
Abstract
Simple sequence repeats (SSRs) are highly polymorphic and co-dominant markers, providing an important genomic resource for genetic research. Recently, large-scale transcriptome sequencing has become a reliable and efficient approach for the identification and development of new genic-SSR markers and has been successfully conducted in a few important plant species. However, SSR development based on transcriptome sequencing remains limited in radish (Raphanus sativus L.). In the present study, from a total of 73,084 unigenes and 150,455 contigs which were assembled from 71.95 million Illumina sequence reads of a radish taproot library, a collection of 11,928 genic-SSR loci were successfully identified in 11,311 unigene sequences. Trinucleotide repeats were the most abundant repeat units, as in many other plants, with a frequency of 52 %. Furthermore, a total of 5,503 genic-SSR primers were developed, from which 1,052 SSR primers were synthesized, and a subset of 823 (78.23 %) primers could generate stable bands. Moreover, 67 selected informative genic-SSR markers were used to determine the genetic diversity of 32 radish genotypes, in which the polymorphism information content values ranged from 0.49 to 0.89. For effective cultivar identification, a novel strategy called manual cultivar identification diagram was employed. Thirty-two radish accessions were clearly separated by six genic-SSR markers. Additionally, the cross-species/genera transferability of these SSRs was further validated in nine relatives in Brassicaceae. These results suggested that the novel genic-SSR markers, as a basis for future genetic linkage and gene tagging analysis, could be very valuable in facilitating genetic mapping, marker-assisted selection and comparative genome analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microsatellites or simple sequence repeats (SSRs) are tandem repeat sequences of short units of 2–6 nucleotides that occur frequently in all prokaryotic and eukaryotic genomes studied to date (Koelling et al. 2012). Due to their hyper-variability, reproducibility, high abundance and transferability, multi-allelic nature and co-dominant inheritance, SSR markers have become valuable and reliable tools for genetic diversity analysis, genetic mapping, gene tagging and comparative mapping in plants (Li et al. 2012; Silva et al. 2013). Additionally, SSR markers can be isolated from both conserved coding regions and non-coding nucleotide sequences of all higher organisms (Sraphet et al. 2011). However, the development of genomic SSRs is relatively laborious and time-consuming; alternatively, SSRs have been mined from the public sequence databases of expressed sequence tags (ESTs) (Huang et al. 2011; Koelling et al. 2012; Sraphet et al. 2011; Zeng et al. 2010) or coding sequences by RNA-seq technology (Kaur et al. 2012; Li et al. 2012). Nevertheless, compared with the SSR markers derived from transcriptome sequences, the development of SSRs from the large-scale cloning and sequencing of DNA or insufficient public EST libraries would yield multiple sets of markers at the same locus due to sequence redundancy. To circumvent the problem of redundancy in EST databases, a non-redundant unigene EST data set obtained by de novo transcriptome sequencing technologies should be used.
In recent years, transcriptome sequencing using the next-generation sequencing (NGS) technology platforms of SOLiD, Illumina and 454 has become increasingly popular in many crop species (Li et al. 2010; Lu and Lu 2010). The NGS technology is a tremendously efficient approach for the large-scale generation of reliable and robust transcript sequences (Blanca et al. 2011). Even in model species, such as Arabidopsis thaliana, this deep sequencing is desirable for identifying new transcripts not present in previous EST collections (Blanca et al. 2011). This technology offers a simple, direct and reliable approach for identification and development of massive unigene-based microsatellite markers (UGMS) with diverse motifs through data mining by bioinformatic methods. Additionally, the genic-SSRs target the transcribed regions specifically and increase the potential for linkage to loci that contribute to agronomic phenotypes. Because of their target coding domains that are more likely to be conserved between relatives, the markers can also facilitate better cross-genome comparisons (Dutta et al. 2011). In recent decades, the comprehensive transcriptome of EST sequences by NGS technology has become one of the significant SSR detection resources. To date, a large number of SSR markers based on transcriptome sequences has been developed and utilized in diverse species, such as rice (Yu et al. 2012), cotton (Wang et al. 2012a), orange (Song et al. 2012), linseed (Kale et al. 2012) and orchid (Fu et al. 2011). Recent advances in next-generation sequencing technologies have generated a huge wealth of sequence information, which offers the opportunity to develop genic-SSR markers on a large scale for molecular breeding, genetic mapping and identification of plant species and their varieties with the manual cultivar identification diagram (MCID) (Korir et al. 2013).
Radish, Raphanus sativus L. (2n = 2x = 18), is a major root vegetable crop of the Brassicaceae family. Several molecular markers have been utilized in radish including universal sequence-related amplified polymorphism (SRAP), random amplified polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP) markers (Kong et al. 2011; Shao et al. 2011) and species-specific SSR markers (Liu et al. 2008). The existence of informative molecular markers can provide valuable insights into genetic and genomic studies in radish (Zhai et al. 2013). Recently, a number of SSRs have been developed from the published EST database (Jiang et al. 2012; Shirasawa et al. 2011; Zhai et al. 2013). However, in comparison with other members of the Brassicaceae family, the development and application of genic-SSR markers from transcriptome sequences in radish are still largely limited. Wang et al. (2012b) identified a total of 14,641 potential EST–SSRs from the unigenes generated by high-throughput transcriptomic sequencing. However, these markers were not further detected and utilized. Thus, developing genic-SSRs by de novo RNA sequencing technology is needed in radish.
In this study, we developed a comprehensive set of genic-SSRs based on de novo transcriptome sequencing, and we characterized uni-transcript sequences of R. sativus (an advanced inbred line, NAU-RG) obtained by Illumina paired-end sequencing technology. A total of 11,928 genic-SSRs from 11,311 unigenes were generated in the de novo transcriptome. Moreover, a set of 5,503 novel genic-SSR markers were developed, and the application of genic-SSR markers was well demonstrated in the genetic diversity analysis of 32 genotypes in radish. All radish genotypes could be quickly distinguished by a combination of six genic-SSR primers with MCID, which was a new strategy making both morphological descriptors and molecular markers easy, referable and practical to use (Korir et al. 2013). Furthermore, for SSRs located in conserved coding regions, they showed high transferability across nine related species in the Brassicaceae family, which makes them beneficial for comparative analysis across relatives in Brassicaceae. These newly developed SSR markers will rapidly enrich the number of functional molecular markers directly related to expressed regions of the genes in radish and be very valuable in facilitating genetic mapping and comparative genome analysis in radish.
Materials and methods
Plant materials
Plants of a radish advanced inbred line, NAU-RG, with a white global taproot, were grown in a temperature-controlled greenhouse (25 °C) with a relative humidity of 75 % and 14 h of light daily. The taproots for RNA extraction were harvested and placed in liquid nitrogen immediately and then stored at −80 °C. A total of 32 radish genotypes [Electronic Supplementary Material (ESM) Table S1] with different root colors and origins were selected for genetic diversity and cultivar identification, and nine related species in Brassicaceae for transferability studies.
RNA isolation and cDNA library construction
For Illumina sequencing, the total RNA was extracted from the taproots at seedling, taproot thickening, and mature stages of NAU-RG with Trizol® Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions, and was stored at −80 °C. The cDNA library construction and sequencing with an Illumina HiSeq™ 2000 were performed at the Beijing Genomics Institute (BGI; Shenzhen, China). The unigenes assembled were annotated using BLASTX against the nr (non-redundant) protein database in the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). The unigenes were allocated to the corresponding functional categories based on gene ontology (GO) terms by Blast2GO (Conesa et al. 2005) with GO weight 2 (Huang et al. 2011).
Novel genic-SSR identification and primer design
All the uni-transcript sequences generated by deep transcriptome sequencing in radish were screened for SSRs by SSR Locator software. In this study, SSRs were considered to contain di-, tri-, tetra-, penta- and hexa-nucleotides with minimum repeat numbers of 6, 4, 3, 3 and 3, respectively (Iorizzo et al. 2011; Kaur et al. 2012). Primer pairs flanking the SSRs were designed using Primer3 in accordance with the core criteria of predicted product size ranging from 100 to 500 bp, a GC percentage between 40 and 60 %, optimum primer length of 22 bp and melting temperature between 50 and 60 °C.
DNA extraction, PCR amplification and detection
Genomic DNA from 32 accessions of radish with different root colors and origins, and from 10 related species of Brassica were extracted from young leaves using a modified CTAB protocol (Liu et al. 2003).
PCR amplifications were performed on a Thermal Cycler (SensoQuest) with a 15-μl final reaction volume containing 2.0 mM MgCl2, 0.2 mM dNTPs, 0.75 U Taq DNA polymerase (TaKaRa Bio Inc., Dalian, China), 0.1 μM of each primer and 10 ng of template DNA. The PCR conditions comprised an initial denaturation at 94 °C for 2 min; 35 cycles of 94 °C for 40 s, 55–60 °C (varying with the T m of the different primer sets) of annealing for 45 s, and 72 °C for 1 min; and a final extension of 72 °C for 7 min. PCR products were separated on 8 % non-denaturing polyacrylamide gels at 160 V for 2–2.5 h and visualized with a rapid silver staining method (Liu et al. 2008).
Partial amplification products with the expected sizes were recovered from PAGE gels using the AxyPrep DNA gel extraction kit (Axygen Bio Inc., Hangzhou, China). Extracted products were subcloned with the T-A cloning kit (TaKaRa), and the positive clones were sequenced with ABI 3730 at the BGI.
Survey of polymorphism and genetic diversity analysis
To validate the polymorphism of these loci and further study the application of these genic-SSR markers, 32 different radish genotypes consisting of R. sativus and its wild relatives (ESM Table S1) were employed and clustered based on the estimated genetic distance. For dendrogram analysis, only the data for the polymorphic alleles were entered for all DNA samples, scored for their presence (1) or absence (0). Through the similarity matrix based on Jaccard’s coefficient, a dendrogram was calculated using NTSYS-pc2.10e software by the un-weighted pair group method using the arithmetic averages (UPGMA) methodology in the SAHN module, which could illustrate the genetic relationships among the samples (Zhai et al. 2013; Zhu et al. 2012).
For primers that produced the predicted fragments after PCR reactions, the number of alleles was noted, and the polymorphism information content (PIC) value (Smith et al. 1997) of an SSR locus was calculated according to the formula PIC = ∑p 2 i , where p i is the frequency of the ith allele out of the total number of alleles at each SSR locus. Moreover, a new strategy called manual cultivar identification diagram (MCID) was adopted (Zhang et al. 2012c; Korir et al. 2013), which facilitates the efficient use of the primers and was easily operated. In this method, the clear specific bands were chosen and manually scored for cultivar identification. The cultivars with a specific band in the fingerprint generated from one primer were separated singly, and those sharing the same banding pattern were clustered into the same subgroup (Wang et al. 2011). Based on additional genic-SSR markers employed with specific band sizes, the 32 radish genotypes were completely separated from each other.
Detection of the transferability of genic-SSR primers
To assess the transferability of the SSR markers, 37 novel primer pairs were amplified from 10 related species of crops in the Brassicaeae family using the above PCR conditions. Subsequently, DNA bands with the expected size fragments in the non-denaturing polyacrylamide gels were recovered and directly cloned into the pMD 18 TA cloning vector (TaKaRa). The sequences of positive clones were analyzed with ClustalX software (http://www.seekbio.com/DownloadShow.asp?id=2247) (Jiang et al. 2012; Zhai et al. 2013).
Results
Uni-transcript sequences and gene ontology analysis
The radish cDNA library was sequenced by the Solexa system and yielded a total of 71.95 million raw reads. After removing the short and low-quality reads and trimming off the adapter sequences and poly-A tails, approximately 66.11 million high-confidence reads were obtained, which were assembled into a total of 73,084 unigenes with an average length of 763 nucleotides and selected for further analyses.
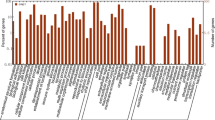
According to the BLASTX results against NCBI databases, in total 3,935 unigenes were successfully associated with GO terms out of 9,876 unique unigene sequences containing SSR loci. The GO-annotated unigenes belonged to the cellular components, molecular functions and biological processes clusters and were classified into 37 categories at process level 2 (Fig. 1). Among the cellular component classification class, cell part (8.70 %), cell (8.70 %), organelle (3.50 %) and macromolecular complex (2.30 %) were the most represented categories. Moreover, the metabolic process (16.60 %) and cellular process (15.90 %) of the biological processes classification contributed the largest proportion of all annotations, followed by biological regulation (5.00 %), pigmentation (4.90 %), localization (2.70 %) and establishment of localization (2.70 %). In the molecular function classification class, binding (25.90 %) and catalytic activity (15.10 %) constituted the two major categories, followed by transcription regulator activity (2.30 %) and transporter activity (1.00 %). Other components were represented at proportions less than 1 % of the total (Fig. 1). These results suggested that those unigenes take part in various biological processes of regulation, growth, development, metabolism and apoptosis in radish.

Gene Ontology (GO) classification of unigene sequences containing SSR loci from radish taproot. The results are summarized in three main categories: cellular component, molecular function and biological process
Characteristics of genic-SSRs in the radish transcriptome
SSRs were highly abundant in the assembled unigene dataset. In total, 11,928 potential SSRs with a minimum of four repetitions for all motifs were identified from 150,455 contigs generated by Illumina sequencing. The frequency of occurrence of SSR loci was one in every 4.93 kb of unigene sequence. Among all repeat types, the length of SSRs was distributed from 12 to 114 bp, with an average of 16.29 bp. Incidences of different repeat types were determined. SSRs existed mainly as dinucleotide repeats (DNR) and trinucleotide repeats (TNR), accounting for 91.83 %. TNR, comprising 52 % of the total SSRs, were the most abundant repeat unit, followed by di-(40 %), tetra-(5 %), hexa-(2 %) and penta-nucleotides (1 %), with the repeat unit number of SSR loci ranging from four to 23 (Fig. 2a, b). Most (98.09 %) of the DNRs and TNRs had 4–10 repeat units, while motifs with more than 10 reiterations were rare, with a frequency of <5 %. Within the identified SSRs, 400 motif sequence types were searched, and the maximum motif with TTCC (6.47 %), GAA (7.04 %) and TC (28.13 %) was found in tetra-, tri- and dinucleotides, respectively (Fig. 2c).

Characterization of SSRs in radish taproot transcriptome. a Distribution of different SSR repeat motif types; b number of different repeat motifs; c frequency distribution of major SSRs based on main motif sequence type
The 20 types of major motifs and the frequencies of individual SSR units are shown in Fig. 2c. In this study, the most common types of all the motifs detected in radish contig sequences were AG/CT (1,970; 16.52 %) and GA/TC (15.81 %), followed by GAA/TTC (5.42 %), AGA/TCT (4.71 %), AAG/TCC (4.38 %), CTC/GAG (3.63 %), GGA/TCC (3.49 %) and ATC/GAT (2.95 %), reflecting the AG/CT-rich nature of the Raphanus genome. However, other motifs shown in Fig. 2c made up less than 2.5 % of the total SSRs, and the GC motif was not detected.
Development and detection of genic-SSR markers
From 73,084 unigenes of the radish taproot, a total of 11,311 SSR-containing sequences were employed for primer design, from which 5,503 (44.67 %) sequences containing SSRs could be successfully used for SSR primer development. A total of 1,052 SSR primers (ESM Table S2) were synthesized randomly and tested for initial validation, and amplified using three radish DNA templates of YB, DY13 and ZQH. Of these SSR primers, 823 (78.23 %) pairs of primers exhibited stable and repeatable amplification. To further confirm the reality and positivity of the polymorphic microsatellite-containing sequences, 20 co-dominant segregation segments were recovered and sequenced after T-A cloning. The results showed that the sequences were consistent with the original unigene ones, indicating that these developed SSR primers were highly specific.
Genetic diversity analysis of radish genotypes
A sample of 67 genic-SSR primers was selected to assess the genetic diversity and to differentiate a set of 32 radish accessions from different countries (ESM Table S1). Eleven primers resulted in weak amplifications or non-specific amplicons, and the remaining primers could generate distinct bands with expected size (Fig. 3a–c). Out of these 56 primers, 17 genic-SSR markers generated unified and poor polymorphic bands. A total of 39 analyzed primer pairs showed allelic polymorphisms, and 211 alleles were detected in total. The number of alleles per locus ranged from 1 to 10 with an average of 3.6. The PIC values of the primers with polymorphisms varied from 0.49 to 0.89, with a mean value of 0.66. The sequences of these informative genic-SSR primers and other major information including core motif, annealing temperature, PIC value and expected size of the PCR products are shown in Table 1.

Genetic diversity analysis of 32 radish genotypes with SSR markers. Polyacrylamide gel electrophoresis patterns amplified with SSR primers UgRsr-6 (a), UgRsr-56 (b) and UgRsr-18 (c), and the UPGMA dendrogram constructed from SSR marker analysis (d). M: DNA ladder 100 bp; numbers (1–32) represent radish genotypes listed in ESM Table S1
Based on the genetic similarity results, a dendrogram placed the 32 radish accessions into three main clusters with similarity coefficients ranging from 0.59 to 0.90 (Fig. 3d). The first main cluster (I) consisted of 11 accessions collected from China, most of which had a different color root skin. In this group, there were three subgroups at a cut-off point of 0.67 similarity coefficient primarily according to the color of the xylem and phloem of the taproot. Additionally, ZQH showed a high similarity with WH, both of which had red phloem and white xylem of taproot in medium maturity. WXQ, BJXLM, QTBJQ and YZH were distinctly divergent from the other subgroups in cluster I with a different colored phloem taproot. The second main cluster (II) comprised 15 accessions and further separated into five sub-clusters mainly based on geographical sources, color of taproot and the number of chromosomes. The cultivars in the subgroup IIb mainly originated from America and Korea, while the others were collected from China. In comparison to all accessions of radish in this study, only Lw23 was grouped into subgroup IIe with nine chromosomes. The five wild relatives showed a strong relationship, some of them having purple long siliques (more than 20 cm) or deep purple flowers, which was very different from the local radish cultivars. In addition, all accessions in the group IIIb were early maturing with a white skin root.
MCID of cultivar identification with genic-SSR markers
A total of six genic-SSR primers with reproducible and polymorphic bands were successfully employed to identify the 32 radish cultivars. Of the six primers used, primer UgRsr-6 was the first to be screened and used in the identification (Fig. 4). The electrophoresis results show that primer UgRsr-6 (Fig. 3a) generated two polymorphic bands in 32 cultivars, which could separate these radish genotypes into three groups by the presence or absence of distinct 300- and 310-bp bands. Subsequently, primers UgRsr-28 and UgRsr-18 further separated the three groups of cultivars into smaller groups or singly, such as NPZ, XBY and YZH. Following this cultivar identification procedure, the other five primers (Fig. 4; Table 1) were screened step by step and chosen to differentiate the radish cultivars. By primer UgRsr-11, all the 32 cultivars were completely separated in the MCID (Fig. 4). It should be emphasized that only the clear polymorphic bands generated with each primer were used to differentiate the cultivars. In addition, the sizes and the presence/absence of polymorphic bands used in the MCID (Fig. 4) can make the diagram clear and referable. These observations revealed that the MCID is a valuable and efficient strategy for cultivar identification in radish.

MCID analysis of the 32 radish genotypes with the DNA fingerprints of six genic-SSR primers. Number above each horizontal line in the diagram denotes the size of the polymorphic bands used to separate the genotypes following the line, reported in bp; (+) or (−) denotes the presence or absence of the polymorphic band
Applicability of genic-SSR primers to other species in Brassicaceae
In order to inspect the potential transferability between cross-species, we randomly selected 45 stable and reliable SSR primers for amplification on nine different species of the Brassicaceae family (ESM Fig. S1). Overall, 39 of the 45 (86.67 %) genic-SSR primers showed transferability to one or more of the nine related Brassica species tested; among these, a total of 14 primers yielded distinct and stable bands in all nine species. The specific polymorphic markers were further sequenced. The DNA sequences were subjected to alignment analysis and SSR motifs with different copies were identified (ESM Fig. S1), suggesting that the regions flanking the SSRs were sufficiently conserved across genera of the same family. The relatively high level homology of genic-SSRs were observed in the coding regions, which demonstrated that these newly developed radish SSR markers were applicable and reliable in some related species of the Brassicaceae family.
Discussion
Genic-SSR markers are considered to have strong potential for diversity analysis and genetic mapping in crop species due to their specificity and high degree of conservation (Zhang et al. 2012a; Dutta et al. 2011). Increasing number of studies demonstrated that the identification of SSR-marker-based NGS is an efficient and cost-effective strategy. In order to develop more genic-SSR markers, we sequenced the transcriptome of radish taproots and identified 5,503 genic-SSR primer pairs, out of which 1,052 SSR markers were selected and validated. Functional markers were used in genetic diversity analysis and cultivar identification among 32 different genotypes of radish and in transferability analysis across nine relatives in the Brassicaceae.
Due to the steady decrease in cost per sequenced nucleotide and increase in throughput data, NGS technologies have become a powerful approach for the high-throughput discovery of genes and generate a large amount of sequence data for molecular marker identification (Silva et al. 2013; Iorizzo et al. 2011). Recently, de novo transcriptome assemblies using Illumina sequences have been successfully developed and widely applied in a few important plant species, including rice, wheat and maize (Duan et al. 2012; Li et al. 2010; Lu and Lu 2010). Large-scale root-specific transcriptome analysis could provide useful reference data to profile systemic gene expression and clarify the genetic mechanisms underlying radish taproot formation. To provide a better assembly and coverage of the transcriptome in radish roots, in this study a large collection of longer reads was generated for further novel specific gene discovery in taproot and novel genic-SSR marker development (Wang et al. 2010, 2012c).
In recent years, transcriptome sequencing has proven to be one of the most powerful sources for SSR development in many important plant species, and a large number of transcriptome-based SSRs have been extensively used in genetic diversity analysis, molecular mapping and gene-based association studies (Zhang et al. 2012b; Zeng et al. 2010). Many studies showed that the de novo transcriptome could provide high-quality flanking regions for SSR primer design (Silva et al. 2013). In this study, the repeat sequences are also distributed throughout the genome; about 7.93 % of the radish transcriptome dataset possessed at least one SSR, which is consistent with the range of frequencies reported previously for other dicotyledonous species (2.65–16.82 %). The abundance of SSRs in radish was 4.93 kbp/SSR on average, which was much lower than that of 5.4 in wheat (Peng and Lapitan 2005), 14.0 in poplar and Arabidopsis (Cardle et al. 2000), 20.0 in cotton (Jena et al. 2011) and 23.8 in soybean (Gao et al. 2003). Differences in SSR frequency and abundance might be partially due to the genome composition, dataset size, SSR detection tools and microsatellite mining criteria (Biswas et al. 2012). Nevertheless, our results showed that the SSRs with trinucleotide repeats (52 %) were predominant, which is consistent with previous reports on radish (Wang et al. 2012c; Shirasawa et al. 2011) and other plant species, including rice, peanut, field pea and faba bean (Liang et al. 2009; Zhang et al. 2012b; Kaur et al. 2012).
Among trinucleotide repeats in coding regions, the GAA/TTC motif was the most frequent in radish; this finding was in accordance with other reports in pea (Gong et al. 2010), sesame (Zhang et al. 2012a) and peanut (Liang et al. 2009). Moreover, the dinucleotide motif AG/CT showed the highest frequency (16.52 %), followed by GA/TC, similar to the recent results in radish and other species (Wang et al. 2012c; Zhang et al. 2012a). Although the functional significance of SSRs in plant transcript regions is not quite clear, the motif AG/CT, a homopurine–homopyrimidine stretch with high frequency in the 5′ untranslated region, has been reported to play a role in the regulation of gene expression and nucleic acid metabolism in plants (Martienssen and Colot 2001; Scaglione et al. 2009; Wöhrmann and Weising 2011). Furthermore, it seems to be a common feature in eukaryotic genomes that (GC) n repeats are extremely rare (Biswas et al. 2012; Feng et al. 2009).
Functional genetic markers like EST–SSRs, which were attractive for multi-allelic detection, high cross-species transferability and reproducibility, have increasingly become a powerful tool for gaining insight into genetic studies (Kaur et al. 2012). To date, with the advent of NGS technologies, a large-scale superior resource for gene-based SSR marker discovery has directly or indirectly enabled rapid progress in marker-assisted breeding (Iorizzo et al. 2011; Parchman et al. 2010). A great number of genic-SSR markers have been extensively identified in many plants (Kale et al. 2012; Hiremath et al. 2011) by analysis of the transcriptome sequence data generated. In the present study, a very large number of SSR primer sets were successfully designed from transcriptome sequences of radish taproot, which was discriminative to a certain degree as compared to the previous transcriptome report in radish (Wang et al. 2012c). Most of the primers yielded specific bands, while several markers amplified much larger bands, which was attributable to the presence of a small intron between each pair of primer pairs or variation in the repeat numbers (Gasic et al. 2009; Zeng et al. 2010). Furthermore, in spite of various tests under multiple amplification conditions, the remainder of the primers still did not produce PCR fragments, probably due to assembly errors in cDNA contigs, the existence of null alleles or large introns and primer pairs designed across a splice site, as previously described (Dutta et al. 2011).
Recently, the application of genic-SSR markers for diversity analyses and fingerprinting has been reported in several plant species (Barchi et al. 2011; Triwitayakorn et al. 2011). In this study, genic-SSR markers were demonstrated to be efficient at differentiating 32 radish cultivars, and the distribution was not completely based on their geographical sources, which is consistent with some previous studies (Zhang et al. 2012a; Hernan and Petr 2006). Most of the varieties released in China were clustered in the same subgroup in the dendrogram, suggesting the limited genetic diversity and narrow basis of Chinese radish cultivars (Fig. 3d). To enlarge the genetic basis, more exotic accessions should be used in future radish breeding programs. In conclusion, the genic-SSR markers from the transcriptome sequence were appropriate and superior markers for the discrimination of cultivated landraces and wild species (Dutta et al. 2011).
Manual cultivar identification diagram (MCID) is a new strategy which is more effective, economical and practical for identifying plant cultivars using fewer primers (Zhang et al. 2012c). It can enhance the power of the markers developed in this study in radish cultivar identification and gives reliable information for future rapid identification work. In MCID, only the genic-SSR markers with clear polymorphic bands are selected to gradually distinguish the individual samples, and this method creates a readable and recordable flow chart (Wang et al. 2011), making plant cultivar identification much easier than before (Korir et al. 2013). In this study, six genic-SSR primers were sufficient to distinguish all 32 radish cultivars using the MCID strategy. This is the first report, to our knowledge, that assesses the use of genic-SSR markers for the complete identification of radish cultivars by the MCID strategy.
It is generally accepted that polymorphic SSR markers are invaluable in marker-assisted genetic studies. Moreover, the PIC values, indicating substantial genetic information, can be used to assess the degrees of polymorphism of informative SSR markers in radish. According to the criterion previously described where the three categories were defined as high (PIC > 0.5), moderate (0.25 < PIC < 0. 5) and low (PIC < 0.25) (Yadav et al. 2011; Xu et al. 2012), approximately two-thirds of primers reported herein exhibited moderate or high levels of PIC (Table 1). Generally, genic-SSR markers show a lower level of polymorphism than genomic-SSR markers, but, in contrast, in this study it was a little higher or the same as the previous studies (Dutta et al. 2011; Wang et al. 2012d). In addition, the number of alleles produced may be correlated with heterozygosity level and microsatellite polymorphism.
As the gene-based genetic markers generated in the current study were designed in transcript regions, they could be highly conserved and transferable to closely related species (Kaur et al. 2012). Recently, a considerable degree of transferability of microsatellite markers from one species to other species or genera has been demonstrated within several plant families including cereals (Ince et al. 2010), Leguminosae (Gutierrez et al. 2005) and Rosaceae (Gasic et al. 2009). In this study, similar to EST–SSRs, the genic-SSR markers designed from the transcriptome sequences and representing putative function were observed with a relatively high level of transferability from radish to some related Brassicaceae species. The success of highly efficient amplification was also observed in other research studies, where 93 of 108 SSR primers (86.1 %) adopted from other Prunus species were transferable to chokecherry (Wang et al. 2012b). While a lower transferability rate was observed within Medicago truncatula microsatellites to chickpea (36.3 %) and pea (37.6 %) (Gutierrez et al. 2005), the rate of genic-SSR marker transferability in different species is related to the genetic distance between the species from which the genic-SSR markers were developed and other species. Therefore, these novel genic-SSR markers developed in the present study with a relatively high level of transferability will help advance the investigation of comparative mapping analyses in the Brassicaceae family.
In summary, the genic-SSR marker prediction and development in non-model organisms by Illumina paired-end sequencing technology is a much faster and more cost-effective approach. With their polymorphism, reproducibility and transferability, these newly developed genic-SSR markers will be valuable molecular tools for germplasm identification, genetic mapping, gene tagging, comparative mapping and genetic diversity analyses in radish and relative species.
References
Barchi L, Lanteri S, Portis E, Acquadro A, Valè G, Toppino L, Rotino GL (2011) Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genomics 12:304
Biswas MK, Chai LJ, Mayer C, Xu Q, Guo WW, Deng XX (2012) Exploiting BAC-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol Biol Rep 39:5373–5386
Blanca J, Cañizares J, Roig C, Ziarsolo P, Nuez F, Picó B (2011) Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics 12:104
Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Duan JL, Xia C, Zhao GY, Jia JZ, Kong XY (2012) Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics 13:392
Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, VivekDogra V, Gaikwad K, Sharma TR, Raje RS, Bandhopadhya TK, Datta S, Singh MN, Bashasab F, Kulwal P, Wanjari KB, Varshney RK, Cook DR, Singh NK (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol 11:17
Feng SP, Li WG, Huang HS, Wang JY, Wu YT (2009) Development, characterization and cross-species/genera transferability of EST–SSR markers for rubber tree (Hevea brasiliensis). Mol Breed 23:85–97
Fu CH, Chen YW, Hsiao YY, Pan ZJ, Liu ZJ, Huang YM, Tsai WC, Chen HH (2011) Orchid Base: a collection of sequences of transcriptome derived from orchids. Plant Cell Physiol 52:238–243
Gao LF, Tang JF, Li HW (2003) Analysis of microsatellites in major crops assessed by computational and experimental approaches. Mol Breed 12:245–261
Gasic K, Han YP, Kertbundi S, Shulaev V, Iezzoni AF, Stover EW (2009) Characteristics and transferability of new apple EST-derived SSRs to other Rosaceae species. Mol Breed 23:397–411
Gong YM, Xu SC, Mao WH, Hui QZ, Zhang GW, Ding J, Li YD (2010) Developing new SSR markers from ESTs of pea (Pisum sativum L.). J Zhejiang Univ Sci B 11:702–707
Gutierrez MV, Vaz Patto MC, Huguet T, Cubero JI, Moreno MT, Torres AM (2005) Cross-species amplification of Medicago truncatula microsatellites across three major pulse crops. Theor Appl Genet 110:1210–1217
Hernan EL, Petr K (2006) Genetic relationship and diversity in a sesame (Sesamum indicum L.) germplasm collection using amplified fragment length polymorphism (AFLP). BMC Genet 7:10
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, Bhanuprakash A, Mulaosmanovic B, Gujaria N, Krishnamurthy L, Gaur PM, Kavikishor PB, Shah T, Srinivasan R, Lohse M, Xiao Y, Town CD, Cook DR, May GD, Varshney RK (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J 9:922–931
Huang H, Lu J, Ren Z, Hunter W, Dowd SE, Dang P (2011) Mining and validating grape (Vitis L.) ESTs to develop EST–SSR markers for genotyping and mapping. Mol Breed 28:241–254
Ince AG, Karaca M, Onus AN (2010) Polymorphic microsatellite markers transferable across Capsicum species. Plant Mol Biol Rep 28:285–291
Iorizzo M, Senalik DA, Grzebelus D, Bowman M, Cavagnaro PF, Matvienko M, Ashrafi H, Deynze AV, Simon PW (2011) De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12:389
Jena SN, Srivastava A, Rai KM, Ranjan A, Singh SK, Nisar T, Srivastava M, Bag SK, Mantri S, Asif MH, Yadav HK, Tuli R, Sawant SV (2011) Development and characterization of genomic and expressed SSRs for levant cotton (Gossypium herbaceum L.). Theor Appl Genet 124:565–576
Jiang LN, Wang LJ, Liu LW, Zhu XW, Zhai LL, Gong YQ (2012) Development and characterization of cDNA library based novel EST–SSR marker in radish (Raphanus sativus L.). Sci Hort 140:164–172
Kale SM, Pardeshi VC, Kadoo NY, Ghorpade PB, Jana MM, Gupta VS (2012) Development of genomic simple sequence repeat markers for linseed using next-generation sequencing technology. Mol Breed 30:597–606
Kaur S, Pembleton LW, Cogan NO, Savin KW, Leonforte T, Paull J, Materne M, Forster JW (2012) Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13:104
Koelling J, Coles MC, Matthews PD, Schwekendiek A (2012) Development of new microsatellite markers (SSRs) for Humulus lupulus. Mol Breed 30:479–484
Kong QS, Li XX, Xiang CP, Wang HP, Song JP, Zhi HY (2011) Genetic diversity of radish (Raphanus sativus L.) germplasm resources revealed by AFLP and RAPD markers. Plant Mol Biol Rep 29:217–223
Korir NK, Han J, Shangguan LF, Wang C, Kayesh E, Zhang YY, Fang JG (2013) Plant variety and cultivar identification: advances and prospects. Crit Rev Biotechnol 33:111–125
Li PH, Ponnala L, Gandotra N, Wang L, Si YQ, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, Reidel EJ, Turgeon R, Liu P, Sun Q, Nelson T, Brutnell TP (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42:1060–1069
Li DJ, Deng Z, Qin B, Liu XH, Men ZH (2012) De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST–SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13:192
Liang XQ, Chen XP, Hong YB, Liu HY, Zhou GY, Li SX, Guo BZ (2009) Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species. BMC Plant Biol 9:35
Liu L, Guo W, Zhu X, Zhang T (2003) Inheritance and fine mapping of fertility-restoration for cytoplasmic male sterility in Gossypium hirsutum L. Theor Appl Genet 106:461–469
Liu LW, Zhao LP, Gong YQ, Wang MX, Chen LM, Yu FM, Wang LZ (2008) DNA fingerprinting and genetic diversity analysis of late-bolting radish cultivars with RAPD, ISSR and SRAP markers. Sci Hort 116:240–247
Lu TT, Lu GJ (2010) Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res 20:1238–1249
Martienssen RA, Colot V (2001) DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293:1070–1074
Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA (2010) Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11:180
Peng JH, Lapitan NV (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR markers. Funct Integr Genomics 5:80–96
Scaglione D, Acuadro A, Portis E, Taylor CA, Lanteri S, Knapp SJ (2009) Ontology and diversity of transcript-associated microsatellites mined from a globe artichoke EST database. BMC Genomics 10:454
Shao YC, Xu L, Chen FS (2011) Genetic diversity analysis of Monascus strains using SRAP and ISSR markers. Mycoscience 52:224–233
Shirasawa K, Oyama M, Hirakawa H, Sato S, Tabata S, Fujioka T (2011) An EST–SSR linkage map of Raphanus sativus and comparative genomics of the Brassicaceae. DNA Res 18:1–12
Silva PI, Martins AM, Gouvea EG, Pessoa-Filho M, Ferreira ME (2013) Development and validation of microsatellite markers for Brachiaria ruziziensis obtained by partial genome assembly of Illumina single-end reads. BMC Genomics 14:17
Smith JSC, Chin ECL, Shu H, Smith OS, Wall SJ, Senior ML, Mitchell SE, Kresovich S, Ziegle J (1997) An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.) comparisons with data from RFLPs and pedigree. Theor Appl Genet 95:163–173
Song J, Sun RJ, Li DZ, Tan FJ, Li X, Jiang PP, Huang XJ, Lin L, Deng ZN, Deng Y (2012) An improvement of shotgun proteomics analysis by adding next-generation sequencing transcriptome data in orange. PLoS One 7:e39494
Sraphet S, Boonchanawiwa A, Thanyasiriwat T, Boonseng O, Tabata S, Sasamoto S, Shirasawa K, Isobe S, Lightfoot DA, Tangphatsornruang S, Triwitayakorn K (2011) SSR and EST–SSR based genetic linkage map of cassava (Manihot esculenta Crantz). Theor Appl Genet 122:1161–1170
Triwitayakorn K, Chatkulkawin P, Kanjanawattanawong S, Sraphet S, Yoocha T, Sangsrakru D, Chanprasert J, Ngamphiw C, Jomchai N, Therawattanasuk K, Tangphatsornruang S (2011) Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res 18:471–482
Wang YZ, Fang BP, Chen JY, Zhang XG, Luo ZX, Huang LF, Chen XL, Li YJ (2010) De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 11:726
Wang YJ, Li XY, Han J, Fang WM, Li XD, Wang SS, Fang JG (2011) Analysis of genetic relationships and identification of flowering-mei cultivars using EST–SSR markers developed from apricot and fruiting-mei. Sci Hort 132:12–17
Wang G, Zhu QG, Meng QW, Wu CG (2012a) Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol Plant 34:107–115
Wang HX, Walla JA, Zhong SB, Huang DQ, Dai WH (2012b) Development and cross-species genera transferability of microsatellite markers discovered using 454 genome sequencing in chokecherry (Prunus virginiana L.). Plant Cell Rep 31:2047–2055
Wang SF, Wang XF, He QW, Liu XX, Xu WL, Li LB, Gao JW, Wang FD (2012c) Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST–SSR markers in radish. Plant Cell Rep 31:1437–1447
Wang YX, Zeng X, Iyer NJ, Bryant DW, Mockler TC, Mahalingam R (2012d) Exploring the switchgrass transcriptome using second-generation sequencing technology. PLoS One 7:e34225
Wöhrmann T, Weising K (2011) In silico mining for simple sequence repeat loci in a pineapple expressed sequence tag database and cross-species amplification of EST–SSR markers across Bromeliaceae. Theor Appl Genet 123:635–647
Xu W, Yang Q, Huai HY, Liu AZ (2012) Development of EST–SSR markers and investigation of genetic relatedness in tung tree. Tree Genet Genomes 8:933–940
Yadav HK, Ranjan A, Asif MH, Mantri S, Sawant SV, Tuli R (2011) EST-derived SSR markers in Jatropha curcas L.: development, characterization, polymorphism, and transferability across the species/genera. Tree Genet Genomes 7:207–219
Yu LJ, Luo YF, Liao B, Xie LJ, Chen L, Xiao S, Li JT, Hu SN, Shu WS (2012) Comparative transcriptome analysis of transporters, phytohormone and lipid metabolism pathways in response to arsenic stress in rice (Oryza sativa). New Phytol 195:97–112
Zeng S, Xiao G, Guo J, Fei Z, Xu Y, Roe BA, Wang Y (2010) Development of a EST dataset and characterization of EST–SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics 11:94
Zhai LL, Liu LL, Zhu XW, Xu L, Jiang LN, Gong YQ (2013) Development, characterization and application of novel expressed sequence tag-simple sequence repeat (EST–SSR) markers in radish (Raphanus sativus L.). Afr J Biotechnol 12:921–935
Zhang HY, Wei LB, Miao HM, Zhang T, Wang CY (2012a) Development and validation of genic-SSR markers in sesame by RNA-seq. BMC Genomics 13:316
Zhang JN, Liang S, Duan JL, Wang J, Chen SL, Cheng ZS, Zhang Q, Liang XQ, Li TR (2012b) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13:90
Zhang YP, Tan HH, Cao SY, Wang XC, Yang G, Fang JG (2012c) A novel strategy for identification of 47 pomegranate (Punica granatum) cultivars using RAPD markers. Genet Mol Res 11:3032–3041
Zhu XC, Raman H, Wu HW, Lemerle D, Burrows GE, Stanton R (2012) Development of SSR markers for genetic analysis of silverleaf nightshade (Solanum elaeagnifolium) and related species. Plant Mol Biol Rep 31:248–254
Acknowledgments
This work was in part supported by grants from the Program for the National Key Technologies R&D Program of China (2012BAD02B01), the NSFC (31171956, 31372064), Key Technology R&D Program of Jiangsu Province (BE2010328, BE2013429), JASTIF [CX (12)2006, CX(13)2007] and the PAPD. We thank Dr. X. Zhu at North Dakota State University for his critical review and helpful comments during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhai, L., Xu, L., Wang, Y. et al. Novel and useful genic-SSR markers from de novo transcriptome sequencing of radish (Raphanus sativus L.). Mol Breeding 33, 611–624 (2014). https://doi.org/10.1007/s11032-013-9978-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-013-9978-x