
Abstract
Ghrelin has a protective effect on diabetic encephalopathy. To expound the protective mechanism, we investigated the effects of ghrelin on high glucose-induced cell apoptosis and intracellular signaling in cultured PC12, which is a suitable model for studying neuronal cell death. High glucose-induced PC12 apoptosis was significantly inhibited by co-treatment of ghrelin. Sustaining inflammatory response is one of the molecular mechanisms of diabetic encephalopathy and TLR4 signaling has close relationship with inflammatory response. But there is no report about the biologic role of toll-like receptor 4/nuclear factor-κB (TLR4/NF-κB) signaling in controlling high glucose-induced PC12 apoptosis by ghrelin. In this study, we found that TLR4/NF-κB pathway was activated by high glucose stimulation in PC12 and significantly alleviated by the co-treatment of ghrelin. From these findings, we made the conclusion that ghrelin could attenuate the symptoms of diabetic encephalopathy, which alleviates inflammatory reaction of diabetic encephalopathy by regulating TLR4/NF-κB pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Diabetic encephalopathy, which is first proposed by Nielon in 1965, is an increasingly recognized complication of diabetes [1]. It appears to be correlated with age of onset, duration of diabetes, and quality of glycemic control and leads to impaired learning abilities, memory, and intelligence in affected patients [2]. Experimental rodent models display impaired spatial learning and memory associated with impaired hippocampal long-term potentiation. Although the mechanisms underlying diabetic encephalopathy are not fully understood, the consequences of hyperglycemia and impaired insulin signaling are likely to play important pathophysiological roles [3–5].
Ghrelin, a novel gastrointestinal hormone, was first identified in 1999 as an endogenous ligand for the growth hormone secretagogue receptor type 1a (i.e., ghrelin receptor). In addition to its growth hormone-releasing properties, ghrelin has now been proved to possess other endocrine and non-endocrine activities including anti-inflammation [6, 7]. Ghrelin can cross the blood–brain barrier and its receptors are expressed at a high density in the brain [8], especially in the dorsal motor nucleus and nucleus tractus solitarius [9, 10].
Emerging evidence suggests that inflammatory mechanisms play an important role in the pathogenesis and progression of diabetes mellitus and its complications [11]. And Toll-like receptor (TLR) 4 is one of the most popular regulatory factors in the process of inflammation. After combining with corresponding ligand, TLR4 can activate nuclear factor kappa B (NF-κB). And then, a series of cytokines are upregulated to activate the immune response and inflammatory reaction. The TLR is one of the key transmembrane proteins that transfer the information of cell antigen recognition and induce inflammation in mammalians. It is maintained that TLR is the main component in the process of mediating immune response and inflammation. More and more attention has been paid to the role of TLR4 in cerebral diseases. For example, it was reported that TLR4-mediated NF-κB signaling participates in the pathogenesis of intracerebral hemorrhage [12].
The pheochromocytoma cells (PC12), which are derived from a catecholamine-secreting adrenal chromaffin tumor in rats, are a suitable model for studying neuronal cell death [13]. The aim of the present study was to investigate the protective effects of ghrelin on high glucose-induced cell apoptosis in cultured PC12. We further examined whether ghrelin could attenuate high glucose-induced activation of the TLR4/NF-κB signaling pathway.
MATERIALS AND METHODS
Materials
FBS and horse serum were purchased from Gibco BRL (Grand Island, NY, USA). The Dulbecco’s modified Eagle’s medium (DMEM) medium and trypsin-EDTA (0.25 %) were supplied by Hyclone. Ghrelin was purchased from Phoenix Pharmaceuticals. 3-[4,5-Dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide (MTT), phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO), DEPC, sodium dodecyl sulfate (SDS), Tris/HCl, and D-lys-3-GHRP-6 (an inhibitor of ghrelin) were obtained from Sigma (St. Louis, MO). Trizol reagent, PrimeScript RT reagent kit, and PCR reagent kit were obtained from TaKaRa (TaKaRa Biotechnology, Dalian, China). The primers were synthesized by Invitrogen Life Tech (Carlsbad, CA, USA). The whole protein extraction kit, anti-β-actin monoclonal antibody, and reagents for western blot were obtained from Beyotime Biotechnology (Beyotime, Jiangsu, China). The mouse monoclonal antibody to TLR4, myeloid differentiation primary response gene (MyD) 88, and human tumor necrosis factor receptor-associated factor (TRAF) 6 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Electrochemiluminescence (ECL) reagent kit and annexin V reagent kit were purchased from Keygen (Nanjing, China).
Cell Culture
PC12 cells were obtained from ATCC. Cells were maintained at 37 °C in a 90 % humidified atmosphere containing 5 % CO2. Cells were grown in DMEM with 5 % (v/v) fetal bovine serum, 10 % (v/v) horse serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. The concentration of glucose in DMEM was 4.5 mg/ml. According to the manufacturer’s instructions, PC12 cells were maintained with culture medium containing 4.5 mg/ml glucose. Thus, a threefold concentration of glucose (13.5 mg/ml) was considered as a high glucose medium. Cells were detached by using cold 0.25 % trypsin in PBS.
Cell Viability
After two passages, PC12 cells were plated at the density of 5,000 per well in a 96-microplate well for the MTT assay. Control cells were grown in DMEM with 4.5 mg/ml glucose and the other cells grown in DMEM with 13.5 mg/ml glucose. Cell cultures that were exposed to 13.5 mg/ml mannitol in MTT assay were used as osmolar control. The viability and proliferation of the cells were assessed by the MTT assay, which is based on the reduction of MTT by the mitochondrial dehydrogenase of intact cells to form a purple formazan product [14]. In brief, 50 μl MTT solution (5 mg/ml in DMEM) was added to the 96-well plates and the cells were allowed to incubate for 4 h at 37 °C. After removing of medium, the cells and dye crystals were solubilized by adding 150 μl of DMSO. The amount of formazan was determined by measuring the absorbance (A) value at a wavelength of 570 nm by using a microplate reader (Bio-Rad, CA, USA).
Transmission Electron Microscopy
We trained the control group in normal condition and the experiment group with 13.5 mg/ml high glucose DMEM for 72 h. Fresh specimens of PC12 cells were fixed by immersing them immediately in 2.5 % glutaraldehyde fixative for 24 h. Semithin sections were obtained by using an ultramicrotome. We observed the ultrastructural changes of PC12 cells under transmission electron microscopy (TEM).
Annexin V Assay
For cell apoptosis analysis, 4 × 105 cells/well were seeded in six-well multidishes and incubated until they reached the logarithmic phase. The cells were rinsed twice with PBS and divided into four groups: (1) the control group; (2) the high glucose (HG) group (13.5 mg/ml high glucose DMEM); (3) the HG (13.5 mg/ml) + ghrelin (100 ng/μL) group; and (4) the HG (13.5 mg/ml) + ghrelin (100 ng/μL) + D-lys-3-GHRP-6 (30 g/L) group. After cultivating for 72 h, cells were subsequently trypsinized and centrifuged at 1,000×g at 4 °C for 5 min. The supernatant was removed, and the cells were resuspended in 1 ml of PBS. And then, we removed the supernatants, added RNase, water bathed for 1 h at 37 °C, and ice bathed with 0.5 mg/L PI and annexin V. Finally, we detected cell apoptosis rate by flow cytometry assay.
RNA Isolation and RT-PCR
The cells were divided into four groups: (1) the control group; (2) the HG group (13.5 mg/ml high glucose DMEM); (3) the HG (13.5 mg/ml) + ghrelin group (100 ng/μL); and (4) the HG (13.5 mg/ml) + ghrelin (100 ng/μL) + D-lys-3-GHRP-6 (30 g/L) group. Total RNA of each group was extracted from PC12 by using Trizol, and quality was verified by resolving samples using 1 % agarose gel electrophoresis. The following primers were used: TLR4, forward 5′-CAA GCA GTT AGA TGA AAG GGA A-3′, reverse 5′-CAG ACG ACG ATG ATG ATG GA-3′; MyD88, forward 5′-GCT GAT TTG ATG GAG TTG GAC ATG G-3′, reverse-5′-GCC AAA CGC TGG ACA TTA GTG G-3′; TRAF6, forward-5′ ATG CCA ACC TCC TCA ACG ACC-3′, reverse 5′-TGG CAC AGA GGG CAA CGA AGG-3′; and GAPDH, forward 5′-ACA GCC TCA AGA TCA TCA GCA-3′, reverse 5′-TGA GTC CTT CCA CGA TAC CAA-3′. PCR conditions are as follows: denatured at 94 °C for 5 min, 94 °C for 30 s, 58 °C for 30 s, 72 °C for 20 s, target genes for 34 cycles and GAPDH for 28 cycles, and 72 °C for 10 min. PCR products were resolved by using 2 % agarose gel electrophoresis for 40 min. We observed the results under an ultraviolet lamp, took photos, and analyzed images by using UVIMap software. And then, we compared the data of each lane of TLR4, MyD88, and TRAF6 absorbency respectively, as the relative contents of TLR4, MyD88, and TRAF6 mRNA.
Western Blot Analysis
The cells were divided into four groups: (1) the control group; (2) the HG group (13.5 mg/ml high glucose DMEM); (3) the HG (13.5 mg/ml) + ghrelin (100 ng/μL) group; and (4) the HG (13.5 mg/ml) + ghrelin (100 ng/μL) + D-lys-3-GHRP-6 (30 g/L) group. Then, the cells were lysed in RIPA buffer that was composed of 40 mM Tris–HCl (pH 7.4) containing 150 mM NaCl and 1 % (v/v) Triton X-100, supplemented with a cocktail of protease inhibitors. Equal amounts of protein were resolved on 10 % SDS-PAGE gels and then transferred to a PVDF membrane. Blots were blocked and then probed with antibodies against TLR4, MyD88, TRAF6 (1:1,000, 1:1,500, 1:2,500), and β-actin (1:1,000). After washing, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies and visualized by ECL kit. Protein band intensities were evaluated by using Scion Image software (Scion Corporation, Frederick, MD, USA).
Statistical Analysis
Values are reported as the means ± SD from experiments performed on six different occasions. One-way ANOVA was used to determine the significance among groups, after which a modified t test with Bonferroni’s post hoc test was used for comparisons between individual groups. A value of p < 0.05 was considered to be statistically significant.
RESULTS
Effect of High Glucose on PC12 Cell Viability
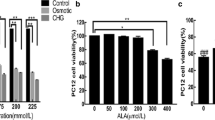
The effects of high concentration of glucose on PC12 cell viability were examined by using the MTT assay. After the initial 24 h cultivation, the cells were exposed to glucose at the concentration of 13.5 mg/ml for 24, 48, 72, and 96 h. As shown in Fig. 1, glucose (13.5 mg/ml) could decrease the viability of PC12 cells after 48 h and reach IC50 at 72 h. This toxicity was time-dependently increased, so increasing the incubation time caused higher toxicity. And the inhibiting effect of high glucose also displayed in the results of TEM (Fig. 2) and the annexin V assay (Fig. 3a, b).

Effect of high glucose on PC12 cell viability. High glucose induces PC12 apoptosis in a time-dependent manner. In the control group, cells were grown in DMEM with 4.5 mg/ml glucose and cells were grown in DMEM with 13.5 mg/ml glucose as high glucose (HG) group. Cell cultures that were exposed to 13.5 mg/ml mannitol were used as osmolar control. PC12 were treated for the indicated time, and cell number was calculated. (n = 8 for each condition) *p < 0.05 versus control group.

The ultrastructure of PC12 cells by TEM. a The control group. b, c The 13.5-mg/ml high glucose group at 72 h. The sign of apoptosis, blebbing, cell shrinkage, nuclear fragmentation, and chromatin condensation can be seen from the high glucose group. *p < 0.05 versus control group.

Results of annexin V cell apoptosis assay. a Control group. b High glucose group. c High glucose + ghrelin group. d High glucose + ghrelin + D-lys-3-GHRP-6 group. Apoptosis ratio was increased in b (p < 0.05 versus a). Ghrelin decreased the apoptosis ratio significantly in c (p > 0.05 versus a). However, D-lys-3-GHRP-6 reverses the effects of ghrelin in d (p < 0.05 versus a). Values are the means ± SD, n = 6. *p < 0.05 versus control group.
Ghrelin Inhibited High Glucose-Induced PC12 Apoptosis
To examine whether ghrelin affects high glucose-induced PC12 apoptosis, PC12 cells were treated with 100 ng/μL ghrelin and 13.5 mg/ml high glucose for 72 h. As shown in Fig. 3c, ghrelin treatment significantly suppressed high glucose-induced PC12 apoptosis (p < 0.05 versus high glucose group). Co-treatment with D-lys-3-GHRP-6 (30 g/L), the inhibitory effect was reversed (Fig. 3d).
Ghrelin Attenuated High Glucose-Induced Activation of TLR4/NF-κB Signaling Pathway
In view of the confirmed inhibitory effects of ghrelin on high glucose-induced PC12 apoptosis, we questioned whether ghrelin suppressed high glucose-induced PC12 apoptosis via the TLR4/NF-κB pathway. As shown in Figs. 4 and 5, exposure with ghrelin and high glucose had significant inhibitory effects on the TLR4/NF-κB pathway compared with that of only high glucose treatment group. From these results, it is suggested that the inhibitory effect of ghrelin on high glucose-induced PC12 apoptosis was involved in the suppression of TLR4/NF-κB pathway. The suppression of TLR4/NF-κB pathway induced by ghrelin was abolished by D-lys-3-GHRP-6, an antagonist of ghrelin.

RT-PCR detected TLR4, MyD88, and TRAF6 mRNA expression of PC12 cells in control group, high glucose (HG) group, HG + ghrelin group, and HG + ghrelin + D-lys-3-GHRP-6 group. TLR4 mRNA expression was decreased in the HG group (p < 0.05 versus control group). Ghrelin promoted TLR4 mRNA expression in the HG + ghrelin group. However, D-lys-3-GHRP-6 reverses the effects of ghrelin in the HG + ghrelin + D-lys-3-GHRP-6 group (p < 0.05 versus control group). MyD88 mRNA expression was decreased in HG group (p < 0.05 versus control group). Ghrelin promoted MyD88 mRNA expression in HG + ghrelin group. However, D-lys-3-GHRP-6 reverses the effects of ghrelin in HG + ghrelin + D-lys-3-GHRP-6 group (p < 0.05 versus control group). TRAF6 mRNA expression was decreased in HG group (p < 0.05 versus control group). Ghrelin promoted TRAF6 mRNA expression in HG + ghrelin group. However, D-lys-3-GHRP-6 reverses the effects of ghrelin in HG + ghrelin + D-lys-3-GHRP-6 group (p < 0.05 versus control group). *p < 0.05 versus control group.

Western blot analysis detected TLR4, MyD88, and TRAF6 expression of PC12 cells in control group, high glucose (HG) group, HG + ghrelin group, and HG + ghrelin + D-lys-3-GHRP-6 group. TLR4 expression was decreased in HG group (p < 0.05 versus control group). Ghrelin promoted TLR4 expression in the HG + ghrelin group. However, D-lys-3-GHRP-6 reverses the effects of ghrelin in HG + ghrelin + D-lys-3-GHRP-6 group (p < 0.05 versus control group). MyD88 expression was decreased in HG group (p < 0.05 versus control group). Ghrelin promoted MyD88 expression in HG + ghrelin group. However, D-lys-3-GHRP-6 reverses the effects of ghrelin in HG + ghrelin + D-lys-3-GHRP-6 group (p < 0.05 versus control group). TRAF6 expression was decreased in HG group (p < 0.05 versus control group). Ghrelin promoted TRAF6 expression in HG + ghrelin group. However, D-lys-3-GHRP-6 reverses the effects of ghrelin in HG + ghrelin + D-lys-3-GHRP-6 group (*p < 0.05 versus control group).
DISCUSSION
Despite numerous investigations, the precise mechanism of diabetic encephalopathy is still not clearly known yet. An increasing number of researches demonstrated that inflammation and activated innate immunity is one of the pathogenesis of diabetes and its complications [15]. It has been reported that C-reactive protein and IL-6 can be the predictors of diabetes mellitus [16, 17]. Lontchi-Yimagou et al. [18] pointed out that inflammatory response likely contributes to diabetes occurrence by causing insulin resistance and is in turn intensified in the presence of hyperglycemia to promote long-term complications of diabetes. He emphasized that targeting inflammatory pathways could possibly be a component of the strategies to prevent and control diabetes and related complications [18]. Meanwhile, some studies showed that all of the levels of inflammatory mediators are abnormal [19]. Sima et al. [20] discovered that activation of innate immune responses contributes to the development of diabetic encephalopathy and that it can be effectively prevented by replacement of insulinomimetic C-peptide.
In this study, we demonstrated that high concentration of glucose could induce neuronal cell death by using PC12, for it is a suitable model for studying neuronal cell death. Based on the previous data, the optimal glucose concentration for PC12 cell culture (in vitro) was 4.5 mg/ml, and 13.5 mg/ml glucose was used as a corresponding high glucose concentration for PC12 cells, which is previously being shown by Sharifi et al. [21]. To exclude the disturbance of hypertonic culture medium, cell cultures that were exposed to 13.5 mg/ml mannitol were used as osmolar control. The present results showed that the viability of PC12 cells was significantly decreased after 48 h of incubation with 13.5 mg/ml of glucose in the culture medium and continued to decrease in a time-dependent manner. The apoptosis ratio can reach IC50 at 72 h in the atmosphere of 13.5 mg/ml high glucose. The apoptosis of PC12 which is induced by high glucose is reflected directly in the results of TEM. PC12 cells in the treatment group, which is cultured in 13.5 mg/ml high glucose culture medium for 72 h, had the changes of blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, apoptotic bodies, and other signs of apoptosis.
Ghrelin is a stomach-derived 28-amino acid peptide which has been shown to exert various effects in both physiological and pathological conditions. Some of the actions of ghrelin include energy homeostasis, stimulation of growth hormone release, and inhibition of cell death. Ghrelin has also been found to inhibit apoptosis in cerebral ischemia [22]. A previous study of our team proved that ghrelin improved cognitive ability in diabetic rats by improving the expression of BDNF and CREB and by attenuating hippocampus neuronal apoptosis [23]. However, the whole mechanisms that ghrelin improves cognitive ability are still unclear yet and few reports are correlated to the effect of improving cognitive ability in vitro. It is well known that ghrelin has a significant effect of anti-inflammation at present. Some reports have demonstrated that both systemic and tissue-associated ghrelin and GHS-R protein and mRNA expression are regulated during states of acute and chronic inflammation [24]. Madison et al. [25] provided further support for the potential role of ghrelin as a therapeutic agent in inflammatory illnesses. Dixit et al. [26] have previously reported that that ghrelin is expressed in T cells and exerts anti-inflammation effects. Several researches declared that ghrelin inhibits proinflammation cytokine production and nuclear factor-κB activation in human endothelial cells in vitro and endotoxin-induced cytokine production in vivo [27]. Cletus et al. [28] found that human ghrelin is a neuroprotective agent which inhibits inflammation and apoptosis in focal cerebral ischemia through a vagal pathway. Talat et al. [29] demonstrated that ghrelin has significant anti-inflammatory effects through modulation of secretion of both pro- and anti-inflammatory cytokines from LPS-stimulated macrophages through distinct signaling cascades. Hence, the anti-inflammatory effects of ghrelin may be one of the mechanisms in treating diabetic encephalopathy, the inflammatory disease.
In the annexin V assay, we found that ghrelin significantly attenuated the apoptosis of PC12 cells induced by high glucose. The apoptosis ratio of control group, HG group, and HG + ghrelin group are 2.96 ± 0.85, 31.65 ± 1.23, and 3.79 ± 0.19 respectively. There was a statistical significance between control group and high glucose group which proved that 13.5 mg/ml glucose can induce a large amount apoptosis of PC12 cells. There was also a statistical significance between high glucose group and high glucose + ghrelin group which signifies that ghrelin can attenuate the apoptosis of PC12 induced by high glucose. Nevertheless, such effect was demolished by D-lys-3-GHRP-6, the inhibitor of ghrelin, with an apoptosis ratio of 30.28 ± 0.72 and there is no statistical significance versus HG group.
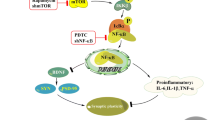
And then, we began to verify the specific pathway of the anti-inflammation effect of ghrelin. TLRs are a family of signal transduction molecules known to activate the innate immune response following systemic bacterial infection and cerebral injury [30]. Among the TLRs, TLR4 has been shown to play an important role in initiating the inflammatory response in the damaged brain. TLR4-mediated signaling pathways mainly stimulate the activation of NF-κB. This critical nuclear transcription factor regulates many pro-inflammatory genes such as cytokines. TLR4-mediated NF-κB signaling plays a vital role in the initiation of cerebral inflammation in several central nervous system diseases, such as inflammatory or autoimmune CNS diseases, and cerebral ischemic injury [31]. The TLR4/NF-κB signaling has two different pathways, the MyD88-dependent pathway and the MyD88-independent pathway, and our study object is the former. To study this inflammatory pathway, we chose three key proteins in this pathway, which were TLR4, MyD88, and TRAF6, respectively.
In the RT-PCR assay and western blot experiment, we found that ghrelin down-regulated TLR4/NF-κB pathway which has a close relationship with inflammation. For all of those detected proteins, there was a statistical significance between control group and HG group which proved that high glucose can activate the TLR4/NF-κB pathway. There was also a statistical significance between HG group and HG + ghrelin group which signifies that ghrelin can attenuate the activation of this pathway. Nevertheless, such effect was demolished by D-lys-3-GHRP-6, the inhibitor of ghrelin, and there is no statistical significance versus HG group.
In conclusion, our findings indicate that ghrelin can inhibit high glucose-induced PC12 apoptosis. More researches are required to clarify the changes of cytokines and other inflammatory mediators subsequently. The present studies suggest that ghrelin could be a potential therapeutic for diabetic encephalopathy. And the mechanisms may be that ghrelin alleviates the inflammatory response of diabetic encephalopathy by down-regulating TLR4/NF-κB signaling pathway, which may be a useful strategy for treating diabetic encephalopathy.
Abbreviations
- TLR4/NF-κB:
-
Toll-like receptor 4/nuclear factor-κB
- HG:
-
High glucose
- TEM:
-
Transmission electron microscopy
- FBS:
-
Fetal bovine serum
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- DEPC:
-
Diethylpyrocarbonate
- SDS:
-
Sodium dodecyl sulfate
- ECL:
-
Electrochemiluminescence
- ATCC:
-
American Type Culture Collection
- PBS:
-
Phosphate-buffered saline
- MyD88:
-
Myeloid differentiation primary response gene 88
- TRAF6:
-
Human tumor necrosis factor receptor-associated factor 6
REFERENCES
Biessels, G.J., I.J. Deary, and C.M. Ryan. 2008. Cognition and diabetes: a life span perspective. Lancet Neurology 7: 184–190.
Ferguson, S.C., A. Blane, J. Wardlaw, et al. 2005. Influence of early onset age of type 1 diabetes on cerebral structure and cognitive function. Diabetes Care 28: 1431–1437.
Perantie, D.C., J. Wu, J.M. Koller, et al. 2007. Regional brain volume differences associated with hyperglycemia and severe hypoglycemia in youth with type 1 diabetes. Diabetes Care 30: 2331–2337.
Hoffman, W.H., C.M. Artlett, W. Zhang, et al. 2008. Receptor for advanced glycation end products and neuronal deficit in fatal brain edema of diabetic ketoacidosis. Brain Research 1238: 154–162.
Francis, G.J., J.A. Martinez, W.Q. Liu, et al. 2008. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type 1 diabetic encephalopathy. Brain 131(Pt 12): 3311–3334.
Wu, R., M. Zhou, P. Das, et al. 2007. Ghrelin inhibits sympathetic nervous activity in sepsis. American Journal of Physiology. Endocrinology and Metabolism 293: E1697–E1702.
Wu, R., W. Dong, M. Zhou, et al. 2007. Ghrelin attenuates sepsis-induced acute lung injury and mortality in rats. American Journal of Respiratory and Critical Care Medicine 176: 805–813.
Cowley, M.A., R.G. Smith, S. Diano, et al. 2003. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37: 649–661.
Lin, Y., K. Matsumura, M. Fukuhara, et al. 2004. Ghrelin acts at the nucleus of the solitary tract to decrease arterial pressure in rats. Hypertension 43: 977–982.
Zhang, W., T.R. Lin, Y. Hu, et al. 2004. Ghrelin stimulates neurogenesis in the dorsal motor nucleus of the vagus. The Journal of Physiology 559: 729–737.
Navarro-González, J.F., and C. Mora-Fernández. 2008. The role of inflammatory cytokines in diabetic nephropathy. Journal of the American Society of Nephrology 19: 433–442.
Teng, W., L. Wang, W. Xue, and C. Guan. 2009. Activation of TLR4-mediated NFkappaB signaling in hemorrhagic brain in rats. Mediators of Inflammation 2009: 473276.
Martin, T.F., and R.N. Grishanin. 2003. PC12 cells as a model for studies of regulated secretion in neuronal and endocrine cells. Methods in Cell Biology 71: 267–286.
Abid-Essefi, S., I. Baudrimont, W. Hassen, et al. 2003. DNA fragmentation, apoptosis and cell cycle arrest induced by zearalenone in cultured DOK, Vero and Caco-2 cells: prevention by vitamin E. Toxicology 192: 237–248.
Pickup, J.C. 2004. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 27(3): 813–823.
Spranger, J., A. Kroke, M. Mohlig, et al. 2003. Inflammatory cytokines and the risk to develop type2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam study. Diabetes 52: 812–817.
Thorand, B., H. Lowel, A. Schneider, et al. 2003. C-reactive protein as a predictor for incident diabetes mellitus among middle-aged men: results from the MONICA Augsburg cohort study. Archives of Internal Medicine 163: 93–99.
Lontchi-Yimagou, Eric, et al. 2013. Diabetes mellitus and inflammation. Current Diabetes Reports 13(3): 435–444.
Kempf, Kerstin, et al. 2006. Inflammation in metabolic syndrome and type 2 diabetes impact of dietary glucose. Annals of the New York Academy of Sciences 1084: 30–48.
Sima, Anders A.F., Weixian Zhang, Christian W. Kreipke, et al. 2009. Inflammation in diabetic encephalopathy is prevented by C-peptide. Rev Diabet Stud 6(1): 37–42.
Sharifi, Ali M., Seyed Hadi Mousavi, and Mona Farhadi. 2007. Study of high glucose-induced apoptosis in PC12 cells: role of Bax protein. J Pharmacol Sci 104: 258–262.
Miao, Y., Q. Xia, Z. Hou, et al. 2007. Ghrelin protects cortical neuron against focal ischemia/reperfusion in rats. Biochemical and Biophysical Research Communications 359: 795–800.
Ma, Louyan, Dongmin Zhang, Yong Tang, et al. 2011. Ghrelin-attenuated cognitive dysfunction in streptozotocin-induced diabetic rats. Alzheimer Disease and Associated Disorders 25: 352–363.
Dolgor, B., P. Kalpesh, and D.T. Dennis. 2011. The effects of ghrelin on inflammation and the immune system. Molecular and Cellular Endocrinology 340: 44–58.
Madison, L.D., J.M. Scarlett, P. Levasseur, et al. 2008. Prostacyclin signaling regulates circulating ghrelin during acute inflammation. Journal of Endocrinology 196: 263–273.
Dixit, V.D., et al. 2009. Reduction of T cell-derived ghrelin enhances proinflammatory cytokine expression: implications for age-associated increases in inflammation. Blood 113(21): 5202–5205.
Wei, G.L., et al. 2004. Ghrelin inhibits proinflammatory responses and nuclear factor-κB activation in human endothelial cells. Circulation 109: 2221–2226.
Cletus, C., et al. 2011. Ghrelin suppresses inflammation and neuronal nitric oxide synthase in focal cerebral ischemia via the vagus nerve. Shock 35(3): 258–265.
Talat, W., et al. 2008. Exogenous ghrelin modulates release of pro- and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery 143(3): 334–342.
Downes, C.E., and P.J. Crack. 2010. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS. British Journal of Pharmacology 160: 1872–1888.
Racke, M.K., and P.D. Drew. 2009. Toll-like receptors in multiple sclerosis. Current Topics in Microbiology and Immunology 336: 155–168.
ACKNOWLEDGMENTS
Our work was supported by grants from both the Key Projects of Medical Science sponsored by Chongqing Municipal Health Bureau in the People’s Republic of China (2010-1-8) and the Natural Science Foundation of Chongqing in the People’s Republic of China (CSTC, 2010BB5396). The study is also supported by the National Foundation of Natural Science of China (no. 81170752).
Conflict of interest
The authors declare no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, X., Xiao, Q., Zhao, K. et al. Ghrelin Inhibits High Glucose-Induced PC12 Cell Apoptosis by Regulating TLR4/NF-κB Pathway. Inflammation 36, 1286–1294 (2013). https://doi.org/10.1007/s10753-013-9667-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-013-9667-2