
Abstract
Genome-wide gene expression profiling was conducted by Solexa sequencing in order to gain insight into the transcriptome dynamics that are associated with salt stress of cotton seedlings. A total of 145,794 and 138,518 clean tags were generated from the control and salinity libraries, respectively. Of these, 75,500 (51.8%) and 72,077 (52.0%) tags were matched to the reference genes. The most differentially regulated tags with a log2ratio >2 or >−2 (P < 0.001) were analyzed further, representing 125 up- and 171 down-regulated genes except for unknown transcripts, which were classified into ten functional categories. The most enriched categories were those of metabolism, signaling pathway, environmental response and transcription. Many genes or biological pathways were found to be commonly shared between salt and other abiotic stresses in plants such as genes participating in environmental response, ABA signaling JA signaling, etc. Furthermore, the expression patterns of 12 genes were assessed by quantitative real-time PCR, and the results obtained showed general agreement with the Solexa data. Further analysis indicated the important roles of selected genes in salt tolerance by comparison with the mRNA levels in salt-tolerant cotton cultivar ZM3 with that in salt-sensitive cultivar LM6. Overall, we reveal the complex changes at the transcriptional level during salt stress of cotton seedlings and provide useful starting points for more in-depth analyses of cotton’s salt tolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton (Gossypium hirsutum L.) is not only the world’s leading textile fiber and oilseed crop, but also a crop that is of significance for foil energy and bioengergy production, and model system for studies of many biological processes. However, the growth and productivity of cotton are adversely affected by salt stress, especially at germination and young seedling stage. The salt-tolerant mechanism of cotton has been investigated physiologically, including ion homeostasis, accumulation of osmolytes and antioxidants (Ashraf 2001). Consistently, independent studies revealed some molecular adaptation of cotton to salt stress. For instance, the ethylene-responsive element-binding factors, GhERF2, GhERF3, GhERF6 (Jin et al. 2010), GhERF4 (Jin and Liu 2008) and GhERF5 (Champion et al. 2009) were induced by ethylene, abscisic acid (ABA), salt, cold and drought stresses. A cotton DRE-binding transcription factor gene (GhDREB) conferred enhanced tolerance to drought, high salt and freezing stresses in transgenic wheat (Gao et al. 2009). We characterized the salt, drought, cold and fungi-induced genes of cotton, including a cotton tonoplast Na+/H+ antiporter (GhNHX1) (Wu et al. 2004), a novel CCCH-type zinc finger (GhZFP1) (Guo et al. 2009) and a cotton metallothionein GhMT3a (Xue et al. 2009). Large-scale approaches including microarrays (Walia et al. 2006; Buchanan et al. 2005; Ueda et al. 2004; Rabbani et al. 2003) and whole-genome tiling arrays (Zeller et al. 2009) have been employed to identify genes responding to salinity in plants. One motivation for expression profiling in a reference plant such as Arabidopsis is to exploit the understanding of conserved stress-signaling networks to improve the stress tolerance of crops (Denby and Gehring 2005; Valliyodan and Nguyen 2006). Therefore, it is interesting to determine whether the cotton salt response shares the same regulatory mechanisms with that in other plants. To obtain comprehensive and unbiased transcript profiles during salt stress, we performed deep sequencing analysis using Solexa digital gene expression system. This system is an improved tag-based method that can sequence in parallel millions of total cDNA for the derivation of an accurate measure of gene expression and the discovery of novel transcriptional regions, dramatically changing the way that the functional complexity of transcriptome can be studied (Ford et al. 2008; Li et al. 2011).
In the present study, an overall impression of gene profiles during the salt stress of cotton seedlings was acquired by deep sequencing. We have comprehensively characterized the molecular basis of the physiological processes during cotton salt stress and provided useful information for further research.
Materials and methods
Plant materials and RNA extraction
A salt-tolerant upland cotton cultivar ZM3 was mainly used in the present study, except for an experiment to verify the expression of 12 selected genes in a salt-sensitive cultivar LM6. Seeds of ZM3 and LM6 were pre-germinated in distilled deionized water in a plant growth chamber at 60% humidity with 300 μmol m−2 s−1 light intensity and day/night temperature of 28/20°C for 48 h. Then germinated seeds were planted into soil and cultured in the same chamber with the same conditions for 20 days. The 20 uniformly developed seedlings were transferred into mediums containing 250 mM NaCl for indicated times. The true leaves and roots were harvested directly into liquid nitrogen and stored at −80°C. Then, total RNA was prepared by RNeasy Plant Mini Kit (QIAGEN, USA) in accordance with the manufacturer’s instruction.
Determination of total antioxidant capacity and MDA content
The total antioxidant capacity (T-AOC) of the protective system and MDA content was measured using commercial assay kits (supplied by Nanjing Jian Cheng Bioengineering Institute, China) according to the manufacturer’s instructions. Briefly, for analysis of T-AOC, 0.5 g of samples was ground in 4.5 ml of phosphate buffer (pH 7.0) and centrifuged at 4°C and 12,000g for 15 min. The supernatant was used for T-AOC measurement according to the manufacturer’s instruction. The method was based on the reduction of Fe3+ to Fe2+ by antioxidant, and then Fe2+ reacted with the phenanthroline to form a complex. The mixture was measured at 520 nm. Increase of 0.01 unit at A520 in 1 min/mg protein was considered as one unit of antioxidant capacity, and T-AOC was expressed as unit/mg protein. For MDA content measurement, 0.5 g of samples was ground in 4.5 ml of 10% thiobarbituric acid and centrifuged at 22°C and 12,000g for 15 min. The supernatant was used for MDA measurement according to the manufacturer’s instruction. The absorbance of the mixture was measured at 532 nm. MDA was expressed as nmol/mg protein.
Tag library construction and sequencing
Sequence tag preparation and sequencing were done with the Digital Gene Expression Tag Profiling Kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s protocol (version 2.1B). Twenty micrograms of total RNA were used for mRNA purification using biotin-Oligo (dT) magnetic bead adsorption. Then first- and second-strand cDNA was synthesized. While on the beads, double-strand cDNA was digested with NlaIII endonuclease to produce a bead-bound cDNA fragment containing sequence from the 3′-most CATG to the poly (A)-tail. These 3′ cDNA fragments were purified using magnetic bead precipitation, and the Solexa adapter 1 was added to the new 5′ end. The junction of Solexa adapter 1 and CATG site was recognized by MmeI. The enzyme cuts and produces 17 bp cDNA sequence tags with adapter 1. Then, the Solexa adapter 2 was ligated to the 3′ end of the cDNA tag. The adapter-ligated cDNA tags were enriched using PCR primers that anneal to the adaptor ends. The resulting 85 base PCR fragments were purified from a 6% acrylamide gel and subjected to the Solexa sequencing system (Illumina Inc., San Diego, CA, USA).
Tag-to-gene assignment and functional categorization
Sequencing quality evaluation and data summarization were performed using Solexa pipeline software after sequencing. “Clean Tags” were obtained by filtering off adaptor-only tags and low-quality tags (containing ambiguous bases). Comparison of the sequences by BLASTN against the nucleotide sequences downloaded from the Institute for Genome Research (TIGR) (http://plantta.jcvi.org/) was carried out. All clean tags were annotated based on the reference genes. For conservative and precise annotation, only sequences with perfect homology or 1 nt mismatch were considered further. The number of annotated clean tags for each gene was calculated and then normalized to TPM (number of transcripts per million clean tags) (Audic and Claverie 1997; Poole et al. 2008). Sequences were manually assigned to functional categories based on the analysis of scientific literature.
Reverse transcriptase PCR (RT-PCR) analysis
Total RNA isolated above was treated with DNase I to remove genomic DNA contamination. The first-strand cDNA synthesis and the qRT-PCR were carried out using the PrimeScript™ RT reagent Kit (perfect Real Time) and SYBR PrimeScript™ RT-PCR Kit (perfect Real Time), respectively. The qRT-PCR was performed on an Opticon II system (MJ Research/Bio-Rad) with the following cycling parameters: 94°C for 30 s, indicated cycles of 94°C for 12 s, 58°C for 30 s, 72°C for 30 s, and 1 s at 80°C for plate reading. The ubiquitin (GenBank: EU604080) gene was used for the normalization of reactions. The expression levels of the genes were calculated from the threshold cycle using the delta–delta C t method (Livak and Schmittgen 2001). Semiquantitative RT-PCR was also used for ten unknown function genes with the cycling parameters: 94°C for 30 s, indicated cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and 1 min at 72°C for plate reading. All reactions were performed with at least three replicates.
Results
Effects of salinity on T-AOC and MDA content of ZM3
To select a suitable time point of 250 mM NaCl treatment, we measured the total antioxidant capacity (T-AOC) of the protective system and the malondialdehyde (MDA) content, which is the decomposition product of polyunsaturated fatty acids of biomembrane. The changes in T-AOC and MDA are shown in Fig. 1. T-AOC increased to a higher level at 24 h of NaCl treatment and then decreased at 48 and 72 h of salt treatments (Fig. 1a). In contrast, the content of MDA increased slowly before 48 h of NaCl treatment, but increased rapidly at 72 h (Fig. 1b). These data indicated that the imbalance of ROS production and scavenging occurred at 12 h of NaCl treatment. However, a stronger ROS scavenging was built up at 24 h of NaCl treatment. Therefore, we carried out the next experiment by using 250-mM NaCl treatment within 24 h.

Effects of salt stress on the total activities of antioxidant enzymes (T-AOC) a and MDA contents b in the true leaves of 20-day cotton seedlings at different time points
Characterization of the sequenced Solexa libraries
To identify genes involved in salinity, two cotton Solexa libraries were constructed from 20-day-old seedlings treated without or with 250 mM NaCl for 24 h. Sequencing depths of 3,583,646 and 3,694,330 tags were achieved in the two libraries, including 395,874 and 389,356 distinct tags, respectively. To make the libraries meaningful, tags recorded only once were wiped off, leaving 145,794 and 138,518 clean tags in each library that were detected multiple times. The frequency of these tags are shown in Table 1, in which the majority of clean tags (about 86% from each) were present at low copy numbers (<10 copies), and approximately 9% tags from each library were counted between 11 and 100 times. Only approximately 1.4% tags were detected more than 100 times.
To identify the genes corresponding to the 145,794 and 138,518 meaningful tags in each library, an essential dataset containing 70,667 reference genes expressed in the cotton from the http://plantta.jcvi.org/plant TA search was used. Altogether, 62,029 genes (87.8%) have the CATG sites, resulting in a total number of 121,721 (79.5%) unambiguous reference tags. By assigning the eperimental Solexa tags to the virtual reference ones (Table1), we observed that 54,937 (37.7%) and 50,493 36.5%) tags were perfectly matched to the reference genes n control and salinity libraries, respectively. Out of the tags, approximately 20 and 34% tags in control and salinity libraries were mapped to multiple locations. Further sequence analysis revealed that some of them were mapped to highly conserved domains shared by different genes. For instance, CATGTGATCACCGTAGCAGAG could be matched to CO499433, CD486297, TA21047_3635, DN801579, CD486180, TA21046_3635, DT050874, TA21044_3635, TA21045_3635, DT054118, DT050861, DV850220, TA21048_3635 and CD485873 with 95–100% identities to late embryogenesis abundant protein LEA5-A of cotton. In addition, approximately 18% tags in the two libraries were mapped to the antisense strands, demonstrating that those regions might be bidirectionally transcribed. However, for the discrepancy between the reference tags and experimental tags [25], approximately 14% of 1-bp mismatched tags were present in the two libraries. Overall, 75,500 (51.8%) and 72,077 (52.0%) tags in the Solexa libraries treated without or with NaCl were matched to the reference genes, respectively. As a result of the significant sequencing depth of Solexa technology and incomplete genomic data of the cotton, however, approximately 14% unmatched tags in each library were observed.
Identification of differentially expressed transcripts
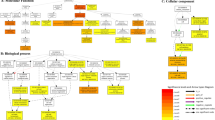
By comparing two Solexa libraries, a great number of differentially expressed transcripts were identified. The distribution of fold changes in tag number between the two libraries is shown in Appendix S1. The great majority of transcripts were expressed at similar levels in the two libraries: 98.9% tags showed a <5-fold difference in expression regulated by salinity, while tags with expressional changes in the range 5- to 200-fold only accounted for 1.1%. Scatter plot analysis also presented a broader scope of differentially expressed tags than annotated genes, demonstrating that a great number of unknown transcripts were revealed (Appendix S2). To study a subset of genes that were associated with salinity, we analyzed the most differentially regulated tags with a log2ratio >2 or <−2 using a greater statistically significant value (P < 0.001) as well as false discovery rates (FDR < 0.01), representing 223 up- and 317 down-regulated transcripts. Apart from the unknown transcripts (45%), 125 up- and 171 down-regulated genes were classified into ten categories according to their putative or known functions (Fig. 2, Appendix S3 and S4). Of these, the most enriched functional categories are those of metabolism (35.5%), signaling pathway (15.2%), environmental response (13.9%) and transcription (10.8%). Gene categories showed an obvious increase in transcript abundance for genes involved in environmental response. In contrast, transcriptional abundance for genes participating in protein synthesis, metabolism and cytoskeleton were reduced.

Functional classification of genes differentially expressed during salt stress of the cotton plants
In detail, we characterized some significant regulated pathways (Appendix S3 and S4). For example, out of the 15 differentially expressed genes involved in cell growth and cytoskeleton, 9 transcripts encoding for expansins and actins were down-regulated significantly. Many auxin and gibberellin-associated genes were also reduced in the present study, such as SAUR protein and GASA2-like protein, indicating their negative regulation in salt stress. Moreover, a total of 25 phosphorylation/de phosphorylation cascades and 32 transcription factors (TFs) were detected with different changes. Of these, 6 protein phosphatase genes were up-regulated, while 12 protein kinase genes were repressed, indicating lower phosphorylation regulation during salt stress. Among the TFs, NAC (petunia NAM and Arabidopsis ATAF1, ATAF2 and CUC2) (4 genes), zinc finger (4 genes), homodomain leucine zipper (HD zip) (4 genes) and AREB (ABA responsive element-binding protein) (2 genes) TFs were mostly up-regulated, while MYB (3 genes) and ERF (ethylene-responsive element-binding) (3 genes) TFs were mainly down-regulated. This implied the importance of signal transduction during the salt adaptation process. In addition, many transcripts encoding for heat shock proteins (HSPs), late embryogenesis abundant proteins (LEAs) and compatible solutes were greatly induced in the salt library. Similarly, genes encoding for organic molecule transporters were also significantly induced. On the contrary, genes encoding for cation/metal transporters were significantly repressed. The over representation of these categories strongly suggested their regulating roles in the salinity adaptation in plants.
Furthermore, with the benefit of Solexa sequencing, numerous novel transcripts with unclear functions were also detected (Appendix S5).
Quantitative real-time PCR (qRT-PCR) confirmation
To evaluate the validity of Solexa analysis and to further asses the patterns of differential gene expression, seven known and five unknown transcripts with high abundances were selected and detected by real-time RT-PCR (qRT-PCR) (Table 2) with gene-specific primers (Appendix S6), including four LEAs and two trehalose-6-phosphate synthases (TPSs). The log2ratios for the four LEA genes of M19406 (D-113), M19379 (D-11), X15086 (D-7) and TA24166_3635 (LEA14-A) were 12.1, 9.7, 4.9 and 2.7, and those for the two TPS genes of TA26597_3635 and TA30732_3635 were 2.5 and 2.9, respectively. Different from the Solexa data, the highest up-regulation of M19379 was observed with almost 2,000-fold in salinity, while the transcript abundance of M19406 was induced by approximately 520-fold, lower than that of M19379 (Fig. 3). The induction of the transcript abundance of the two TPS genes was much lower, while the three significantly down-regulated genes of CD486436, DW231112 and DT567567 were also detected with lower changes by qRT-PCR than by Solexa sequencing. Furthermore, another ten unknown transcripts were selected and detected by relative RT-PCR (Table 2) with gene-specific primers (Appendix S6). As shown in Fig. 4, nine unknown transcripts were induced in root or leaves or both at 6 and 12 h of salinity treatment, while one transcript was repressed during salt stress. Importantly, the transcription levels of 12 of the genes mentioned above in a salt-sensitive cotton cultivar LM6 showed almost no differences or slightly inducible patterns after salt treatment than the control (Fig. 5). The mRNA levels of TA22164_3635 and TA32043_3635 were repressed in the salt-sensitive cotton cultivar LM6 (Fig. 5). These data further indicated the important role of these genes in cotton salt tolerance. Taken together, the expression patterns of these genes in ZM3 showed agreement with the Solexa data. The apparent discrepancies should be attributed to the essentially different algorithms determined by the two techniques (Ekman et al. 2003). The deep sequencing method generates absolute rather than relative expression measurements. However, these results basically confirmed the reliability of our transcriptome analysis.

Relative expression of the 12 genes detected by qRT-PCR in different conditions of the cotton seedlings. The transcript levels were normalized to that of ubiquitin, and the level of each gene in the control samples (CK) was set at 1.0. Error bars represent the SE for three independent experiments. The gene expression in Solexa experiment and the descriptions are listed in Table 2. The primers used for each gene are listed in Appendix S6

The expression profiles of another ten selected unknown function genes detected by relative RT-PCR. The plants used in this experiment were 20-day-old cotton seedlings that were treated with 250 mM NaCl for 0, 6 and 12 h. The roots and leaves of cotton seedlings were harvested, respectively, for this experiment. A total of three independent experiments were carried out and the results were similar in expression changes. The gene expression in the Solexa experiment, their descriptions and primers are also listed in Table 2 and in Appendix S6, respectively

The expression profiles of 12 selected genes in salt-sensitive cultivar LM6 detected by relative RT-PCR. The plants used in this experiment were 20-day-old cotton seedlings that were treated with 250 mM NaCl for 24 h. The whole seedlings were harvested for this experiment. The results were illustrated by one of three independent experiments because of the similar expression changes. The gene descriptions and their primers are also listed in Table 2 and in Appendix S6, respectively
Discussion
The major goal of the present study is to preliminarily explore transcripts involved in salinity response of cotton seedlings, as well as to provide groundwork for investigating their regulating mechanisms. Several batches of cotton microarrays have been fabricated. The most number of gene elements are derived from fibers (Arpat et al. 2004) or ovales (Shi et al. 2006; Wu et al. 2006). To our knowledge, the results represent the first large-scale investigation for the transcriptional changes during salt stress in cotton seedlings by the Solexa deep sequencing technology. Recently, Wang et al. (2009) revealed the epigenetic modifications in maize shoots and roots by Solexa sequencing. Our work revealed maize brace root development by Solexa sequencing (Li et al. 2011). This demonstrated that Solexa sequencing analysis had emerged as an efficient and economical method for sampling transcript profiles under specific experimental conditions. In total, we could map approximately 52% of tags to unique or non-unique positions. We consider this as the limited amount of genomic sequences available for cotton and related species in GenBank. In the present study, only 87.8% of reference genes from the cotton EST library have the CATG sites, leaving 8,538 genes unanalyzed, which means that a number of the related genes involved in cotton salt responses would be neglected.
Solexa sequencing could provide a comprehensive and unbiased dataset in the global analysis of gene expression. In the present study, 2,239 differently expressed annotated and novel transcripts (P < 0.01) were explored. Except for unknown transcripts, the most differentially expressed genes with a log2ratio >2 or <−2 (P < 0.001) also participate in various biological pathways, such as environmental response, macromolecule metabolism, signal transduction and transcriptional regulation. Interestingly, many transcripts detected at very low copy numbers in the control library were significantly up-regulated in the salt library (Appendix S3), implying that these genes might be specifically expressed in salt stress. For example, the presence of LEA protein D-113, secretory laccase, putative dehydration-induced myb-related protein and class IV chitinase genes only in the salinity stress indicates that they begin to take on functions to survive under salt stress. Osmoprotection, detoxication and turgor maintenance processes also showed different changes, explaining the different cell responses to salt stress, such as galactinol and trehalose accumulation due to the induced galactinol synthase and TPSs. These changes are common in abiotic stresses. It has been reported that an A. thaliana mutant overexpression of a galactinol synthase gene was more tolerant to desiccation, cold and salinity stress (Taji et al. 2002; Ottow et al. 2005). Overexpression of TPS and d-pyrroline-5-carboxylase synthetase genes conferred tolerance to desiccation and other abiotic stresses in A. thaliana plants and rice (Ashraf and Foolad 2007). In addition, a number of LEA genes responsive to low temperature, drought, high salinity and ABA have been reported (Xiong et al. 2002; Rorat 2006; Hundertmark and Hincha 2008).
Previous studies have reported that the transcripts involved in the regulation of these stress responsive genes were regulated by phosphorylation/dephosphorylation cascades and specific transcription factors (Chinnusamy et al. 2004). In the present study, 18 protein phosphatase/protein kinase genes were regulated by salt stress out of 25 phosphorylation/dephosphorylation cascades (Appendix S3 and S4), indicating the lower phosphorylation regulation during salt stress. Moreover, large-scale transcriptome analysis using the areb1 areb2 abf3 triple mutant revealed downstream genes in response to water stress, including many LEA class and group-Ab PP2C genes (Yoshid et al. 2010). This indicated that ABA might participate in the phosphorylation/dephosphorylation pathway in salt stress. In concordance with this, we observed the differential expression of many transcriptional factors conserved in salt and/or ABA responses, such as NAC [4 genes (Fujita et al. 2004; Tran et al. 2007; Ohnishi et al. 2005; Nakashim et al. 2007], zinc finger (4 genes) (Tran et al. 2007; Guo et al. 2009), AREB (2 genes) (Fujita et al. 2005; Yoshid et al. 2010), ERFs (Qiao et al. 2008; Jin et al. 2010; Champion et al. 2009). Hence, the crosstalk between salt stress and ethylene and ABA responses could possibly be mediated by ERFs in cotton. Moreover, auxin-related genes were also regulated in the salt library (Appendix S3 and S4), including down-regulated auxin-related genes, implying that: the auxin signaling was negatively involved in salinity responses; and genes encoding for jasmonate synthesis and responses, such as allene oxide synthase, lipoxygenases, lipase, alcohol dehydrogenase and chitinases, were regulated under salinity. The increased levels of JA in barley plants under osmotic stress (Lehmann et al. 1995), and rice (Moons et al. 1997) and Iris hexagona (Wang et al. 2001) under salt stress have been reported. Recently, Walia et al. (2007) reported that the JA mediated adaptation of barley to salinity stress and that 38% of salt-induced genes were induced by JA. We found that 36 and 39% of salt-regulated genes were regulated by ABA and MeJA, respectively (unpublished data). These indicated the crosstalk of salt stress with auxin and the JA signaling pathway. Taken together, salt stress crosstalks with multiple phytohormones, such as ABA, ethylene, JA and auxin in cotton, in the present study, and the regulatory mechanisms appear to be commonly shared with respect to cotton salinity-regulated genes and other plant abiotic responsive genes.
References
Arpat A, Waugh M, Sullivan JP et al (2004) Functional genomics of cell elongation in developing cotton fibers. Plant Mol Biol 54:911–929
Ashraf M (2001) Salt tolerance of cotton: some new advances. Crit Rev Plant Sci 21:1–32
Ashraf M, Foolad M (2007) Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ Exp Bot 59:206–216
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7(10):986–995
Buchanan CD, Lim S, Salzma RA, Kagiampakis I, Morishige DT, Weers BD, Klein RR, Prat LH, Cordonnier-Pratt MM, Klei PE, Mullet JE (2005) Sorghum bicolor’s transcriptome response to dehydration, high salinity and ABA. Plant Mol Biol 58:699–720
Champion A, Hebrard E, Parra B, Bournaud C, Marmey P, Tranchant C, Nicole M (2009) Molecular diversity and gene expression of cotton ERF transcription factors reveal that group IXa members are responsive to jasmonate, ethylene and xanthomonas. Mol Plant Pathol 10(4):471–485
Chinnusamy V, Schumaker K, Zhu JK (2004) Molecular genetic perspectives on cross-talk and specificity in abiotic stress signalling in plants. J Exp Bot 55(395):225–236
Denby K, Gehring C (2005) Engineering drought and salinity tolerance in plants: lessons from genome-wide expression profiling in Arabidopsis. Trends Biotech 23:547–552
Ekman DR, Lorenz WW, Przybyla AE, Wolfe NL, Dean JF (2003) SAGE analysis of transcriptome responses in Arabidopsis roots exposed to 2, 4, 6-trinitrotoluene. Plant Physiol 133:1397–1406
Ford GD, Ford BD, Steele EC Jr, Gates A, Hood D, Matthews MAB, Mirza S, MacLeish PR (2008) Analysis of transcriptional profiles and functional clustering of global cerebellar gene expression in PCD3J mice. Biochem Biophys Res Com 377:556–561
Fujita M, Fujita Y, Maruyama K, Seki M, Hiratsu K, Ohme-Takagi M, Tran LS, Yamaguchi-Shinozaki K, Shinozaki K (2004) A dehydration-induced NAC protein, RD26, is involved in a novel ABA-dependent stress-signaling pathway. Plant J 39:863–876
Fujita Y, Fujita M, Satoh R, Maruyama K, Parvez MM, Seki M, Hiratsu K, Ohme-Takagi M, Shinozaki K, Yamaguchi-Shinozaki K (2005) AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 17:3470–3488
Gao SQ, Chen M, Xia LQ, Xiu HJ, Xu ZS, Li LC, Zhao CP, Cheng XG, Ma YZ (2009) A cotton (Gossypium hirsutum) DRE-binding transcription factor gene, GhDREB, confers enhanced tolerance to drought, high salt, and freezing stresses in transgenic wheat. Plant Cell Rep 28(2):301–311
Guo YH, Yu YP, Wang D, Wu CA, Yang GD, Huang JG, Zheng CC (2009) Fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol 183(1):62–75
Hundertmark M, Hincha DK (2008) LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC genomics 9:118
Jin LG, Liu JY (2008) Molecular cloning, expression profile and promoter analysis of a novel ethylene responsive transcription factor gene GhERF4 from cotton (Gossypium hirstum). Plant Physiol Biochem 46(1):46–53
Jin LG, Li H, Liu JY (2010) Molecular characterization of three ethylene responsive element binding factor genes from cotton. J Integr Plant Biol 52(5):485–495
Lehmann J, Atzorn R, Bruchner C, Reinbothe S, Leoponld J, Wasternack C, Parthier B (1995) Accumulation of jasmonate, abscisic acid, specific transcripts and proteins in osmotically stressed barley leaf segments. Planta 197:156–162
Li YJ, Fu YR, Huang JG, Wu CA, Zheng CC (2011) Transcript profiling during the early development of the maize brace root via Solexa sequencing. FEBS J 278:156–166
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2[-Delta Delta C(T)] method. Methods 25:402–408
Moons A, Prinsen E, Bauw G, Van Montagu M (1997) Antagonistic effects of abscisic acid and jasmonates on salt stress-inducible transcripts in rice roots. Plant cell 9(12):2243–2259
Nakashim K, Tran LSP, Nguyen DV, Maruyamal K, Todaka D, Ito Y, Hayashi N, Shinozak K, Yamaguchi-Shinozak K (2007) Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J 51:617–630
Ohnishi T, Sugahara S, Yamada T, Kikuchi K, Yoshiba Y, Hirano HY, Tsutsumi N (2005) OsNAC6, a member of the NAC gene family, is induced by various stresses in rice. Genes Genet Syst 80:135–139
Ottow EA, Brinker M, Teichmann T, Fritz E, Kaiser W, Brosche M, Kangasjarvi J, Jiang X, Polle A (2005) Populus euphratica displays apoplastic sodium accumulation, osmotic adjustment by decreases in calcium and soluble carbohydrates, and develops leaf succulence under salt stress. Plant Physiol 139(4):1762–1772
Poole RL, Barker GL, Werner K, Biggi GF, Coghill J, Gibbings JG, Berry S, Dunwell JM, Edwards KJ (2008) Analysis of wheat SAGE tags reveals evidence for widespread antisense transcription. BMC Genomics 9:475
Qiao ZX, Huang B, Liu JY (2008) Molecular cloning and functional analysis of an ERF gene from cotton (Gossypium hirsutum). Biochimica et Biophysica Acta 1779:122–127
Rabbani MA, Maruyama K, Abe H, Khan MA, Katsura K, Ito Y, Yoshiwara K, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid spplication using cDNA microarray and RNA gel-blot analyses. Plant Physiol 133:1755–1767
Rorat T (2006) Plant dehydrins—tissue location, structure and function. Cell Mol Biol Lett 11(4):536–556
Shi YH, Zhu SW, Mao XZ et al (2006) Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 18(3):651–664
Taji T, Ohsumi C, Iuchi S, Seki M, Kasuga M, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K (2002) Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J 29(4):417–426
Tran LS, Nakashima K, Sakuma Y, Osakabe Y, Qin F, Simpson SD, Maruyama K, Fujita Y, Shinozaki K, Yamaguchi-Shinozaki K (2007) Co-expression of the stress-inducible zinc finger homeodomain ZFHD1 and NAC transcription factors enhances expression of the ERD1 gene in Arabidopsis. Plant J 49:46–63
Ueda A, Kathiresan A, Inada M, Narita Y, Nakamura T, Shi W, Takabe T, Bennett J (2004) Osmotic stress in barley regulates expression of a different set of genes than salt stress does. J Exp Bot 55:2213–2218
Valliyodan B, Nguyen HT (2006) Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr Opin Plant Biol 9:189–195
Walia H, Wilson C, Wahid A, Condamine P, Cui XP, Close TJ (2006) Expression analysis of barley (Hordeum vulgare L.) during salinity stress. Funct Integr Genomics 6:143–156
Walia H, Wilson C, Condamine P, Liu X, Ismail AM, Close TJ (2007) Large-scale expression profiling and physiological characterization of jasmonic acid-mediated adaptation of barley to salinity stress. Plant Cell Environ 30(4):410–421
Wang Y, Mopper S, Hasenstein KH (2001) Effects of salinity on endogenous ABA, IAA, JA, AND SA in Iris hexagona. J Chem Eco 27(2):327–342
Wang X, Elling AA, Li X, Li N, Peng Z, He G, Sun H, Qi Y, Liu XS, Deng XW (2009) Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 21:1053–1069
Wu CA, Yang GD, Meng QW, Zheng CC (2004) The cotton GhNHX1 gene encoding a novel putative tonoplast Na+/H+ antiporter plays an important role in salt stress. Plant Cell Physiol 45(5):600–607
Wu Y, Machado AC, White RG, Llewellyn DJ, Dennis ES (2006) Expression profiling identifies gene expressed early during lint fiber initiation in cotton. Plant Cell Physiol 18:651–664
Xiong L, Schumaker KS, Zhu JK (2002) Cell signaling during cold, drought, and salt stress. Plant Cell Environ 14:S165–S183
Xue T, Li X, Zhu W, Wu C, Yang G, Zheng C (2009) Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J Exp Bot 60(1):339–349
Yoshid T, Fujit Y, Sayam H, Kidokoro S, Maruyama F, Mizoi J, Shinozak K, Yamaguchi-Shinozaki K (2010) AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J 61:672–685
Zeller G, Henz SR, Widme CK, Sachsenber T, Rätsch G, Weige D, Laubinge S (2009) Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J 58:1068–1082
Acknowledgments
This work was financially supported by the National Natural Science Foundation (31071339) and the Genetically Modified Organisms Breeding Major Projects in China (2009ZX08009-092B).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Y. Wang.
The two authors, G. Wang and Q. Zhu, contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Wang, G., Zhu, Q., Meng, Q. et al. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol Plant 34, 107–115 (2012). https://doi.org/10.1007/s11738-011-0809-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11738-011-0809-6