
Abstract
Single nucleotide polymorphisms (SNPs) have rapidly become the molecular marker of choice in plant and animal association mapping (AM) studies. In this work, a genome-wide association study (GWAS) and candidate quantitative trait loci (cQTL) approaches were used to identify SNP markers associated with seed quality traits, in a Brassica napus L. association panel composed of 89 adapted winter oilseed rape accessions. Six seed quality traits (oil and protein content, linolenic acid, total glucosinolates, hemicellulose and cellulose content) were evaluated in two different locations for two seasons. For GWAS, 4025 SNP markers evenly distributed along the B. napus genome were genotyped using a 6K Illumina array platform. For cQTL, 100 SNP markers previously discovered in genomic regions underlying seed quality QTL were genotyped using a competitive allele-specific PCR (KASPar). Analysis of the population structure revealed the presence of two weakly differentiated subpopulations (F ST = 0.037), with 82 % of the pairwise kinship comparisons ranging from 0 to 0.1. The GWAS approach resulted in the identification of 17 and 5 significant associations for seed glucosinolate content and seed hemicellulose content, respectively. The cQTL approach identified 4 significant associations for seed glucosinolate content and 6 significant associations for seed hemicellulose content. The associated SNPs were consistently identified across environments and were mapped to previously reported QTL. These results illustrate the suitability of AM to identify SNP markers associated with seed quality traits in B. napus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Association mapping (AM) is a valuable tool for the dissection of quantitative trait loci (QTL) controlling complex traits in crop plants. This approach detects associations between phenotypes and markers on the basis of linkage disequilibrium (LD). AM decreases the time and cost associated with the generation and maintenance of biparental populations (often used in linkage mapping), using association panels (Zhu et al. 2008). In addition, the use of association panels provides a higher mapping resolution since it exploits all the ancestral recombination events present in a plant species (Soto-Cerda and Cloutier 2012).
One prerequisite for AM resides in the evaluation of the extent of LD as well as any population and family structures that are present in a supposedly genetically diverse panel (Sorkheh et al. 2008). There have been attempts to characterize the extent of LD and define the number of markers required for suitable mapping resolution in B. napus (Ecke et al. 2010; Bus et al. 2011; Xiao et al. 2012). Recently, a more extensive study (Delourme et al. 2013) generated a high-density SNP-based genetic map (2.56 SNP per cM) using 5764 SNP markers and 1603 PCR markers in four segregating double haploid (DH) populations. The extent of LD varied from 0.7 to 1.2 cM, and a faster LD decay was observed in spring compared to winter oilseed rape lines. A wide variation in the extent of LD was also observed along the different linkage groups. The authors concluded that a relatively low number (few thousands) of evenly spaced SNP markers would be necessary to perform genome-wide association studies (GWAS) in oilseed rape (Delourme et al. 2013). Similarly, Bus et al. (2011) elegantly analyzed a genetically diverse panel composed of 509 B. napus accessions, concluding that a minimum of 1.5–2 markers (~2000 markers) every cM were needed for a detailed GWAS in this species.
Assessing the population structure of an association panel is necessary to minimize the occurrence of spurious or false-positive associations that might arise as a consequence of an unbalanced allele frequency distribution among individuals of diverse geographic origin or breeding history. There are several algorithms to estimate population structure. One of the most commonly used algorithms is STRUCTURE (Pritchard et al. 2000), which uses allele frequency differences between populations to infer ancestry of individuals. Principal coordinates (PCoA) and phylogenetic analyses are usually used as complementary approaches to confirm the results obtained using STRUCTURE.
Oilseed rape (B. napus; 2n = 4x = 38, genome AACC) is the world’s second most important oilseed crop after soybean (Glycine max) (www.fao.org). The seeds are principally used for edible oil production, and the high-value protein-rich meal is used to feed livestock. Particularly in Europe, an alternative non-food use is biofuel production (Rashid and Anwar 2008). Understanding the biology and genetics of characters related to seed quality (e.g., seed oil and protein content, fatty acid profile and antinutritional factors) is crucial for oilseed rape breeding programs worldwide. Several studies have identified regions (QTL) of the B. napus genome associated with seed quality traits (Ecke et al. 1995; Toroser et al. 1995; Uzunova et al. 1995; Thormann et al. 1996; Zhao and Meng 2003; Marwede et al. 2005; Zhao et al. 2005, 2006; Delourme et al. 2006; Qiu et al. 2006; Radoev 2007; Amar et al. 2008; Zhao et al. 2008). However, most of these studies have relied on genetic marker technologies that have been difficult to implement in breeding programs. With the advent of cost-effective genotyping technologies, markers such as SNPs associated with traits of interest could aid marker-assisted selection and trait introgression during the process of oilseed rape improvement. A reasonable approach to link SNP markers to traits of interest is to conduct AM analysis in diversity panels or sets of B. napus.
The short domestication period (400–500 years) and a double bottleneck created during the selection for low erucic acid and low glucosinolates while breeding for modern oilseed rape (Kimber and McGregor 1995, reviewed in Iniguez-Luy and Federico 2011) has led to narrow genetic diversity in modern B. napus with canola seed quality (Bus et al. 2011). For this reason, it is beneficial to use genetically diverse panels of oilseed rape from different geographic origins and breeding status in AM studies to identify useful new genetic variation for seed quality traits.
AM has been widely used to identify significant marker–trait associations in several crops including maize (Poland et al. 2011; Kump et al. 2011; Tian et al. 2011), wheat (Rousset et al. 2011), barley (Pasam et al. 2012), rice (Huang et al. 2010) and bean (Galeano et al. 2012). Two main approaches have been used for AM in plant species: (1) GWAS and (2) candidate gene (CG). GWAS requires the use of a large number of genome-wide markers. Nowadays, next-generation sequencing platforms dramatically reduce the cost and time of obtaining large number of markers, making GWAS an efficient approach for the discovery of QTL in plant genomes. On the other hand, CG is often used to interrogate well-known biosynthetic pathways and typically uses a low number of markers for the search for significant associations. Both types of AM approaches have been used in genetic studies in oilseed rape. For instance, GWAS has been used to identify markers associated with phenological and morphological traits (Honsdorf et al. 2010) and seed quality traits (Zou et al. 2010; Rezaeizad et al. 2011; Harper et al. 2012; Li et al. 2014). CG has been used to identify polymorphisms in genes involved in glucosinolate biosynthesis (Hasan et al. 2008), genes associated with tocopherol content and composition (Fritsche et al. 2012) and allelic resistance to Leptosphaeria maculans (Jestin et al. 2011) in B. napus.
In this study, two different AM approaches, GWAS and candidate QTL (cQTL), were used to identify SNP markers associated with six B. napus seed quality traits. More than 4000 SNP markers, evenly distributed along the oilseed rape genome, were used for the GWAS analysis. In the case of cQTL, a set of 100 SNP markers previously developed by resequencing areas of the B. napus genome harboring QTL for seed quality characters (Clarke et al. 2013) was used for the analysis. This kind of cQTL approach has been previously reported in B. napus (Rezaeizad et al. 2011); however, a bigger population, considerably more markers, additional seed quality traits and more restrictive association mapping models were assessed in the present work. Both mapping approaches were used to analyze seed quality traits obtained in a B. napus winter oilseed rape association panel (89 inbred lines), using seed quality trait data collected during two seasons across two locations in Germany.
Materials and methods
Plant material and field trials
A set of 89 genetically diverse winter oilseed rape accessions, representing cultivars and breeding lines from different European breeding programs, were sown following standard agronomic practices at two locations in Germany, Groβ Gerau (GG) and Rauischholzhausen (R), during two seasons (2010 and 2011). Environments were named as follows: GG2010, GG2011, R2010 and R2011, respectively. The experimental design used was a complete randomized design (CDR) with 3-row plots (2 m long) separated by 35 cm each. Seeds were sown in winter, and mature plants were harvested in the following summer. Three subsamples were harvested from each field for analytical measurements. Biparental mapping was conducted in a subset of informative individuals from the double haploid (DH) population Express 617 × V8 (ExV8-DH; Basunanda et al. 2010). This was conducted in order to anchor SNP markers that were previously discovered in areas of the genome that underlie traits of agronomical and nutritional interest. The DH population was grown in the greenhouse to generate plant tissue for DNA extraction using standard potting mix (Kekkilä Garden, horticultural substrate), standard photoperiod conditions (16 h light) and 70 % humidity.
Seed quality trait measurements
A total of six seed quality traits were evaluated after harvest using near-infrared spectroscopy (NIR), in a NIRSystem 6500 spectrometer (FOSS NIRSystems Inc., Silver Spring, MD) calibrated for each target trait. The six traits studied were: (1) seed oil content (SOC), (2) seed protein content (SPC), (3) seed linolenic acid content (SLAC), (4) seed glucosinolate content (SGC), (5) seed cellulose content (SCC) and (6) seed hemicellulose content (SHC). SOC, SPC, SCC and SHC were calculated as percentage (%) of seed dry weight, SLAC as % of total seed oil, and SGC as μmol/g of seed dry weight. The average values for the four environments for each trait are shown in Online resource 1, Table S1.
Statistical analysis
Variance components, means, ranges and phenotypic correlations were estimated using GenStat v16. In order to estimate the variance components, a restricted maximum likelihood (REML) analysis was conducted for seven sources of variation for the six seed quality traits analyzed. Genotype (G), genotype by location (G × L), genotype by season (G × S) and genotype by location by season (G × L × S) were considered as random terms, while location (L), season (S) and location by season (L × S) were considered as fixed terms. The significance for fixed terms was evaluated using a Wald statistic test. For REML analysis, data expressed in percentages (i.e., all traits except SGC which is expressed in μmol/g) were normalized using the logit transformation procedure in GenStat v16. The mean values of three subsamples by plot were used for the REML analysis. Broad-sense heritability (H) of the evaluated traits was calculated according to the formula H = σ 2g /(σ 2g + σ 2gl + σ 2gs + σ 2gls ), where σ 2g is the genotypic variance, σ 2gl is the genotype by location interaction variance, σ 2gs is the genotype by season interaction variance, and σ 2gls is the genotype by location by season interaction variance.
DNA extraction and SNP genotyping
Nucleic DNA was extracted from flower buds and leaf tissue using the CTAB procedure (Doyle and Doyle 1990) scaled to 1.7 ml volume. DNA was quantified using the Quant-iT PicoGreen dsDNA reagent (Invitrogen Inc., CA) and adjusted to a final concentration of 5 ng/µl. For the GWAS analysis, a total of 5506 SNP markers were assayed against a selection of B. napus accessions (Online resource 1, Table S2) using a Brassica 6K iSelect 24x1HD Custom Genotyping Beadchip (http://aafc-aac.usask.ca/ASSYST/). The samples were prepared and assayed using the Infinium HD Assay Ultra Protocol (Infinium HD Ultra User Guide 11328087_RevB, Illumina, Inc. San Diego, CA). The Brassica 6K beadchips were imaged using an Illumina HiScan system, and the SNP alleles were called using the Genotyping Module v1.9.4, within the Genome Studio software suite v2011.1 (Illumina, Inc. San Diego, CA).
For cQTL, KASPar assay version 4.0 (LGC Genomics UK, Cuppen 2007) was used to genotype 100 additional SNP markers (Online resource 3) previously discovered by sequence capture technology in genomic regions underlying seed quality traits (Clarke et al. 2013). These QTL were previously reported in the A genome of B. napus and were principally composed of QTL for SOC and SGC (Clarke et al. 2013). A total of 300 primers were designed using Primer Picker (KBiocience) default parameters (Online resource 3). KASPar reactions were conducted according to the manufacturer’s instructions in an 8-µl final volume reaction. GWAS and cQTL analyses were carried out in the same B. napus association panel composed of 89 winter accessions (Online resource 1, Table S2). In addition, the 100 SNP markers used for the cQTL analysis were genotyped in 29 individuals from the ExV8-DH mapping population (Online resource 1, Table S3). This was conducted in order to obtain an estimate of the genomic position (linkage map location) and confirm previously identified QTL positions and newly discovered trait–marker associations.
Population structure, relative kinship and diversity analysis
To elucidate the patterns of population and family structure in the association panel selected for AM, three different approaches were applied: (1) a Bayesian-based analysis, (2) a principal coordinates analysis (PCoA) and (3) a phylogenetic analysis. A subset of 1000 SNP markers randomly distributed through the A and C B. napus genomes were chosen from a total of 5506 SNP markers originally genotyped in the association panel. The Bayesian-based analysis was run in the software STRUCTURE v2.3.4 (Pritchard et al. 2000) using the admixture ancestry and correlated allele frequency models (Falush et al. 2003), with a burn-in period of 10,000 and Markov chain Monte Carlo iterations of 50,000 across ten independent reps for each k (number of populations) from 1 to 12. The most likely number of populations (k) in the association panel was calculated with the Evanno method (Evanno et al. 2005) using STRUCTURE Harvester (Earl and von Holdt 2012). The Q matrix with the lowest variance for the most likely number of k populations was selected and used as the fixed covariate in AM models. PCoA was performed in GENALEX v.6.5 (Peakall and Smouse 2006) using the codominant genotypic distance and the covariance-standardized methodology. A PCoA plot was illustrated using the first two coordinates. To assess the genetic relationships between lines present in the association panel, a dendrogram was generated using a neighbor-joining (NJ) algorithm based on the Nei minimum genetic distance method implemented in Power Maker v3.25 (Liu and Muse 2005) and displayed by MEGA 5. This analysis was performed with 1000 SNP markers, and the reliability of the dendrogram topology was confirmed with 1000 bootstraps with replacement.
The pairwise kinship values (kinship matrix) for the association panel were calculated using the software SpaGEdi v1.4 (Loiselle et al. 1995; Hardy and Vekemans 2002). This kinship matrix was used for the subsequent association analysis in TASSEL 3.0 as a random factor. The kinship coefficient was calculated and plotted versus its frequency in the association panel.
To determine the level of genetic diversity present in the B. napus association panel, the information generated by the 1000 SNP markers was used to estimate the genetic distance between the lines. GENALEX V.6.5 was used to estimate the following: number of alleles, number of effective alleles, Shannon’s information index, observed heterozygosity, expected heterozygosity, unbiased expected heterozygosity, fixation index and F statistics.
Association analysis
The association analysis was performed in TASSEL 3.0 (Bradbury et al. 2007) using the information from 89 lines genotyped with 5506 and 100 SNP markers for the GWAS and cQTL, respectively. SNPs exhibiting minor allele frequencies (MAF) <0.05 were considered as missing data for the analysis (Breseghello and Sorrells 2006). The general linear model (GLM + Q) and two mixed linear models (MLM + K and MLM + Q + K) were used to search for SNP associations with seed quality traits. The GLM + Q model takes into account the population structure (Q matrix), MLM + K considers the relative kinship (K matrix), and MLM + K + Q considers both population structure and relative kinship effects. The Q matrix was obtained from STRUCTURE v2.3.4, and the K matrix was obtained with SpaGedi v1.4. Association analysis was performed for each trait in each environment as well as considering their overall mean for all of the environments evaluated. Correction for multiple testing was performed using the qFDR value which is an extension of the false discovery rate (FDR) method (Benjamini and Hochberg 1995). Values for q were calculated with the QVALUE R package using the smoother method (Storey and Tibshirani 2003). Markers with FDR q < 0.05 were considered significant. In addition, the estimated effects for each allelic class were obtained directly from the mixed linear model. The allelic class effect was defined as the allelic class 1 effect over the allelic class 2 (reference allelic class for TASSEL). Since the allelic class genotype order obtained from the output of TASSEL is arbitrary, the corresponding genotype (A:A or B:B) for each allelic class is included in Table 2.
Biparental mapping of SNP markers used for cQTL approach
To estimate and corroborate the genomic location of the SNP markers used for the cQTL approach, 29 DH lines from the ExV8-DH mapping population (Basunanda et al. 2010) were genotyped with the set of 100 SNP markers described above. Using this genotypic information, the SNP markers were incorporated into a previously developed linkage map from the above-mentioned population using JoinMap® v4.0 (Van Ooijen 2006). Linked loci were grouped using a LOD threshold of 5–8 and a maximum recombination fraction of 0.4. Grouped marker loci, including the newly mapped SNP markers, were arranged into a scoring matrix using JoinMap® v4.0. Map distances were given in centimorgans (cM) and were calculated using the Kosambi mapping function. Seed quality trait QTL confidence intervals were positioned according to a previously reported study (Clarke et al. 2013).
Characterization of significantly associated SNP markers
Significantly associated SNP markers were used to blast existing Brassica species databases (http://brassicadb.org/brad/, Cheng et al. 2011) to search for annotated genes in the Brassica A (Brassica rapa genome, Wang et al. 2011) or C (Brassica oleracea genome, Liu et al. 2014) genomes. SNPs found to be located in a coding sequence (CDS) were classified as genic, whereas all other SNPs were considered intergenic. Additionally, SNPs were localized by a homology search approach via a BLAST analysis of their flanking sequences onto a B. napus ‘Darmor-Bzh’ reference genome (http://www.genoscope.cns.fr/brassicanapus/, Chalhoub et al. 2014). Finally, SNPs were classified as transitions and transversions depending on the type of base substitutions.
Results
Statistical analysis
The descriptive analysis of the residuals showed a normal distribution for SOC, SPC and SHC. An approximately normal distribution of the residuals was observed for SLAC and SCC, and a non-normal distribution of the residuals was observed for SGC (Online resource 2, Fig. S1). Furthermore, when the SGC expressed in μmol/g data distribution was examined (data not shown), it showed a clear bimodal distribution. Variance components for random terms for each of the six seed quality traits studied were calculated using a REML analysis (Table 1). The values of variance components for the random terms suggest an important genetic effect (G) for all the traits analyzed. In addition, significant effect of location (L), season (S) and their interactions (L × S) was observed for all traits, except for SHC and SGC where S and L × S interaction were not significant, respectively. Additionally, mean, range and broad-sense heritability (H) for each of the six seed quality traits studied are also included in Table 1. Moderate to high broad-sense heritability was calculated for all traits, ranging from 0.39 for SCC to 0.97 for SGC (Table 1). Table S4 (Online resource 1) shows phenotypic correlations for all assessed traits. Significant and high negative correlations were observed for SOC–SPC, SOC–SCC, SOC–SHC and SLAC–SCC. On the other hand, positive correlations were observed for SPC–SGC and SCC–SHC.
Population structure, relative kinship and diversity analysis
The Bayesian approach implemented in STRUCTURE revealed the presence of two subpopulations (Fig. 1a) with the highest likelihood for K = 2 (Online resource 2, Fig. S2) and Ln P (D) = −65,329.34 with a variance value of 7.57. The population structure between the two subpopulations was weak but significant (F ST 0.037; p < 0.01, Online resource 1, Table S5b). The PCoA analysis confirmed the existence of the two subpopulations detected by STRUCTURE, with the first two coordinates accounting for 16.7 % of the genotypic variation (Fig. 1b). A phylogenetic analysis partitioned the 89 lines into two principal groups composed of 44 and 45 lines each, similar to the results obtained with STRUCTURE and PCoA analyses (Online resource 2, Fig. S3). Subpopulation 1 is mainly composed of lines from Germany (20), France (11), UK (7) and Moldova (1), whereas subpopulation 2 included accessions of a more diverse origin including accessions originating from Sweden (5), Poland (5), Ukraine (1), Czech Republic (3), Russia (1), North Korea (1), Japan (2), Germany (13) and France (5) (Online resource 1, Table S2) .

Population structure and relative kinship analyses. a Model-based Bayesian clustering performed by STRUCTURE for K = 2 subpopulations. Each of the 89 B. napus winter oilseed rape accessions is represented by a single column broken into colored segments with lengths proportional to each of the K inferred ancestral populations (red subpopulation 1, green subpopulation 2). b Principal coordinates analysis (PCoA) performed in GENALEX for the association panel, based on information provided by 1000 SNP markers. The accessions were colored in accordance with the previously assigned group in STRUCTURE, red subpopulation 1 and green subpopulation 2. c Distribution of relative kinship coefficient across the 89 accessions. (Color figure online)
In addition, the relative kinship between all the accessions was evaluated. The average of pairwise kinship comparisons between any two genotypes was low (0.04). Approximately 82 % of the pairwise kinship comparisons ranged from 0 to 0.1 (Fig. 1c).
Genetic diversity parameters for both subpopulations were calculated using the software GENALEX based on the 1000 SNP markers used for the population structure analysis (Online resource 1, Table S5a and S5b). The results suggest a low heterozygosity and low genetic differentiation within and between subpopulations.
Associations of SNPs to seed quality traits using GWAS and cQTL approaches
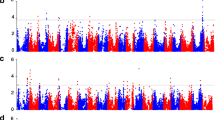
After examining the entire SNP data set for minor allele frequency (MAF < 0.05), a total of 4025 SNPs were retained for the GWAS analysis. The results of the three tested AM models are presented in Fig. 2. As depicted in the cumulative probability–probability (PP) plots, numerous false-positive associations were obtained for all the traits using the GLM + Q model (Fig. 2). When the kinship effect (MLM + K) was incorporated, potential spurious associations were filtered out and the p values were closer to the expected distribution for all the traits analyzed (Fig. 2). However, the most adjusted model was observed when both population structure and relative kinship matrices were incorporated into a mixed lineal model (MLM + K + Q). Therefore, both models (MLM + K and MLM + Q + K) were used to search for SNP–trait associations in the six seed quality traits evaluated. Using the MLM + K model, we found five significant associations for SHC after multiple comparison adjustments (qFDR, q value <0.05), explaining between 20 and 31 % of the phenotypic variation. On the other hand, using the most conservative model (MLM + K + Q), we found seventeen significant associations for SGC after multiple comparison adjustments (qFDR, q value <0.05), explaining 6–26 % of the phenotypic variation (Table 2). The estimated allelic class effect values obtained from MLM models showed positive values ranging from 16.3 to 41.4 μmol/g of seed dry weight and negative values ranging from −28.6 to −21.5 μmol/g of seed dry weight for SGC. For SHC, the estimated allelic class effects values were all negatives, ranging from −0.26 to −0.41 % of seed dry weight (Table 2).

Evaluation of three associative mapping (AM) statistical models (from less to strongly conservative) used for the GWAS approach in each of the six seed quality traits analyzed. Cumulative probability–probability (P–P) plots of the observed −Log10 (P) values against the expected distribution of −Log10 (P) values (x axes, dashed black line) for three AM models GLM + Q, MLM + K and MLM + Q + K using a GWAS approach. SOC seed oil content, SPC seed protein content, SLAC seed linolenic acid content, SGC seed glucosinolate content, SCC seed cellulose content, SHC seed hemicellulose content. (Color figure online)
With regard to cQTL analysis, 100 SNP markers chosen specifically from QTL regions controlling seed quality traits (Clarke et al. 2013) were genotyped in the 89 winter-type inbred lines using the KASPar assay. After filtering the SNP data (i.e., allele frequency <0.05) in TASSEL, we obtained a total of 94 SNP markers for the association analysis. Similar to GWAS, three statistical models were evaluated (GLM + Q, MLM + K and MLM + Q+K) in order to minimize the occurrence of false-positive associations. As depicted in Fig. 3, for the cQTL approach the best adjusted model to the expected p value distribution varied in accordance with the trait analyzed. Whereas for SPC and SLAC the MLM + K model showed the best fit to the expected distribution, for SOC and SGC the MLM + K and MLM + K + Q models showed similar results. On the other hand, GLM + Q and MLM + K + Q exhibited the best fit to the expected distribution of p values for SHC, while for SCC the GLM + Q model showed the best adjustment to the expected distribution. To minimize possible false-positive associations, we selected the MLM + Q + K model to analyze SOC and SGC, MLM + K to analyze SPC and SLAC, and GLM + Q to analyze SHC and SCC. As shown in Table 2, we observed a total of four SNP markers significantly associated with SGC and six SNP markers significantly associated with SHC after multiple comparison adjustments (qFDR, q value <0.05). These associations were consistent in at least three environments and explained 5–15 % of the phenotypic variation for SGC (Table 2). For SHC, the seven associated SNP markers were significant only in one environment (including the means for the entire set of environments) and explained 8–13 % of the phenotypic variation (Table 2). Finally, for SGC, the estimated allelic effect values obtained from the statistical model showed positive values for the allelic class 1 over the allelic class 2 in two of the associated SNPs (SNP99 and SNP 100), and negative values for the allelic class 1 over the allelic class 2 in the other two remaining SNPs (SNP9 and SNP19). For SHC, the estimates of allelic class effects values ranged from 0.18 to 0.27 % of seed dry weight (Table 2).

Evaluation of three associative mapping (AM) statistical models (from less to strongly conservative) used for the cQTL approach in each of the six seed quality traits analyzed. Cumulative probability–probability (P–P) plots of the observed −Log10 (P) values against the expected distribution of −Log10 (P) values (x axes, dashed black line) for three AM models GLM + Q, MLM + K and MLM + Q + K using cQTL approach. SOC seed oil content, SPC seed protein content, SLAC seed linolenic acid content, SGC seed glucosinolate content, SCC seed cellulose content, SHC seed hemicellulose content. (Color figure online)
Position confirmation of significantly associated SNP markers identified in cQTL approach
In order to confirm SNP positions, previously discovered to target-specific regions associated with seed quality traits (Snowdon and Iniguez-Luy 2012; Clarke et al. 2013), and eventually identify new genomic locations associated with seed quality traits, the SNP markers evaluated in cQTL AM approach were mapped in a linkage map derived from a DH population (ExV8-DH). A total of 60 SNP markers were mapped, and their genomic locations were identified (Online resource 2, Fig. S4). From these 60 SNP markers, a total of 49 were positioned in the corresponding SNP discovery genomic position (81.6 %). For instance, SNP99 and SNP100 (significantly associated with SGC) were mapped to a QTL interval previously reported for SGC on chromosome A9 (Udall et al. 2006). Similarly, SNP9 and SNP19 (also significantly associated with SGC) were mapped to a QTL interval previously reported for SOC on chromosome A5 (Udall et al. 2006). Lastly, SNP93, SNP94, SNP95, SNP96 and SNP97 significantly associated with SHC were also mapped to a QTL interval previously reported for SOC (Delourme et al. 2006) on chromosome A8 (Online resource 2, Fig. S4).
Characterization of SNP markers significantly associated with SGC and SHC
SNP markers associated with SGC (identified by both GWAS and cQTL approaches) were mapped via alignment to the reference genome sequence of B. napus (http://www.genoscope.cns.fr/brassicanapus/) in order to characterize their specific genomic location. The results from the alignment analysis are summarized in Online resource 1, Table S6. From a total of 21 SNP markers significantly associated with SGC, one SNP marker was localized on chromosome A2, 1 SNP marker on chromosome A3, 4 SNP markers on chromosome A9, 3 SNP markers on chromosome C2, 1 SNP marker on chromosome C4, 2 SNP markers on chromosome C6 and 4 SNP markers on chromosome C9. Also, 3 SNP markers showed a match position in more than one chromosome, 1 SNP marker on A2-C2, 1 SNP marker on A9-C9 and 1 SNP marker on A9-C9-C3 chromosomes. For a total of 11 SNP markers associated with SHC, 1 SNP marker was localized on chromosome A5, 3 SNP markers on chromosome A8 and 4 SNP markers on chromosome C3. In addition, 3 SNP markers showed a match position on the chromosomes A8 and C3 at the same time. With regard to genic or non-genic classification, a total of 15 SNP markers presented a genic localization and 15 SNP markers exhibited a non-genic localization. Finally, the type of substitution (e.g., transition or transversions) was analyzed in order to classify the significantly associated SNP markers. Transitions were the predominant mutation, representing 59.37 % of the total SNP markers (19 out of 32) with significant associations. Thirteen transversions were observed, corresponding to 40.62 % of SNP markers significantly associated (Online resource 1, Table S6).
Discussion
Phenotypic analysis in the association panel
The variance components analysis for random terms showed that genotype had the main effects over all the traits evaluated. This result agrees with the association analysis where the associated SNPs were consistent across the different environments evaluated. However, the significant effect observed for the fixed terms (L, S and L × S) suggests a significant environment influence over the traits. This influence was reflected in the moderate broad-sense heritability values observed for most of the traits assessed. A different performance was observed for SGC, which registered the highest broad-sense heritability, suggesting an important genetic control for this trait. In addition, SGC presented a non-normal distribution of residuals (Online resource 2, Fig. S1); instead, a bimodal distribution of the raw trait values was observed (data not shown), probably due to the extensive selection history for high or low SGC content in oilseed rape breeding (Snowdon et al. 2007). This may also be the reason why this trait showed a higher phenotypic variability in comparison with the other traits analyzed. Correlations between the analyzed traits coincide with those observed in previous B. napus studies. Negative correlations observed for SOC–SPC, SOC–SCC and SOC–SHC, and positive correlations for SPC–SGC and SCC–SHC have been reported using AM and biparental mapping (Zhang et al. 2006; Wittkop et al. 2009; Honsdorf et al. 2010; Ma et al. 2013).
Population structure, diversity and relative kinship analysis
In order to conduct the AM analysis, we first determined the population structure and relative kinship present in the association panel. The population structure analyses demonstrated that the B. napus panel is composed of two subpopulations (Fig. 1). The F ST value (Online resource 1, Table S5b), a measurement of the genetic differentiation between subpopulations, suggested a weak population structure where the individuals in both subpopulations share a high number of alleles. Different studies using worldwide oilseed rape collections (Bus et al. 2011; Gyawali et al. 2013) have reported low F ST values among subpopulations (0.176 and 0.09, respectively) differentiated principally for growth habit. For a winter oilseed rape population characterized by Bus et al. (2011), however, a low intra-population F ST value was observed (0.087). This agrees with the results presented in this work. In addition, the F IS and F IT values suggested low heterozygosity for the individuals within subpopulations and individuals within the total population, respectively (Online resource 1, Table S5b). The weak stratification and low heterozygosity could result from different factors as geographic origin, flowering behavior (winter lines), mating system (open pollinated) and breeding history. The global effect of these factors has led to a comparatively narrow genetic base in current oilseed rape breeding lines or germplasm (Becker et al. 1995; Hasan et al. 2006).
The subdivision observed in the association panel details further the diversity found in winter oilseed rape germplasm. Previous studies of this kind were conducted using diversity sets that include spring oilseed rape germplasm which hinder the ability to further dissect the winter-type germplasm (Hasan et al. 2006; Ecke et al. 2010; Bus et al. 2011; Gyawali et al. 2013). In this respect, the current study increases our understanding of the genetic composition of winter-type oilseed rape and expands on previous work in this field. Approximately 80 % of the lines exhibit low relative kinship, a phenomenon that has also been observed in other B. napus association panels (Xiao et al. 2012; Fritsche et al. 2012). The evaluation of both, population structure and kinship, suggested a favorable scenario to conduct AM (Soto-Cerda and Cloutier 2012).
Association mapping using GWAS and cQTL approaches
Recently, several AM studies have been conducted in B. napus. These have helped to identify and/or confirm QTL underlying seed quality traits (Hasan et al. 2008; Honsdorf et al. 2010; Fritsche et al. 2012; Harper et al. 2012; Li et al. 2014), demonstrating the power of AM to discover marker–trait associations in genetically diverse panels or populations (reviewed in Gupta et al. 2005; Soto-Cerda and Cloutier 2012). Taking into account the low extension of LD and rapid LD decay observed in different B. napus populations (Bus et al. 2011; Xiao et al. 2012; Delourme et al. 2013), the use of 4025 evenly spaced SNP markers provided an adequate genome coverage to conduct GWAS.
False-positive associations were examined using three different AM models. These models showed significant corrections when both K and Q matrices were incorporated in GWAS and cQTL analyses (Figs. 2, 3). The incorporation of both Q and K matrices into a mixed lineal model (MLM) has been successfully used previously in B. napus and others species (Yu et al. 2006; Yang et al. 2010; Pasam et al. 2012; Upadhyaya et al. 2013; Li et al. 2014). When the MLM + K + Q model was evaluated in the GWAS approach, 17 SNP markers were identified to be significantly associated with SGC, explaining 6–26 % of the phenotypic variation for the trait. Moreover, when the MLM + K model was evaluated in the GWAS approach, 5 SNP markers were identified to be significantly associated with SHC, explaining 20–31 % of the phenotypic variation for the trait. The estimated effect value of allelic class 1 over allelic class 2 for both SGC and SHC showed the presence of higher estimated effects for SGC than for SHC due to the range relation of each trait. These results suggest the availability of these SNP markers to assist a breeding B. napus program.
Additionally, 66 SNPs associated with other seed quality traits (SOC, SLAC and SHC) were identified using the less restrictive GLM + Q model (Online resource 1, Table S7). These SNP markers were consistent in at least three of the four environments evaluated. Although these SNP markers showed significant association (under a less restrictive statistical model, GLM + Q), further analysis should be undertaken to test for the presence of false-negative associations.
Candidate QTL (cQTL) association mapping was conducted using 100 SNP markers that targeted QTL regions previously reported to harbor seed quality traits. As a result, 4 SNP markers significantly associated with SGC and 6 SNP markers significantly associated with SHC were identified. Even though a higher number of significant associations were expected for the cQTL AM approach, no significant associations were detected for four of the traits evaluated: SOC, SPC, SLAC and SCC. This result could in part be due to the low SNP coverage (10–20 SNP markers per QTL interrogated) used to survey the QTL regions of interest, which may not be sufficient to encompass the differences in the size of LD blocks between biparental populations and association panels. Moreover, the QTL intervals used to choose the SNP markers only control seed oil, erucic acid and glucosinolate content (Clarke et al. 2013), explaining why no SNP markers were associated with SOC, SPC, SLAC and SCC. In addition, the polygenic control reported for SOC (Delourme et al. 2006; Qiu et al. 2006), SPC, SCC and SHC (Ma et al. 2013) may also decrease the power of the SNP–QTL coverage.
The nutritional importance of SGC and SHC for oilseed rape meal quality increases the necessity of providing good quality markers for breeding purposes. Glucosinolates are a class of secondary metabolites derived from amino acids that are present in the seeds and leaves of all Brassica species. Glucosinolates and their breakdown products have different beneficial functions in Brassica species (Tierens et al. 2001; Brader et al. 2001; Gruber et al. 2009); however, they are considered as antinutritional compounds for the livestock feed industry. Although commercial B. napus lines with low seed glucosinolate concentrations (10 μmol/g) are available (Khajali and Slominski 2012), introgression breeding to obtain useful variation for other traits often involves crosses with high-glucosinolate germplasm. Molecular markers for cost-effective screening of breeding lines could therefore be extremely useful in commercial breeding to recover elite backgrounds when exotic sources are used to widen the genetic diversity. On the other hand, SHC is a biochemically structural fiber that requires carbon sources produced by photosynthesis and therefore has a negative correlation with seed oil content (Online resource 1, Table S4). Marker-assisted selection for reduced SHC could therefore be utilized as a novel strategy to improve oil content in the seed by redirecting photosynthetic assimilates from hemicellulose biosynthesis into seed oil biosynthesis (Liu et al. 2013). Therefore, identifying markers linked to SHC could help to design seeds that combine increased oil content with a reduction in antinutritional fiber in the seed meal.
Position confirmation of SNP markers significantly associated with SGC and SHC identified by cQTL approach
A total of 60 out of 100 SNP markers evaluated using the cQTL approach were mapped to their corresponding QTL intervals (Clarke et al. 2013) in the B. napus genome (Online resource 2, Fig. S4). As expected, two of the three SNPs significantly associated with SGC (SNP99 and SNP100) were mapped to a previously reported SGC–QTL on chromosome A9 (Udall et al. 2006; Howell et al. 2003; Zhao and Meng 2003; Quijada et al. 2006), corroborating the power of AM to identify marker–trait associations (Online resource 2, Fig. S4). The other pair of SNP markers associated with SGC (SNP9 and SNP19) did not localize to a previously reported QTL for SGC. SNP9 and SNP19 mapped to chromosome A5 (Online resource 2, Fig. S4). This could be considered as a new putative association for SGC in B. napus, not previously reported.
In addition, SNP markers significantly associated with SHC mapped to a previously reported QTL for SOC on chromosome A8 (Delourme et al. 2006; Qiu et al. 2006; Wang et al. 2008). The negative correlation between these two traits suggests a pleiotropic role for this QTL. Due to the dependency of both oil and hemicellulose biosynthesis for carbon, this interaction is not unexpected; however, further analysis is required to confirm a causal relationship of this QTL on both traits.
Characterization of the SNP markers significantly associated with SGC and SHC
BLAST analysis using the B. napus genome showed that the majority of SNP markers associated with SGC localize to chromosome A9 and C2 (Online resource 1, Table S6), which is consistent with a previous study (Feng et al. 2012). Additional SNPs associated with SGC found on chromosomes A2, A3, C4, C6 and C9 could represent a novel genomic variation for this trait; however, additional studies are necessary to confirm this finding. Similarly, the SNPs associated with SHC (located on chromosomes A8 and C3) did not coincide with the positions found in a previous work (chromosomes A3 and A9, Liu et al. 2013) and could represent novel genomic regions controlling this trait.
Genic SNPs associated with the seed quality traits examined in this study were located on CDS exhibiting high identities with genes involved in transmembrane transport processes, transcription regulation and transferase activity (Online resource 1, Table S6). No genes directly involved in glucosinolate or hemicellulose biosynthesis were identified. Since SGC in B. napus is controlled by different metabolic pathways that regulate the synthesis of aliphatic, aromatic and indole glucosinolates (Feng et al. 2012), the challenge of identifying individual causative genes has been previously acknowledged (Hasan et al. 2008).
Lastly, the type of nucleotide substitutions indicated the presence of 19 transitions and 13 transversions, a similar proportion observed in other reports for plants and other organisms (Collins and Jukes 1994; Ebersberger et al. 2002; Park et al. 2010; Barchi et al. 2011; Bus et al. 2012) including B. napus (Clarke et al. 2013).
Final remarks
The SNP–trait associations identified in this study were highly significant and consistent across all environments evaluated. The targeted cQTL approach resulted in a more efficient methodology to identify SNP associations with seed quality traits compared to GWAS. Therefore, the associated SNP markers identified in the present work have a good potential to assist the selection of lines with reduced levels of seed glucosinolates and lower hemicellulose content (contributing to improved oil content) in winter B. napus breeding programs.
References
Amar S, Ecke W, Becker HC, Möllers C (2008) QTL for phytosterol and sinapate ester content in Brassica napus L. collocate with the two erucic acid genes. Theor Appl Genet 116:1051–1061
Barchi L, Lanteri S, Portis E, Acquardo A, Vale G, Toppino L, Rotino GL (2011) Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genom 12:304
Basunanda P, Radoev M, Ecke W, Friedt W, Becker H, Snowdon RJ (2010) Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). Theor Appl Genet 120:271–281
Becker HC, Engqvist GM, Karlsson B (1995) Comparison of rapeseed cultivars and resynthesized lines based on allozyme and RFLP markers. Theor Appl Genet 91:62–67
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bradbury PJ, Zhang Z, Kroon DE, Casstevens RM, Ramdoss Y, Buckler ES (2007) TASSELL software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brader G, Tas E, Palva ET (2001) Jasmonate-dependent induction of indol glucosinolates in Arabidopsis by culture filtrates of the nonspecific pathogen Erwinia carotovora. Plant Physiol 126:849–860
Breseghello F, Sorrells M (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Bus A, Körber N, Snowdon RJ, Stich B (2011) Patterns of molecular variation in a species-wide germplasm set of Brassica napus. Theor Appl Genet 123:1413–1423
Bus A, Hecht J, Huettel B, Reinhardt R, Stich B (2012) High-throughput polymorphism detection and genotyping in Brassica napus using next-generation RAD sequencing. BMC Genom 13:281
Chalhoub et al (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953
Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B, Li P, Hua W, Wang X (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11:136
Clarke WE, Parkin IA, Gajardo HA, Gerhardt DJ, Higgins E, Sidebottom C, Sharpe AG, Snowdon RJ, Federico ML, Iniguez-Luy FL (2013) Genomic DNA enrichment using sequence capture microarrays: a novel approach to discover sequence nucleotide polymorphisms (SNP) in Brassica napus L. PLoS ONE 8(12):e81992
Collins DW, Jukes TH (1994) Rates of transition and transversion in coding sequence since the human-rodent divergence. Genomics 20:386–396
Cuppen E (2007) Genotyping by Allele-Specific Amplification (KASPar). Protoc, Cold Spring Harb. doi:10.1101/pdb.prot4841
Delourme R, Falentin C, Huteau V, Clouet V, Horvais R, Gandon B, Specel S, Hanneton L, Dheu JE, Deschamps M, Margale E, Vincourt P, Renard M (2006) Genetic control of oil content in oilseed rape (Brassica napus L.). Theor Appl Genet 113:1331–1345
Delourme R, Falentin C, Fopa Fomeju B, Boillot M, Lassalle G, André I, Duarte J, Gauthier V, Lucante N, Marty A, Pauchon M, Pichon J-P, Ribière N, Trotoux G, Blanchard P, Rivière N, Martinant J-P, Pauquet J (2013) High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genom 14:120
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl Dent A, von Holdt Bridgett M (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ebersberger I, Metzler D, Scwarz C, Paabo S (2002) Genomewide comparison of DNA between humans and chimpanzees. Am J Hum Genet 70:1490–1497
Ecke W, Uzunova M, Weißleder K (1995) Mapping the genome of rapeseed (Brassica napus L.). II. Localization of genes controlling erucic acid synthesis and seed oil content. Theor Appl Genet 91:972–977
Ecke W, Clemens R, Honsdorf N, Becker HC (2010) Extent and structure of linkage disequilibrium in canola quality winter rapeseed (Brassica napus L.). Theor Appl Genet 120:921–931
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Feng J, Long Y, Shi L, Shi J, Barker G, Meng J (2012) Characterization of metabolite quantitative trait loci and metabolic networks that control glucosinolate concentration in the seeds and leaves of Brassica napus. New Phytol 193:96–108
Fritsche S, Wang X, Li J, Stich B, Kopisch-Obuch FJ, Endrigkeit J, Leckband G, Dreyer F, Friedt W, Meng J, Jung C (2012) A candidate gene-based association study of tocopherol content and composition in rapeseed (Brassica napus). Front Plant Sci 3:129
Galeano C, Cortés A, Fernández A, Soler A, Franco-Herrera N, Makunde G, Vanderleyden J, Blair M (2012) Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet 13:48
Gruber MY, Xu N, Grenkow LF, Onyilagha J, Soroka JJ, Westcott ND, Hugedus DD (2009) Responses of the crucifer flea beetle to Brassica volatiles in an olfactometer. Environ Entomol 38:1467–1479
Gupta P, Rustgi S, Kulwal P (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Gyawali S, Hegedus DD, Parkin I, Poon J, Higgins E, Horner K, Bekkaoui D, Coutu C, Buchwaldt L (2013) Genetic diversity and population structure in a world collection of Brassica napus accessions with emphasis on South Korea, Japan, and Pakistan. Crop Sci 53:1537–1545
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I (2012) Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat Biotech 30:798–802
Hasan M, Seyis F, Badani AG, Pons-Kühnemann J, Friedt W, Lühs W, Snowdon RJ (2006) Analysis of genetic diversity in the Brassica napus L. gene pool using SSR markers. Genet Resour Crop Evol 53:793–802
Hasan M, Friedt W, Pons-Kühnemann J, Freitag NM, Link K, Snowdon RJ (2008) Association of gene-linked SSR markers to seed glucosinolate content in oilseed rape (Brassica napus ssp. napus). Theor Appl Genet 116:1035–1049
Honsdorf N, Becker HC, Ecke W (2010) Association mapping for phenological, morphological, and quality traits in canola quality winter rapeseed (Brassica napus L.). Genome 53:899–907
Howell PM, Sharpe AG, Lydiate DJ (2003) Homoeologous loci control the accumulation of seed glucosinolates in oilseed rape (Brassica napus). Genome 46:454–460
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler E, Qian Q, Zhang Q, Li J, Han B (2010) Genome-wide studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
Iniguez-Luy FL, Federico ML (2011) The genetics of Brassica napus L. In: Schmidt R, Bancroft I (ed) Genetic and Genomics of the Brassicaceae, New York, pp 291–322
Jestin C, Lodé M, Vallée P, Domin C, Falentin C, Horvais R, Coedel S, Manzanares-Dauleux MJ, Delourme R (2011) Association mapping of quantitative resistance for Leptosphaeria maculans in oilseed rape (Brassica napus L.). Mol Breed 27:271–287
Khajali F, Slominski BA (2012) Factors that affect the nutritive value of canola meal for poultry. Poult Sci 91:2564–2575
Kimber DS, McGregor DI (1995) The species and their origin, cultivation and world production. In: Kimber D, McGregor DI (eds) Brassica oilseeds: production and utilization. CABI Publishing, Wallingford, pp 1–9
Kump K, Bradbury P, Wisser R, Buckler E, Belcher A, Oropeza-Rosas M, Zwonitzer J, Kresovich S, McMullen M, Ware D, Balint-Kurti P, Holland J (2011) Genome wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet 43:163–168
Li F, Chen B, Xu K, Wu J, Song W, Bancroft I, Harper A, Trick M, Liu S, Gao G, Wang N, Yan G, Qiao J, Li J, Li H, Xiao X, Zhang T and Wu X (2014) Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Research, pp 1–13
Liu K, Muse S (2005) PowerMarker: an integrative analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu L, Qu C, Witttkop B, Yi B, Xiao Y, He Y, Snowdon R, Li J (2013) A high-density SNP map for accurate mapping of seed fiber QTL in Brassica napus L. PLoS ONE 8(12):e83052
Liu S et al (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploidy genomes. Nat Commun 5:3930
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Ma ZZ, Li JN, Wittkop B, Frauen M, Yan XY, Liu LZ, Xiao Y (2013) QTL mapping for oil, protein, cellulose and hemicellulose content in seeds of Brassica napus L. Acta Agron Sin 39:1214–1222
Marwede V, Gül MK, Becker HC, Ecke W (2005) Mapping of QTL controlling tocopherol contents in winter oilseed rape. Plant Breed 124:20–26
Park S, Yu HJ, Mun JH, Lee SC (2010) Genome-wide discovery of DNA polymorphism in Brassica rapa. Mol Genet Genomics 283:135–145
Pasam R, Sharma R, Malosetti M, van Eeuwijk F, Haseneyer G, Kilian B, Graner A (2012) Genome-wide association studies for agronomical traits in a worldwide spring barley collection. BMC Plant Biol 12:16
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Poland J, Bradbury P, Buckler E, Nelson R (2011) Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. PNAS 108:6893–6898
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qiu D, Morgan C, Shi J, Long Y, Liu J, Li R, Zhuang X, Wang Y, Tan X, Dietrich E, Weihmann T, Everett C, Vanstraelen S, Beckett P, Fraser F, Trick M, Barnes S, Wilmer J, Schmidt R, Li J, Li D, Meng J, Brancroft I (2006) A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet 114:67–80
Quijada PA, Udall JA, Lambert B, Osborn TC (2006) Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 1. Identification of genomic regions from winter germplasm. Theor Appl Genet 113:549–561
Radoev M (2007) Genetic analysis of heterosis in rapeseed (B. napus L.) by QTL mapping [online]. Ph.D. thesis, Faculty of Agriculture, University Göttingen. http://webdoc.sub.gwdg.de/diss/2007/radoev/radoev.pdf
Rashid U, Anwar F (2008) Production of biodiesel through optimized alkaline-catalyzed transesterification of rapeseed oil. Fuel 87:265–273
Rezaeizad A, Wittkop B, Snowdon R, Hasan M, Mohammadi V, Zali A, Friedt W (2011) Identification of QTLs for phenolic compounds in oilseed rape (Brassica napus L.) by association mapping using SSR markers. Euphytica 177:335–342
Rousset M, Bonnin I, Remoué C, Falque M, Rhoné B, Veyrieras J, Madur D, Murigneux A, Balfourier F, Le Gouis J, Santoni S, Goldringer I (2011) Deciphering the genetics of flowering time by an association study on candidate genes in bread wheat (Triticum aestivum L.). Theor Appl Genet 123:907–926
Snowdon RJ, Iniguez-Luy FL (2012) Potential to improve oilseed rape and canola breeding in the genomics era. Plant Breed 131:351–360
Snowdon RJ, Luhs W, Friedt W (2007) Oilseed rape. In: Kole C (ed) Genome mapping and molecular breeding in plants, vol 2. Springer, Heidelberg, pp 55–114
Sorkheh K, Malysheva-Otto LV, Wirthensohn MG, Tarkesh-Esfahani S, Martínez-Gómez P (2008) Linkage disequilibrium, genetic association mapping and gene localization in crop plants. Genet Mol Biol 31:805–814
Soto-Cerda BJ, Cloutier S (2012) Association mapping in plant genomes. In: Caliskan M (ed) Genetic diversity in plants. InTech, Rijeka, pp 29–54
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. PNAS 100:9440–9445
Thormann CE, Romero J, Mantet J, Osborn TC (1996) Mapping loci controlling the concentrations of erucic and linolenic acids in seed oil of Brassica napus L. Theor Appl Genet 93:282–286
Tian F, Bradbury P, Brown P, Hung H, Sun Q, Flint-Garcia S, Rocheford T, McMullen M, Holland J, Buckler E (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43:159–162
Tierens KF, Thomma BP, Brouwer M, Schmidt J, Kistner K, Porzel A, Mauch-Mani B, Cammue BP, Broekaert WF (2001) Study of the role of antimicrobial glucosinolate-derived isothiocyanates in resistance of Arabidopsis to microbial pathogens. Plant Physiol 125:1688–1699
Toroser D, Thormann CE, Osborn TC, Mithen R (1995) RFLP mapping of quantitative trait loci controlling seed aliphatic-glucosinolate content in oilseed rape (Brassica napus L.). Theor Appl Genet 91:802–880
Udall JA, Quijada PA, Lambert B, Osborn TC (2006) Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 2. identification of alleles from unadapted germplasm. Theor Appl Genet 113:597–609
Upadhyaya HD, Wang YH, Gowda CLL, Sharma S (2013) Association mapping of maturity and plant height using SNP markers with the sorghum mini core collection. Theor Appl Genet 126:2003–2015
Uzunova M, Ecke W, Weißleder K, Röbbelen G (1995) Mapping the genome of rapeseed (Brassica napus L.) I. construction of an RFLP linkage map and localization of QTLs for seed glucosinolate content. Theor Appl Genet 90:194–204
Van Ooijen JW (2006) Join Map ® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma B.V., Wageningen, Netherlands.
Wang N, Wang Y, Tian F, King GJ, Zhang C, Long Y, Shi L, Meng J (2008) A functional genomics resource for Brassica napus: development of an EMS mutagenized population and discovery of FAE1 point mutations by TILLING. New Phytol 180:751–765
Wang X et al (2011) The genome of the mesopolyploi crop species Brassica rapa. Nat Genet 43:1035–1039
Wittkop B, Snowdon R, Friedt W (2009) Status and perspectives of breeding for enhanced yield and quality of oilseed crops for Europe. Euphytica 170:131–140
Xiao Y, Cai D, Yang W, Ye W, Younas M, Wu J, Liu K (2012) Genetic structure and linkage disequilibrium pattern of a rapeseed (Brassica napus L.) association mapping panel revealed by microsatellites. Theor Appl Genet 125:437–447
Yang X, Yan J, Shah T, Warburton ML, Li Q, Li L, Gao Y, Chai Y, Fu Z, Zhou Y, Xu S, Bai G, Meng Y, Zheng Y, Li J (2010) Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor Appl Genet 121:417–431
Yu J, Pressoir G, Briggs W, Bi IV, Yamasaki M, Doebley J, McMullen M, Gaut B, Nielsen D, Holland J, Kresovich S, Buckler E (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhang GQ, He Y, Xu L, Tang GX, Zhou WJ (2006) Genetic analyses of agronomic and seed quality traits of doubled haploid population in Brassica napus through microspore culture. Euphytica 149:169–177
Zhao J, Meng J (2003) Detection of loci controlling seed glucosinolate content and their association with Sclerotinia resistance in Brassica napus. Plant Breed 122:19–23
Zhao JY, Becker HC, Zhang DQ, Zhang YF, Ecke W (2005) Oil content in a European Chinese rapeseed population: QTL with additive and epistatic effects and their genotype-environment interactions. Crop Sci 45:51–59
Zhao J, Becker HC, Zhang D, Zhang Y, Ecke W (2006) Conditional QTL mapping of oil content in rapeseed with respect to protein content and traits related to plant development and grain yield. Theor Appl Genet 113:33–38
Zhao J, Dimov Z, Becker HC, Ecke W, Möllers C (2008) Mapping QTL controlling fatty acid composition in a doubled haploid rapeseed population segregating for oil content. Mol Breed 21:115–125
Zhu Ch, Gore M, Buckler E, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Zou J, Jiang C, Cao Z, Li R, Long Y, Chen S, Meng J (2010) Association mapping of seed oil content in Brassica napus and comparison with quantitative trait loci identified from linkage mapping. Genome 53:908–916
Acknowledgments
The authors would like to thank Katherine Andara for her technical assistance. We acknowledge Fondecyt 1100732, Proyecto Fortalecimiento R13F1001, Comisión Nacional de Investigación Científica y Tecnológica (CONICYT) Regional Program and the Araucania Regional Government/CGNA/R10C1001 and INIA for its support providing laboratory infrastructure. HG was supported by Becas de Magíster Nacional—CONICYT No: 22121770.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gajardo, H.A., Wittkop, B., Soto-Cerda, B. et al. Association mapping of seed quality traits in Brassica napus L. using GWAS and candidate QTL approaches. Mol Breeding 35, 143 (2015). https://doi.org/10.1007/s11032-015-0340-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0340-3