xanthine oxidase is a superoxide-producing enzyme; therefore, allopurinol (xanthine oxidase inhibitor) or releasing xanthine oxidase from the surface of ECs significantly diminishes superoxide production in aortic rings from diabetic rats [211]. Also, hyperglycemia-induced mitochondrial ROS is inhibited by PPARγ coactivator-1α (PGC-1α), which increases the expression of VEGF in bovine retinal capillary ECs [212].
Diabetic EPCs have high ROS concentration, probably due to excessive activation of mitochondrial ROS generation [213]. Diminished anti-oxidant capacity of EPCs in diabetes could result from impaired AMPK activation secondary to elevated protein phosphatase 2A (PP2A), and subsequently downregulation of MnSOD expression (the mitochondrial isoform of SOD) [214]. High glucose-induced PKC in cell culture significantly increases EPCs apoptosis through upregulating p66Shc protein expression and inducing ROS generation by the mitochondria [215]. Moreover, EPCs are characterized by significantly diminished mitochondrial MnSOD expression (but not cytosolic CuZnSOD) in db/db mice. Interestingly, MnSOD gene therapy in diabetic EPCs before transplantation in db/db mice significantly augmented and accelerated wound healing [213]. Taken together, these findings imply the pivotal role of mitochondrial ROS in ECs and EPCs dysfunction in the setting of diabetes.
Adipokines and endothelial dysfunction
Adipose tissue is now widely recognized as not only a storage organ for lipids in the form of TGs but also an active endocrine organ which produces and secretes a wide range of protein hormones, cytokines, and mediators, collectively known as adipokines or adipocytokines. Adipokines are involved in many vital processes in different body organs including glucose and lipid metabolism, IR, inflammation, atherogenesis, apoptosis, and regulation of appetite and body weight [216]. Therefore, adipokines are relatively new and pivotal tools to understand the molecular pathophysiology of metabolic and cardiovascular diseases. Adipokines might exert their action on ECs and EPCs directly or indirectly, through modulation of metabolic pathways, inflammatory mediators, and even through modulating the effect of other adipokines, autacoids, and hormones. In this section, we will discuss recent research investigating the association between certain adipokines and ED.
Adiponectin
Generally, adiponectin exerts insulin-sensitizing, antidiabetic, antioxidant, anti-inflammatory, anti-atherogenic, vasoprotective, and anti-apoptotic actions on different cell types through adiponectin receptors, e.g., AdipoR1 and AdipoR2 [217]. Diabetes, IR, CVD, and metabolic syndrome are associated with reduced adiponectin level in animal models [218, 219] and in human patients [220, 221]. Also, hypoadiponectinemia is associated with ED and impaired vascular reactivity [222].
The association between adiponectin serum level and oxidative stress markers has been investigated in a cross-sectional study by Gustafsson et al. It has been shown that higher levels of adiponectin were associated with more favorable oxidative stress profile [223]. Adiponectin stimulates vascular NO production through phosphorylation of eNOS at Ser1177 and inhibits ROS production in ECs mainly through suppression of NADPH oxidase activity [221, 224]. In support with this, Antonopoulos et al. depict that reduced adiponectin level stimulates vascular NADPH oxidase in T2DM subjects [225]. In humans with CVD and atherogenic mice, adiponectin improves endothelial function by reducing ROS production, serum LDL-cholesterol, and TGs as well as increasing BH4 bioavailability and restoring eNOS coupling [226–228]. Moreover, adiponectin prevents hyperglycemia-induced ROS production in ECs by suppressing ROS/p38 MAPK/p16INK4A and/or activating cAMP/PKA signaling pathways [229, 230]. Also, adiponectin reduces ox-LDL, angiotensin II (AG II), and hyperglycemia-induced ROS production in cultured HUVECs, thus protecting ECs against ox-LDL, fluctuating glucose concentration, and AG II-induced endothelial damage and apoptosis [224, 231–233]. Interestingly, Chen et al. showed that the inflammatory effect of microRNA-221 (miR-221) on HUVECs is mediated by targeting AdipoR1 receptors and abolishing the anti-inflammatory effect of adiponectin [234].
Adiponectin promotes proliferation, survival, differentiation, migration, and vascular recruitment of EPCs through activation of both PI3k/Cdc42/Rac1 and AMPK signaling pathways [235–237]. Also, it has been proposed that pretreatment of angiogenic stem cells with adiponectin before transplantation enhances their survival and proliferation activities [238]. Therefore, the number of circulating EPCs is reduced in adiponectin knockout mice and administration of adiponectin restores the number of EPCs [229]. Moreover, plasma levels of adiponectin were positively associated with EPCs number in patients with coronary artery disease [239] and atherosclerotic stroke in large arteries [240]. On the other hand, Li et al. showed that in diabetic patients, hypoadiponectinemia was not associated with reduced level of EPCs; however, they confirmed the beneficial effects of adiponectin on EPCs in vitro [241]. This controversy could be, in part, due to the variance in the degree of adiponectin deficiency, diabetic state and other disease conditions, and other concurrent metabolic dysregulations affecting EPCs activity, e.g., insulin and leptin sensitivity.
Collectively, diabetic hypoadiponectinemia may play a pivotal role in the development of ED and CVD, particularly through enhancing production of ROS and pro-inflammatory mediators, promoting IR, NO inactivation, eNOS uncoupling, and impairment of EPCs migration and function as well. Therefore, adiponectin could be used as a potential marker for predicting CVD and a measure for the risk of metabolic disorders [242]. In addition, adiponectin could be used as a target for treating metabolic disorders and its associated CVD via increasing endogenous production or mimicking the actions of adiponectin on its signaling pathways. For example, rosiglitazone (PPARγ agonist) has been shown to restore normal vascular function in diabetic and obese mice through adiponectin-mediated mechanism [243, 244].
Leptin
Leptin is involved in obesity, IR, CVD, inflammation, and diabetes, partly due to its regulatory action on satiety and energy homeostasis, production of inflammatory mediators, and on lipid and carbohydrate metabolism [216]. IR is associated with increased levels of leptin (hyperleptinemia), with exclusively augmented leptin effects on some organs (or pathways) and diminished actions on others, reflecting a state of leptin resistance [245], which may be analogous to IR in its selectivity.
Leptin is the adipokine of controversy, with a myriad of discordant results. Although several studies show positive association between leptin concentration and high incidence and severity of cardiovascular events [246], Martin et al. demonstrated that no such association exists in a multi-ethnic cohort study [247]. Also, the association between serum leptin concentration and vascular endothelial function in the setting of T2DM and obesity is controversial; while some results indicate positive association [248], others showed inverse relationship between leptin levels and endothelial function [249] and even enhanced vascular function in severe obese people with high levels of leptin compared with obese subjects [250]. While the difference in subject properties (e.g., age, sex, body mass index, disease status, drugs used, and degree of hyperleptinemia) and the technique used in assessing endothelial function could be possible explanations for these inconsistent data [248], it could be further explained in the context of leptin resistance. First, the effect of leptin on ECs and generally on cardiovascular system may depend on the degree of leptin sensitivity. Second, it is not clear when leptin resistance starts in the context of metabolic disorders. Third, the exact cut-off value at which leptin sensitivity ends and resistance begins cannot be determined. Last but not least, the selective leptin resistance affecting certain signaling pathways could be another powerful perspective.
Endothelium-dependent vasodilatation induced by leptin is mediated by NO and endothelium-derived hyperpolarizing factor (EDHF). Also, leptin enhances endothelium-dependent relaxation through induction of endothelial neuronal NOS (nNOS) expression via JAK2/STAT3 in mice lacking eNOS, HFD mice, and in HUVECs [251]. Hyperleptinemia and obesity may result in impaired leptin-induced NO production but enhanced EDHR-induced action, and both actions are diminished by leptin antagonist [252].
Leptin upregulates eNOS expression in ECs and thereby enhances NO production [253]. Short-term exposure to leptin failed to enhance superoxide and peroxynitrite production in HUVECs. In contrast, long term exposure to leptin (or hyperleptinemia) enhances superoxide, peroxynitrite and ET-1 production, and consequently inactivates bioavailable NO, in spite of the overexpression of eNOS [253, 254]. This could partly explain that hyperleptinemia could trigger eNOS uncoupling from NO-synthesizing to superoxide-producing enzyme. Therefore, hyperleptinemia inversely correlates with NO bioavailability and cGMP in the aortic endothelium and the aortic wall, respectively, and directly correlates with ROS in ECs of obese mice [253]. This is also why hyperleptinemia in obese subjects is associated with enhanced ROS generation, poor oxidative stress profile and lower level of NO metabolites [255, 256], resulting in ED.
In addition, it has been shown that chronic leptin administration results in ED in mice via stimulation of the pressor effect of the sympathetic nervous system [257], and these effects were confirmed in healthy subjects [258]. Moreover, pretreatment of mice with TEMPOL (superoxide scavenger) completely diminishes the pressor effect of chronic leptin administration on endothelial function [257], re-emphasizing the role of ROS in mediating hyperleptinemia-induced ED.
It has been suggested that leptin plays an important role in mediating subclinical inflammatory state associated with obesity-related conditions. Hyperleptinemia is associated with low-grade inflammation and metabolic dysregulation manifested by high levels of CRP, TNF-α, IL-10, IL-12, glucose, insulin, and TGs and high IR values in obese mice and humans [259]. On the contrary, it has been shown that leptin does not underlie the pro-inflammatory condition in obese human subjects [260, 261]. Leptin enhances the secretion of IL-18 (implicated in T2DM complications) from monocytes via activation of caspase-1 inflammasome [262]. Leptin enhances ERK1/2 phosphorylation and NF-κB activation as well as the production of TNF-α in HUVECs [263].
Leptin through its receptors on EPCs stimulates tube formation of EPCs at physiological concentration, while in hyperleptinemic states, tube formation and EPCs migration are inhibited, with no effect in both conditions on EPCs proliferation [264]. It has been demonstrated that leptin stimulates the homing potential and adhesive activities of EPCs, thus enhancing its repair activity in vivo and in vitro [265]. Also, leptin enhances the mobilization of vascular angiogenic ECs (sca-1+, flk-1+) from bone marrow in mice through leptin-mediated activation of Nox2, enhanced ROS generation, and stimulation of MMP9 expression [266]. These effects could explain the enhanced vascular effects in hyperleptinemic obese subjects [250]. Taken together, we thought that the state of leptin resistance has no or little effect on the beneficial effects of leptin on EPCs.
In a nutshell, although there is an open debate over whether leptin possesses beneficial or detrimental effects on cardiovascular system, generally clinical studies support the notion that leptin in a cardiovascular risk factor, thus reducing leptin production could have beneficial effects in the clinical settings.
Resistin
Generally, resistin is a diabetogenic, inflammatory, and angiogenic adipokine which plays a pivotal role in the pathogenesis of diabetes and its complications. Although some studies indicate that resistin is not associated with IR and metabolic syndrome [267–270], large body of evidence supports the notion that elevated resistin levels are implicated in IR, diabetes, and metabolic syndrome and could be an important risk factor for CVD [271–276].
Endothelial function is shown to be inversely affected by resistin level [277]. Resistin impairs bradykinin-induced relaxation (but not ACh), and this effect is shown to be not mediated via modulating superoxide, NO, and PGI2 production [278]. Also, resistin inhibits insulin-mediated vasodilatation via altering IRS-1 phosphorylation and subsequent inhibition of Akt/eNOS phosphorylation [279].
In ECs, resistin causes imbalance in redox enzymes and mitochondrial dysfunction; therefore, it enhances the generation of ROS and activation of p38 MAPK/JNK pathways, which eventually leads to eNOS downregulation and decreased NO bioavailability [280, 281]. Not surprisingly, hyperresistinemia interferes with endothelial PI3K/Akt signaling pathway and consequently reduces eNOS phosphorylation/activation and NO production [282].
In obese children and hypertensive T2DM patients, resistin serum levels were correlated positively with markers of endothelial activation (ICAM-1, VCAM-1 and E-selectin), homocysteine, and oxidative/nitrosative stress [283, 284]. Furthermore, serum resistin level is associated with CRP in T2DM patients [285].
Recently, Ying et al. have shown that the protective effects of miR-492 against IR/hyperglycemia-induced ED are mainly due to its downregulating effect on resistin production [286]. Therefore, we thought that decreasing resistin levels will be of great importance in the context of T2DM and its associated CVD. To the best of our knowledge, resistin effect on EPCs is still uncovered yet.
Visfatin
Another controversial adipokine is visfatin or extracellular nicotinamide phosphoribosyltransferase (Nampt), an essential enzyme in the pathway of NAD biosynthesis [287]. Although visfatin is mainly synthesized and secreted from visceral adipose tissue in rats [216], leukocytes are the major visfatin source in humans [288], suggesting an inflammatory role of visfatin in humans rather than a metabolic role.
Serum visfatin concentrations are increased in obesity, T2DM, metabolic syndrome, CVD, and gestational diabetes [289, 290]; however, these results are still controversial [291, 292]. Serum visfatin levels negatively correlate with endothelial function in T2DM [293], but this association do not appear in prediabetic obese patients [294]. Moreover, it has been shown that serum visfatin levels correlate with inflammatory markers rather than markers of IR [295]. This results also contradicts with other research which shows that serum visfatin positively correlates with systemic IR and hyperlipidemia [296], suggesting that visfatin levels could change primarily according to the degree of inflammation associated with IR, obesity, and T2DM. Therefore, visfatin could be used as a marker for systemic inflammation in T2DM [297].
Visfatin induces the expression of inflammatory and adhesion molecules in ECs, and this mechanism could be mediated through ROS-dependent NF-κB activation [298, 299]. Also, inflammed ECs (exposed to pro-inflammatory mediators, e.g., TNF-α, IL-1β, or AG II) are considered a source of visfatin [300].
Circulating visfatin concentrations are positively correlated with coronary artery disease in patients with T2DM [301], suggesting a deleterious effect of high concentrations of visfatin on ECs. In human endothelial and vascular smooth muscle cells, visfatin enhances NF-κB signaling pathways [302, 303], and these inflammatory activities are thought to be mediated by its intrinsic Nampt activity [303].
Consistently, Xia et al. demonstrated that visfatin induces ED in coronary ECs via stimulating p47phox translocation, activation of NADPH oxidase, and therefore increases superoxide production [304]. Vallejo et al. reported similar visfatin activities in HUVECs and mesenteric microvessels of rats and humans; however, they showed that visfatin-induced ED through activation of NADPH oxidase is dependent on its Nampt activity [305]. Recently, Villalobos et al. further investigated the detrimental effects of visfatin on ECs and showed that visfatin-induced ROS production causes DNA destruction, p53 upregulation, and telomere dysfunction in HUVECs [306]. Nevertheless, recent study by Karataş et al. showed no such association between visfatin serum levels and ED [307].
Intriguingly, Borradaile et al. demonstrate that enhanced expression of visfatin in human aortic ECs protects the cells against ROS-induced senescence caused by hyperglycemia and thus enhance the replicative longevity and angiogenic capacity of ECs [308]. Consistent with this, Visfatin enhances the activity of eNOS as well as increase production of NO and cGMP in HUVECs and coronary artery ECs via activation of PI3K/Akt pathway, consequently exerting vasodilating effect [309, 310]. These contradictions require further investigation focusing on the role of visfatin concentration and the disease context at which visfatin exerts its actions.
Regarding the effect of visfatin on EPCs function, Chen et al. have shown that increased visfatin levels along with enhanced oxidative stress reduce EPCs number in obese patients [311]. Also, visfatin adversely affects the migration and adhesion capacities of EPCs through activation of NF-κB [312]. These effects are consistent with visfatin-induced ED in the setting of metabolic syndrome.
Omentin
Omentin is an adipokine with insulin sensitizing, antidiabetic, anti-inflammatory, and anti-atherogenic activities. It positively correlates with HDL and adiponectin concentrations. Its serum concentration is decreased in IR, obesity, and T2DM [313]. Reduced circulating omentin level is independent risk factor for CVD in T2DM patients [314]. Administration of omentin in rats increases adiponectin levels, reduces IL-6 concentration, and enhances the production of NO [315]. Moreover, circulating omentin levels could be used as a useful marker for evaluation of endothelial function [316].
Interestingly, omentin enhances HUVECs survival and differentiation through activation/phosphorylation of Akt/eNOS pathway [317]. Moreover, omentin inhibits TNF-α-induced COX-2 activation and production of adhesion molecules via inhibition of the JNK activation and diminishing ERK/NF-κB pathway in HUVECs [318, 319]. To the best of our knowledge, the effect of omentin on EPCs has not been investigated yet.
Vaspin
Vaspin is one of the most recently discovered adipokines, exerting mainly insulin sensitizing and anti-atherogenic actions. Circulating vaspin levels are significantly elevated in IR, obesity, and T2DM [320], suggesting a compensatory role of vaspin in the setting of IR.
In ECs, Jung et al. demonstrated that vaspin enhances NO production and alleviates NEFA-induced ED. Mechanistically, vaspin enhances eNOS activity mainly by reducing asymmetric dimethyl arginine [inhibitor of eNOS] (ADMA) level through induction of dimethylarginine dimethylaminohydrolase (DDAH II) expression—the enzyme responsible for ADMA degradation. These effects are mediated via STAT3 pathway [321]. Also, vaspin protects ECs against NEFA-induced apoptosis via activation of PI3k/Akt signaling pathway [322]. Consistently, vaspin diminishes methylglyoxal-induced apoptotic activity in ECs by inhibiting caspase-3 activation through inhibition of Nox-mediated ROS generation [323]. Taken together, vaspin could have powerful anti-apoptotic and anti-oxidant actions particularly on ECs.
While Fu et al. study showed that vaspin could not protect HUVECs from TNF-α-induced inflammatory damage [324], Jung et al. and Liu et al. experiments indicate that vaspin attenuates TNF-α/IL-1-induced NF-κB activation through stimulation of AMPK in ECs [325, 326], confirming the anti-inflammatory role of vaspin on ECs.
Treatment of EPCs with vaspin elevates the number and enhances proliferation and migration capacities of EPCs in a dose-dependent manner. Moreover, vaspin mitigates hyperglycemia-induced EPCs dysfunction. These beneficial effects are shown to be mediated through activation of PI3K/Akt/eNOS pathway [327].
Chemerin
Chemerin is involved in the regulation of glucose homeostasis and insulin sensitivity. High-plasma chemerin levels are associated with metabolic syndrome, IR, obesity, and T2DM [328, 329]. In obese patients, chemerin positively associates with oxidative stress and inflammation as well as leptin concentration, whereas it shows a negative association with anti-oxidant status and adiponectin level [330]. Interestingly, chemerin levels were not only elevated in metabolic syndrome patients with coronary artery disease compared with patients without coronary artery disease but also correlated positively with the severity of CVD [331]. Overexpression of chemerin and its receptors ChemR23 is associated with diabetic microvascular complications, e.g., diabetic nephropathy, and reducing the expression of chemerin, and ChemR23 could be a possible way to ameliorate diabetic complications [332]. Collectively, chemerin could be a possible link between T2DM and its cardiovascular complications.
Chemerin potentiates the vasoconstrictor responses to phenylephrine and ET-1 via activation of ERK1/2 pathway [333]. Also, chemerin decreases NO production, enhances NO breakdown, and also decreases NO-dependent cGMP signaling, particularly via inducing eNOS uncoupling, enhancing superoxide production and diminishing GC activity [334, 335]. Moreover, treatment of human aorta ECs with chemerin induces ROS generation with enhanced expression of the autophagy-related genes. Knockdown of ChemR23 or treatment with the antioxidant (Mito-TEMPO) diminishes the chemerin-associated ROS generation and abrogates the upregulation of autophagy-related genes [336]. These results support the idea that chemerin induces ED and could be a cardiovascular risk factor, particularly in T2DM and obesity.
While chemerin levels are associated with systemic inflammation in obese and T2DM patients [337], Yamawaki et al. demonstrated the protective anti-inflammatory role of chemerin in ECs against TNF-α-induced inflammatory response, via inhibition of NF-κB pathway [338]. This discrepancy between in vivo and in vitro experiments needs further elucidation. Also, the effect chemerin on EPCs remains to be investigated.
Apelin
Apelin has a role in energy metabolism. Elevated apelin levels are associated with obesity, hyperinsulinemia, IR, and T2DM in humans and mice. Apelin exerts a powerful glucose-lowering effect concomitant with enhanced glucose disposal in skeletal muscle and adipose tissues [339], suggesting that elevated apelin levels could be a compensatory mechanism to enhance insulin sensitivity in case of IR-related disorders.
Apelin is a vasoactive peptide. In healthy volunteers, apelin exhibits arterial NO-mediated vasodilatation [340]. Apelin abolishes the vasoconstricting action of AG II and potentiates the vasodilator response to ACh via inducing phosphorylation of Akt and eNOS in diabetic mice [341].
Interestingly, single-nucleotide polymorphism at the apelin gene is associated with arterial stiffness. This observation led Liao et al. to overexpress miR-765—which interferes with apelin gene expression—in ECs to further elucidate the underlying mechanism of arterial stiffness. They found that overexpression of miR-765 reduces mRNA and protein levels of apelin, consequently inhibiting the phosphorylation of eNOS and ERK/Akt/AMPK signaling pathways [342].
However, Nagano et al. showed that apelin exerts a vaso-constricting effect in mice treated with L-NAME, suggesting that apelin induces ED under pathological conditions [343]. In addition, Lu et al. experiments depict that treatment of HUVECs with apelin induces the expression of adhesion molecules and chemokines via NF-κB/JNK signal pathway, implying a role of apelin in endothelial inflammation [344].
Role of incretins and gut hormones in endothelial dysfunction
Diabetes is associated with incretin defect; that is a reduction or even loss of incretin secretion/action [345]. Recently, Ravassa et al. showed that T2DM patients with decreased GLP-1 levels have enhanced oxidative stress profile, with increased risk for cardiovascular events [346].
In the last 15 years, incretins have been extensively studied in cell cultures, animals, and human, and now there are many incretin-based therapies used in the treatment of diabetes and associated obesity. Incretin-based therapies include (1) GLP-1 agonists or analogs such as exendin-4, liraglutide, and exenatide that mimics the action of GLP-1 on its receptors (GLP-1R) and (2) dipeptidyl peptidase-4 (DPP-4) inhibitors or gliptins such as sitagliptin, vildagliptin, and linagliptin which inhibit the catalytic activity of DDP-4, the enzyme that degrades incretins [347]. Herein, we will discuss the beneficial effects of GLP-1, incretin-based therapies and other gut hormones on endothelial function.
Glucagon-like peptide-1 (GLP-1)
Generally, GLP-1 exerts vasodilator, anti-inflammatory, anti-oxidant, and anti-apoptotic actions on ECs. GLP-1 and vildagliptin improve endothelial function in T2DM and T1DM patients [348–350]. In obese patients, alogliptin (DPP-4 inhibitor) improves postprandial ED [351].
In a clinical trial (NCT01181986), exenatide protects T2DM patients from hyperglycemia and lipemia-induced ED via GLP-1R and AMPK-dependent mechanism [352]. However, the clinical trial (NCT01931982) failed to show a beneficial effect of 10 weeks liraglutide treatment on coronary and peripheral microvascular function in T2DM patients [353]. More clinical research and meta-analysis of data are needed to confirm these results.
GLP-1 and its analogs enhance the phosphorylation/activity and the protein expression of eNOS in HUVECs through a GLP-1R-dependent mechanism and downstream PKA signaling pathway [354, 355]. GLP-1 and its analogs abolish TNF-α mediated inflammatory injury and thus exerting a potent anti-inflammatory effect on ECs, presumably via activation of PPARγ [356–358].
GLP-1 protects ECs from ROS-induced senescence via activation of PKA pathway and enhancing the expression of antioxidant genes [359]. GLP-1 protects HUVECs from apoptosis induced by AGEs mainly by inhibiting ROS generation and suppressing the expression of RAGE [360, 361]. GLP-1 protects ECs from ox-LDL-induced apoptosis via inhibiting PARP-1/iNOS pathway [362]. The DPP-4 inhibitor linagliptin and GLP-1R agonist exendin-4 improve endothelium-dependent relaxation of rat mesenteric arteries pretreated with high glucose, partly by inhibition of superoxide generation caused by hyperglycemia [363]. Liraglutide abolishes hyperglycemia-induced ROS in human aortic ECs by inhibiting PKC/NAD(P)H oxidase pathway and diminishing Endoplasmic reticulum stress [364, 365].
GLP-1 enhances EPCs proliferation and differentiation via increasing VEGF expression [366]. Sitagliptin increases circulating EPCs in T2DM patients and experimental animals through upregulation of SDF-1α, which is a target for DPP-4 action, via eNOS pathway [367, 368]. These results may further elucidate the improvement in endothelial function in patients treated with incretin-based therapies.
Ghrelin
Ghrelin is an orexigenic and hyperglycemic peptide. Ghrelin is post-translationally modified to exist in two forms: unacylated and acylated, where acylated form being in excess compared to unacylated one in conditions of IR [369].
Although there are some contradictory results, T2DM and obesity are associated with decreased levels of ghrelin, suggesting a compensatory mechanism in response to IR and may be a cause in inducing hyperinsulinemia [370]. Lower ghrelin concentrations are associated with oxidative stress in obese patients [371] and also could be a risk factor for cardiovascular events [372].
Ghrelin enhances endothelial function and restores the balance between ET-1 and NO in patients with metabolic syndrome [373, 374]. It diminishes homocysteine-induced ED via enhancing the eNOS expression, abolishing the ROS generation, and inhibition of NF-κB activation in porcine coronary artery cardiac microvascular ECs and human coronary artery ECs [375, 376]. Unacylated ghrelin protect against oxidative stress in ECs by increasing SIRT1 and SOD-2 expression in mouse model [377].
In addition, it diminishes AG II and NEFA-induced apoptosis via activation of the PI3K/Akt pathway and inhibiting the NEFA-induced ROS generation in ECs [378, 379]. Also, it inhibits hyperglycemia, AG II, and TNF-α-induced endothelial injury in HUVECs via suppressing NF-κB activation, thus exerting anti-inflammatory and immunomodulatory effects [380–382]. Also, recent research shows that ghrelin protects HUVECs from hyperglycemia-induced apoptosis through activation of mTOR/P70S6K signaling pathway [383].
Regarding ghrelin effect on EPCs, Chen et al. have shown that ghrelin induces EPCs migratory capacity via activating PI3K/Akt/eNOS/NO signaling pathway [384]. Consistently, Togliatto et al. showed that unacylated ghrelin enhances EPCs vasculogenic activity, stimulates EPCs mobilization through activating Akt/eNOS/NO pathway, and inhibits EPCs senescence via modulating Rac1, the regulatory protein in NADPH oxidase [385].
Overall, ghrelin has been proved to exert many beneficial and protecting effects on ECs; however, its hyperglycemic and orexigenic effects might limit its clinical application. Therefore, developing ghrelin agonists which specifically mimic ghrelin effects on ECs and are devoid from its effects on food, insulin, and glucose metabolism would be a promising way to address ED.
Obestatin
Lower levels of the gut hormone obestatin are associated with obesity, T2DM, prediabetics and IR [386, 387]. Low level of plasma obestatin could be associated with early atherosclerosis in T2DM patients [388]. Obestatin exerts vascular relaxation via activation of specific endothelium-dependent NO signaling pathways [389]. Obestatin actions are still to be fully elucidated in many organ tissues.
Conclusion
In conclusion, the etiology of diabetic vascular dysfunction is complex. Furthermore, metabolic disturbances such as IR, compensatory hyperinsulinemia, inflammation, hyperlipidemia, hyperglycemia, as well as the defect in secretion/action of adipokines and incretins contribute independently to ED, at least in part, through activation of ROS generating mechanisms. The complex metabolic disturbances that characterize diabetes contribute to elevated oxidative stress to various extents. Hyperglycemia-induced ED mediated by multiple biochemical pathways converge into increasing ROS generation in ECs and EPCs. While in dyslipidemia, a sole pathway (Ox-LDL) is the major contributor to oxidative stress-induced ED. Moreover, some metabolic pathways increase oxidative stress by potentiating the effect of other pathways rather than having a direct effect. Furthermore, it appears that in the majority of metabolic disturbances discussed, an intricate interplay exists between metabolic intermediates of distinct pathways that lead to ED either directly or by promoting oxidative stress, making the distinction between causes and consequences somewhat blurred. Thus, given the current research methodologies that isolate the effect of a certain biochemical parameter, the elucidation of the complex interplay that characterizes the role of oxidative stress in ED will require novel research protocols that harness contemporary computational advances, which will allow the analysis of such complex experiments.
However, the amount of evidence on the detrimental effects of oxidative stress on endothelial function and its link to the pathophysiological pathways underlying diabetic complications is compelling. This notion led us and many other researchers to suggest that the oxidative stress is the common soil, which transmits signals from other metabolic conditions leading to ED, and consequently CVD. An improved understanding of the mechanisms of ED in this setting could provide new approaches for patient management. Intriguingly, a main source of discrepancy seen in many studies may be attributed to (1) differences in ECs and EPCs isolation methods, (2) culture techniques between different labs, and (3) differences between ECs from microvascular versus macrovascular tissues, which show wide variability in gene expression profiles.
The use of antioxidants as adjuvant to standard diabetic treatment has long yielded controversial results in clinical trials, probably due to emphasizing short-term effects of these agents. We discussed in this review the long-term effects of oxidative stress on ECs and EPCs; therefore, we suggest that a better assessment of long-term effects of antioxidants would elucidate the potential clinical benefits of these agents—if any. Finally, more research is needed to characterize targets and develop more powerful anti-oxidants, paving the way for efficient management of ED, and hopefully therefore, reducing the risk of diabetic cardiovascular events.
Abbreviations
- ACh:
-
Acetylcholine
- ADMA:
-
Asymmetric dimethylarginine
- AMPK:
-
Adenosine monophosphate-dependent kinase
- AGEs:
-
Advanced glycation end products
- AG II:
-
Angiotensin II
- AR:
-
Aldose reductase
- ARI:
-
Aldose reductase inhibitors
- BH4 :
-
Tetrahydrobiopterin
- BAD:
-
Bcl-2-associated death promoter
- CRP:
-
C-reactive protein
- CXCR-4:
-
Chemokine receptor type-4
- CVD:
-
Cardiovascular disease
- cGMP:
-
Cyclic guanosine monophosphate
- COX:
-
Cyclooxygenase
- CMKLR1:
-
Chemokine-like receptor 1
- DPP-4:
-
Dipeptidyl-peptidase-4
- DAG:
-
Diacyl glycerol
- ET-1:
-
Endothelin-1
- ED:
-
Endothelial dysfunction
- DDAH:
-
Dimethylarginine dimethylaminohydrolase
- ECs:
-
Endothelial cells
- EPCs:
-
Endothelial progenitor cells
- ERK:
-
Extracellar signal-regulated kinases
- EDHF:
-
Endothelium-derived hyperpolarizing factor
- eNOS:
-
Endothelial NO synthase
- FAD:
-
Flavin adenine dinucleotide
- FMN:
-
Flavin mononucleotide
- FOXO3a:
-
Forkhead transcription factor3a
- GTP:
-
Guanosine triphosphate
- GC:
-
Guanylate cyclase
- GSH:
-
Glutathione
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GA-3P:
-
Glyceraldehyde-3-phosphate
- G-6-Pase:
-
Glucose-6-phosphatase
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- HDL:
-
High density lipoprotein
- HIF-1α:
-
Hypoxia-inducible factor-1α
- HFD:
-
High fat diet
- HUVECs:
-
Human umbilical vein endothelial cells
- HO-1:
-
Heme oxygenase-1
- HSP:
-
Hexosamine synthetic pathway
- HSL:
-
Hormone sensitive lipase
- IR:
-
Insulin resistance
- IKK:
-
Inhibitor of κB kinase
- IRS:
-
Insulin receptor substrate
- IGF-I:
-
Insulin growth factor
- ICAM-1:
-
Intercellular adhesion molecule-1
- JNK:
-
c-Jun N-terminal kinases
- JAM-A:
-
Junction adhesion molecule A
- LDL:
-
Low density lipoprotein
- L-NAME:
-
Nitro-l-arginine methyl ester
- LOX-1:
-
Lecithin-like ox-LDL receptor-1
- LC:
-
Lactosyl ceramide
- MMP-9:
-
Matrix metallopeptidase-9
- MAPK:
-
Mitogen activated protein kinase
- MCP-1:
-
Monocyte chemotactic protein-1
- MnSOD:
-
Mn superoxide dismutase
- MPO:
-
Myeloperoxidase
- MDA:
-
Malondialdehyde
- NF-κB:
-
Nuclear factor κB
- NO:
-
Nitric oxide
- NEFA:
-
Non-esterified fatty acids
- Nox:
-
NADPH oxidase
- NADPH:
-
Reduced nicotinamide adenine dinucleotide phosphate
- NIK:
-
NF-kB inducing kinase
- Nampt:
-
Nicotinamide phosphoribosyltransferase
- PTEN:
-
Phosphatase and tensin homolog
- PK:
-
Protein kinase
- PI3K/Akt:
-
Phosphatidylinositol triphosphate kinase/Akt (protein kinase B)
- PKC:
-
Protein kinase C
- PTPs:
-
Protein tyrosine phosphatases
- PON-1:
-
Paraoxonase-1
- PGI2 :
-
Prostacyclin
- PSGL:
-
P-selectin glycoprotein ligand
- PPARγ:
-
Peroxisome proliferator activated receptor
- PARP:
-
Poly (ADP-ribose) polymerase
- PDGF:
-
Platelet-derived growth factor
- PLC:
-
Phospholipase C
- PAI:
-
Plasminogen activator inhibitor
- PGC:
-
Peroxisome proliferator activated receptor γ coactivator
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- ROS:
-
Reactive oxygen species
- RAGE:
-
Receptors for advanced glycated end products
- STAT:
-
Signal transducer and activator of transcription
- SDF-1:
-
Stromal cell-derived factor-1
- S1P:
-
Sphingosine-1-phosphate
- SK:
-
Sphingosine kinase
- SOD:
-
Superoxide dismutase
- SDH:
-
Sorbitol dehydrogenase
- SOCS-3:
-
Supressor of cytokine signaling 3
- SHP:
-
Src-homology-2 domain-containing phosphatase
- T1DM:
-
Type 1 diabetes mellitus
- T2DM:
-
Type 2 diabetes mellitus
- TNF-α:
-
Tumor necrosis factor-α
- TNFR:
-
Tumor necrosis factor receptor
- TRAF:
-
TNFR-associated factor
- TRADD:
-
TNF receptor-associated death domain
- TGF:
-
Transforming growth factor
- TXA2 :
-
Thromboxane A2
- tPA:
-
Tissue plasminogen activator
- UDP-GlcNAc:
-
UDP-N-acetylglucosamine
- UCP:
-
Uncoupling protein
- VLDL:
-
Very low density lipoprotein
- VSMCs:
-
Vascular smooth muscle cells
- VEGF:
-
Vascular endothelial growth factor
- VCAM:
-
Vascular cell adhesion molecule
- XRCC1:
-
X-ray repair cross-complementing protein-1
- ZO:
-
Zucker obese rats
- ZL:
-
Zucker lean rats
References
Association AD, Diagnosis and classification of diabetes mellitus. Diabetes care 36(Supplement 1), S67–S74 (2013)
P.Z. Zimmet, D.J. Magliano, W.H. Herman, J.E. Shaw, Diabetes: a 21st century challenge. Lancet Diabetes Endocrinol 2(1), 56–64 (2014)
R.A. Defronzo, Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58(4), 773–795 (2009)
Y. Lin, Z. Sun, Current views on type 2 diabetes. J. Endocrinol. 204(1), 1–11 (2010)
E. Hopps, D. Noto, G. Caimi, M. Averna, A novel component of the metabolic syndrome: the oxidative stress. Nutr Metab Cardiovasc Dis 20(1), 72–77 (2010)
J.S. Johansen, A.K. Harris, D.J. Rychly, A. Ergul, Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol 4(1), 5 (2005)
G. Cohen, Y. Riahi, E. Alpert, A. Gruzman, S. Sasson, The roles of hyperglycaemia and oxidative stress in the rise and collapse of the natural protective mechanism against vascular endothelial cell dysfunction in diabetes. Arch. Physiol. Biochem. 113(4–5), 259–267 (2007)
S. Ansar, J. Koska, P.D. Reaven et al., Postprandial hyperlipidemia, endothelial dysfunction and cardiovascular risk: focus on incretins. Cardiovasc. Diabetol. 10(1), 61 (2011)
D. Versari, E. Daghini, A. Virdis, L. Ghiadoni, S. Taddei, Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes care 32(Suppl 2), S314–S321 (2009)
R.J. Esper, R.A. Nordaby, J.O. Vilariño, A. Paragano, J.L. Cacharrón, R.A. Machado et al., Endothelial dysfunction: a comprehensive appraisal. Cardiovasc Diabetol. 5(4), 4–22 (2006)
C.N. Marti, M. Gheorghiade, A.P. Kalogeropoulos, V.V. Georgiopoulou, A.A. Quyyumi, J. Butler, Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 60(16), 1455–1469 (2012)
S. Hamed, B. Brenner, A. Aharon, D. Daoud, A. Roguin, Nitric oxide and superoxide dismutase modulate endothelial progenitor cell function in type 2 diabetes mellitus. Cardiovasc Diabetol. 8(56), 666–674 (2009)
F. Timmermans, J. Plum, M.C. Yöder, D.A. Ingram, B. Vandekerckhove, J. Case, Endothelial progenitor cells: identity defined? J. Cell Mol. Med. 13(1), 87–102 (2009)
D.P. Basile, M.C. Yoder, Circulating and tissue resident endothelial progenitor cells. J. Cell. Physiol. 229(1), 10–16 (2014)
R.M. Cubbon, A. Rajwani, S.B. Wheatcroft, The impact of insulin resistance on endothelial function, progenitor cells and repair. Diabetes Vasc. Dis. Res. 4(2), 103–111 (2007)
M.C. Yoder, Human endothelial progenitor cells. Cold Spring Harbor perspectives in medicine. Cold Spring: Harbor Laboratory Press; 2012;2(7):a006692 (2012). doi:10.1101/cshperspect.a006692
M. Zhang, A.B. Malik, J. Rehman, Endothelial progenitor cells and vascular repair. Curr. Opin. Hematol. 21(3), 224–228 (2014)
P. Staller, J. Sulitkova, J. Lisztwan, H. Moch, E.J. Oakeley, W. Krek, Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 425(6955), 307–311 (2003)
D.J. Ceradini, A.R. Kulkarni, M.J. Callaghan, O.M. Tepper, N. Bastidas, M.E. Kleinman et al., Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10(8), 858–864 (2004)
A. Avogaro, G.P. Fadini, A. Gallo, E. Pagnin, S. de Kreutzenberg, Endothelial dysfunction in type 2 diabetes mellitus. Nutr. Metab. Cardiovasc. Dis. 16, S39–S45 (2006)
M. Gili, A. Orsello, S. Gallo, M.F. Brizzi, Diabetes-associated macrovascular complications: cell-based therapy a new tool? Endocrine 44(3), 557–575 (2013)
T. Thum, D. Fraccarollo, M. Schultheiss, S. Froese, P. Galuppo, J.D. Widder et al., Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 56(3), 666–674 (2007)
L. Bruyndonckx, V.Y. Hoymans, G. Frederix, A. Guchtenaere, H. Franckx, D.K. Vissers et al., Endothelial progenitor cells and endothelial microparticles are independent predictors of endothelial function. J. Pediatr. 165(2), 300–305 (2014)
M.F. Lombardo, P. Iacopino, M. Cuzzola, E. Spiniello, C. Garreffa, F. Ferrelli et al., Type 2 diabetes mellitus impairs the maturation of endothelial progenitor cells and increases the number of circulating endothelial cells in peripheral blood. Cytom. Part A 81(10), 856–864 (2012)
M. Frisard, E. Ravussin, Energy metabolism and oxidative stress. Endocrine 29(1), 27–32 (2006)
J. Tanaka, L. Qiang, A.S. Banks, C.L. Welch, M. Matsumoto, T. Kitamura et al., Foxo 1 links hyperglycemia to LDL oxidation and endothelial nitric oxide synthase dysfunction in vascular endothelial cells. Diabetes 58(10), 2344–2354 (2009)
K. Tsuchiya, J. Tanaka, Y. Shuiqing, C.L. Welch, R.A. DePinho, I. Tabas et al., FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. 15(3), 372–381 (2012)
F. Yan, Y. Wang, X. Wu, H.M. Peshavariya, G.J. Dusting, M. Zhang et al., Nox4 and redox signaling mediate TGF-β-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 5, e1010 (2014). doi:10.1038/cddis.2013.551
V. Romaschenko, R. Zinovkin, I. Galkin, V. Zakharova, A. Panteleeva, A. Tokarchuk et al., Low concentrations of uncouplers of oxidative phosphorylation prevent inflammatory activation of endothelial cells by tumor necrosis factor. Biochemistry (Moscow) 80(5), 610–619 (2015)
M.T. Mathews, B.C. Berk, PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler. Thromb. Vasc. Biol. 28(4), 711–717 (2008)
L. Gong, F. Liu, J. Wang, X. Wang, X. Hou, Y. Sun et al., yperglycemia induces apoptosis of pancreatic islet endothelial cells via reactive nitrogen species-mediated Jun N-terminal kinase activation. Mol. Cell Res. 1813(6), 1211–1219 (2011)
A. Vikram, Y.-R. Kim, S. Kumar, A. Naqvi, T.A. Hoffman, A. Kumar et al., Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arterioscler. Thrombos. Vasc. Biol. 34(10), 2301–2309 (2014)
J. Case, D.A. Ingram, L.S. Haneline, Oxidative stress impairs endothelial progenitor cell function. Antioxid. Redox Signal. 10(11), 1895–1907 (2008)
D. Sukmawati, S. Fujimura, S. Jitsukawa, R. Ito-Hirano, T. Ishii, T. Sato et al., Oxidative stress tolerance of early stage diabetic endothelial progenitor cell. Regener. Therapy 1, 38–44 (2015)
F. Wang, Y.-Q. Wang, Q. Cao, J.-J. Zhang, L.-Y. Huang, T.-T. Sang et al., Hydrogen peroxide induced impairment of endothelial progenitor cell viability is mediated through a FoxO3a dependant mechanism. Microvasc. Res. 90, 48–54 (2013)
A. Rosso, A. Balsamo, R. Gambino, P. Dentelli, R. Falcioni, M. Cassader et al., p53 Mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. J. Biol. Chem. 281(7), 4339–4347 (2006)
J. Song, Y. Lee, Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 373, 845–853 (2003)
D.A. Ingram, T.R. Krier, L.E. Mead, C. McGuire, D.N. Prater, J. Bhavsar et al., Clonogenic endothelial progenitor cells are sensitive to oxidative stress. Stem Cells 25(2), 297–304 (2007)
J. Wei, Y. Liu, M. Chang, C.-L. Sun, D.-W. Li, Z.-Q. Liu et al., Proteomic analysis of oxidative modification in endothelial colony-forming cells treated by hydrogen peroxide. Int. J. Mol. Med. 29(6), 1099–1105 (2012)
N. Musi, L.J. Goodyear, Insulin resistance and improvements in signal transduction. Endocrine 29(1), 73–80 (2006)
H.C. Lam, Role of endothelin in diabetic vascular complications. Endocrine 14(3), 277–284 (2001)
K. Cusi, K. Maezono, A. Osman, M. Pendergrass, M.E. Patti, T. Pratipanawatr et al., Insulin resistance differentially affects the PI 3-kinase-and MAP kinase-mediated signaling in human muscle. J. Clin. Investig. 105(3), 311 (2000)
E.C. Eringa, C.D.A. Stehouwer, G.P. van Nieuw Amerongen, L. Ouwehand, N. Westerhof, P. Sipkema, Vasoconstrictor effects of insulin in skeletal muscle arterioles are mediated by ERK1/2 activation in endothelium. Am. J. Physiol. Heart Circ. Physiol. 287(5), H2043–H2048 (2004)
W.A. Hsueh, R.E. Law, Insulin signaling in the arterial wall. Am. J. Cardiol. 84(1A), 21J–24J (1999)
P.V. Katakam, J.A. Snipes, M.M. Steed, D.W. Busija, Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J. Cereb. Blood Flow Metab. 32(5), 792–804 (2012)
U. Förstermann, T. Münzel, Endothelial nitric oxide synthase in vascular disease from marvel to menace. Circulation 113(13), 1708–1714 (2006)
M. Yokoyama, K. Hirata, Endothelial nitric oxide synthase uncoupling: is it a physiological mechanism of endothelium-dependent relaxation in cerebral artery? Cardiovasc. Res. 73(1), 8–9 (2007)
J. Vásquez-Vivar, B. Kalyanaraman, P. Martásek, N. Hogg, B.S. Masters, H. Karoui et al., Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl. Acad. Sci. U.S.A. 95(16), 9220–9225 (1998)
T. Sugiyama, B.D. Levy, T. Michel, Tetrahydrobiopterin recycling, a key determinant of endothelial nitric-oxide synthase-dependent signaling pathways in cultured vascular endothelial cells. J. Biol. Chem. 284(19), 12691–12700 (2009)
J.G. San, J. Bidegain, P.A. Robador, J. Díez, A. Fortuño, G. Zalba, Insulin-induced NADPH oxidase activation promotes proliferation and matrix metalloproteinase activation in monocytes/macrophages. Free Radic. Biol. Med. 46(8), 1058–1067 (2009)
A. Görlach, R. Brandes, K. Nguyen, M. Amidi, F. Dehghani, R. Busse, A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ. Res. 87(1), 26–32 (2000)
G. Douglas, J.K. Bendall, M.J. Crabtree, A.L. Tatham, E.E. Carter, A.B. Hale et al., Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE-/- mice. Cardiovasc. Res. 94(1), 20–29 (2012)
T. Ago, T. Kitazono, H. Ooboshi, T. Iyama, Y.H. Han, J. Takada et al., Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 109(2), 227–233 (2004)
S. Furukawa, T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima et al., Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 114(12), 1752 (2004)
G. Ceolotto, M. Bevilacqua, I. Papparella, E. Baritono, L. Franco, C. Corvaja et al., Insulin generates free radicals by an NAD (P) H, phosphatidylinositol 3′-kinase-dependent mechanism in human skin fibroblasts ex vivo. Diabetes 53(5), 1344–1351 (2004)
D. Zhuang, A.-C. Ceacareanu, Y. Lin, B. Ceacareanu, M. Dixit, K.E. Chapman et al., Nitric oxide attenuates insulin-or IGF-I-stimulated aortic smooth muscle cell motility by decreasing H2O2 levels: essential role of cGMP. Am. J. Physiol.-Heart Circ. Physiol. 286(6), H2103–H2112 (2004)
K. Shinozaki, T. Okamura, Y. Nishio, A. Kashiwagi, R. Kikkawa, N. Toda, Evaluation of endothelial free radical release by vascular tension responses in insulin-resistant rat aorta. Eur. J. Pharmacol. 394(2–3), 295–299 (2000)
C. Murphy, G.S. Kanaganayagam, B. Jiang, P.J. Chowienczyk, R. Zbinden, M. Saha et al., Vascular dysfunction and reduced circulating endothelial progenitor cells in young healthy UK South Asian men. Arterioscler. Thromb. Vasc. Biol. 27(4), 936–942 (2007)
P.M. Humpert, Z. Djuric, U. Zeuge, D. Oikonomou, Y. Seregin, K. Laine et al., Insulin stimulates the clonogenic potential of angiogenic endothelial progenitor cells by IGF-1 receptor-dependent signaling. Mol. Med. 14(5–6), 301 (2008)
N.Y. Yuldasheva, S.T. Rashid, N.J. Haywood, P. Cordell, R. Mughal, H. Viswambharan et al., Haploinsufficiency of the insulin-like growth factor-1 receptor enhances endothelial repair and favorably modifies angiogenic progenitor cell phenotype. Arterioscler. Thrombos. Vasc. Biol. 34(9), 2051–2058 (2014)
L. Benboubker, H. Watier, A. Carion, M.-T. Georget, I. Desbois, P. Colombat et al., Association between the SDF1-3′ A allele and high levels of CD34+ progenitor cells mobilized into peripheral blood in humans. Br. J. Haematol. 113(1), 247–250 (2001)
A. Sengupta, H. Viswambharan, N. Yuldasheva, B. Mercer, S. Galloway, N. Ali et al., Endothelial insulin sensitisation enhances vascular repair in systemic insulin resistance and improves endothelial function by restoring nitric oxide bioavailability. Circulation 130(2), A13829 (2014)
M.B. Kahn, N.Y. Yuldasheva, R.M. Cubbon, J. Smith, S.T. Rashid, H. Viswambharan et al., Insulin resistance impairs circulating angiogenic progenitor cell function and delays endothelial regeneration. Diabetes 60(4), 1295–1303 (2011)
M.M. Hartge, T. Unger, U. Kintscher, The endothelium and vascular inflammation in diabetes. Diabetes Vasc. Dis. Res. 4(2), 84–88 (2007)
R.B. Goldberg, Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J. Clin. Endocrinol. Metab. 94(9), 3171–3182 (2009)
T. Yang, C.-H. Chu, P.-C. Hsieh, C.-H. Hsu, Y.-C. Chou, S.-H. Yang et al., C-reactive protein concentration as a significant correlate for metabolic syndrome: a Chinese population-based study. Endocrine 43(2), 351–359 (2013)
S. Ma, Y. Jin, W. Xu, W. Hu, F. Bai, X. Wu, Increased serum levels of ischemia-modified albumin and C-reactive protein in type 1 diabetes patients with ketoacidosis. Endocrine 42(3), 570–576 (2012)
A.D. Pradhan, J.E. Manson, N. Rifai, J.E. Buring, P.M. Ridker, C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286(3), 327–334 (2001)
D.J. Freeman, J. Norrie, M.J. Caslake, A. Gaw, I. Ford, G.D. Lowe et al., C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes 51(5), 1596–1600 (2002)
S. Devaraj, P.R. Kumaresan, I. Jialal, C-reactive protein induces release of both endothelial microparticles and circulating endothelial cells in vitro and in vivo: further evidence of endothelial dysfunction. Clin. Chem. 57(12), 1757–1761 (2011)
Y. Fujita, S. Yamaguchi, A. Kakino, S. Iwamoto, R. Yoshimoto, T. Sawamura, Lectin-like oxidized LDL receptor 1 is involved in CRP-mediated complement activation. Clin. Chem. 57(10), 1398–1405 (2011)
K. Tanigaki, C. Mineo, I.S. Yuhanna, K.L. Chambliss, M.J. Quon, E. Bonvini et al., C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of Fc$\gamma$RIIB and SHIP-1. Circ. Res. 104(11), 1275–1282 (2009)
X. Wang, W. Liu, Y. Wu, X. Liu, X. Liang, Z. Wan et al., C-reactive protein reduces protein S-nitrosylation in endothelial cells. Mol. Cell. Biochem. 375(1–2), 131–138 (2013)
Y. Zhong, C. Cheng, Y. Luo, C. Tian, H. Yang, B. Liu et al., C-reactive protein stimulates RAGE expression in human coronary artery endothelial cells in vitro via ROS generation and ERK/NF κB activation. Acta Pharmacol. Sinica 36, 440–447 (2015)
J. Chen, J. Jin, M. Song, H. Dong, G. Zhao, L. Huang, C-reactive protein down-regulates endothelial nitric oxide synthase expression and promotes apoptosis in endothelial progenitor cells through receptor for advanced glycation end-products. Gene 496(2), 128–135 (2012)
H. Fujii, S.-H. Li, P.E. Szmitko, P.W. Fedak, S. Verma, C-reactive protein alters antioxidant defenses and promotes apoptosis in endothelial progenitor cells. Arterioscler. Thrombos. Vasc. Biol. 26(11), 2476–2482 (2006)
K.A. Fasing, B.J. Nissan, J.J. Greiner, B.L. Stauffer, C.A. DeSouza, Influence of elevated levels of C-reactive protein on circulating endothelial progenitor cell function. Clin. Transl. Sci. 7(2), 137–140 (2014)
K. Yamagata, N. Tanaka, H. Matsufuji, M. Chino, β-Carotene reverses the IL-1βmediated reduction in paraoxonase-1 expression via induction of the CaMKKII pathway in human endothelial cells. Microvasc. Res. 84(3), 297–305 (2012)
S. Vallejo, E. Palacios, T. Romacho, L. Villalobos, C. Peiró, C.F. Sánchez-Ferrer, The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 13(1), 158 (2014)
A. Rosell, K. Arai, J. Lok, T. He, S. Guo, M. Navarro et al., Interleukin-1β augments angiogenic responses of murine endothelial progenitor cells in vitro. J. Cereb. Blood Flow Metab. 29(5), 933–943 (2009)
L. Yang, X.-G. Guo, C.-Q. Du, J.-X. Yang, D.-M. Jiang, B. Li et al., Interleukin-1 beta increases activity of human endothelial progenitor cells: involvement of PI3K-Akt signaling pathway. Inflammation 35(4), 1242–1250 (2012)
A. Mao, C. Liu, Y. Guo, D. Su, T. Luo, W. Fu et al., Modulation of the number and functions of endothelial progenitor cells by interleukin 1$\beta$ in the peripheral blood of pigs: involvement of p38 mitogen-activated protein kinase signaling in vitro. J. Trauma Acute Care Surg. 73(5), 1145–1151 (2012)
M. Lagman, J. Ly, T. Saing, S.M. Kaur, T.E. Vera, D. Morris et al., Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 10(3), e0118436 (2015). doi:10.1371/journal.pone.0118436
B. Thorand, H. Kolb, J. Baumert, W. Koenig, L. Chambless, C. Meisinger et al., Elevated levels of interleukin-18 predict the development of type 2 diabetes results from the MONICA/KORA Augsburg study, 1984-2002. Diabetes 54(10), 2932–2938 (2005)
M. Mishra, H. Kumar, S. Bajpai, R.K. Singh, K. Tripathi, Level of serum IL-12 and its correlation with endothelial dysfunction, insulin resistance, proinflammatory cytokines and lipid profile in newly diagnosed type 2 diabetes. Diabetes Res. Clin. Pract. 94(2), 255–261 (2011)
M.-C. Durpès, C. Morin, J. Paquin-Veillet, R. Beland, M. Paré, M.-O. Guimond et al., PKC-$\beta$ activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc. Res. 106(2), 303–313 (2015)
B. Chandrasekar, W.H. Boylston, K. Venkatachalam, N.J. Webster, S.D. Prabhu, A.J. Valente, Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-$\kappa$B/PTEN suppression. J. Biol. Chem. 283(36), 24889–24898 (2008)
J.M. Kahlenberg, S.G. Thacker, C.C. Berthier, C.D. Cohen, M. Kretzler, M.J. Kaplan, Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J. Immunol. 187(11), 6143–6156 (2011)
X. Zhang, L. Ma, F. Peng, Y. Wu, Y. Chen, L. Yu et al., The endothelial dysfunction in patients with type 2 diabetes mellitus is associated with IL-6 gene promoter polymorphism in Chinese population. Endocrine 40(1), 124–129 (2011)
D.Y. Yuen, R.M. Dwyer, V.B. Matthews, L. Zhang, B.G. Drew, B. Neill et al., Interleukin-6 attenuates insulin-mediated increases in endothelial cell signaling but augments skeletal muscle insulin action via differential effects on tumor necrosis factor-$\alpha$ expression. Diabetes 58(5), 1086–1095 (2009)
F. Andreozzi, E. Laratta, C. Procopio, M.L. Hribal, A. Sciacqua, M. Perticone et al., Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol. Cell. Biol. 27(6), 2372–2383 (2007)
Y. Fan, J. Ye, F. Shen, Y. Zhu, Y. Yeghiazarians, W. Zhu et al., Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J. Cereb. Blood Flow Metab. 28(1), 90–98 (2008)
Y. Yin, W. Liu, G. Ji, Y. Dai, The essential role of p38 MAPK in mediating the interplay of oxLDL and IL-10 in regulating endothelial cell apoptosis. Eur. J. Cell Biol. 92(4), 150–159 (2013)
O. Huet, E. Laemmel, Y. Fu, L. Dupic, A. Aprico, K.L. Andrews et al., Interleukin 10 antioxidant effect decreases leukocytes/endothelial interaction induced by tumor necrosis factor $\alpha$. Shock 39(1), 83–88 (2013)
Y. Wang, Q. Chen, Z. Zhang, F. Jiang, X. Meng, H. Yan, Interleukin-10 overexpression improves the function of endothelial progenitor cells stimulated with TNF-$\alpha$ through the activation of the STAT3 signaling pathway. Int. J. Mol. Med. 35(2), 471–477 (2015)
B.B. Aggarwal, Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3(9), 745–756 (2003)
G. Chen, D.V. Goeddel, TNF-R1 signaling: a beautiful pathway. Science 296(5573), 1634–1635 (2002)
B.B. Aggarwal, S.C. Gupta, J.H. Kim, Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 119(3), 651–665 (2012)
P.J. Naudé, J.A. Boer, P.G. Luiten, U.L. Eisel, Tumor necrosis factor receptor cross-talk. FEBS J. 278(6), 888–898 (2011)
N. Makino, T. Maeda, M. Sugano, S. Satoh, R. Watanabe, N. Abe, High serum TNF-$\alpha$ level in Type 2 diabetic patients with microangiopathy is associated with eNOS down-regulation and apoptosis in endothelial cells. J. Diabetes Complic. 19(6), 347–355 (2005)
K. Zorena, J. Mysliwska, M. Mysliwiec, A. Balcerska, P. Lipowski, K. Raczynska, Relationship between serum levels of tumor necrosis factor-alpha and interleukin-6 in diabetes mellitus type 1 children. Central Eur J Immunol. 32(3), 124 (2007)
X. Gao, S. Belmadani, A. Picchi, X. Xu, B.J. Potter, N. Tewari-Singh et al., Tumor necrosis factor-$\alpha$ induces endothelial dysfunction in Leprdb mice. Circulation 115(2), 245–254 (2007)
H. Zhang, J. Zhang, Z. Ungvari, C. Zhang, Resveratrol improves endothelial function role of TNF$\alpha$ and vascular oxidative stress. Arterioscler. Thrombos. Vasc. Biol. 29(8), 1164–1171 (2009)
A. Picchi, X. Gao, S. Belmadani, B.J. Potter, M. Focardi, W.M. Chilian et al., Tumor necrosis factor-$\alpha$ induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ. Res. 99(1), 69–77 (2006)
S. Basuroy, D. Tcheranova, S. Bhattacharya, C.W. Leffler, H. Parfenova, Nox4 NADPH oxidase-derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-$\alpha$-induced apoptosis. Am. J. Physiol.-Cell Physiol. 300(2), C256–C265 (2011)
H.M. Peshavariya, C.J. Taylor, C. Goh, G.-S. Liu, F. Jiang, E.C. Chan et al., Annexin peptide Ac2-26 suppresses TNF $\alpha$-induced inflammatory responses via inhibition of Rac1-dependent NADPH oxidase in human endothelial cells. PLoS ONE 8(4), e60790 (2013). doi:10.1371/journal.pone.0060790
S. Basuroy, S. Bhattacharya, C.W. Leffler, H. Parfenova, Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-$\alpha$ in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 296(3), C422–C432 (2009)
J.-M. Li, L.M. Fan, M.R. Christie, A.M. Shah, Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol. 25(6), 2320–2330 (2005)
J. van Buul, M. Fernandez-Borja, E. Anthony, P. Hordijk, Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid. Redox Signal. 7(3–4), 308–317 (2005)
C. Papaharalambus, W. Sajjad, A. Syed, C. Zhang, M.O. Bergo, R.W. Alexander et al., Tumor necrosis factor alpha stimulation of Rac1 activity. Role of isoprenylcysteine carboxylmethyltransferase. J. Biol. Chem. 280(19), 18790–18796 (2005)
D.N. Meijles, L.M. Fan, B.J. Howlin, J.-M. Li, Molecular insights of p47phox phosphorylation dynamics in the regulation of NADPH oxidase activation and superoxide production. J. Biol. Chem. 289(33), 22759–22770 (2014)
M. Tesauro, F. Schinzari, V. Rovella, D. Melina, N. Mores, A. Barini et al., Tumor necrosis factor-$\alpha$ antagonism improves vasodilation during hyperinsulinemia in metabolic syndrome. Diabetes Care 31(7), 1439–1441 (2008)
H. Dominguez, H. Storgaard, C. Rask-Madsen, H.T. Steffen, N. Ihlemann, N.D. Baunbjerg et al., Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J. Vasc. Res. 42(6), 517–525 (2004)
C.V. Desouza, F.G. Hamel, K. Bidasee, K. O’Connell, Role of inflammation and insulin resistance in endothelial progenitor cell dysfunction. Diabetes 60(4), 1286–1294 (2011)
T.-G. Chen, Z.-Y. Zhong, G.-F. Sun, Y.-X. Zhou, Y. Zhao, Effects of tumour necrosis factor-alpha on activity and nitric oxide synthase of endothelial progenitor cells from peripheral blood. Cell Prolif. 44(4), 352–359 (2011)
A. Hoffmann, D. Baltimore, Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 210, 171–186 (2006)
M.S. Hayden, S. Ghosh, Signaling to NF-$\kappa$B. Genes Dev. 18(18), 2195–2224 (2004)
A. Bierhaus, S. Schiekofer, M. Schwaninger, M. Andrassy, P.M. Humpert, J. Chen et al., Diabetes-associated sustained activation of the transcription factor nuclear factor-$\kappa$B. Diabetes 50(12), 2792–2808 (2001)
Y. Hasegawa, T. Saito, T. Ogihara, Y. Ishigaki, T. Yamada, J. Imai et al., Blockade of the nuclear factor-$\kappa$B pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125(9), 1122–1133 (2012)
G.L. Pierce, L.A. Lesniewski, B.R. Lawson, S.D. Beske, D.R. Seals, Nuclear factor-$\kappa$B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 119(9), 1284–1292 (2009)
G. Guan, H. Han, Y. Yang, Y. Jin, X. Wang, X. Liu, Neferine prevented hyperglycemia-induced endothelial cell apoptosis through suppressing ROS/Akt/NF-$\kappa$B signal. Endocrine 47(3), 764–771 (2014)
S. Brouard, P.O. Berberat, E. Tobiasch, M.P. Seldon, F.H. Bach, M.P. Soares, Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-$\kappa$B to protect endothelial cells from tumor necrosis factor-$\alpha$-mediated apoptosis. J. Biol. Chem. 277(20), 17950–17961 (2002)
T. Kisseleva, L. Song, M. Vorontchikhina, N. Feirt, J. Kitajewski, C. Schindler, NF-$\kappa$B regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Investig. 116(11), 2955 (2006)
A. Pfosser, C. El-Aouni, I. Pfisterer, M. Dietz, F. Globisch, G. Stachel et al., NF $\kappa$B activation in embryonic endothelial progenitor cells enhances neovascularization via PSGL-1 mediated recruitment: novel role for LL37. Stem Cells 28(2), 376–385 (2010)
K. Ji, C. Xing, F. Jiang, X. Wang, H. Guo, J. Nan et al., Benzo [a] pyrene induces oxidative stress and endothelial progenitor cell dysfunction via the activation of the NF-$\kappa$B pathway. Int. J. Mol. Med. 31(4), 922–930 (2013)
S. Parthasarathy, A. Raghavamenon, M.O. Garelnabi, N. Santanam, Oxidized low-density lipoprotein. Free Radic. Antioxid. Protoc. 610, 403–417 (2010)
M.T. Marin, P.S. Dasari, J.B. Tryggestad, C.E. Aston, A.M. Teague, K.R. Short, Oxidized HDL and LDL in adolescents with type 2 diabetes compared to normal weight and obese peers. J. Diabetes Complic. 29(5), 679–685 (2015)
G. Maiolino, G. Rossitto, P. Caielli, V. Bisogni, G.P. Rossi, L.A. Calò, The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediat. Inflamm. (2013). doi:10.1155/2013/714653
J. Galle, R. Schneider, B. Winner, C. Lehmann-Bodem, R. Schinzel, G. Münch et al., Glyc-oxidized LDL impair endothelial function more potently than oxidized LDL: role of enhanced oxidative stress. Atherosclerosis 138(1), 65–77 (1998)
Y. Shi, F. Cosentino, G.G. Camici, A. Akhmedov, P.M. Vanhoutte, F.C. Tanner et al., Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-$\beta$, and c-Jun N-terminal kinase in human endothelial cells. Arterioscler. Thrombos. Vasc. Biol. 31(9), 2090–2097 (2011)
Y. Shi, T.F. Lüscher, G.G. Camici, Dual role of endothelial nitric oxide synthase in oxidized LDL-induced, p66Shc-mediated oxidative stress in cultured human endothelial cells. PLoS ONE 9(9), e107787 (2014). doi:10.1371/journal.pone.0107787
K. Yamamoto, A. Kakino, H. Takeshita, N. Hayashi, L. Li, A. Nakano et al. Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin-like oxLDL receptor. FASEB J. fj–15 (2015). doi:10.1096/fj.15-271627
C.-S. Chu, Y.-C. Wang, L.-S. Lu, B. Walton, H.R. Yilmaz, R.Y. Huang et al., Electronegative low-density lipoprotein increases C-reactive protein expression in vascular endothelial cells through the LOX-1 receptor. PLoS ONE 8(8), e70533 (2013). doi:10.1371/journal.pone.0070533
S. Hamed, B. Brenner, Z. Abassi, A. Aharon, D. Daoud, A. Roguin, Hyperglycemia and oxidized-LDL exert a deleterious effect on endothelial progenitor cell migration in type 2 diabetes mellitus. Thromb. Res. 126(3), 166–174 (2010)
T. Imanishi, T. Hano, T. Sawamura, I. Nishio, Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin. Exp. Pharmacol. Physiol. 31(7), 407–413 (2004)
K.-T. Ji, L. Qian, J.-L. Nan, Y.-J. Xue, S.-Q. Zhang, G.-Q. Wang et al., Ox-LDL induces dysfunction of endothelial progenitor cells via activation of NF-κB. Biomed. Res. Int. (2015). doi:10.1155/2015/175291
F.X. Ma, B. Zhou, Z. Chen, Q. Ren, S.H. Lu, T. Sawamura et al., Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 47(6), 1227–1237 (2006)
B. Shao, J.W. Heinecke, Impact of HDL oxidation by the myeloperoxidase system on sterol efflux by the ABCA1 pathway. J. Proteomics 74(11), 2289–2299 (2011)
B. Pan, Y. Ma, H. Ren, Y. He, Y. Wang, X. Lv et al., Diabetic HDL is dysfunctional in stimulating endothelial cell migration and proliferation due to down regulation of SR-BI expression. PLoS ONE 7(11), e48530 (2012). doi:10.1371/journal.pone.0048530
A. Undurti, Y. Huang, J.A. Lupica, J.D. Smith, J.A. DiDonato, S.L. Hazen, Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 284(45), 30825–30835 (2009)
T. Matsunaga, S. Hokari, I. Koyama, T. Harada, T. Komoda, NF-$\kappa$B activation in endothelial cells treated with oxidized high-density lipoprotein. Biochem. Biophys. Res. Commun. 303(1), 313–319 (2003)
O. Van Oostrom, M. Nieuwdorp, P. Westerweel, I. Hoefer, R. Basser, E. Stroes et al., Reconstituted HDL increases circulating endothelial progenitor cells in patients with type 2 diabetes. Arterioscler. Thrombos. Vasc. Biol. 27(8), 1864–1865 (2007)
V. Petoumenos, G. Nickenig, N. Werner, High-density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J. Cell Mol. Med. 13(11–12), 4623–4635 (2009)
C.-Y. Huang, F.-Y. Lin, C.-M. Shih, H.-K. Au, Y.-J. Chang, H. Nakagami et al., Moderate to high concentrations of high-density lipoprotein from healthy subjects paradoxically impair human endothelial progenitor cells and related angiogenesis by activating Rho-associated kinase pathways. Arterioscler. Thrombos. Vasc. Biol. 32(10), 2405–2417 (2012)
J. Wu, Z. He, X. Gao, F. Wu, R. Ding, Y. Ren et al., Oxidized high-density lipoprotein impairs endothelial progenitor cells’ function by activation of CD36-MAPK-TSP-1 pathways. Antioxid. Redox Signal. 22(4), 308–324 (2015). doi:10.1089/ars.2013.5743
C. Schmitz-Peiffer, Targeting ceramide synthesis to reverse insulin resistance. Diabetes 59(10), 2351–2353 (2010)
Q.-J. Zhang, W.L. Holland, L. Wilson, J.M. Tanner, D. Kearns, J.M. Cahoon et al., Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 61(7), 1848–1859 (2012)
V.C. Mehra, E. Jackson, X.M. Zhang, X.-C. Jiang, L.W. Dobrucki, J. Yu et al., Ceramide-activated phosphatase mediates fatty acid-induced endothelial VEGF resistance and impaired angiogenesis. Am. J. Pathol. 184(5), 1562–1576 (2014)
L. Chun, Z. Junlin, W. Aimin, L. Niansheng, C. Benmei, L. Minxiang, Inhibition of ceramide synthesis reverses endothelial dysfunction and atherosclerosis in streptozotocin-induced diabetic rats. Diabetes Res. Clin. Pract. 93(1), 77–85 (2011)
D.X. Zhang, A.-P. Zou, P.-L. Li, Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 284(2), H605–H612 (2003)
M. Fu, Z. Li, T. Tan, W. Guo, N. Xie, Q. Liu et al., Akt/eNOS signaling pathway mediates inhibition of endothelial progenitor cells by palmitate-induced ceramide. Am. J. Physiol. Heart Circ. Physiol. 308(1), H11–H17 (2015)
P. Zhu, G. Chen, T. You, J. Yao, Q. Jiang, X. Lin et al., High FFA-induced proliferation and apoptosis in human umbilical vein endothelial cell partly through Wnt/$\beta$-catenin signal pathway. Mol. Cell. Biochem. 338(1–2), 123–131 (2010)
X.L. Wang, L. Zhang, K. Youker, M.-X. Zhang, J. Wang, S.A. LeMaire et al., Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 55(8), 2301–2310 (2006)
W.-Y. Zhang, E. Schwartz, Y. Wang, J. Attrep, Z. Li, P. Reaven, Elevated concentrations of nonesterified fatty acids increase monocyte expression of CD11b and adhesion to endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26(3), 514–519 (2006)
M. Artwohl, A. Lindenmair, V. Sexl, C. Maier, G. Rainer, A. Freudenthaler et al., Different mechanisms of saturated versus polyunsaturated FFA-induced apoptosis in human endothelial cells. J. Lipid Res. 49(12), 2627–2640 (2008)
K. Staiger, H. Staiger, C. Weigert, C. Haas, H.-U. Häring, M. Kellerer, Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-$\kappa$B activation. Diabetes 55(11), 3121–3126 (2006)
Y. Lu, L. Qian, Q. Zhang, B. Chen, L. Gui, D. Huang et al., Palmitate induces apoptosis in mouse aortic endothelial cells and endothelial dysfunction in mice fed high-calorie and high-cholesterol diets. Lifesciences 92(24), 1165–1173 (2013)
Z. Gao, H. Zhang, J. Liu, C.W. Lau, P. Liu, Z.Y. Chen et al., Cyclooxygenase-2-dependent oxidative stress mediates palmitate-induced impairment of endothelium-dependent relaxations in mouse arteries. Biochem. Pharmacol. 91(4), 474–482 (2014)
Q. Wang, X.-L. Cheng, D.-Y. Zhang, X.-J. Gao, L. Zhou, X.-Y. Qin et al., Tectorigenin attenuates palmitate-induced endothelial insulin resistance via targeting ROS-associated inflammation and IRS-1 pathway. PLoS ONE 8(6), e66417 (2013). doi:10.1371/journal.pone.0066417
A. Trombetta, G. Togliatto, A. Rosso, P. Dentelli, C. Olgasi, P. Cotogni et al., Increase of palmitic acid concentration impairs endothelial progenitor cell and bone marrow-derived progenitor cell bioavailability role of the STAT5/PPAR$\gamma$ transcriptional complex. Diabetes 62(4), 1245–1257 (2013)
W.-X. Guo, Q.-D. Yang, Y.-H. Liu, X.-Y. Xie, M. Wang, R.-C. Niu, Palmitic and linoleic acids impair endothelial progenitor cells by inhibition of Akt/eNOS pathway. Arch. Med. Res. 39(4), 434–442 (2008)
X. Gao, H. Zhang, A.M. Schmidt, C. Zhang, AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 295(2), H491–H498 (2008)
M.-P. Wautier, O. Chappey, S. Corda, D.M. Stern, A.M. Schmidt, J.-L. Wautier, Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 280(5), E685–E694 (2001)
K.-C. Lan, C.-Y. Chiu, C.-W. Kao, K.-H. Huang, C.-C. Wang, K.-T. Huang et al., Advanced glycation end-products induce apoptosis in pancreatic islet endothelial cells via NF-$\kappa$B-activated cyclooxygenase-2/prostaglandin E 2 up-regulation. PLoS ONE 8(6), e66781 (2013). doi:10.1371/journal.pone.0066781
S.-J. Yoon, Y.W. Yoon, B.K. Lee, H.M. Kwon, K.-C. Hwang, M. Kim et al., Potential role of HMG CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp. Mol. Med. 41(11), 802–811 (2009)
Y. Ishibashi, T. Matsui, S. Maeda, Y. Higashimoto, S. Yamagishi, Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc Diabetol. 12, 125 (2013)
G. Togliatto, A. Trombetta, P. Dentelli, A. Rosso, M. Brizzi, MIR221/MIR222-driven post-transcriptional regulation of P27KIP1 and P57KIP2 is crucial for high-glucose-and AGE-mediated vascular cell damage. Diabetologia 54(7), 1930–1940 (2011)
C. Shen, Q. Li, Y. Zhang, G. Ma, Y. Feng, Q. Zhu et al., Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed. Pharmacother. 64(1), 35–43 (2010)
J. Chen, M. Song, S. Yu, P. Gao, Y. Yu, H. Wang et al., Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol. Cell. Biochem. 335(1–2), 137–146 (2010)
Q. Chen, L. Dong, L. Wang, L. Kang, B. Xu, Advanced glycation end products impair function of late endothelial progenitor cells through effects on protein kinase Akt and cyclooxygenase-2. Biochem. Biophys. Res. Commun. 381(2), 192–197 (2009)
S. Ueda, S. Yamagishi, T. Matsui, Y. Noda, S. Ueda, Y. Jinnouchi et al., Serum levels of advanced glycation end products (AGEs) are inversely associated with the number and migratory activity of circulating endothelial progenitor cells in apparently healthy subjects. Cardiovasc. Ther. 30(4), 249–254 (2012)
H. Li, X. Zhang, X. Guan, X. Cui, Y. Wang, H. Chu et al., Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc. Diabetol. 11(1), 46 (2012)
A.D. Bhatwadekar, J.V. Glenn, G. Li, T.M. Curtis, T.A. Gardiner, A.W. Stitt, Advanced glycation of fibronectin impairs vascular repair by endothelial progenitor cells: implications for vasodegeneration in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 49(3), 1232–1241 (2008)
S.F. Travis, A.D. Morrison, R.S. Clements, A.I. Winegrad, F.A. Oski, The sorbitol pathway of the human erythrocyte. Pediatr. Res. 4(5), 466 (1970)
S.S. Chung, E.C. Ho, K.S. Lam, S.K. Chung, Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 14(suppl 3), S233–S236 (2003)
H.M. Cheng, R.G. González, The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metab., Clin. Exp. 35((4 Suppl 1)), 10–14 (1986)
Y.C. Hwang, S. Bakr, C.A. Ellery, P.J. Oates, R. Ramasamy, Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. FASEB J. 17(15), 2331–2333 (2003)
J.R. Williamson, K. Chang, M. Frangos, K.S. Hasan, Y. Ido, T. Kawamura et al., Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42(6), 801–813 (1993)
T.J. Guzik, S. Mussa, D. Gastaldi, J. Sadowski, C. Ratnatunga, R. Pillai et al., Mechanisms of increased vascular superoxide production in human diabetes mellitus role of NAD (P) H oxidase and endothelial nitric oxide synthase. Circulation 105(14), 1656–1662 (2002)
M. Takeuchi, M. Iwaki, J. Takino, H. Shirai, M. Kawakami, R. Bucala et al., Immunological detection of fructose-derived advanced glycation end-products. Lab. Invest. 90(7), 1117–1127 (2010)
S. Lightman, Does aldose reductase have a role in the development of the ocular complications of diabetes? Eye 7(2), 238–241 (1993)
S. Yagihashi, S.I. Yamagishi, R. Wada Ri, M. Baba, T.C. Hohman, C. Yabe-Nishimura et al., Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain 124((Pt 12)), 2448–2458 (2001)
A.M. Gonzalez, M. Sochor, P. McLean, The effect of an aldose reductase inhibitor (sorbinil) on the level of metabolites in lenses of diabetic rats. Diabetes 32(5), 482–485 (1983)
M. Mira, M. Azevedo, C. Manso, The neutralization of hydroxyl radical by silibin, sorbinil and bendazac. Free Radic. Res. 4(2), 125–129 (1987)
X. Du, T. Matsumura, D. Edelstein, L. Rossetti, Z. Zsengellér, C. Szabó et al., Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 112(7), 1049 (2003)
P.J. Thornalley, The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 269(1), 1 (1990)
D.J. Ceradini, D. Yao, R.H. Grogan, M.J. Callaghan, D. Edelstein, M. Brownlee et al., Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J. Biol. Chem. 283(16), 10930–10938 (2008)
H. Thangarajah, D. Yao, E.I. Chang, Y. Shi, L. Jazayeri, I.N. Vial et al., The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc. Natl. Acad. Sci. U.S.A. 106(32), 13505–13510 (2009)
T.-S. Lee, L.C. MacGregor, S.J. Fluharty, G.L. King et al., Differential regulation of protein kinase C and (Na, K)-adenosine triphosphatase activities by elevated glucose levels in retinal capillary endothelial cells. J Clin. Investig. 83(1), 90–94 (1989)
T.-S. Lee, K.A. Saltsman, H. Ohashi, G.L. King, Activation of protein kinase C by elevation of glucose concentration: proposal for a mechanism in the development of diabetic vascular complications. Proc. Natl. Acad. Sci. 86(13), 5141–5145 (1989)
T. Inoguchi, R. Battan, E. Handler, J.R. Sportsman, W. Heath, G.L. King, Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc. Natl. Acad. Sci. 89(22), 11059–11063 (1992)
T. Inoguchi, F. Ueda, F. Umeda, T. Yamashita, H. Nawata, Inhibition of intercellular communication via gap junction in cultured aortic endothelial cells by elevated glucose and phorbol ester. Biochem. Biophys. Res. Commun. 208(2), 492–497 (1995)
J.J. Lynch, T.J. Ferro, F.A. Blumenstock, A.M. Brockenauer, A.B. Malik, Increased endothelial albumin permeability mediated by protein kinase C activation. J Clin. Invest. 85(6), 1991–1998 (1990)
N.S. Harhaj, E.A. Felinski, E.B. Wolpert, J.M. Sundstrom, T.W. Gardner, D.A. Antonetti, VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest. Ophthalmol. Vis. Sci. 47(11), 5106–5115 (2006)
F. Cosentino, M. Eto, P. De Paolis, B. van der Loo, M. Bachschmid, V. Ullrich et al., High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells role of protein kinase C and reactive oxygen species. Circulation 107(7), 1017–1023 (2003)
L. Quagliaro, L. Piconi, R. Assaloni, L. Martinelli, E. Motz, A. Ceriello, Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells the role of protein kinase C and NAD (P) H-oxidase activation. Diabetes 52(11), 2795–2804 (2003)
P. Geraldes, J. Hiraoka-Yamamoto, M. Matsumoto, A. Clermont, M. Leitges, A. Marette et al., Activation of PKC-$\delta$ and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 15(11), 1298–1306 (2009)
F. Cosentino, P. Francia, G.G. Camici, P.G. Pelicci, M. Volpe, T.F. Lüscher, Final common molecular pathways of aging and cardiovascular disease role of the p66Shc protein. Arterioscler. Thrombos. Vasc. Biol. 28(4), 622–628 (2008)
G.M. Pieper et al., Activation of nuclear factor-$\kappa$B in cultured endothelial cells by increased glucose concentration: prevention by calphostin C. J. Cardiovasc. Pharmacol. 30(4), 528–532 (1997)
Y. Hou, Y. Wu, S.M. Farooq, X. Guan, S. Wang, Y. Liu et al., A critical role of CXCR2 PDZ-mediated interactions in endothelial progenitor cell homing and angiogenesis. Stem Cell Res. 14(2), 133–143 (2014)
M.G. Buse, Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab. 290(1), E1–E8 (2006)
C. Weigert, K. Brodbeck, M. Sawadogo, H.U. Häring, E.D. Schleicher, Upstream stimulatory factor (USF) proteins induce human TGF-$\beta$1 gene activation via the glucose-response element-1013/-1002 in mesangial cells up-regulation of USF activity by the hexosamine biosynthetic pathway. J. Biol. Chem. 279(16), 15908–15915 (2004)
X.-L. Du, D. Edelstein, L. Rossetti, I.G. Fantus, H. Goldberg, F. Ziyadeh et al., Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. 97(22), 12222–12226 (2000)
M. Horal, Z. Zhang, R. Stanton, A. Virkamäki, M.R. Loeken, Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth Defects Res. A 70(8), 519–527 (2004)
B. Musicki, M.F. Kramer, R.E. Becker, A.L. Burnett, Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. U.S.A. 102(33), 11870–11875 (2005)
B.L. Trumpower, The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 265(20), 11409–11412 (1990)
S.S. Korshunov, V.P. Skulachev, A.A. Starkov, High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416(1), 15–18 (1997)
M. Brownlee, The pathobiology of diabetic complications a unifying mechanism. Diabetes 54(6), 1615–1625 (2005)
X.L. Du, D. Edelstein, S. Dimmeler, Q. Ju, C. Sui, M. Brownlee, Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 108(9), 1341 (2001)
F. Giacco, M. Brownlee, Oxidative stress and diabetic complications. Circ. Res. 107(9), 1058–1070 (2010)
M.-C. Desco, M. Asensi, R. Márquez, J. Martínez-Valls, M. Vento, F.V. Pallardó et al., Xanthine oxidase is involved in free radical production in type 1 diabetes protection by allopurinol. Diabetes 51(4), 1118–1124 (2002)
K. Guo, J. Lu, Y. Huang, M. Wu, L. Zhang, H. Yu et al., Protective role of PGC-1$\alpha$ in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS ONE 10(4), e0125176 (2015). doi:10.1371/journal.pone.0125176
E.J. Marrotte, D.-D. Chen, J.S. Hakim, A.F. Chen, Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J. Clin. Investig. 120(12), 4207 (2010)
X.-R. Wang, M.-W. Zhang, D.-D. Chen, Y. Zhang, A.F. Chen, AMP-activated protein kinase rescues the angiogenic functions of endothelial progenitor cells via manganese superoxide dismutase induction in type 1 diabetes. Am. J. Physiol.-Endocrinol. Metab. 300(6), E1135–E1145 (2011)
V. Di Stefano, C. Cencioni, G. Zaccagnini, A. Magenta, M.C. Capogrossi, F. Martelli, p66ShcA modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc. Res. (2009). doi:10.1093/cvr/cvp082
B. Antuna-Puente, B. Feve, S. Fellahi, J.-P. Bastard, Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 34(1), 2–11 (2008)
K. Brochu-Gaudreau, C. Rehfeldt, R. Blouin, V. Bordignon, B.D. Murphy, M.-F. Palin, Adiponectin action from head to toe. Endocrine 37(1), 11–32 (2010)
M.I. Saad, M.A. Kamel, M.Y. Hanafi, Modulation of adipocytokines production and serum NEFA level by metformin, glimepiride, and sitagliptin in HFD/STZ diabetic rats. Biochem. Res. Int. (2015). doi:10.1155/2015/138134
S.H. Tawfik, B.F. Mahmoud, M.I. Saad, M. Shehata, M.A. Kamel, M.H. Helmy, Similar and additive effects of ovariectomy and diabetes on insulin resistance and lipid metabolism. Biochem. Res. Int. (2015). doi:10.1155/2015/567945
S.R. Kashyap, L.J. Roman, L. Mandarino, R. DeFronzo, M. Bajaj, Hypoadiponectinemia is closely associated with impaired nitric oxide synthase activity in skeletal muscle of type 2 diabetic subjects. Metab. Syndr. Rel. Disord. 8(5), 459–463 (2010)
M. Ebrahimi-Mamaeghani, S. Mohammadi, S.R. Arefhosseini, P. Fallah, Z. Bazi, Adiponectin as a potential biomarker of vascular disease. Vasc. Health Risk Manag. 11, 55–70 (2015). doi:10.2147/VHRM.S48753
K. Tan, A. Xu, W. Chow, M. Lam, V. Ai, S. Tam et al., Hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation. J. Clin. Endocrinol. Metab. 89(2), 765–769 (2004)
S. Gustafsson, L. Lind, S. Söderberg, M. Zilmer, J. Hulthe, E. Ingelsson, Oxidative stress and inflammatory markers in relation to circulating levels of adiponectin. Obesity 21(7), 1467–1473 (2013)
X. Xiao, Y. Dong, J. Zhong, R. Cao, X. Zhao, G. Wen et al., Adiponectin protects endothelial cells from the damages induced by the intermittent high level of glucose. Endocrine 40(3), 386–393 (2011)
A.S. Antonopoulos, M. Margaritis, P. Coutinho, C. Shirodaria, C. Psarros, L. Herdman et al., Adiponectin as a link between type 2 diabetes mellitus and vascular NADPH-oxidase activity in the human arterial wall: the regulatory role of perivascular adipose tissue. Diabetes 64, 2207–2219 (2015). doi:10.2337/db14-1011
M. Margaritis, A.S. Antonopoulos, J. Digby, R. Lee, S. Reilly, P. Coutinho et al., Interactions between vascular wall and perivascular adipose tissue reveal novel roles for adiponectin in the regulation of eNOS function in human vessels. Circulation 127, 2209–2221 (2013). doi:10.1161/CIRCULATIONAHA.112.001133
X. Wang, H. Pu, C. Ma, T. Jiang, Q. Wei, M. Duan et al., Adiponectin abates atherosclerosis by reducing oxidative stress. Med. Sci. Monit.: Int. Med. J. Exp. Clin. Res. 20, 1792–1800 (2014). doi:10.12659/MSM.892299
X. Cai, X. Li, L. Li, X.-Z. Huang, Y.-S. Liu, L. Chen et al., Adiponectin reduces carotid atherosclerotic plaque formation in ApoE-/-mice: roles of oxidative and nitrosative stress and inducible nitric oxide synthase. Mol. Med. Rep. 11(3), 1715–1721 (2015)
J. Chang, Y. Li, Y. Huang, K.S. Lam, R.L. Hoo, W.T. Wong et al., Adiponectin prevents diabetic premature senescence of endothelial progenitor cells and promotes endothelial repair by suppressing the p38 MAP kinase/p16INK4A signaling pathway. Diabetes 59(11), 2949–2959 (2010)
R. Ouedraogo, X. Wu, S.-Q. Xu, L. Fuchsel, H. Motoshima, K. Mahadev et al., Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells evidence for involvement of a cAMP signaling pathway. Diabetes 55(6), 1840–1846 (2006)
Z. Zhi, Z. Pengfei, T. Xiaoyi, M. Genshan, Adiponectin ameliorates angiotensin II-induced vascular endothelial damage. Cell Stress Chaperones 19(5), 705–713 (2014)
H. Motoshima, X. Wu, K. Mahadev, B.J. Goldstein, Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem. Biophys. Res. Commun. 315(2), 264–271 (2004)
S. Plant, B. Shand, P. Elder, R. Scott, Adiponectin attenuates endothelial dysfunction induced by oxidised low-density lipoproteins. Diabetes Vasc. Dis. Res. 5(2), 102–108 (2008)
C.-F. Chen, J. Huang, H. Li, C. Zhang, X. Huang, G. Tong et al., MicroRNA-221 regulates endothelial nitric oxide production and inflammatory response by targeting adiponectin receptor 1. Gene 565(2), 246–251 (2015). doi:10.1016/j.gene.2015.04.014
A. Xu, P.M. Vanhoutte, Adiponectin and adipocyte fatty acid binding protein in the pathogenesis of cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 302(6), H1231–H1240 (2012)
V. Lavoie, A.-E. Kernaleguen, G. Charron, N. Farhat, M. Cossette, A.M. Mamarbachi et al., Functional effects of adiponectin on endothelial progenitor cells. Obesity 19(4), 722–728 (2011)
S.F. Leicht, T.M. Schwarz, P.C. Hermann, J. Seissler, A. Aicher, C. Heeschen, Adiponectin pretreatment counteracts the detrimental effect of a diabetic environment on endothelial progenitors. Diabetes 60(2), 652–661 (2011)
P. Eren, S. Camus, G. Matrone, T.G. Ebrahimian, D. François, A. Tedgui et al., Adiponectinemia controls pro-angiogenic cell therapy. Stem Cells 27(11), 2712–2721 (2009)
Y. Matsuo, T. Imanishi, A. Kuroi, H. Kitabata, T. Kubo, Y. Hayashi et al., Effects of plasma adiponectin levels on the number and function of endothelial progenitor cells in patients with coronary artery disease. Circ. J. 71(9), 1376–1382 (2007)
X. Zhang, Z. Huang, Y. Xie, X. Chen, J. Zhang, Z. Qiu et al., Lower levels of plasma adiponectin and endothelial progenitor cells are associated with large artery atherosclerotic stroke. Int. J. Neurosci. 0, 1–23 (2014). doi:10.3109/00207454.2014.994624
M. Li, J.C. Ho, K.W. Lai, K.K. Au, A. Xu, B.M. Cheung et al., The decrement in circulating endothelial progenitor cells (EPCs) in type 2 diabetes is independent of the severity of the hypoadiponectemia. Diabetes/Metab. Res. Rev. 27(2), 185–194 (2011)
C.-Y. Chen, M. Asakura, H. Asanuma, T. Hasegawa, J. Tanaka, N. Toh et al., Plasma adiponectin levels predict cardiovascular events in the observational Arita Cohort Study in Japan: the importance of the plasma adiponectin levels. Hypertens. Res. 35(8), 843–848 (2012)
W.T. Wong, X.Y. Tian, A. Xu, J. Yu, C.W. Lau, R.L.C. Hoo et al., Adiponectin is required for PPARγ-mediated improvement of endothelial function in diabetic mice. Cell Metab. 14(1), 104–115 (2011)
M.I. Yilmaz, A. Sonmez, K. Caglar, D.E. Gok, T. Eyileten, M. Yenicesu et al., Peroxisome proliferator-activated receptor $\gamma$ (PPAR-$\gamma$) agonist increases plasma adiponectin levels in type 2 diabetic patients with proteinuria. Endocrine 25(3), 207–214 (2004)
A.C. Könner, J.C. Brüning, Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 16(2), 144–152 (2012)
S. Guzel, A. Seven, A. Kocaoglu, B. Ilk, E.C. Guzel, G.V. Saracoglu et al., Osteoprotegerin, leptin and IL-6: Association with silent myocardial ischemia in type 2 diabetes mellitus. Diabetes Vasc. Dis. Res. 10(1), 25–31 (2013)
S.S. Martin, M.J. Blaha, E.D. Muse, A.N. Qasim, M.P. Reilly, R.S. Blumenthal et al., Leptin and incident cardiovascular disease: the multi-ethnic study of atherosclerosis (MESA). Atherosclerosis 239(1), 67–72 (2015)
T. Morioka, M. Emoto, Y. Yamazaki, N. Kawano, S. Imamura, R. Numaguchi et al., Leptin is associated with vascular endothelial function in overweight patients with type 2 diabetes. Cardiovasc Diabetol. 13, 10 (2014)
T. Kazumi, A. Tsuboi, K. Fukuo, Inverse association of serum leptin with flow-mediated dilatation independent of body fat distribution, insulin resistance, oxidative stress and inflammation in young women. J. Endocrinol. Diabetes Mellit. 2(1), 21–25 (2014)
L.M. Biasucci, F. Graziani, V. Rizzello, G. Liuzzo, C. Guidone, A.R. De Caterina et al., Paradoxical preservation of vascular function in severe obesity. Am. J. Med. 123(8), 727–734 (2010)
S. Benkhoff, A.E. Loot, I. Pierson, A. Sturza, K. Kohlstedt, I. Fleming et al., Leptin potentiates endothelium-dependent relaxation by inducing endothelial expression of neuronal NO synthase. Arterioscler. Thromb. Vasc. Biol. 32(7), 1605–1612 (2012)
A. Jamroz-Wi’sniewska, A. Gertler, G. Solomon, M.E. Wood, M. Whiteman, J. Beltowski, Leptin-induced endothelium-dependent vasorelaxation of peripheral arteries in lean and obese rats: role of nitric oxide and hydrogen sulfide. PLoS ONE 9(1), e86744 (2014). doi:10.1371/journal.pone.0086744
M. Korda, R. Kubant, S. Patton, T. Malinski, Leptin-induced endothelial dysfunction in obesity. Am. J. Physiol. Heart Circ. Physiol. 295(4), H1514–H1521 (2008)
J.D. Knudson, U.D. Dincer, C. Zhang, A.N. Swafford, R. Koshida, A. Picchi et al., Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 289(1), H48–H56 (2005)
D. Konukoglu, O. Serin, M.S. Turhan, Plasma leptin and its relationship with lipid peroxidation and nitric oxide in obese female patients with or without hypertension. Arch. Med. Res. 37(5), 602–606 (2006)
G. Pandey, M.S. Shihabudeen, H.P. David, E. Thirumurugan, K. Thirumurugan, Association between hyperleptinemia and oxidative stress in obese diabetic subjects. J. Diabetes Metab. Disord. 14(1), 24 (2015)
J. Wang, H. Wang, W. Luo, C. Guo, J. Wang, Y.E. Chen et al., Leptin-induced endothelial dysfunction is mediated by sympathetic nervous system activity. J Am Heart Assoc. 2(5), e000299 (2013). doi:10.1161/JAHA.113.000299
F. Machleidt, P. Simon, A.F. Krapalis, M. Hallschmid, H. Lehnert, F. Sayk, Experimental hyperleptinemia acutely increases vasoconstrictory sympathetic nerve activity in healthy humans. J. Clin. Endocrinol. Metab. 98(3), E491–E496 (2013)
S. Leon-Cabrera, L. Solís-Lozano, K. Suárez-Álvarez, A. González-Chávez, Y.L. Béjar, G. Robles-Díaz et al., Hyperleptinemia is associated with parameters of low-grade systemic inflammation and metabolic dysfunction in obese human beings. Front. Integr. Neurosci. 7, 62 (2013). doi:10.3389/fnint.2013.00062
C.J. Hukshorn, J.H.N. Lindeman, K.H. Toet, W.H.M. Saris, P.H.C. Eilers, M.S. Westerterp-Plantenga et al., Leptin and the proinflammatory state associated with human obesity. J. Clin. Endocrinol. Metab. 89(4), 1773–1778 (2004)
D. Spruijt-Metz, B. Adar Emken, M.R. Spruijt, J.M. Richey, L.J. Berman, B.R. Belcher et al., CRP is related to higher leptin levels in minority peripubertal females regardless of adiposity levels. Obesity 20(3), 512–516 (2012)
P. Jitprasertwong, K.M. Jaedicke, C.J. Nile, P.M. Preshaw, J.J. Taylor, Leptin enhances the secretion of interleukin (IL)-18, but not IL-1β, from human monocytes via activation of caspase-1. Cytokine 65(2), 222–230 (2014)
M.R. Indra, S. Karyono, R. Ratnawati, S.G. Malik, Quercetin suppresses inflammation by reducing ERK1/2 phosphorylation and NF kappa B activation in leptin-induced human umbilical vein endothelial cells (HUVECs). BMC Res. Notes 6(1), 275 (2013)
R. Wolk, A. Deb, N.M. Caplice, V.K. Somers, Leptin receptor and functional effects of leptin in human endothelial progenitor cells. Atherosclerosis 183(1), 131–139 (2005)
M.R. Schroeter, M. Leifheit, P. Sudholt, N.-M. Heida, C. Dellas, I. Rohm et al., Leptin enhances the recruitment of endothelial progenitor cells into neointimal lesions after vascular injury by promoting integrin-mediated adhesion. Circ. Res. 103(5), 536–544 (2008)
M.R. Schroeter, S. Stein, N.-M. Heida, M. Leifheit-Nestler, I.-F. Cheng, R. Gogiraju et al., Leptin promotes the mobilization of vascular progenitor cells and neovascularization by NOX2-mediated activation of MMP9. Cardiovasc. Res. (2011). doi:10.1093/cvr/cvr275
K.M. Utzschneider, D.B. Carr, J. Tong, T.M. Wallace, R.L. Hull, S. Zraika et al., Resistin is not associated with insulin sensitivity or the metabolic syndrome in humans. Diabetologia 48(11), 2330–2333 (2005)
G. Hasegawa, M. Ohta, Y. Ichida, H. Obayashi, M. Shigeta, M. Yamasaki et al., Increased serum resistin levels in patients with type 2 diabetes are not linked with markers of insulin resistance and adiposity. Acta Diabetol. 42(2), 104–109 (2005)
G. Hoefle, C.H. Saely, L. Risch, L. Koch, F. Schmid, P. Rein et al., Relationship between the adipose-tissue hormone resistin and coronary artery disease. Clin. Chim. Acta 386(1), 1–6 (2007)
H. Ye, H.J. Zhang, A. Xu, R.L.C. Hoo, Resistin production from adipose tissue is decreased in db/db obese mice, and is reversed by rosiglitazone. PLoS ONE 8(6), e65543 (2013). doi:10.1371/journal.pone.0065543
H. Raff, E.D. Bruder, Adiponectin and resistin in the neonatal rat. Endocrine 29(2), 341–344 (2006)
Z. Luo, Y. Zhang, F. Li, J. He, H. Ding, L. Yan et al., Resistin induces insulin resistance by both AMPK-dependent and AMPK-independent mechanisms in HepG2 cells. Endocrine 36(1), 60–69 (2009)
B.H. Chen, Y. Song, E.L. Ding, C.K. Roberts, J.E. Manson, N. Rifai et al., Circulating levels of resistin and risk of type 2 diabetes in men and women: results from two prospective cohorts. Diabetes Care 32(2), 329–334 (2009)
Y. Takata, H. Osawa, M. Kurata, M. Kurokawa, J. Yamauchi, M. Ochi et al., Hyperresistinemia is associated with coexistence of hypertension and type 2 diabetes. Hypertension 51(2), 534–539 (2008)
C. Menzaghi, S. Bacci, L. Salvemini, C. Mendonca, G. Palladino, A. Fontana et al., Serum resistin, cardiovascular disease and all-cause mortality in patients with type 2 diabetes. PLoS ONE 8(6), e64729 (2013). doi:10.1371/journal.pone.0064729
E.D. Muse, D.I. Feldman, M.J. Blaha, Z.A. Dardari, R.S. Blumenthal, M.J. Budoff et al., The association of resistin with cardiovascular disease in the multi-ethnic study of atherosclerosis. Atherosclerosis. 239(1), 101–108 (2015)
G. Lupattelli, S. Marchesi, T. Ronti, R. Lombardini, S. Bruscoli, R. Bianchini et al., Endothelial dysfunction in vivo is related to monocyte resistin mRNA expression. J. Clin. Pharm. Ther. 32(4), 373–379 (2007)
G.M. Dick, P.S. Katz, M. Farias, M. Morris, J. James, J.D. Knudson et al., Resistin impairs endothelium-dependent dilation to bradykinin, but not acetylcholine, in the coronary circulation. Am. J. Physiol. Heart Circ. Physiol. 291(6), H2997–H3002 (2006)
M.T. Gentile, C. Vecchione, G. Marino, A. Aretini, A. Di Pardo, G. Antenucci et al., Resistin impairs insulin-evoked vasodilation. Diabetes 57(3), 577–583 (2008)
C. Chen, J. Jiang, J.-M. Lü, H. Chai, X. Wang, P.H. Lin et al., Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 299(1), H193–H201 (2010)
M.S. Jamaluddin, S. Yan, J. Lü, Z. Liang, Q. Yao, C. Chen, Resistin increases monolayer permeability of human coronary artery endothelial cells. PLoS ONE 8(12), e84576 (2013). doi:10.1371/journal.pone.0084576
Y. Li, Y. Wang, Q. Li, Y. Chen, S. Sun, W. Zhang et al., Effect of resistin on vascular endothelium secretion dysfunction in rats. Endothelium 14(4–5), 207–214 (2007)
P. Codoñer-Franch, S. Tavárez-Alonso, M. Porcar-Almela, M. Navarro-Solera, Á. Arilla-Codoñer, E. Alonso-Iglesias, Plasma resistin levels are associated with homocysteine, endothelial activation, and nitrosative stress in obese youths. Clin. Biochem. 47(1–2), 44–48 (2014)
J.J. Lozano-Nuevo, T. Estrada-Garcia, H. Vargas-Robles, B.A. Escalante-Acosta, A.F. Rubio-Guerra, Correlation between circulating adhesion molecules and resistin levels in hypertensive type-2 diabetic patients. Inflamm. Allergy Drug Targets 10(1), 27–31 (2011)
N. Al-Daghri, R. Chetty, P.G. McTernan, K. Al-Rubean, O. Al-Attas, A. Jones et al., Serum resistin is associated with C-reactive protein and LDL-cholesterol in type 2 diabetes and coronary artery disease in a Saudi population. Cardiovasc. Diabetol. 4(1), 10 (2005)
C. Ying, L. Sui-xin, X. Kang-ling, Z. Wen-liang, D. Lei, L. Yuan et al., MicroRNA-492 reverses high glucose-induced insulin resistance in huvec cells through targeting resistin. Mol. Cell. Biochem. 391(1–2), 117–125 (2014)
J.R. Revollo, A. Körner, K.F. Mills, A. Satoh, T. Wang, A. Garten et al., Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6(5), 363–375 (2007)
D. Friebe, M. Neef, J. Kratzsch, S. Erbs, K. Dittrich, A. Garten et al., Leucocytes are a major source of circulating nicotinamide phosphoribosyltransferase (NAMPT)/pre-B cell colony (PBEF)/visfatin linking obesity and inflammation in humans. Diabetologia 54(5), 1200–1211 (2011)
K. Krzyzanowska, W. Krugluger, F. Mittermayer, R. Rahman, D. Haider, N. Shnawa et al., Increased visfatin concentrations in women with gestational diabetes mellitus. Clin. Sci. 110, 605–609 (2006)
Y.-H. Chang, D.-M. Chang, K.-C. Lin, S.-J. Shin, Y.-J. Lee, Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: a meta-analysis and systemic review. Diabetes/Metab. Res. Rev. 27(6), 515–527 (2011)
T.-F. Chan, Y.-L. Chen, C.-H. Lee, F.-H. Chou, L.-C. Wu, S.-B. Jong et al., Decreased plasma visfatin concentrations in women with gestational diabetes mellitus. J. Soc. Gynecol. Investig. 13(5), 364–367 (2006)
P. Wang, M.M.J. van Greevenbroek, F.G. Bouwman, M.C.G.J. Brouwers, C.J.H. van der Kallen, E. Smit et al., The circulating PBEF/NAMPT/visfatin level is associated with a beneficial blood lipid profile. Pflugers Arch. 454(6), 971–976 (2007)
K. Takebayashi, M. Suetsugu, S. Wakabayashi, Y. Aso, T. Inukai, Association between plasma visfatin and vascular endothelial function in patients with type 2 diabetes mellitus. Metabolism 56(4), 451–458 (2007)
A. Kamińska, E. Kopczyńska, M. Bieliński, A. Borkowska, R. Junik, Visfatin concentrations in obese patients in relation to the presence of newly diagnosed glucose metabolism disorders. Endokrynol. Polska. 66(2), 108–113 (2015)
K. Oki, K. Yamane, N. Kamei, H. Nojima, N. Kohno, Circulating visfatin level is correlated with inflammation, but not with insulin resistance. Clin. Endocrinol. 67(5), 796–800 (2007)
Y.-C. Chang, T.-J. Chang, W.-J. Lee, L.-M. Chuang, The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism 59(1), 93–99 (2010)
Y.S. Kang, H.K. Song, M.H. Lee, G.J. Ko, D.R. Cha, Plasma concentration of visfatin is a new surrogate marker of systemic inflammation in type 2 diabetic patients. Diabetes Res. Clin. Pract. 89(2), 141–149 (2010)
W. Lee, C. Wu, H. Lin, I. Lee, C. Wu, J. Tseng et al., Visfatin-induced expression of inflammatory mediators in human endothelial cells through the NF-$\kappa$B pathway. Int. J. Obes. 33(4), 465–472 (2009)
S.-R. Kim, Y.-H. Bae, S.-K. Bae, K.-S. Choi, K.-H. Yoon, T.H. Koo et al., Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta 1783(5), 886–895 (2008)
T. Romacho, L.A. Villalobos, E. Cercas, R. Carraro, C.F. Sánchez-Ferrer, C. Peiró, Visfatin as a novel mediator released by inflamed human endothelial cells. PLoS ONE 8(10), e78283 (2013). doi:10.1371/journal.pone.0078283
P. Saddi-Rosa, C. Oliveira, F. Crispim, F. Giuffrida, V. De Lima, J. Vieira et al., Association of circulating levels of nicotinamide phosphoribosyltransferase (NAMPT/Visfatin) and of a frequent polymorphism in the promoter of the NAMPT gene with coronary artery disease in diabetic and non-diabetic subjects. Cardiovasc. Diabetol. 12(1), 119 (2013)
R. Adya, B.K. Tan, J. Chen, H.S. Randeva, Nuclear factor-$\kappa$B induction by visfatin in human vascular endothelial cells its role in MMP-2/9 production and activation. Diabetes Care 31(4), 758–760 (2008)
T. Romacho, V. Azcutia, M. Vazquez-Bella, N. Matesanz, E. Cercas, J. Nevado et al., Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia 52(11), 2455–2463 (2009)
M. Xia, C. Zhang, K.M. Boini, A.M. Thacker, P.-L. Li, Membrane raft-lysosome redox signaling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc. Res. (2010). doi:10.1093/cvr/cvq286
S. Vallejo, T. Romacho, J. Angulo, L.A. Villalobos, E. Cercas, A. Leivas et al., Visfatin impairs endothelium-dependent relaxation in rat and human mesenteric microvessels through nicotinamide phosphoribosyltransferase activity. PLoS ONE 6(11), e27299 (2011). doi:10.1371/journal.pone.0027299
L.A. Villalobos, A. Uryga, T. Romacho, A. Leivas, C.F. Sánchez-Ferrer, J.D. Erusalimsky et al., Visfatin/Nampt induces telomere damage and senescence in human endothelial cells. Int. J. Cardiol. 175(3), 573–575 (2014)
Ö.F. Karataş, M. Yıldırım, H. Celik, H. Badem, M. Çaviş, E. Çimentepe, The association between plasma visfatin levels and ED. Int. J. Impot. Res. 27(4), 157–160 (2015). doi:10.1038/ijir.2015.1
N.M. Borradaile, J.G. Pickering, Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 8(2), 100–112 (2009)
F. Lovren, Y. Pan, P.C. Shukla, A. Quan, H. Teoh, P.E. Szmitko et al., Visfatin activates eNOS via Akt and MAP kinases and improves endothelial cell function and angiogenesis in vitro and in vivo: translational implications for atherosclerosis. Am. J. Physiol. Endocrinol. Metab. 296(6), E1440–E1449 (2009)
H. Yamawaki, N. Hara, M. Okada, Y. Hara, Visfatin causes endothelium-dependent relaxation in isolated blood vessels. Biochem. Biophys. Res. Commun. 383(4), 503–508 (2009)
S. Chen, L. Sun, H. Gao, L. Ren, N. Liu, G. Song, Visfatin and oxidative stress influence endothelial progenitor cells in obese populations. Endocr. Res. 40, 1–5 (2014)
Y. Sun, S. Chen, G. Song, L. Ren, L. Wei, N. Liu et al., Effect of visfatin on the function of endothelial progenitor cells in high-fat-fed obese rats and investigation of its mechanism of action. Int. J. Mol. Med. 30(3), 622–628 (2012)
J.-Y. Zhou, L. Chan, S.-W. Zhou, Omentin: linking metabolic syndrome and cardiovascular disease. Curr. Vasc. Pharmacol. 12(1), 136–143 (2014)
H.J. Yoo, S.Y. Hwang, H.C. Hong, H.Y. Choi, S.J. Yang, K.W. Lee et al., Implication of circulating omentin-1 level on the arterial stiffening in type 2 diabetes mellitus. Endocrine 44(3), 680–687 (2013)
L. Brunetti, S. Leone, G. Orlando, C. Ferrante, L. Recinella, A. Chiavaroli et al., Hypotensive effects of omentin-1 related to increased adiponectin and decreased interleukin-6 in intra-thoracic pericardial adipose tissue. Pharmacol. Rep. 66(6), 991–995 (2014)
J.M. Moreno-Navarrete, F. Ortega, A. Castro, M. Sabater, W. Ricart, J.M. Fernández-Real, Circulating omentin as a novel biomarker of endothelial dysfunction. Obesity 19(8), 1552–1559 (2011)
S. Maruyama, R. Shibata, R. Kikuchi, Y. Izumiya, T. Rokutanda, S. Araki et al., Fat-derived factor omentin stimulates endothelial cell function and ischemia-induced revascularization via endothelial nitric oxide synthase-dependent mechanism. J. Biol. Chem. 287(1), 408–417 (2012)
H. Yamawaki, J. Kuramoto, S. Kameshima, T. Usui, M. Okada, Y. Hara, Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem. Biophys. Res. Commun. 408(2), 339–343 (2011)
X. Zhong, X. Li, F. Liu, H. Tan, D. Shang, Omentin inhibits TNF-α-induced expression of adhesion molecules in endothelial cells via ERK/NF-κB pathway. Biochem. Biophys. Res. Commun. 425(2), 401–406 (2012)
M. Blüher, Vaspin in obesity and diabetes: pathophysiological and clinical significance. Endocrine 41(2), 176–182 (2012)
C.H. Jung, W.J. Lee, J.Y. Hwang, M.J. Lee, S.M. Seol, Y.M. Kim et al., Vaspin increases nitric oxide bioavailability through the reduction of asymmetric dimethylarginine in vascular endothelial cells. PLoS ONE 7(12), e52346 (2012). doi:10.1371/journal.pone.0052346
C.H. Jung, W.J. Lee, J.Y. Hwang, S.M. Seol, Y.M. Kim, Y. La Lee et al., Vaspin protects vascular endothelial cells against free fatty acid-induced apoptosis through a phosphatidylinositol 3-kinase/Akt pathway. Biochem. Biophys. Res. Commun. 413(2), 264–269 (2011)
S. Phalitakul, M. Okada, Y. Hara, H. Yamawaki, Vaspin prevents methylglyoxal-induced apoptosis in human vascular endothelial cells by inhibiting reactive oxygen species generation. Acta Physiol. 209(3), 212–219 (2013)
B.-D. Fu, H. Yamawaki, M. Okada, Y. Hara, Vaspin can not inhibit TNF-alpha-induced inflammation of human umbilical vein endothelial cells. J. Vet. Med. Sci. 71(9), 1201–1207 (2009)
C.H. Jung, M.J. Lee, Y.M. Kang, Y.L. Lee, H.K. Yoon, S.-W. Kang et al., Vaspin inhibits cytokine-induced nuclear factor-kappa B activation and adhesion molecule expression via AMP-activated protein kinase activation in vascular endothelial cells. Cardiovasc. Diabetol. 13, 41 (2014)
S. Liu, Y. Dong, T. Wang, S. Zhao, K. Yang, X. Chen et al., Vaspin inhibited proinflammatory cytokine induced activation of nuclear factor-kappa B and its downstream molecules in human endothelial EA. hy926 cells. Diabetes Res. Clin. Pract. 103(3), 482–488 (2014)
N. Sun, H. Wang, L. Wang, Vaspin alleviates dysfunction of endothelial progenitor cells induced by high glucose via PI3K/Akt/eNOS pathway. Int. J. Clin. Exp. Pathol. 8(1), 482 (2015)
I. Jialal, S. Devaraj, H. Kaur, B. Adams-Huet, A.A. Bremer, Increased chemerin and decreased omentin-1 in both adipose tissue and plasma in nascent metabolic syndrome. J. Clin. Endocrinol. Metab. 98(3), E514–E517 (2013)
A.A. Roman, S.D. Parlee, C.J. Sinal, Chemerin: a potential endocrine link between obesity and type 2 diabetes. Endocrine 42(2), 243–251 (2012)
P. Fülöp, I. Seres, H. Lőrincz, M. Harangi, S. Somodi, G. Paragh, Association of chemerin with oxidative stress, inflammation and classical adipokines in non-diabetic obese patients. J. Cell Mol. Med. 18(7), 1313–1320 (2014)
Q. Yan, Y. Zhang, J. Hong, W. Gu, M. Dai, J. Shi et al., The association of serum chemerin level with risk of coronary artery disease in Chinese adults. Endocrine 41(2), 281–288 (2012)
W. Hu, Q. Yu, J. Zhang, D. Liu, Rosiglitazone ameliorates diabetic nephropathy by reducing the expression of chemerin and ChemR23 in the kidney of streptozotocin-induced diabetic rats. Inflammation 35(4), 1287–1293 (2012)
N.S. Lobato, K.B. Neves, F.P. Filgueira, Z.B. Fortes, M.H.C. Carvalho, R.C. Webb et al., The adipokine chemerin augments vascular reactivity to contractile stimuli via activation of the MEK-ERK1/2 pathway. Life Science 91(13–14), 600–606 (2012)
K.B. Neves, N.S. Lobato, R.A.M. Lopes, F.P. Filgueira, C.Z. Zanotto, A.M. Oliveira et al., Chemerin reduces vascular nitric oxide/cGMP signalling in rat aorta: a link to vascular dysfunction in obesity? Clin. Sci. 127(2), 111–122 (2014)
L. Wang, T. Yang, Y. Ding, Y. Zhong, L. Yu, M. Peng, Chemerin plays a protective role by regulating human umbilical vein endothelial cell-induced nitric oxide signaling in preeclampsia. Endocrine 48(1), 299–308 (2015)
W. Shen, C. Tian, H. Chen, Y. Yang, D. Zhu, P. Gao et al., Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic. Biol. Med. 55, 73–82 (2013)
J. Weigert, M. Neumeier, J. Wanninger, M. Filarsky, S. Bauer, R. Wiest et al., Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin. Endocrinol. 72(3), 342–348 (2010)
H. Yamawaki, S. Kameshima, T. Usui, M. Okada, Y. Hara, A novel adipocytokine, chemerin exerts anti-inflammatory roles in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 423(1), 152–157 (2012)
I. Castan-Laurell, C. Dray, C. Attané, T. Duparc, C. Knauf, P. Valet, Apelin, diabetes, and obesity. Endocrine 40(1), 1–9 (2011)
A.G. Japp, N.L. Cruden, D.A. Amer, V.K. Li, E.B. Goudie, N.R. Johnston et al., Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 52(11), 908–913 (2008)
J.-C. Zhong, X.-Y. Yu, Y. Huang, L.-M. Yung, C.-W. Lau, S.-G. Lin, Apelin modulates aortic vascular tone via endothelial nitric oxide synthase phosphorylation pathway in diabetic mice. Cardiovasc. Res. 74(3), 388–395 (2007)
Y.-C. Liao, Y.-S. Wang, E. Hsi, M.-H. Chang, Y.-Z. You, S.-H.H. Juo, MicroRNA-765 influences arterial stiffness through modulating apelin expression. Mol. Cell. Endocrinol. 411, 11–19 (2015). doi:10.1016/j.mce.2015.04.006
K. Nagano, J. Ishida, M. Unno, T. Matsukura, A. Fukamizu, Apelin elevates blood pressure in ICR mice with L-NAME-induced endothelial dysfunction. Mol. Med. Rep. 7(5), 1371–1375 (2013)
Y. Lu, X. Zhu, G.-X. Liang, R.-R. Cui, Y. Liu, S.-S. Wu et al., Apelin-APJ induces ICAM-1, VCAM-1 and MCP-1 expression via NF-κB/JNK signal pathway in human umbilical vein endothelial cells. Amino Acids 43(5), 2125–2136 (2012)
D.J. Drucker, M.A. Nauck, The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368(9548), 1696–1705 (2006)
S. Ravassa, J. Beaumont, A. Huerta, J. Barba, I. Coma-Canella, A. González et al., Association of low GLP-1 with oxidative stress is related to cardiac disease and outcome in patients with type 2 diabetes mellitus: a pilot study. Free Radic. Biol. Med. (2015). doi:10.1016/j.freeradbiomed.2015.01.002
B. Gallwitz, Extra-pancreatic effects of incretin-based therapies. Endocrine 47(2), 360–371 (2014)
A. Ceriello, A. Novials, E. Ortega, S. Canivell, L. La Sala, G. Pujadas et al., Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care 36(8), 2346–2350 (2013)
P.C. Van Poppel, M.G. Netea, P. Smits, C.J. Tack, Vildagliptin improves endothelium-dependent vasodilatation in type 2 diabetes. Diabetes Care 34(9), 2072–2077 (2011)
A. Ceriello, L. La Sala, V. De Nigris, G. Pujadas, R. Testa, A. Uccellatore et al., GLP-1 reduces metalloproteinase-14 and soluble endoglin induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Endocrine. 2015;1–4. doi:10.1007/s12020-015-0565-2
Y. Noda, T. Miyoshi, H. Oe, Y. Ohno, K. Nakamura, N. Toh et al., Alogliptin ameliorates postprandial lipemia and postprandial endothelial dysfunction in non-diabetic subjects: a preliminary report. Cardiovasc. Diabetol. 12(8), 8–12 (2013)
J. Koska, M. Sands, C. Burciu, K.M. D’Souza, K. Raravikar, J. Liu et al., Exenatide protects against glucose and lipid-induced endothelial dysfunction: evidence for direct vasodilation effect of GLP-1 receptor agonists in humans. Diabetes (2015). doi:10.2337/db14-0976
R. Faber, M. Zander, A. Pena, M.M. Michelsen, N.D. Mygind, E. Prescott, Effect of the glucagon-like peptide-1 analogue liraglutide on coronary microvascular function in patients with type 2 diabetes-a randomized, single-blinded, cross-over pilot study. Cardiovasc. Diabetol. 14(1), 41 (2015)
L. Ding, J. Zhang, Glucagon-like peptide-1 activates endothelial nitric oxide synthase in human umbilical vein endothelial cells. Acta Pharmacol. Sin. 33(1), 75–81 (2012)
Z. Dong, W. Chai, W. Wang, L. Zhao, Z. Fu, W. Cao et al., Protein kinase A mediates glucagon-like peptide 1-induced nitric oxide production and muscle microvascular recruitment. Am. J. Physiol. Endocrinol. Metab. 304(2), E222–E228 (2013)
H. Liu, Y. Hu, R.W. Simpson, A.E. Dear, Glucagon-like peptide-1 attenuates tumour necrosis factor-alpha-mediated induction of plasminogen [corrected] activator inhibitor-1 expression. J. Endocrinol. 196(1), 57–65 (2008)
H. Liu, A.E. Dear, L.B. Knudsen, R.W. Simpson, A long-acting glucagon-like peptide-1 analogue attenuates induction of plasminogen activator inhibitor type-1 and vascular adhesion molecules. J. Endocrinol. 201(1), 59–66 (2009)
N.M. Krasner, Y. Ido, N.B. Ruderman, J.M. Cacicedo, Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS ONE 9(5), e97554 (2014). doi:10.1371/journal.pone.0097554
H. Oeseburg, R.A. de Boer, H. Buikema, P. van der Harst, W.H. van Gilst, H.H. Silljé, Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler. Thromb. Vasc. Biol. 30(7), 1407–1414 (2010)
Y. Zhan, H. Sun, H. Chen, H. Zhang, J. Sun, Z. Zhang et al., Glucagon-like peptide-1 (GLP-1) protects vascular endothelial cells against advanced glycation end products (AGEs)-induced apoptosis. Ann. Transpl. 18(7), BR286–BR291 (2012)
Y. Ishibashi, T. Matsui, M. Takeuchi, S. Yamagishi, Glucagon-like peptide-1 (GLP-1) inhibits advanced glycation end product (AGE)-induced up-regulation of VCAM-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression. Biochem. Biophys. Res. Commun. 391(3), 1405–1408 (2010)
F. Liu, X. Zhang, L. Gong, X. Wang, J. Wang, X. Hou et al., Glucagon-like peptide 1 protects microvascular endothelial cells by inactivating the PARP-1/iNOS/NO pathway. Mol. Cell. Endocrinol. 339(1–2), 25–33 (2011)
S. Salheen, U. Panchapakesan, C. Pollock, O. Woodman, The DPP-4 inhibitor linagliptin and the GLP-1 receptor agonist exendin-4 improve endothelium-dependent relaxation of rat mesenteric arteries in the presence of high glucose. Pharmacol. Res. 94, 26–33 (2015)
B. Batchuluun, T. Inoguchi, N. Sonoda, S. Sasaki, T. Inoue, Y. Fujimura et al., Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD (P) H oxidase pathway in human aortic endothelial cells. Atherosclerosis 232(1), 156–164 (2014)
B. Schisano, A.L. Harte, K. Lois, P. Saravanan, N. Al-Daghri, O. Al-Attas et al., GLP-1 analogue, Liraglutide protects human umbilical vein endothelial cells against high glucose induced endoplasmic reticulum stress. Regul. Pept. 174(1), 46–52 (2012)
X. Xiao-Yun, M. Zhao-Hui, C. Ke, H. Hong-Hui, X. Yan-Hong, Glucagon-like peptide-1 improves proliferation and differentiation of endothelial progenitor cells via upregulating VEGF generation. Ann. Transpl. 17(2), BR35–BR41 (2011)
G.P. Fadini, E. Boscaro, M. Albiero, L. Menegazzo, V. Frison, S. De Kreutzenberg et al., The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes possible role of stromal-derived factor-1$\alpha$. Diabetes Care 33(7), 1607–1609 (2010)
C.-Y. Huang, C.-M. Shih, N.-W. Tsao, Y.-W. Lin, P.-H. Huang, S.-C. Wu et al., Dipeptidyl peptidase-4 inhibitor improves neovascularization by increasing circulating endothelial progenitor cells. Br. J. Pharmacol. 167(7), 1506–1519 (2012)
L. Pulkkinen, O. Ukkola, M. Kolehmainen, M. Uusitupa, Ghrelin in diabetes and metabolic syndrome. Int. J. Pept. (2010). doi:10.1155/2010/248948
D. Perez-Tilve, R. Nogueiras, F. Mallo, S.C. Benoit, M. Tschoep, Gut hormones ghrelin, PYY, and GLP-1 in the regulation of energy, balance and metabolism. Endocrine 29(1), 61–71 (2006)
M. Suematsu, A. Katsuki, Y. Sumida, E.C. Gabazza, S. Murashima, K. Matsumoto et al., Decreased circulating levels of active ghrelin are associated with increased oxidative stress in obese subjects. Eur. J. Endocrinol. 153(3), 403–407 (2005)
O. Gruzdeva, E. Uchasova, E. Belik, Y. Dyleva, E. Shurygina, O. Barbarash, Lipid, adipokine and ghrelin levels in myocardial infarction patients with insulin resistance. BMC Cardiovasc. Disord. 14(1), 7 (2014)
M. Tesauro, F. Schinzari, M. Iantorno, S. Rizza, D. Melina, D. Lauro et al., Ghrelin improves endothelial function in patients with metabolic syndrome. Circulation 112(19), 2986–2992 (2005)
M. Tesauro, F. Schinzari, V. Rovella, N. Di Daniele, D. Lauro, N. Mores et al., Ghrelin restores the endothelin 1/nitric oxide balance in patients with obesity-related metabolic syndrome. Hypertension 54(5), 995–1000 (2009)
N. Hedayati, S. Annambhotla, J. Jiang, X. Wang, H. Chai, P.H. Lin et al., Growth hormone-releasing peptide ghrelin inhibits homocysteine-induced endothelial dysfunction in porcine coronary arteries and human endothelial cells. J. Vasc. Surg. 49(1), 199–207 (2009)
D. Wang, H. Wang, P. Luo, A. Hwang, D. Sun, Y. Wang et al., Effects of ghrelin on homocysteine-induced dysfunction and inflammatory response in rat cardiac microvascular endothelial cells. Cell Biol. Int. 36(6), 511–517 (2012)
G. Togliatto, A. Trombetta, P. Dentelli, S. Gallo, A. Rosso, P. Cotogni et al., Unacylated ghrelin (UnAG) induces oxidative stress resistance in a glucose intolerance mouse model and peripheral artery disease by restoring endothelial cell miR-126 expression. Diabetes (2014). doi:10.2337/db14-0991
D. Zhang, W. Wang, Y. Zhou, Y. Chen, L. Han, Y. Liu et al., Ghrelin inhibits apoptosis induced by palmitate in rat aortic endothelial cells. Med. Sci. Monit. Basic Res. 16(12), 396–403 (2010)
Y. Xiang, Q. Li, M. Li, W. Wang, C. Cui, J. Zhang, Ghrelin inhibits AGEs-induced apoptosis in human endothelial cells involving ERK1/2 and PI3K/Akt pathways. Cell Biochem. Funct. 29(2), 149–155 (2011)
W.G. Li, D. Gavrila, X. Liu, L. Wang, S. Gunnlaugsson, L.L. Stoll et al., Ghrelin inhibits proinflammatory responses and nuclear factor-$\kappa$B activation in human endothelial cells. Circulation 109(18), 2221–2226 (2004)
B. Deng, F. Fang, T. Yang, Z. Yu, B. Zhang, X. Xie, Ghrelin inhibits AngII-induced expression of TNF-$\alpha$, IL-8, MCP-1 in human umbilical vein endothelial cells. Int. J. Clin. Exp. Med. 8(1), 579 (2015)
X. Liu, Q. Xiao, K. Zhao, Y. Gao, Ghrelin inhibits high glucose-induced PC12 cell apoptosis by regulating TLR4/NF-κB pathway. Inflammation 36(6), 1286–1294 (2013)
J. Zhu, C. Zheng, J. Chen, J. Luo, B. Su, Y. Huang et al., Ghrelin protects human umbilical vein endothelial cells against high glucose-induced apoptosis via mTOR/P70S6K signaling pathway. Peptides 52, 23–28 (2014)
X. Chen, Q. Chen, L. Wang, G. Li, Ghrelin induces cell migration through GHSR1a-mediated PI3K/Akt/eNOS/NO signaling pathway in endothelial progenitor cells. Metab., Clin. Exp. 62(5), 743–752 (2013)
G. Togliatto, A. Trombetta, P. Dentelli, A. Baragli, A. Rosso, R. Granata et al., Unacylated ghrelin rescues endothelial progenitor cell function in individuals with type 2 diabetes. Diabetes 59(4), 1016–1025 (2010)
L. Trovato, D. Gallo, F. Settanni, I. Gesmundo, E. Ghigo, R. Granata, Obestatin: is it really doing something. 42:175–85 (2014). doi:10.1159/000358346
R. Granata, D. Gallo, R.M. Luque, A. Baragli, F. Scarlatti, C. Grande et al., Obestatin regulates adipocyte function and protects against diet-induced insulin resistance and inflammation. FASEB J. 26(8), 3393–3411 (2012)
P. Gu, D. Kang, W. Wang, Y. Chen, Z. Zhao, H. Zheng et al., Relevance of plasma obestatin and early arteriosclerosis in patients with type 2 diabetes mellitus. J. Diabetes Res. (2013). doi:10.1155/2013/563919
A.J. Agnew, E. Robinson, C.M. McVicar, A.P. Harvey, I.H.A. Ali, J.E. Lindsay et al., The gastrointestinal peptide obestatin induces vascular relaxation via specific activation of endothelium-dependent NO signalling. Br. J. Pharmacol. 166(1), 327–338 (2012)
Acknowledgments
The authors would like to thank Dr. Maha Mahmoud Samy for her valuable assistance.
Authors’ Contribution
MIS, MAK, and HD designed and edited this review. MIS, TMA, and MMS wrote this review. MY and SHT critically revised this review and helped in literature search and data collection. All the authors have read and approved the final version of this review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Electronic supplementary material
Below is the link to the electronic supplementary material.








Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Saad, M.I., Abdelkhalek, T.M., Saleh, M.M. et al. Insights into the molecular mechanisms of diabetes-induced endothelial dysfunction: focus on oxidative stress and endothelial progenitor cells. Endocrine 50, 537–567 (2015). https://doi.org/10.1007/s12020-015-0709-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0709-4