
Abstract
The WUSCHEL-related homeobox (WOX) gene family is a hotspot for diverse functions in development biology. Recently available whole-genome sequences allowed a more comprehensive analysis of WOX genes in watermelon (Citrullus lanatus). The results of this study provide a genomic framework for further research of watermelon WOX genes and contribute to understanding of the evolutionary mode of WOX genes in Cucurbitaceae crops. The qRT-PCR analysis demonstrated active expression of 11 WOX genes in watermelon tissues, which brings new evidence for WOX genes acting as conserved factors during watermelon development. Moreover, the distinct expression profiles of WOX genes during shoot initiation might lead to different shoot regeneration abilities. This work gives an overview of the differentially expressed WOX genes during shoot regeneration in watermelon. The interrelations of WOX genes, phytohormones and other transcription factors during the process will be the focus of future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The WUSCHEL-related homeobox (WOX) gene family is a subgroup of the plant homeodomain (HB) transcription factors that regulate cell differentiation and cell fate. The WOX genes contain a family featured helix-loop-helix-turn-helix structure compared with other HB transcription factors (Gehring et al. 1990; Mukherjee et al. 2009). Recent studies indicate that plant WOX genes contribute to various physiological and developmental processes. The 15 WOX members of the model eudicot plant Arabidopsis thaliana are grouped into three clades (van der Graaff et al. 2009) and several were shown to function in stem-cell maintenance (Aichinger et al. 2012), embryonic patterning (Haecker et al. 2004) and organ formation (Lin et al. 2013b; Sarkar et al. 2007). Arabidopsis WUSCHEL (AtWUS) was shown to be essential for shoot apical meristem maintenance and promotes vegetative-to-embryonic transition (Mayer et al. 1998; Zuo et al. 2002). Overexpression of AtWOX1 disrupts normal meristem development (Zhang et al. 2011). AtWOX2, 8 and 9 are expressed during zygote development (Haecker et al. 2004; Ueda et al. 2011), and AtWOX2 and 8 promote cotyledon boundary formation (Lie et al. 2012). AtWOX3 (PRS1) recruits founder cells that form lateral domains of vegetative and floral organs (Shimizu et al. 2009). AtWOX4 and 14 both function in regulating vascular meristem development (Etchells et al. 2013; Hirakawa et al. 2010; Ji et al. 2010; Wang et al. 2013a). The function of AtWOX5 is to maintain the root apical meristem by mediating cellular auxin response (Sarkar et al. 2007; Tian et al. 2014). AtWOX6 (PRETTY FEW SEEDS2) regulates ovule development (Park et al. 2005). AtWOX11 and 12 are involved in root organogenesis (Liu et al. 2014b) and AtWOX13 promotes replum formation in fruit (Romera-Branchat et al. 2013). The functional diversity of WOX genes makes it a research hotspot in development biology.
Watermelon (Citrullus lanatus) is an important cucurbit crop with high production about 90 million tons in the world (http://faostat.fao.org/). Most consumers enjoy watermelon for the important nutritional compounds it contains, including sugars, lycopene and healthy amino acids (Collins et al. 2007; Perkins-Veazie et al. 2006). Therefore, watermelon studies have mainly focused on fruit development (Grassi et al. 2013; Guo et al. 2011) and disease resistance (Ouibrahim et al. 2014; Wu et al. 2014), with the aim of improving quality and production. In contrast, there are no reports on the mechanisms regulating shoot regeneration, although regeneration systems and genetic transformation have been successfully applied in watermelon for decades (Choi et al. 1994; Compton et al. 2004; Wang et al. 2013b). Shoot regeneration is affected by the expression of WOX genes (Liu et al. 2014a; Sarkar et al. 2007), especially WUS (Ikeda et al. 2009; Leibfried et al. 2005; Mayer et al. 1998). The completed watermelon genome allows the genome-wide analysis of WOX genes (Guo et al. 2013).
In the present study, we successfully identified 11 WOX genes from the watermelon inbred line 97103 genome. Phylogenetic analysis of WOX proteins was conducted to investigate the evolutionary pattern in Arabidopsis, watermelon, cucumber (Cucumis sativus) and melon (Cucumis melo). We further examined the expression patterns of WOX genes in different watermelon tissues and the process of shoot regeneration. The results provide an overview for the transcription regulation of watermelon shoot regeneration and serve as a guideline for future study.
Materials and methods
Identification of watermelon WOX genes
The 15 sequences of Arabidopsis WOX genes were downloaded from The Arabidopsis Information Resource (http://www.arabidopsis.org) and used as query sequences using TBLASTN (Altschul et al. 1997) to search WOX genes in the watermelon genome database (http://www.icugi.org/cgi-bin/ICuGI/index.cgi) with a cut-off E value of 10−5. Obtained sequences were used as query to search the watermelon genome database again. Redundant sequences with different identification numbers and the same chromosome loci were removed. We also obtained information on chromosome localization for the watermelon WOX genes. The exon/intron structures were analyzed using the online gene structure display server (GSDS, http://gsds.cbi.pku.edu.cn) with coding sequences and genomic sequences (Guo et al. 2007).
Phylogenetic analysis of WOX proteins
A neighbor-joining (NJ) phylogenetic tree was constructed for WOX proteins using MEGA 5.0 software (Tamura et al. 2011). The most parsimonious tree with bootstrap values from 1000 trials was used. Amino acid sequences of WOX protein were aligned using Clustal X (Thompson et al. 1997). The Arabidopsis WOX proteins were from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/) and were selected as the model system for phylogenetic comparisons. The WOX proteins in cucumber and melon were downloaded from National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov). The WOX sequences of orange, soybean, tomato and grape were downloaded from Kyoto Encyclopedia of Genes and Genomes (http://www.kegg.jp).
Plant materials and sampling
Plants of the diploid watermelon inbred line A7 were grown in a temperature-controlled greenhouse at the experimental farm of Wuhan Institute of Agricultural Sciences. Unexpanded leaves, stems, shoot buds and flower buds were collected from the top of flowering plants and roots were collected from the root tip part after 2 months of growth. All samples were performed in triplicate and frozen in liquid nitrogen immediately and stored at −75 °C until RNA isolation.
Regeneration studies were according to published protocols (Compton et al. 2004). Seeds were decoated and surface-sterilized in 10 % NaClO solution for 5 min. After several washings in sterile distilled water, seeds were implanted in culture tubes (25 × 150 mm) containing 20 mL of Murashige and Skoog (MS) medium for germination. Cotyledons from 7-day-old aseptic seedlings were cut into four types (Fig. 1) of segments (0.5 × 0.5 mm) and cultured on MS medium supplemented with 6-BA (1.0 mg/L, Sigma) and NAA (1.0 mg/L, Sigma) in a culture room under cool white fluorescent light with a 16/8 h (light/dark) cycle at 25 ± 2 °C during the day and 20 ± 2 °C at night. Each experiment was repeated three times with 24 cotyledon segments per treatment in each experiment. The adventitious shoot numbers of the four types of cotyledon segments were counted after 21 days and data analyzed using SPSS (Liu et al. 2003).

Adventitious shoot-regeneration frequency and morphological observation for different cotyledon segments of watermelon. A, B Cotyledon segments of A, B and C when incubated for 21 days, respectively; D1–4 cotyledon segments of D when incubated for 7, 14, 21 and 28 days, respectively. The arrows indicated adventitious shoots
RNA was extracted from cotyledon segments grown under the same growth conditions. The samples for type-D segments were collected at 7, 14, 21 and 28 days (Fig. 1, D1–4) and the samples for the four types of segments were collected when incubated for 14 days. Approximately ten explants from the same plate were pooled for RNA extraction, which was repeated three times per treatment.
Quantitative real-time PCR (qRT-PCR) analysis
Watermelon samples at the different growth stages were collected and total RNA was isolated from each sample using a Tiangen®RNA prep Pure Plant Kit (Tiangen Biomart, Beijing). Reverse cDNA for each sample was generated using the GoScript™ Reverse Transcription System (Promega, USA), according to the manufacturer’s instructions. An optical 96-well plate iQ5 multicolor real-time PCR system (Bio-RAD, USA) was used for qRT-PCR analysis. Each reaction contained 1 μL of cDNA template, 10 nM gene-specific primers, 10 μL of iTaq™ Universal SYBR® Green Supermix (Bio-RAD, USA) and 7 μL of ddH2O in a final volume of 20 μL. The watermelon glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) gene was selected as the endogenous control (Kong et al. 2014). Gene-specific primers (Table 1) were designed using Primer 3 (http://primer3.ut.ee/) and commercially synthesized (Sunny Biotech, Shanghai). The thermal cycle used was as follows: 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. Following amplification, a dissociation stage was carried out to detect any complex products. The qRT-PCR was performed in triplicate for each sample. Relative expression levels were calculated as reported previously (Livak and Schmittgen 2001).
Results
Identification of watermelon WOX genes
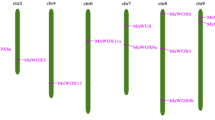
After removing the redundant sequences, 11 WOX genes were obtained and named according to both phylogenetic groups and closely related Arabidopsis homologs (Table 2). The 11 WOX genes were located on one or two ends of eight chromosomes (Fig. 2). The predicted nucleotide and protein sequences of all 11 watermelon WOX genes are provided in Supplementary material 1.

Position of WOX genes on the watermelon chromosomes. The chromosome number is indicated at the top of each chromosome. The arrows next to these numbers show the direction of transcription
The exon/intron positions and phases of the 11 WOX genes in watermelon were further studied. The exon/intron structures were analyzed using GSDS with coding sequences and genomic sequences (Guo et al. 2007). Figure 3 provides a detailed illustration of the relative lengths of the introns and conservation of the corresponding exon sequences within each WOX gene. There were 1–3 introns each in the 11 WOX genes: 1 intron for ClaWOX2, 3, 5, 6, and 10; two for ClaWUS, ClaWOX4, 7 and 8; and three for ClaWOX1 and 9.

Intron/exon configurations of WOX genes in watermelon. Introns and exons are drawn to scale with the full encoding regions of their respective genes. Boxes indicate the exon, and lines indicate the intron. 0 intron phase 0, 1 intron phase 1, 2 intron phase 2
Phylogenetic analysis of WOX proteins in Arabidopsis, Cucurbitaceae crops and other four eudicot plants
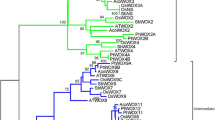
Phylogenetic analysis via the NJ method was conducted in order to determine the relationships among WOX proteins in Arabidopsis, three Cucurbitaceae crops (Table 2) and other four eudicot plants. The Arabidopsis WOX proteins were selected as the model system for phylogenetic comparisons. The Cucurbitaceae sequences were grouped together in each of the subfamilies, respectively. Previous studies showed that the Arabidopsis WOX proteins were divided into three subclades (van der Graaff et al. 2009; Zhang et al. 2010). As a result, WUS, ancient and intermediate clades were composed of 22, six and six Cucurbitaceae sequences, respectively (Fig. 4). The WUS clade contained seven sequences from watermelon, eight from cucumber and seven from melon. Two sequences from each Cucurbitaceae crop were clustered into ancient and intermediate clades.

Phylogenetic analysis of WOX genes in Arabidopsis, orange, soybean, tomato, grape, watermelon, cucumber and melon. Phylogenetic inference was conducted using MEGA 5.0 (Tamura et al. 2011). Branch width corresponds to support values. The numbers on the branches indicate the percentage of 1000 bootstrap replicates that support the node with only values >50 % reported. The Arabidopsis proteins are shown in blue, watermelon in red, cucumber in pink and melon in black. The three subclades are colored: WUS in green, ancient in purple and intermediate in yellow
The WOX family contains the helix-loop-helix-turn-helix domain (Kamiya et al. 2003) and most WOX proteins contain the complete or incomplete WUS box (Lin et al. 2013b). We conducted multiple sequence alignments of the 49 amino acid sequences to investigate features of WOXs (Fig. 5). The proximate Cucurbitaceae sequences were highly conserved. The homeodomain sequences have been shown to contain several conserved amino acids, e.g., Q, L and Y in helix1; P, I and L in helix2; I, N, V, W, F, Q, N, K, R and R in helix3; and G in turn. Interestingly, CsaWOX5-like2 lacked the helix3 region and ClaWOX10 lacked the helix1 region. Furthermore, we found that most WOX proteins in the WUS clade contained a complete WUS box (amino acids, TL × LFP) except AtWOX7, CmeWOX5-like2, CsaWOX1-like, ClaWOX6 and CmeWOX1. Ancient and intermediate clade WOXs contained incomplete WUS boxes with only one conserved amino acid F.

Comparative analysis of the WOX homeodomain and WUS box sequences in Arabidopsis, watermelon, cucumber and melon. The homeodomain of WOX family contains the helix-loop-helix-turn-helix structure (Kamiya et al. 2003). The shaded blocks indicate highly conserved residues by the alignment of homeodomains and WUS boxes in the four angiosperms
Expression of WOX genes in watermelon tissues
To better understand where watermelon WOX genes are active, we analyzed their expression in different tissues from flowering-period plants using qRT-PCR (Fig. 6). All WOX genes were expressed at higher levels in leaf, bud and flower than in stem and extremely low levels in root and fruit. ClaWOX10 was expressed at a much higher level in leaf than other tissues. Additionally, ClaWOX4, 7 and 8 were more highly expressed in root, and ClaWOX8 at much higher levels in fruit, than other WOXs.

Expression patterns of WOX genes in watermelon tissues. The R, L, S, B, FL and FR of X-axis represent root, leaf, stem, bud, flower and fruit, respectively
Expression of WOX genes during adventitious shoot regeneration of watermelon
Cotyledons from the diploid watermelon inbred line A7 were cut into four types of segments (Fig. 1) and incubated on adventitious shoot induction medium, MS with 6-BA (1.0 mg/L) and NAA (1.0 mg/L). After 21 days, we counted adventitious shoot numbers on different types of cotyledon segments (Fig. 1a–c, d3). The result showed that proximal segments had a much higher shoot-regeneration frequency than distal parts of cotyledon (Fig. 1). Therefore, the proximal cotyledon segments (type-D) were selected for qRT-PCR analysis to reveal the expression modulation of WOX genes during watermelon shoot regeneration.
To generate a complete set of observations of WOX genes during shoot regeneration, proximal cotyledon segments (type-D) were sampled when cultured for 7, 14, 21 and 28 days (Fig. 1d1–4). The expression patterns of the 11 watermelon WOX genes showed that most of them were up-regulated at 14 and 28 days (Fig. 7), indicating that expression of WOX genes at 14 days might be related to shoot regeneration.

Expression level of WOX genes in type-D of watermelon cotyledon segments at different development stages
We conducted a further study to examine whether the up-regulation at 14 days of WOX genes affect shoot regeneration. The four types of cotyledon segments were cultured for 14 days and then sampled for qRT-PCR analysis. There were distinct expression patterns of WOX genes in the four cotyledon segment types (Fig. 8). All watermelon WOX genes were expressed at much higher levels in type-A segments. ClaWUS, ClaWOX1, 2, 3, 6 and 9 were expressed at much lower levels in type-B segments than type-C and -D. Additionally, expression of WOX genes differed slightly between type-C and -D segments.

Expression level of WOX genes in four types of cotyledon segments when incubated for 14 days
Discussion
Cucurbitaceae genomes possess smaller scales of WOX genes than Arabidopsis
We successfully identified 11 watermelon WOX genes, which supplement WOX family in the plant kingdom. Previous studies reported a large number of WOX genes in other plants, such as Arabidopsis, poplar, rice, maize and cucumber (Lian et al. 2014; van der Graaff et al. 2009)—on average there were >10 WOX genes in these plants. As we know, the gene family scale in genome was influenced by whole-genome duplication (WGD) (Huang et al. 2009). There were 11 WOX genes in watermelon and melon, while 12 in cucumber because of duplication in AtWOX2 subgroup. We inferred that 11 WOX genes might be the smallest number needed for normal growth and development of Cucurbitaceae plants. There were 1–3 introns for each watermelon WOX gene, consistent with a previous report (Zhang et al. 2010); however, we did not find any watermelon WOX genes without an intron.
Conserved sequence of WOX proteins in Cucurbitaceae crops
In this study, phylogenetic analysis of WOX protein sequences revealed that Cucurbitaceae WOX proteins shared a higher similarity to each other than did those in Arabidopsis or other eudicot plants. The homeodomain of proximate Cucurbitaceae sequences showed high conservation, which indicated a close evolutionary history. Additionally, the ancient clade Cucurbitaceae WOXs were more distinct than those of Arabidopsis, which might indicate a longer evolutionary history than the other clades (Lian et al. 2014). The comparative analysis of homeodomain amino acids further supported this proposal. The conserved amino acids in the homeodomain and WUS box of WUS clade WOXs were consistent with previous studies (Lin et al. 2013b; Zhang et al. 2010). Moreover, the absence of helix1 in ClaWOX10 and helix3 in CsaWOX5-like2 were interesting findings. The reasons for this phenomenon and their functions require further study. The absence of WUS box in CsaWOX1-like, ClaWOX6 and CmeWOX1 suggested that they might share the same function as AtWOX7 (Lin et al. 2013a).
WOX genes were actively expressed in watermelon tissues
The expression patterns of WOX genes in watermelon tissues gave us a better understanding of their functions. Most watermelon WOX genes were actively expressed in root, leaf, stem, bud and flower, respectively. The higher expression of ClaWOX4, 7 and 8 compared to other WOXs in roots indicated that they might have the same function as the key regulation factor AtWOX5 (Sarkar et al. 2007) in the root apical meristem. The high expression of ClaWOX10 barely in leaf suggested it had an important role similar to Medicago truncatula STENOFOLIA (Zhang et al. 2014) in leaf development. However, there is still much work required to identify key WOX regulators during stem, bud and flower development: such as AtWOX4 (Hirakawa et al. 2010; Ji et al. 2010) in the vascular meristem; AtWUS (Mayer et al. 1998; Schoof et al. 2000) in shoot apical meristem; and AtWOX2, 8 and 9 (Breuninger et al. 2008; Haecker et al. 2004; Wu et al. 2007) in embryonic development. Furthermore, ClaWOX8 showed distinct expression in watermelon fruit compared to other WOXs. AtWOX13 was reported to promote replum formation in fruit (Romera-Branchat et al. 2013). Previous studies on watermelon fruit mainly focused on functional genes that regulate metabolite profiles (Grassi et al. 2013; Guo et al. 2011). Studying the dynamics of WOX genes might bring a new perspective to understanding of the mechanisms of watermelon fruit and zygote development.
WOX genes were differentially expressed during adventitious shoot regeneration of watermelon
Watermelon cotyledons are regarded as the best resource of explants for adventitious shoot regeneration (Compton et al. 2004). We cultured cotyledon segments of diploid watermelon inbred line A7 to evaluate adventitious shoot regeneration ability of the genotype. Phytohormones and genotype are reportedly the most important factors affecting shoot regeneration (Akasaka-Kennedy et al. 2005; Landi and Mezzetti 2006; Pourhosseini et al. 2013; Sul and Korban 2005; Wang et al. 2013b). Surprisingly, only proximal cotyledon segments (type-D) showed a high shoot-regeneration frequency under the same culture conditions (Fig. 1). Few molecular studies have attempted to explain this phenomenon although it was discovered a decade ago (Compton 2000). WUS genes were reported to promote shoot regeneration (Chatfield et al. 2013; Li et al. 2011; Rashid et al. 2007) and thus we investigated the expression modulations of watermelon WOX genes during the shoot regeneration process. Most WOX genes were up-regulated when incubated for 14 days, indicating shoot meristem formation at this time point (Motte et al. 2014). The up-regulation of WOX genes at 28 days might be caused by shoot apical meristem maintenance (Mayer et al. 1998; Schoof et al. 2000).
We further analyzed the expression of WOX genes in the four types of cotyledon segments when cultured for 14 days. The results showed distinct expression patterns of WOX genes in type-A and -B segments, both of which generated few adventitious shoots. Overexpression of WUSCHEL could induce somatic embryogenesis and ectopic morphogenesis (Arroyo-Herrera et al. 2008; Bouchabke-Coussa et al. 2013; Xu et al. 2005). These processes might interrupt normal shoot regeneration in type-A segments. In type-B segments, ClaWUS, ClaWOX1, 2, 3, 6 and 9 might be expressed at too low levels to maintain the shoot apical meristem (Carles and Fletcher 2003; Schoof et al. 2000). Watermelon WOX genes had moderate and almost identical expression patterns in type-C and -D segments; however, there was a large difference between shoot-regeneration frequencies of these two types. This phenomenon might be caused by the higher expression of ClaWOX3 in type-D compared to type-C, and more molecular evidence is needed to reveal the functions of WOX genes during shoot regeneration (Cary et al. 2002).
Conclusion
The results of this study provide a genomic framework for further research on watermelon WOX genes and contribute to understanding of the evolutionary mode for WOX genes in Cucurbitaceae crops. The qRT-PCR analysis demonstrates the active expression of the 11 WOX genes in watermelon tissues, and provides new evidence for WOX genes acting as conserved factors during watermelon development. Moreover, the distinct expression profiles of WOX genes during shoot initiation led to different shoot regeneration abilities. This work gives us an overview of how WOX genes regulate shoot regeneration. The interrelations of WOX genes, phytohormone and other transcription factors during this process will be the focus of future studies.
Author contribution statement
Na Zhang, Dingxiang Peng and Xing Huang designed the experiment. Dingxiang Peng and Yuhong Sun provided guidance on the whole study. Na Zhang prepared materials and RNA samples. Xing Huang conducted qRT-PCR validation. Yaning Bao, Lijun Liu and Xia An helped in material preparation. Jie Chen and Lunjin Dai helped in RNA preparation. Na Zhang and Xing Huang carried out the data analysis and wrote the manuscript. Bo Wang revised the manuscript. All authors read and approved the final manuscript.
Abbreviations
- WOX:
-
WUSCHEL-related homeobox
- WUS:
-
WUSCHEL
- MS:
-
Murashige and Skoog
- 6-BA:
-
6-Benzylaminopurine
- NAA:
-
Naphthaleneacetic acid
References
Aichinger E, Kornet N, Friedrich T, Laux T (2012) Plant stem cell niches. Annu Rev Plant Biol 63:615–636. doi:10.1146/annurev-arplant-042811-105555
Akasaka-Kennedy Y, Yoshida H, Takahata Y (2005) Efficient plant regeneration from leaves of rapeseed (Brassica napus L.): the influence of AgNO3 and genotype. Plant Cell Rep 24:649–654. doi:10.1007/s00299-005-0010-8
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Arroyo-Herrera A et al (2008) Expression of WUSCHEL in Coffea canephora causes ectopic morphogenesis and increases somatic embryogenesis. Plant Cell Tiss Org 94:171–180. doi:10.1007/s11240-008-9401-1
Bouchabke-Coussa O, Obellianne M, Linderme D, Montes E, Maia-Grondard A, Vilaine F, Pannetier C (2013) Wuschel overexpression promotes somatic embryogenesis and induces organogenesis in cotton (Gossypium hirsutum L.) tissues cultured in vitro. Plant Cell Rep 32:675–686. doi:10.1007/s00299-013-1402-9
Breuninger H, Rikirsch E, Hermann M, Ueda M, Laux T (2008) Differential expression of WOX genes mediates apical–basal axis formation in the Arabidopsis embryo. Dev Cell 14:867–876. doi:10.1016/j.devcel.2008.03.008
Carles CC, Fletcher JC (2003) Shoot apical meristem maintenance: the art of a dynamic balance. Trends Plant Sci 8:394–401. doi:10.1016/S1360-1385(03)00164-X
Cary AJ, Che P, Howell SH (2002) Developmental events and shoot apical meristem gene expression patterns during shoot development in Arabidopsis thaliana. Plant J 32:867–877. doi:10.1046/j.1365-313X.2002.01479.x
Chatfield SP, Capron R, Severino A, Penttila PA, Alfred S, Nahal H, Provart NJ (2013) Incipient stem cell niche conversion in tissue culture: using a systems approach to probe early events in WUSCHEL-dependent conversion of lateral root primordia into shoot meristems. Plant J 73:798–813. doi:10.1111/Tpj.12085
Choi PS, Soh WY, Kim YS, Yoo OJ, Liu JR (1994) Genetic transformation and plant regeneration of watermelon using Agrobacterium tumefaciens. Plant Cell Rep 13:344–348. doi:10.1007/BF00232634
Collins JK, Wu GY, Perkins-Veazie P, Spears K, Claypool PL, Baker RA, Clevidence BA (2007) Watermelon consumption increases plasma arginine concentrations in adults. Nutrition 23:261–266. doi:10.1016/j.nut.2007.01.005
Compton ME (2000) Interaction between explant size and cultivar affects shoot organogenic competence of watermelon cotyledons. HortScience 35:749–750
Compton ME, Gray DJ, Gaba VP (2004) Use of tissue culture and biotechnology for the genetic improvement of watermelon. Plant Cell Tiss Org 77:231–243. doi:10.1023/B:Ticu.0000018428.43446.58
Etchells JP, Provost CM, Mishra L, Turner SR (2013) WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organisation. Development 140:2224–2234. doi:10.1242/Dev.091314
Gehring WJ et al (1990) The structure of the homeodomain and its functional implications. Trends Genet TIG 6:323–329
Grassi S et al (2013) Comparative genomics reveals candidate carotenoid pathway regulators of ripening watermelon fruit. BMC Genomics. doi:10.1186/1471-2164-14-781 (Artn 781)
Guo AY, Zhu QH, Chen X, Luo JC (2007) GSDS: a gene structure display server. Yi Chuan 29:1023–1026 (in Chinese)
Guo SG et al (2011) Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. BMC Genomics. doi:10.1186/1471-2164-12-454 (Artn 454)
Guo SG et al (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45:U51–U82. doi:10.1038/Ng.2470
Haecker A, Gross-Hardt R, Geiges B, Sarkar A, Breuninger H, Herrmann M, Laux T (2004) Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 131:657–668. doi:10.1242/Dev.00963
Hirakawa Y, Kondo Y, Fukuda H (2010) TDIF peptide signaling regulates vascular stem cell proliferation via the WOX4 homeobox gene in Arabidopsis. Plant Cell 22:2618–2629. doi:10.1105/tpc.110.076083
Huang S et al (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281. doi:10.1038/ng.475
Ikeda M, Mitsuda N, Ohme-Takagi M (2009) Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 21:3493–3505. doi:10.1105/tpc.109.069997
Ji J, Strable J, Shimizu R, Koenig D, Sinha N, Scanlon MJ (2010) WOX4 promotes procambial development. Plant Physiol 152:1346–1356. doi:10.1104/pp.109.149641
Kamiya N, Nagasaki H, Morikami A, Sato Y, Matsuoka M (2003) Isolation and characterization of a rice WUSCHEL-type homeobox gene that is specifically expressed in the central cells of a quiescent center in the root apical meristem. Plant J 35:429–441. doi:10.1046/j.1365-313X.2003.01816.x
Kong Q, Yuan J, Gao L, Zhao S, Jiang W, Huang Y, Bie Z (2014) Identification of suitable reference genes for gene expression normalization in qRT-PCR analysis in watermelon. Plos One 9:e90612. doi:10.1371/journal.pone.0090612
Landi L, Mezzetti B (2006) TDZ, auxin and genotype effects on leaf organogenesis in Fragaria. Plant Cell Rep 25:281–288. doi:10.1007/s00299-005-0066-5
Leibfried A et al (2005) WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 438:1172–1175. doi:10.1038/Nature04270
Li W, Liu H, Cheng ZJ, Su YH, Han HN, Zhang Y, Zhang XS (2011) DNA methylation and histone modifications regulate de novo shoot regeneration in Arabidopsis by modulating WUSCHEL expression and auxin signaling. PLoS Genet. doi:10.1371/journal.pgen.1002243 (ARTN e1002243)
Lian G, Ding Z, Wang Q, Zhang D, Xu J (2014) Origins and evolution of WUSCHEL-related homeobox protein family in plant kingdom. ScientificWorldJournal 2014:534140. doi:10.1155/2014/534140
Lie C, Kelsom C, Wu X (2012) WOX2 and STIMPY-LIKE/WOX8 promote cotyledon boundary formation in Arabidopsis. Plant J 72:674–682. doi:10.1111/j.1365-313X.2012.05113.x
Lin H, Niu LF, McHale NA, Ohme-Takagi M, Mysore KS, Tadege M (2013a) Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc Natl Acad Sci USA 110:366–371. doi:10.1073/pnas.1215376110
Lin H, Niu L, Tadege M (2013b) STENOFOLIA acts as a repressor in regulating leaf blade outgrowth. Plant Signal Behav 8:e24464. doi:10.4161/psb.24464
Liu RX, Kuang J, Gong Q, Hou XL (2003) Principal component regression analysis with SPSS. Comput Methods Prog Biomed 71:141–147. doi:10.1016/S0169-2607(02)00058-5
Liu JC, Sheng LH, Xu YQ, Li JQ, Yang ZN, Huang H, Xu L (2014a) WOX11 and 12 are involved in the first-step cell fate transition during de novo root organogenesis in Arabidopsis. Plant Cell 26:1081–1093. doi:10.1105/tpc.114.122887
Liu BB, Wang L, Zhang J, Li JB, Zheng HQ, Chen J, Lu MZ (2014b) WUSCHEL-related Homeobox genes in Populus tomentosa: diversified expression patterns and a functional similarity in adventitious root formation. BMC Genomics. doi:10.1186/1471-2164-15-296 (Artn 296)
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. doi:10.1006/meth.2001.1262
Mayer KF, Schoof H, Haecker A, Lenhard M, Jurgens G, Laux T (1998) Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95:805–815
Motte H, Vereecke D, Geelen D, Werbrouck S (2014) The molecular path to in vitro shoot regeneration. Biotechnol Adv 32:107–121. doi:10.1016/j.biotechadv.2013.12.002
Mukherjee K, Brocchieri L, Burglin TR (2009) A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol 26:2775–2794. doi:10.1093/molbev/msp201
Ouibrahim L et al (2014) Cloning of the Arabidopsis rwm1 gene for resistance to Watermelon mosaic virus points to a new function for natural virus resistance genes. Plant J 79:705–716. doi:10.1111/tpj.12586
Park SO, Zheng ZG, Oppenheimer DG, Hauser BA (2005) The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development 132:841–849. doi:10.1242/Dev.01654
Perkins-Veazie P, Collins JK, Davis AR, Roberts W (2006) Carotenoid content of 50 watermelon cultivars. J Agric Food Chem 54:2593–2597. doi:10.1021/Jf052066p
Pourhosseini L, Kermani MJ, Habashi AA, Khalighi A (2013) Efficiency of direct and indirect shoot organogenesis in different genotypes of Rosa hybrida. Plant Cell Tiss Org 112:101–108. doi:10.1007/s11240-012-0210-1
Rashid SZ, Yamaji N, Kyo M (2007) Shoot formation from root tip region: a developmental alteration by WUS in transgenic tobacco. Plant Cell Rep 26:1449–1455. doi:10.1007/s00299-007-0342-7
Romera-Branchat M, Ripoll JJ, Yanofsky MF, Pelaz S (2013) The WOX13 homeobox gene promotes replum formation in the Arabidopsis thaliana fruit. Plant J 73:37–49. doi:10.1111/Tpj.12010
Sarkar AK et al (2007) Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature 446:811–814. doi:10.1038/nature05703
Schoof H, Lenhard M, Haecker A, Mayer KF, Jurgens G, Laux T (2000) The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 100:635–644
Shimizu R, Ji J, Kelsey E, Ohtsu K, Schnable PS, Scanlon MJ (2009) Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol 149:841–850. doi:10.1104/pp.108.130765
Sul IW, Korban SS (2005) Direct shoot organogenesis from needles of three genotypes of Sequoia sempervirens. Plant Cell Tiss Org 80:353–358. doi:10.1007/s11240-004-1365-1
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tian HY et al (2014) WOX5-IAA17 feedback circuit-mediated cellular auxin response is crucial for the patterning of root stem cell niches in Arabidopsis. Mol Plant 7:277–289. doi:10.1093/Mp/Sst118
Ueda M, Zhang ZJ, Laux T (2011) Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development (vol 20, pg 264, 2011). Dev Cell 20:408. doi:10.1016/j.devcel.2011.03.009
van der Graaff E, Laux T, Rensing SA (2009) The WUS homeobox-containing (WOX) protein family. Genome Biol. doi:10.1186/Gb-2009-10-12-248 (Artn 248)
Wang XZ, Shang LM, Luan FS (2013a) A highly efficient regeneration system for watermelon (Citrullus lanatus Thunb.). Pak J Bot 45:145–150
Wang JH et al (2013b) The Arabidopsis LRR-RLK, PXC1, is a regulator of secondary wall formation correlated with the TDIF-PXY/TDR-WOX4 signaling pathway. BMC Plant Biol. doi:10.1186/1471-2229-13-94 (Artn 94)
Wu X, Chory J, Weigel D (2007) Combinations of WOX activities regulate tissue proliferation during Arabidopsis embryonic development. Dev Biol 309:306–316. doi:10.1016/j.ydbio.2007.07.019
Wu Z, Wang W, Li Y, Rao X (2014) Development of polyclonal antibodies against nucleocapsid protein of watermelon silver mottle virus and their application to diagnostic. Acta Virol 58:167–172
Xu YY et al (2005) Activation of the WUS gene induces ectopic initiation of floral meristems on mature stem surface in Arabidopsis thaliana. Plant Mol Biol 57:773–784. doi:10.1007/s11103-005-0952-9
Zhang F, Wang YW, Li GF, Tang YH, Kramer EM, Tadege M (2014) STENOFOLIA recruits TOPLESS to repress ASYMMETRIC LEAVES2 at the leaf margin and promote leaf blade outgrowth in Medicago truncatula. Plant Cell 26:650–664. doi:10.1105/tpc.113.121947
Zhang Y, Wu R, Qin G, Chen Z, Gu H, Qu LJ (2011) Over-expression of WOX1 leads to defects in meristem development and polyamine homeostasis in Arabidopsis. J Integr Plant Biol 53:493–506. doi:10.1111/j.1744-7909.2011.01054.x
Zhang X, Zong J, Liu JH, Yin JY, Zhang DB (2010) Genome-wide analysis of WOX gene family in Rice, Sorghum, Maize, Arabidopsis and Poplar. J Integr Plant Biol 52:1016–1026. doi:10.1111/j.1744-7909.2010.00982.x
Zuo J, Niu QW, Frugis G, Chua NH (2002) The WUSCHEL gene promotes vegetative-to-embryonic transition in Arabidopsis. Plant J 30:349–359
Acknowledgments
The authors thank Yuhua Li, Xianfeng Shi and Hongxia Zeng (Wuhan Institute of Agricultural Science) for watermelon cultivation management. This study was supported by the Supporting Program for Science and Technology Research of Hubei Province (2012BBA05001).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Communicated by M. Hajduch.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, N., Huang, X., Bao, Y. et al. Genome-wide identification and expression profiling of WUSCHEL-related homeobox (WOX) genes during adventitious shoot regeneration of watermelon (Citrullus lanatus). Acta Physiol Plant 37, 224 (2015). https://doi.org/10.1007/s11738-015-1964-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-015-1964-y